

Abstract
Tuning interactions at the interfaces in carbon fiber (CF)-reinforced polymer composites necessitates the implementation of CF surface modification strategies that often require destructive environmentally unfriendly chemistries. In this study, interfacial interactions in cellulose-based composites are tailored by means of a mussel-inspired adhesive polydopamine (PDA) coating, being inherently benign for the environment and for the structure of CFs. The step-by-step growth of PDA was followed by increasing treatment time leading to a hydrophilic PDA-coated surface, presumably via surface-based polymerization mechanisms attributed to strong π–π stacking interactions. Although PDA deposition led to an initial increase in the interfacial shear strength (IFSS) (5 h), it decreased at a longer reaction time (24 h), the formation of weakly attached PDA particles on the coated surface can possibly lie behind the latter phenomenon. Nevertheless, the mechanical properties of the prepared short CF-reinforced composite were improved (tensile strength increased ∼12% compared to the unmodified surface) with decreasing IFSS owing to the particular morphological design, resulting in longer fiber segments. Our study underlines the importance of the morphological design at the interface and considers PDA as a promising bioinspired material to tailor interfacial interactions.
1. Introduction
With the growing understanding of many sophisticated biological systems, biomimetic engineering has become of utmost interest to design environmentally benign yet high-performance structural materials.1,2 Although it is rather challenging to reproduce the fine multiscale architecture of several nature-given composites, such as nacre and bone, using contemporary large-scale manufacturing practices,3 several innovative materials have been introduced that are inspired by natural motifs.4−6
The concepts of biomimetism have also been expanded to carbon fiber-reinforced polymers (CFRPs), a dominating class of lightweight advanced composite materials being unquestionably significant for the future.7,8 CFRPs have been designed drawing inspiration from the structure of turtle shells,9 bone,10 nacre,11 the mussel adhesive proteins,12 and carbon fibers (CFs) with butterfly-like colors have also been recently reported.13 The interactions at the interfaces joining the building blocks are essential for the outstanding mechanical characteristics of natural composites,14 as well as of CFRPs.15,16 Therefore, tremendous research efforts have been directed toward CF surface modification strategies to tailor the properties at the interface in these materials. However, the CF surface is notoriously difficult to chemically modify because of the relatively inert nature of the turbostratic carbon, its main structural motif.17 Albeit rather harsh reaction conditions are applied in the early studies on CF surface modification using strong acids,18−20 pioneering work has been done, bringing much milder methods into light using less aggressive oxidants21−24 and electrochemical/chemical grafting methods.25−29
The implementation of mild reaction systems is essential to avoid deterioration in the mechanical properties of single fibers; this phenomenon is clearly outlined in several studies.28,30 A biomimetic approach to modify the CF surface has been recently reported exploiting the exceptional adhesive properties of polydopamine (PDA),12 a polymeric material inspired by the mussel adhesive proteins.31,32 Since its first introduction in 2007,31 PDA has been proven to be a promising multifunctional coating material,33,34 and thus a number of studies have been devoted to investigate its complex structure35 and its deposition mechanism on various substrates.36−39 PDA is prepared via the autoxidation of dopamine at mild alkaline pH, leading to the formation of nanosized particles that can be deposited on a substrate, generating nanostructured thin coating.38,39 The thickness and nanostructural evolution of the coating can be controlled conveniently by changing the dopamine concentration and deposition time.33,38,39 The mild conditions applied during this process are beneficial for preserving crucial single fiber mechanical properties.12 It has also been shown that the nanostructural design of the surface can bring about great improvement in the mechanical properties of CFRPs,11,24,40,41 the importance of a multiscale design has already been proven in natural systems.2
In our previous studies, we have elaborated several CF surface functionalization strategies to tailor the properties of CF-reinforced cellulose-based polymer composites.42−45 Our present study aims at designing a multiscale micro- (CF) to nano(PDA) interface by changing the time of the deposition process during PDA formation, and thus tuning secondary interactions between the cellulosic matrix and PDA at the interface. We anticipate that this green surface modification strategy for tailoring interfacial properties in sustainable cellulose-based composites is of interest to fiber-reinforced composite applications and to other applications where interfacial phenomena occur (e.g., coatings).
2. Results and Discussion
2.1. PDA Deposition and Surface Characterization
PDA was deposited on the surface of unsized short CFs in order to improve the compatibility between the initially hydrophobic pregraphitic carbon (“turbostratic carbon”17) and the relatively hydrophilic cellulosic matrix,46 and thereby establish beneficial interfacial interactions that have a decisive impact on the mechanical performance of CF-reinforced composites.15,16 PDA has been shown to adhere to the surface of CFs12 and to the surface of a range of different carbon-based materials as well.33 In the case of carbon nanotubes, it was suggested that en route to the PDA formation dopamine monomer is first adsorbed on the surface by π–π stacking interactions facilitating the oxidative polymerization to proceed at the surface leading to a fine nanofilm coating.47 Since CF possesses a pregraphitic structure rich in π electrons, a similar mechanism might also apply to the polymerization process at the surface. Previous results reported by Chen et al.12 also support this surface-based polymerization mechanism because the surface morphology of CFs in their study indicates film formation rather than the deposition of aggregates as the reaction proceeds. In order to follow the PDA deposition process on the CF surface, we applied a range of reaction times for the autoxidation.
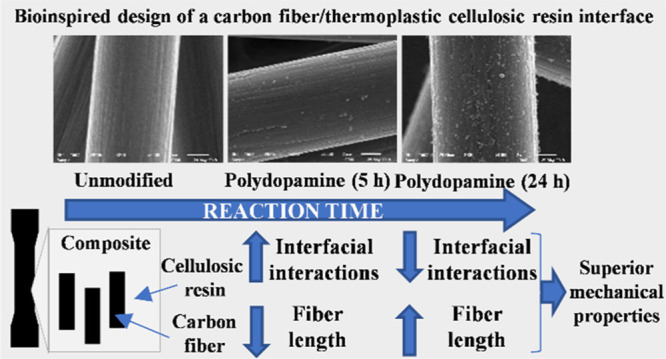
Figure 1 shows scanning electron microscopy (SEM) images of the CF surfaces functionalized with PDA using different reaction times. A control sample was chosen as a reference (Figure 1A), in this case fibers were agitated in Tris buffer for 24 h, without adding dopamine to the system. The striations typical for unsized CFs (e.g., see Woodhead et al.48) are clearly visible on the micrographs, without any observable mechanical damage suggesting the benign nature of the processing conditions. By adding dopamine to the system, at early times (1 and 2 h deposition times, Figure 1C,D, respectively) the formation of submicrometer size spherical particles can be confirmed. A thin layer forms later on the surface as the micrographs taken on CFs following 5 and 10 h deposition times indicate (Figure 1D,E, respectively), the striations are also less visible on these samples. The striations originally present on the CF surface along the fiber axis gradually become less noticeable as the PDA layer grows between the ridges on the surface. This observation indicates the presence of the thin PDA layer, which becomes more visible after 5 h deposition time (Figure 1D). The same topological changes suggesting the formation of the PDA layer were also observed by Chen et al.12 Spherical particles can form and agglomerate in the solution as it was shown previously.37,49 These agglomerates, which might originate from the spherical particles that form in the solution phase, can be seen on the surface of the CF subjected to 1 h deposition time (Figure 1B). This phenomenon is, however, only rarely observable (when many fibers are investigated), and apparently it does not lead to negative effects on the interfacial and tensile properties of the composites (see later in Figures 4 and 5). Interestingly, on the surface of the sample subjected to 24 h deposition time again spherical particles are present, which appear to be located on the thin PDA film (Figure 1F). The formation of the thin PDA layer on the surface might support the surface-based nucleation theory postulated for PDA generation on carbon nanotubes.47 According to this theory, dopamine is immobilized on the surface via π–π stacking interactions owing to the electron-rich graphitic surface as the initial step of PDA growth. π–π stacking interactions are reported to govern the formation and morphology of PDA in the case of carbon nanotubes,47 and also for other systems rich in π electrons.50
Figure 1.

