

Abstract
X-linked scapuloperoneal myopathy (X-SM), one of Four-and-a-half LIM 1 (FHL1) related diseases, is an adult-onset slowly progressive myopathy, often associated with cardiomyopathy. We previously generated a knock-in mouse model that has the same mutation (c.365 G > C, p.W122S) as human X-SM patients. The mutant male mouse developed late-onset slowly progressive myopathy without cardiomyopathy. In this study, we observed that heterozygous (Het) and homozygous (Homo) female mice did not show alterations of skeletal muscle function or histology. In contrast, 20-month-old mutant female mice showed signs of cardiomyopathy on echocardiograms with increased systolic diameter [wild-type (WT): 2.74 ± 0.22 mm, mean ± standard deviation (SD); Het: 3.13 ± 0.11 mm, P < 0.01; Homo: 3.08 ± 0.37 mm, P < 0.05) and lower fractional shortening (WT: 31.1 ± 4.4%, mean ± SD; Het: 22.7 ± 2.5%, P < 0.01; Homo: 22.4 ± 6.9%, P < 0.01]. Histological analysis of cardiac muscle revealed frequent extraordinarily large rectangular nuclei in mutant female mice that were also observed in human cardiac muscle from X-SM patients. Western blot demonstrated decreased Fhl1 protein levels in cardiac muscle, but not in skeletal muscle, of Homo mutant female mice. Proteomic analysis of cardiac muscle from 20-month-old Homo mutant female mice indicated abnormalities of the integrin signaling pathway (ISP) in association with cardiac dysfunction. The ISP dysregulation was further supported by altered levels of a subunit of the ISP downstream effectors Arpc1a in Fhl1 mutant mice and ARPC1A in X-SM patient muscles. This study reveals the first mouse model of FHL1-related cardiomyopathy and implicates ISP dysregulation in the pathogenesis of FHL1 myopathy.
Introduction
Four-and-a-half LIM 1 (FHL1), a protein that is composed of an N-terminus half LIM-domain followed by four complete LIM-domains (1–3). LIM-domains, characterized by eight conserved cysteine and histidine residues that generate a tandem structure of two zinc fingers (4,5), are capable of protein binding, and LIM proteins are thought to function as mediators of protein–protein interactions (5,6). FHL1