SEM images of (A) control CF sample (only Tris buffer, 24 h deposition time), and after PDA deposition using (B) 1, (C) 2, (D) 5, (E) 10 and (F) 24 h reaction time. Arrows point to locations on the surface where the PDA layer or particles are clearly observable.
Figure 4.

Interfacial shear strength (IFSS) determined via microdroplet experiments. CP-cellulose propionate; CF-carbon fiber; and PDA-polydopamine. For “CP control CF”, the fibers were treated the same way as modified samples without dopamine added to the solution (24 h treatment time). The results of the significance analysis are shown with red color; N.S.—not significant, *—significant (P < 0.05), **—significant (P < 0.01). Please note that the red line (the beginning and the end of the line) indicates the two samples that are subjected to statistical significance analysis. Compared to the “CP/CF control”, except the CF sample prepared following 1 h deposition time, all the samples exhibit significantly higher IFSS values (for 2, 5, and 10 h—P < 0.01; for 24 h—P < 0.05).
Figure 5.

Tensile strength and elastic modulus values determined for composites containing CP and 20 wt % unmodified or polydopamine-coated (PDA) CF. Inset shows the strain at break values. “CP unmodified CF” denotes a sample prepared using as-received CFs, while in the case of “CP control CF” the fibers were treated the same way as modified samples without dopamine being added to the solution (24 h treatment time). The results of the significance analysis for the tensile strength data are shown with red color; N.S.—not significant, *—significant (P < 0.05), **—significant (P < 0.01). Please note that red line (the beginning and the end of the line) indicates the two samples that are subjected to statistical significance analysis. Compared to the “CP unmodified CF”, the tensile strength values for the samples containing CFs prepared using 2 h (P < 0.01), 5 h (P < 0.05), and 24 h (P < 0.01) deposition time are significantly higher.
X-ray photoelectron spectroscopy (XPS) was done to confirm the presence of PDA on the surface, the results are shown in Figure 2. In the C 1s spectrum of the control sample (ZOLTEK PX35 as received), a main peak centers at 284.67 eV (66.7% contribution to the total peak area), along with a smaller peak located at lower binding energies (0.6% contribution to the total peak area), these features are typical for the graphitic structure present in CFs.51,52 The C 1s spectrum can be further resolved into several other components indicating the presence of C–N bonds (285.89 eV), C–OH or ether type (C–O–C) linkages (286.82 eV), C=O moieties (288.08 eV), −COOH groups (289.44 eV), and the broad band at higher binding energies (290.89 eV) can be attributed to π–π* shake-up satellites generally observed for graphitic materials.20,51,53 When PDA is deposited on the surface (Figure 2B, sample with 24 h deposition time), a considerable increase can be observed in the intensity of the peak assigned to C–OH or ether type (C–O–C) moieties (286.38 eV; increase from 5.3 to 17.8% contribution to the total peak area for the control and PDA-coated sample, respectively), while the percent contribution of the peak attributed to C–N (285.69 eV) linkages to the total peak area slightly decreases (from 13.4 to 11.4% contribution to the total peak area for the control and PDA-coated sample, respectively) compared to the control sample (Figure 2A). This change is well in agreement with the structural features of PDA rich in −OH groups, similar to other studies.39,53,54 In the O1s spectrum of the sample covered with PDA (Figure 2D), a peak assigned to C–OH moieties (533.04 eV) dominates (72.3% contribution to the total peak area) with minor contribution from signals attributed to C=O groups (531.16 eV; 27.7% contribution to the total peak area), while for the control sample an opposite trend can be noticed (Figure 2C).39,51 Furthermore, the surface of another reference sample was also analyzed (Figure S1), in this case the treatment procedure was conducted without adding dopamine to the system (24 h treatment time). Because the recorded XPS spectra resemble that obtained on an as-received CF sample, it can be concluded that any residual contamination (e.g., Tris buffer) is efficiently eliminated during the washing procedure. The N 1s spectra of the corresponding samples are given in Figure S2. The unmodified CF samples (as received and control, Figure S2A,B, respectively) already have nitrogen in their structure. The CF used in this study is made from polyacrylonitrile precursor, upon the formation of the turbostratic carbon, different nitrogen-containing moieties remain in the structure.17 The N1s spectra of the unmodified samples (Figure S2A,B) are characterized by a broad low-intensity peak, which covers overlapping spectra attributed to several moieties, some of the plausible structures can be found elsewhere.51 When PDA is deposited on the surface (Figure S2C, 24 h deposition time), a distinctly different narrow peak is observable (centered at 400.21 eV) together with smaller peaks centered at 398.77 and 402.24 eV (with 11.1 and 9.5% contribution to the total surface area, respectively). This spectrum clearly resembles that of the PDA reported previously, the latter peaks can be assigned to =N–R (398.77 eV), R–NH2 (402.24 eV), and R–NH–R′ (400.21 eV) functionalities.38 Therefore, the O 1s, N 1s, and the C 1s spectrum in agreement support the deposition of PDA on the CF surface as reported in other studies.39,53,54
Figure 2.

C 1s X-ray photoelectron spectra recorded on (A) unmodified CF (PX35 type, ZOLTEK) samples and (B) on PDA-coated samples using 24 h deposition time, the respective O 1s XPS spectra are shown below the corresponding samples (C and D, respectively). The percentage values (%) in parentheses next to the binding energies indicate the contribution of the corresponding peak area to the total peak area.
Based on the hydrophilic structure of PDA abundant in free hydroxyl groups (see the XPS results, vide supra), it is expected that beneficial secondary interactions can develop between the deposited PDA layer and the cellulosic resin. In order to follow the development of a hydrophilic layer on the originally hydrophobic CF substrate, contact angle measurements were performed (Figure 3). For these experiments, however, a pure graphitic carbon disk (99.98% purity; Nilaco Corporation, Tokyo, Japan) was chosen as a model substrate (due to the difficulties in measuring the contact angle on a single short CF using conventional optical tensiometer). The water contact angle was determined to be 111.9 ± 4.5° on the pure graphitic substrate (Figure 3), this value is very close to that previously reported for CFs (110°).12 Furthermore, the water contact angle was measured to be 81 ± 1.3° on cellulose propionate (CP), which is intended to be used as a polymer matrix to prepare composites. This value is also reasonable if we compare it with previously reported water contact angles on cellulose esters,46 given the high degree of substitution of CP (DS = 2.76).42 PDA deposition makes the surface of the carbon plate hydrophilic even after 1 h of deposition time, as a water contact angle of 57.2 ± 0.5° could be obtained. The water contact angle further decreases to 39.3 ± 2.3° following 2 h reaction time, however, it does not change appreciable after that (35.7 ± 1.3° for 24 h reaction time). Whether this hydrophilic layer on the surface can improve interactions with the surrounding CP matrix will be further investigated by obtaining micrographs of the fracture surfaces after the tensile test is performed on short CF reinforced composites (vide infra).
Figure 3.

Water contact angles measured on a (A) graphitic carbon disk, (B) CP, and graphitic carbon disks after depositing PDA on the surface using (C) 1, (D) 2, and (E) 24 h reaction time.
The amount of PDA on the surface can be predicted by comparing the weight losses of pure PDA and PDA-coated CF after thermogravimetric analysis (TGA, Figure S3). This analysis suggested that ∼3.9 wt % PDA is present on the surface after 24 h reaction time, this value is very similar to the previously reported results (note that our data about TGA appeared to be not reliable for samples prepared using shorter deposition times).12
2.2. Interfacial Interactions and Properties of the Composites
Interfacial interactions at the cellulosic polymer/fiber interfaces were investigated via microdroplet test. This method is a direct way to determine interfacial shear strength (IFSS) by applying pull-out load on a small (∼80–100 μm embedded length) resin droplet prepared on a single fiber until debonding takes place. Figure 4 shows the IFSS values determined using PDA-modified CFs along with a control sample (for explanation see the figure caption). A statistically significant increase in IFSS can be obtained for the sample subjected to a 2 h reaction time, with the maximum value achieved after 5 h (please note that according to the statistical analysis, samples subjected to 2, 5, 10, and 24 h deposition time are significantly different from each other, and compared to the control sample). This increase can be attributed to improving interactions between the resin and the initially hydrophobic CF surface, as a hydrophilic PDA layer is deposited. Furthermore, PDA also has free −OH groups that might make the formation of beneficial hydrogen bonding interactions involving the remaining free −OH groups in CP (DS = 2.76) possible. The IFSS value eventually declines when longer deposition time is applied (Figure 4). The morphology of the CF surface indicates that following a surface-based nucleation that leads to a thin PDA layer clearly seen on the surface of the sample after 5 h deposition time (Figure 1), at longer reaction times, PDA particles are deposited onto this thin PDA layer. It might be postulated that π–π stacking interactions between PDA and the pre-graphitic CF surface are stronger than the interactions between PDA chains, and therefore the particles growing on the initial PDA layer might not be adhered as strongly as the layer attached to the fiber surface. In such a case, it is reasonable to deduce that the particles weakly attached to the PDA surface can eventually be separated from that more easily than the PDA layer itself, facilitating interfacial failure and matrix–fiber separation (a schematic representation is given in Figure S4). This phenomenon would give an explanation for the diminishing IFSS value in the case of the samples treated with longer than 5 h reaction times. Furthermore, it is also possible to attribute the decrease in IFSS to a developing imbalance between PDA-fiber and PDA-resin adhesion, that is, PDA-resin adhesion increases as interactions develop; however, it is not followed by an increase in PDA-fiber adhesion.
Following the initial assessment of interfacial interactions between the surficial PDA layer and the CP resin, short CF-reinforced composites were prepared using the surface-modified CFs, and their tensile properties were evaluated. Figure 5 shows the results of the tensile test. The most prominent change is the abrupt increase in the elastic modulus, as fibers are added to the CP matrix leading to a stiff material, without showing a significant difference after surface modification. It is known that the elastic modulus is mostly determined by the fiber volume fraction, which is constantly 20 wt % in our case, and therefore these results are reasonable.45,55,56 Furthermore, the strain at break value also decreases substantially when fibers are present in the CP matrix indicating the formation of a brittle material, this parameter also remains relatively constant for the surface-modified samples. The rigid fiber segments facilitate microcrack formation and confine polymer chain mobility, which is the underlying reason for the brittleness.45,57 The strain at break value is tightly connected to the number of fiber ends as stress accumulates at these locations, serving as nucleation sites for microcracks. It follows that again fiber volume fraction (which determines the number of fiber ends given that the size of the fibers is relatively similar) influences mostly this parameter.45,55,57,58 Similar results were found in our previous study.45 The parameters influencing the tensile strength of a short CF-reinforced composite are usually discussed using the modified rule of mixture theory according to eq 1(45,59)
| 1 |
where σcu, σl, and σm are the tensile strength of the composite, fiber, and resin, whereas Vf and Vm denotes the volume fraction of the fiber and matrix, respectively. Furthermore, χ1 stands for the fiber orientation factor and χ2 denotes the fiber length factor. Interfacial shear properties are included in the second parameter (χ2), for more details see the work of Fu and Lauke.59 In order to gain insights into the fiber orientation in the composite, microscale X-ray computed tomography images were recorded, which enabled us to investigate the internal microstructure of the composite (Figure S5, Supporting Information). In general, fibers are mostly aligned according to the flow direction during the molding step (which is the same direction as the elongation of the specimen during the tensile test), with more or less randomly aligned segments. Fiber orientation is mostly determined by the processing conditions, which were the same for all of the samples and therefore, there is no reason to connect this property to the changing mechanical properties between the composites. The presence of unmodified fibers (as received) leads to ∼32% improvement in the tensile strength compared to pure CP (Figure 5), and the tensile strength does not differ significantly for the control sample (treatment solution without dopamine added, i.e. no PDA deposition, 24 h treatment time). Interestingly, the tensile strength exhibits a local maximum at shorter reaction times (∼2 h), which one would also expect based on the IFSS values (Figure 4). However, the global maximum in tensile strength is obtained after using longer, 24 h deposition time.
The fracture surfaces of these composites are shown in Figure 6. Interfacial interactions are known to have a decisive impact on the failure mechanism of fiber-reinforced composite materials.59−61 If the interaction is weak between the fiber and matrix, failure can take place at the interface leading to debonding (or fiber pull-out), while in the case of strong interactions the applied load is efficiently transferred through the interface. If the interfacial interaction is stronger than the matrix itself, matrix failure might occur or the fiber itself might break.61 In our previous study,44 it was observed for cellulose-based composites that in the case of strong IFSS matrix failure can take place before interfacial delamination. On the fracture surfaces, starting from the sample containing PDA-coated CFs subjected to 2 h deposition time, compared to the smooth fracture surface of the control sample, the morphology changes appreciably, exhibiting more damaged matrix segments (rough morphology). While most of the fibers remain tightly embedded in the matrix (CF PDA 2–24 h, Figure 6), we can also note that some pull-out eventually takes place for all the samples. Furthermore, the remaining resin on the fibers (which could be expected for a strong IFSS) can be hardly observed (for the sample containing PDA-coated CFs subjected to 24 h deposition time, some locations are indicated in Figure 6). It should be noted that no PDA particles can be observed inside the resin (which might have separated from the fiber surface), which could have a reinforcing effect on the matrix similar to previous studies.49 As a result, the fracture surfaces are not much different for the surface-modified samples and give no clear explanation for the unexpected increase in tensile strength (based on the IFSS values) when surface-modified fibers are applied following 24 h deposition time (Figure 5). Therefore, we also determined the fiber length in these composites because this factor also has a significant impact on the tensile properties of short CF-reinforced composites (see eq 1). Table 1 shows the mean fiber length values determined in the prepared composites. The initially 3 mm long fibers suffer appreciable damage during the extrusion and molding steps, which is a well-known phenomenon when short fiber-reinforced composites are prepared.45,56,58,62 The fiber length becomes shorter when PDA-modified (1, 2, 5, and 10 h deposition time) CF samples are applied compared to the unmodified sample. While the fiber length does not change appreciably between samples PDA 1, 2, and 10 h, a considerable decrease is noticeable for the sample prepared using fibers following 5 h deposition time (Table 1). This decrease agrees well with the maximum IFSS, which can be found in Figure 4. This might allow us to conclude that during the processing steps (extrusion and molding processes), enhanced shear is placed on these fibers because of the improved fiber–matrix interactions eventually leading to shorter fiber segments. It appears that the fiber length increases when 24 h reaction time is used for the surface modification. Based on the fiber length distribution profile (see Figure S6), there are clearly more fibers with lengths longer than 100 μm and less fibers with lengths shorter than 100 μm in this composite compared to the sample containing unmodified CFs (as received). This increase in fiber length is expected to lead to an increase in the tensile strength as indicated in eq 1, giving an explanation for our results. The increasing fiber length can be rationalized on the basis of two phenomena: (a) the decreasing interfacial shear (see Figure 4) between the resin and fibers treated with 24 h reaction time can result in longer fibers after the extrusion and molding steps (less shear stress is placed on the fibers during the processing, i.e., less fiber damage occurs) and (b) the relatively thick PDA coating can have a protecting effect on fibers during the extrusion and molding steps, the protecting effect of a coating resulting in longer fibers after thermal processing was reported previously.62 Because it appears that the mean fiber length values follow the IFSS, that is, an increase in the IFSS results in shorter fibers and a decrease in IFSS leads to longer fibers (see Figure 4 and Table 1), the first (a) phenomenon is expected to dominate the system. Nevertheless, the protecting effect can also be justified based on the fact that although the IFSS values of the unmodified and modified (24 h) samples are similar (Figure 4), the latter has still longer fiber segments (Table 1).
Figure 6.

SEM images of the fracture surfaces after the tensile test. CF—carbon fiber; PDA—polydopamine. Arrows indicate some locations where the better fiber–matrix connections might be observable.
Table 1. Mean Fiber Length in the Composites.
| mean fiber length (μm) | |
|---|---|
| carbon fiber as received | 131.5 |
| PDA 1 h | 118.5 |
| PDA 2 h | 117.5 |
| PDA 5 h | 96.2 |
| PDA 10 h | 117.1 |
| PDA 24 h | 143.1 |
3. Conclusions
Carbon fiber surface functionalization strategies mostly comprise harsh environmentally unfriendly reaction conditions, owing to the relatively chemically inert nature of the pre-graphitic carbon structure. Implementation of biomimetic green approaches for surface modification would bring about a great improvement in the environmental footprint of CF-reinforced composite materials.
Our study is aimed at tailoring interfacial interactions in cellulose-based CF-reinforced composites by applying PDA, a mussel-inspired biomimetic polymer coating. The development of a thin hydrophilic PDA film was observed on the surface with increasing deposition time (1–10 h), suggesting a surface-based polymerization mechanism attributed to strong π–π stacking interactions. A longer reaction time (24 h) resulted in particle formation on the PDA layer, this morphological design although had a diminishing effect on the IFSS, it eventually led to an increase in the mechanical performance of a short CF-reinforced composite. This phenomenon was attributed to the protecting effects against fiber damage during the composite processing steps, resulting in markedly longer fiber segments. By fine-tuning the morphological design of PDA attached to the surface, a ∼12% increase in tensile strength (∼47% increase compared to the CP matrix) could be achieved compared to the unmodified CF-based composite. Our results point out the importance of morphological design at the interface, and through our study it can be recognized that PDA might be a promising biomimetic functional polymer at composite interfaces.
4. Materials and Methods
4.1. Materials
3-Hydroxytyramine hydrochloride (dopamine hydrochloride), tris(hydroxymethyl)aminomethane (Tris), and ethylamine (70% in water) were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). All the other reagents and solvents were obtained from Kanto Chemicals Co., Inc. (Tokyo, Japan).
PX35 type 3 mm long unsized chopped CFs with a nominal diameter of 7.2 μm were provided by ZOLTEK (Toray Group, St. Charles, MO, USA). The fibers were dried at 130 °C for 72 h in a vacuum oven prior to the experiments. Microdroplet tests were conducted on long T700SC-12000-50C type CFs because of the experimental setup, the nominal diameter of these fibers is 7 μm (Toray Industries, Tokyo, Japan). The latter fibers possess ∼1 wt % sizing agent, which was removed according to our previously elaborated procedure.42,63 As a model cellulosic resin, cellulose propionate (Mw ≈ 200,000 g mol–1 according to the producer; Scientific Polymer Products, Inc., Ontario, NY, USA; catalogue number 321), a thermoplastic cellulose ester was chosen exhibiting excellent processability and mechanical characteristics in our previous studies.45 The degree of substitution was determined to be 2.76 elsewhere.42 A carbon disk (99.98% purity; 1 mm thick, 50 mm diameter) for the contact angle measurements was ordered from The Nilaco Corporation (Tokyo, Japan).
4.2. CF Surface Modification—PDA Deposition
Short CFs (6 g) (PX35 type; ZOLTEK, Toray Group, St. Charles, MO, USA) were placed in 1 L aqueous solution containing 2 g of dopamine and 10 mM Tris buffer (pH = 8.5, adjusted using 1 M hydrochloric acid solution). The reaction was conducted at room temperature using a double shaker operating at 80 min–1 speed (Double Shaker NR-30; Taitec Corporation, Koshigaya, Japan), which allows for gentle agitation in order to avoid mechanical damage to the fibers. Since autoxidation of dopamine necessitates the presence of dissolved oxygen in the solution, and oxygen is available at a limited concentration (∼0.27 mM in air-equilibrated solutions at 25 °C and 1 atm according to Henry’s law), the reaction system was covered with perforated Al foil to ensure continuous oxygen supply. After the reaction, the fibers were filtered off and washed exhaustively with around 1 L of distilled water to remove the remaining reactants and undeposited PDA. The fibers were subsequently dried in a vacuum oven at 50 °C for 24 h. A control sample was also prepared and used as a reference, in this case the reaction procedure was repeated without adding dopamine to the system (using 24 h treatment time).
4.3. Surface Characterization
The XPS experiments were conducted with a Thermo Scientific K-Alpha X-ray photoelectron spectrometer system providing monochromated Al Kα radiation (hν = 1486.6 eV, 36 W; Waltham, MA, USA). The analysis area was 400 μm2. The binding energy scale was referenced to the hydrocarbon C 1s peak at 285.0 eV. Survey spectra were collected with a resolution of 1 eV, while high-resolution C 1s, N 1s, and O 1s spectra were obtained at 20 eV pass energy and 0.1 eV resolution. Data analysis was done using Thermo Scientific Avantage Software version 5.89 (Waltham, MA, USA), a built-in Smart algorithm was applied for background correction and Powell method using a Gauss–Lorentz Mix algorithm for peak fitting.
Surface morphologies were analyzed using a JSM-7610F field-emission scanning electron microscope applying 15 kV accelerating voltage (JEOL, Tokyo, Japan). SEM micrographs were recorded on CF samples sputtered with an Au/Pd layer using a Hitachi E1030 type machine and 40 s deposition time (Hitachi, Ltd., Tokyo, Japan).
Water contact angles were measured using a SImage entry 5 model system (Excimer Inc., Yokohama, Japan). Droplets (4 μL) were placed on the surface of the substrate, and contact angles were analyzed with SImage V05 Version 5.001 software. The contact angle was measured right after the drop was placed on the substrate, within less than 1 min (sessile drop). The average contact angle values (and corresponding errors) were based on measurements on three individual droplets.
4.4. Microdroplet Test
IFSS as a quantitative measure of interfacial adhesion was determined via the microdroplet test, using HM410 type testing machine equipped with 1 N load cell (Tohei Sangyo Co., Ltd., Tokyo, Japan). Microdroplets on single CFs were prepared using polymer melts similar to others,64 and in line with our previous report.44 Briefly, CP was kept on a hot plate at 235 °C for 5 min, whereupon a single fiber was gently immersed into the melt and then transferred to another plate heated to 250 °C (3 min treatment time). Upon cooling to room temperature, the formation of separated microdroplets could be confirmed. The microdroplet debonding experiment was conducted with a 0.06 mm min–1 pull-out speed. IFSS (τ) was determined using eq 2
| 2 |
where F stands for the maximum load (N), d is the fiber diameter, and L denotes the embedded length (droplet length). Thirty droplets with sizes ranging between 80 and 100 μm were subjected to the test from each type of modified/unmodified CF samples. Data were subjected to multiple t-test analyses to identify statistically significant differences (assuming equal variances).
4.5. Composite Preparation and Mechanical Test
Short CFRP composites were prepared using Xplore MC5 microcompounder (Xplore Instruments BV, Sittlard, The Netherlands). A rotation speed of 60 rpm (co-rotating twin-screw), 203 °C processing temperature, and 5 min retention time was applied according to our previous reports.45 Flow characteristics of CP can be found elsewhere.45 Standard dumbbell-shaped specimens (JIS K 7161-2:2014 1BA) were made using an Imoto IMC-5705 injection molding machine (Imoto Machinery Co., Ltd., Kyoto, Japan). The following conditions were applied: barrel temperature: 210 °C; mold temperature: 140 °C; and injection pressure: 0.6 MPa.
Tensile test (JIS K7161 standard, in compliance with ISO 527) was conducted on Shimadzu Autograph AG-X Plus machine (Kyoto, Japan) equipped with 5 kN load cell and with a noncontact video type extensometer (TRViewX; Shimadzu, Kyoto, Japan). The samples clamped with 0.5 MPa pressure were elongated applying a crosshead speed of 5 mm min–1. Multiple t-tests were performed on the data set to identify statistically significant differences (assuming equal variances).
The internal microstructure of the composite was examined via X-ray computed tomography (X-ray micro-CT) experiments, carried out on ZEISS Xradia 410 Versa equipment (ZEISS, Oberkochen, Germany).
Fracture surfaces were analyzed using a Hitachi S4500 scanning electron microscope (Hitachi Ltd., Tokyo, Japan), Au/Pd layer was deposited on the surface prior to the experiment as it was mentioned before (vide supra).
The fiber length analysis was performed on microscopy images (Olympus BX50 microscope; Olympus Corporation, Tokyo, Japan) recorded after dissolving the resin in DMF according to our previous studies.45 Images were analyzed using ImageJ program.
Acknowledgments
The authors acknowledge the financial support received from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) and the Japan Science and Technology Agency (JST) through the Center of Innovation Science and Technology based Radical Innovation and Entrepreneurship Program [COI stream, (JPMJCE1315)] for “Construction of next generation infrastructure using innovative materials: Realization of a safe and secure society that can coexist with the Earth for centuries”.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02356.
C 1s and O 1s XPS spectra of a control sample, N 1s spectra of the samples, TGA results, schematic representation explaining the results of the microdroplet experiment, X-ray computed tomography images of the short CF-reinforced composites, and results of the fiber length distribution analysis (PDF)
Author Contributions
The manuscript was written through contributions of all the authors. All the authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Vincent J. F. V.; Bogatyreva O. A.; Bogatyrev N. R.; Bowyer A.; Pahl A.-K. Biomimetics: its practice and theory. J. R. Soc., Interface 2006, 3, 471–482. 10.1098/rsif.2006.0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelat F. Biomimetics for next generation materials. Philos. Trans. R. Soc., A 2007, 365, 2907–2919. 10.1098/rsta.2007.0006. [DOI] [PubMed] [Google Scholar]
- Wegst U. G. K.; Bai H.; Saiz E.; Tomsia A. P.; Ritchie R. O. Bioinspired structural materials. Nat. Mater. 2015, 14, 23–36. 10.1038/nmat4089. [DOI] [PubMed] [Google Scholar]
- Sanchez C.; Arribart H.; Giraud Guille M. M. Biomimetism and bioinspiration as tools for the design of innovative materials and systems. Nat. Mater. 2005, 4, 277–288. 10.1038/nmat1339. [DOI] [PubMed] [Google Scholar]
- Eder M.; Amini S.; Fratzl P. Biological composites - complex structures for functional diversity. Science 2018, 362, 543–547. 10.1126/science.aat8297. [DOI] [PubMed] [Google Scholar]
- Egan P.; Sinko R.; LeDuc P. R.; Keten S. The role of mechanics in biological and bio-inspired systems. Nat. Commun. 2015, 6, 7418. 10.1038/ncomms8418. [DOI] [PubMed] [Google Scholar]
- Das S.; Warren J.; West D.; Schexnayder S. M.. Global carbon fiber supply chain competitiveness analysis; Clean Energy Manufacturing Analysis Center: Denver, May 2016, https://www.nrel.gov/docs/fy16osti/66071.pdf (accessed November 07, 2019).
- Witten E.; Sauer M.; Kühnel M.. Composites Market Report 2019. Market Developments, Trends, Outlook and Challenges; Industrievereinigung Verstärkte Kunststoffe e.V. (Federation of Reinforced Plastics): Frankfurt, September 2019.
- Stojcevski F.; Eykens D. J.; Randall J. D.; Marinovic L. I.; Méric G.; Henderson L. C. Improved out-of-plane strength and weight reduction using hybrid interface composites. Compos. Sci. Technol. 2019, 182, 107730. 10.1016/j.compscitech.2019.107730. [DOI] [Google Scholar]
- Libonati F.; Vellwock A. E.; Lelmini F.; Abliz D.; Ziegmann G.; Vergani L. Bone-inspired enhanced fracture toughness of de novo fiber reinforced composites. Sci. Rep. 2019, 9, 3142. 10.1038/s41598-019-39030-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca F.; Clancy A. J.; Carrero N. R.; Anthony D. B.; De Luca H. G.; Shaffer M. S. P.; Bismarck A. Increasing carbon fiber composite strength with a nanostructured “brick-and-mortar” interphase. Mater. Horiz. 2018, 5, 668–674. 10.1039/c7mh00917h. [DOI] [Google Scholar]
- Chen S.; Cao Y.; Feng J. Polydopamine as an efficient and robust platform to functionalize carbon fiber for high-performance polymer composites. ACS Appl. Mater. Interfaces 2014, 6, 349–356. 10.1021/am404394g. [DOI] [PubMed] [Google Scholar]
- Eyckens D. J.; Arnold C. L.; Randall J. D.; Stojcevski F.; Hendlmeier A.; Stanfield M. K.; Pinson J.; Gengenbach T. R.; Alexander R.; Soulsby L. C.; Francis P. S.; Henderson L. C. Fiber with butterfly wings: creating colored carbon fibers with increased strength, adhesion, and reversible malleability. ACS Appl. Mater. Interfaces 2019, 11, 41617–41625. 10.1021/acsami.9b11826. [DOI] [PubMed] [Google Scholar]
- Barthelat F.; Yin Z.; Buehler M. J. Structure and mechanics of interfaces in biological materials. Nat. Rev. Mater. 2016, 1, 16007. 10.1038/natrevmats.2016.7. [DOI] [Google Scholar]
- Sharma M.; Gao S.; Mäder E.; Sharma H.; Wei L. Y.; Bijwe J. Carbon fiber surfaces and composite interphases. Compos. Sci. Technol. 2014, 102, 35–50. 10.1016/j.compscitech.2014.07.005. [DOI] [Google Scholar]
- Krager-Kocsis J.; Mahmood H.; Pegoretti A. Recent advances in fiber/matrix interphase engineering for polymer composites. Prog. Mater. Sci. 2015, 73, 1–43. 10.1016/j.pmatsci.2015.02.003. [DOI] [Google Scholar]
- Frank E.; Steudle L. M.; Ingildeev D.; Spörl J. M.; Buchmeiser M. R. Carbon fibers: precursor systems, processing, structure, and properties. Angew. Chem., Int. Ed. 2014, 53, 5262–5298. 10.1002/anie.201306129. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Pittman C. U.; Gardner S. D. Nitric acid oxidation of carbon fibers and the effect of subsequent treatment in refluxing aqueous NaOH. Carbon 1995, 33, 597–605. 10.1016/0008-6223(95)00145-4. [DOI] [Google Scholar]
- Pittman C. U.; Wu Z.; Jiang W.; He G.-R.; Wu B.; Li W.; Gardner S. D. Reactivities of amine functions grafted to carbon fiber surfaces by tetraethylenepentamine. Designing interfacial bonding. Carbon 1997, 35, 929–943. 10.1016/s0008-6223(97)00047-x. [DOI] [Google Scholar]
- Zhang G.; Sun S.; Yang D.; Dodelet J.-P.; Sacher E. The surface analytical characterization of carbon fibers functionalized by H2SO4/HNO3 treatment. Carbon 2008, 46, 196–205. 10.1016/j.carbon.2007.11.002. [DOI] [Google Scholar]
- Koutroumanis N.; Manikas A. C.; Pappas P. N.; Petropoulos F.; Sygellou L.; Tasis D.; Papagelis K.; Anagnostopoulos G.; Galiotis C. A novel mild method for surface treatment of carbon fibers in epoxy-matrix composites. Compos. Sci. Technol. 2018, 157, 178–184. 10.1016/j.compscitech.2018.01.048. [DOI] [Google Scholar]
- Yu J.; Meng L.; Fan D.; Zhang C.; Yu F.; Huang Y. The oxidation of carbon fibers through K2S2O8/AgNO3 system that preserves fiber tensile strength. Composites, Part B 2014, 60, 261–267. 10.1016/j.compositesb.2013.12.037. [DOI] [Google Scholar]
- Ma L.; Zhu Y.; Feng P.; Song G.; Huang Y.; Liu H.; Zhang J.; Fan J.; Hou H.; Guo Z. Reinforcing carbon fiber epoxy composites with triazine derivatives functionalized graphene oxide modified sizing agent. Composites, Part B 2019, 176, 107078. 10.1016/j.compositesb.2019.107078. [DOI] [Google Scholar]
- Zhao M.; Meng L.; Ma L.; Ma L.; Yang X.; Huang Y.; Ryu J. E.; Shankar A.; Li T.; Yan C.; Guo Z. Layer-by-layer grafting CNTs onto carbon fibers surface for enhancing the interfacial properties of epoxy resin composites. Compos. Sci. Technol. 2018, 154, 28–36. 10.1016/j.compscitech.2017.11.002. [DOI] [Google Scholar]
- Delamar M.; Désarmot G.; Fagebaume O.; Hitmi R.; Pinsonc J.; Savéant J.-M. Modification of carbon fiber surfaces by electrochemical reduction of aryl diazonium salts: application to carbon epoxy composites. Carbon 1997, 35, 801–807. 10.1016/s0008-6223(97)00010-9. [DOI] [Google Scholar]
- Servinis L.; Beggs K. M.; Gengenbach T. R.; Doeven E. H.; Francis P. S.; Fox B. L.; Pringle J. M.; Pozo-Gonzalo C.; Walsh T. R.; Henderson L. C. Tailoring the fiber-to-matrix interface using click chemistry on carbon fibre surfaces. J. Mater. Chem. A 2017, 5, 11204–11213. 10.1039/c7ta00922d. [DOI] [Google Scholar]
- Randall J. D.; Eyckens D. J.; Stojcevski F.; Francis P. S.; Doeven E. H.; Barlow A. J.; Barrow A. S.; Arnold C. L.; Moses J. E.; Henderson L. C. Modification of carbon fiber surfaces by sulfur-fluoride exchange click chemistry. ChemPhysChem 2018, 19, 3176–3181. 10.1002/cphc.201800789. [DOI] [PubMed] [Google Scholar]
- Randall J. D.; Eyckens D. J.; Servinis L.; Stojcevski F.; O’Dell L. A.; Gengenbach T. R.; Demir B.; Walsh T. R.; Henderson L. C. Designing carbon fiber composite interfaces using ‘graft-to’ approach: surface grafting density versus interphase penetration. Carbon 2019, 146, 88–96. 10.1016/j.carbon.2019.01.064. [DOI] [Google Scholar]
- Arnold C. L.; Beggs K. M.; Eyckens D. J.; Stojcevski F.; Servinis L.; Henderson L. C. Enhancing interfacial shear strength via surface grafting of carbon fibers using the Kolbe decarboxylation reaction. Compos. Sci. Technol. 2018, 159, 135–141. 10.1016/j.compscitech.2018.02.037. [DOI] [Google Scholar]
- Ehlert G. J.; Lin Y.; Sodano H. A. Carboxyl functionalization of carbon fibers through a grafting reaction that preserves fiber tensile strength. Carbon 2011, 49, 4246–4255. 10.1016/j.carbon.2011.05.057. [DOI] [Google Scholar]
- Lee H.; Dellatore S. M.; Miller W. M.; Messersmith P. B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Rho J.; Messersmith P. B. Facile conjugation of biomolecules onto surfaces via mussel adhesive protein inspired coatings. Adv. Mater. 2009, 21, 431–434. 10.1002/adma.200801222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J. H.; Messersmith P. B.; Lee H. Polydopamine surface chemistry: a decade of discovery. ACS Appl. Mater. Interfaces 2018, 10, 7523–7540. 10.1021/acsami.7b19865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhurakkat Perikamana S.K.; Lee J.; Lee Y. B.; Shin Y. M.; Lee E. J.; Mikos A. G.; Shin H. Materials from mussel-inspired chemistry for cell and tissue engineering applications. Biomacromolecules 2015, 16, 2541–2555. 10.1021/acs.biomac.5b00852. [DOI] [PubMed] [Google Scholar]
- Liebscher J. Chemistry of polydopamine - scope, variation, and limitation. Eur. J. Org. Chem. 2019, 4976–4994. 10.1002/ejoc.201900445. [DOI] [Google Scholar]
- Wang J.-l.; Li B.-c.; Li Z.-j.; Ren K.-f.; Jin L.-j.; Zhang S.-m.; Chang H.; Sun Y.-x.; Ji J. Electropolymerization of dopamine for surface modification of complex-shaped cardiovascular stents. Biomaterials 2014, 35, 7679–7689. 10.1016/j.biomaterials.2014.05.047. [DOI] [PubMed] [Google Scholar]
- Vecchia N. F. D.; Luchini A.; Napolitano A.; D’Errico G.; Vitiello G.; Szekely N.; d’Ischia M.; Paduano L. Tris buffer modulates polydopamine growth, aggregation, and paramagnetic properties. Langmuir 2014, 30, 9811–9818. 10.1007/978-3-662-53971-2_182. [DOI] [PubMed] [Google Scholar]
- Zangmeister R. A.; Morris T. A.; Tarlov M. J. Characterization of polydopamine thin films deposited at short times by autoxidation of dopamine. Langmuir 2013, 29, 8619–8628. 10.1021/la400587j. [DOI] [PubMed] [Google Scholar]
- Ball V.; Frari D. D.; Toniazzo V.; Ruch D. Kinetics of polydopamine film deposition as a function of pH and dopamine concentration: insights in the polydopamine deposition mechanism. J. Colloid Interface Sci. 2012, 386, 366–372. 10.1016/j.jcis.2012.07.030. [DOI] [PubMed] [Google Scholar]
- Tsirka K.; Tzounis L.; Avgeropoulos A.; Liebscher M.; Mechtcherine V.; Paipetis A. S. Optimal synergy between micro and nano scale: hierarchical all carbon composite fibers for enhanced stiffness, interfacial shear strength and Raman strain sensing. Compos. Sci. Technol. 2018, 165, 240–249. 10.1016/j.compscitech.2018.07.003. [DOI] [Google Scholar]
- Lavagna L.; Massella D.; Pantano M. F.; Bosia F.; Pugno N. M.; Pavese M. Grafting carbon nanotubes onto carbon fibres doubles their effective strength and the toughness of the composite. Compos. Sci. Technol. 2018, 166, 140–149. 10.1016/j.compscitech.2018.03.015. [DOI] [Google Scholar]
- Szabó L.; Imanishi S.; Kawashima N.; Hoshino R.; Takada K.; Hirose D.; Tsukegi T.; Ninomiya K.; Takahashi K. Carbon fiber reinforced cellulose-based polymers: intensifying interfacial adhesion between the fibre and the matrix. RSC Adv. 2018, 8, 22729–22736. 10.1039/c8ra04299c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó L.; Imanishi S.; Kawashima N.; Hoshino R.; Hirose D.; Tsukegi T.; Ninomiya K.; Takahashi K. Interphase engineering of a cellulose-based carbon fiber reinforced composite by applying click chemistry. ChemistryOpen 2018, 7, 720–729. 10.1002/open.201800180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó L.; Imanishi S.; Tetsuo F.; Hirose D.; Ueda H.; Tsukegi T.; Ninomiya K.; Takahashi K. Lignin as a functional green coating on carbon fiber surface to improve interfacial adhesion in carbon fiber reinforced composites. Materials 2019, 12, 159. 10.3390/ma12010159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó L.; Milotskyi R.; Fujie T.; Tsukegi T.; Wada N.; Ninomiya K.; Takahashi K. Short carbon fiber reinforced polymers: utilizing lignin to engineer potentially sustainable resource-based biocomposites. Front. Chem. 2019, 7, 757. 10.3389/fchem.2019.00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka P. M.; Junior J. A.; Saito R. S. N.; Petri D. F. S.. Thermodynamics of cellulose ester surfaces. In Model Cellulosic Surfaces; Roman M., Ed.; ACS Symposium Series; American Chemical Society: Washington, DC, 2010; pp 223–241. [Google Scholar]
- Shi C.; Deng C.; Zhang X.; Yang P. Synthesis of highly water-dispersible polydopamine-modified multiwalled carbon nanotubes for matrix-assisted laser desorption/ionization mass spectrometry analysis. ACS Appl. Mater. Interfaces 2013, 5, 7770–7776. 10.1021/am4024143. [DOI] [PubMed] [Google Scholar]
- Woodhead A. L.; de Souza M. L.; Church J. S. An investigation into the surface heterogeneity of nitric acid oxidized carbon fiber. Appl. Surf. Sci. 2017, 401, 79–88. 10.1016/j.apsusc.2016.12.218. [DOI] [Google Scholar]
- Xiong S.; Wang Y.; Yu J.; Chen L.; Zhu J.; Hu Z. Polydopamine particles for next-generation multifunctional biocomposites. J. Mater. Chem. A 2014, 2, 7578–7587. 10.1039/c4ta00235k. [DOI] [Google Scholar]
- Chen F.; Xing Y.; Wang Z.; Zheng X.; Zhang J.; Cai K. Nanoscale polydopamine (PDA) meets π-π stacking interactions: an interface-directed coassembly approach for mesoporous nanoparticles. Langmuir 2016, 32, 12119–12128. 10.1021/acs.langmuir.6b03294. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Sherwood P. M. A. X-ray photoelectron spectroscopic studies of carbon fiber surfaces. 10. Valence-band studies interpreted by X-α calculations and the differences between poly(acrylonitrile)- and pitch-based carbon fibers. Chem. Mater. 1989, 1, 427–432. 10.1021/cm00004a009. [DOI] [Google Scholar]
- Bekyarova E.; Itkis M. E.; Ramesh P.; Berger C.; Sprinkle M.; de Heer W. A.; Haddon R. C. Chemical modification of epitaxial graphene: spontaneous grafting of aryl groups. J. Am. Chem. Soc. 2009, 131, 1336–1337. 10.1021/ja8057327. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Fang Y.; Liu X.; Wang X.; Yang B. Mussel-inspired modification of carbon fiber via polyethyleneimine/polydopamine co-deposition for the improved interfacial adhesion. Compos. Sci. Technol. 2017, 151, 164–173. 10.1016/j.compscitech.2017.08.008. [DOI] [Google Scholar]
- Xu L. Q.; Yang W. J.; Neoh K. G.; Kang E. T.; Fu G. D. Dopamine-induced reduction and functionalization of graphene oxide nanosheets. Macromol 2010, 43, 8336–8339. 10.1021/ma101526k. [DOI] [Google Scholar]
- Fu S.-Y.; Lauke B.; Mäder E.; Yue C.-Y.; Hu X. Tensile properties of short-glass-fiber- and short-carbon-fiber-reinforced polypropylene composites. Composites, Part A 2000, 31, 1117–1125. 10.1016/s1359-835x(00)00068-3. [DOI] [Google Scholar]
- Molnár S.; Rosenberger S.; Gulyás J.; Pukánszky B. Structure and impact resistance of short carbon fiber reinforced polyamide 6 composites. J. Macromol. Sci., Part B: Phys. 1999, 38, 721–735. 10.1080/00222349908248134. [DOI] [Google Scholar]
- Karsli N. G.; Aytac A. Effects of maleated polypropylene on the morphology, thermal and mechanical properties of short carbon fiber reinforced polypropylene composites. Mater. Des. 2011, 32, 4069–4073. 10.1016/j.matdes.2011.03.021. [DOI] [Google Scholar]
- Karsli N. G.; Aytac A. Tensile and thermomechanical properties of short carbon fiber reinforced polyamide 6 composites. Composites, Part B 2013, 51, 270–275. 10.1016/j.compositesb.2013.03.023. [DOI] [Google Scholar]
- Fu S.; Lauke B. Effects of fiber length and fiber orientation distributions on the tensile strength of short-fiber-reinforced polymers. Compos. Sci. Technol. 1996, 56, 1179–1190. 10.1016/s0266-3538(96)00072-3. [DOI] [Google Scholar]
- Ehrburger P.; Donnet J. B. Interface in composite materials. Philos. Trans. R. Soc., A 1980, 294, 495–505. 10.1098/rsta.1980.0059. [DOI] [Google Scholar]
- Baxter W. J.The effect of temperature on the strength of composites reinforced with discontinuous fibers. In Proceeding of The Tenth International Conference on Composite Materials, Volume I: Fatigue and Fracture; Poursartip A., Street K., Eds.; Woodhead Publishing Limited: Cambridge, 1995; pp 109–116.
- Ozkan C.; Gamze Karsli N.; Aytac A.; Deniz V. Short carbon fiber reinforced polycarbonate composites: effects of different sizing materials. Composites, Part B 2014, 62, 230–235. 10.1016/j.compositesb.2014.03.002. [DOI] [Google Scholar]
- Szabó L.; Milotskyi R.; Tsukegi T.; Wada N.; Takahashi K. Quantitative analysis of native reactive functional groups on carbon fiber surface: an electrochemical approach. Appl. Surf. Sci. 2019, 494, 315–325. 10.1016/j.apsusc.2019.07.185. [DOI] [Google Scholar]
- Ho K. K. C.; Lamoriniere S.; Kalinka G.; Schulz E.; Bismarck A. Interfacial behaviour between atmospheric-plasma-fluorinated carbon fibers and poly(vinylidene fluoride). J. Colloid Interface Sci. 2007, 313, 476–484. 10.1016/j.jcis.2007.04.076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.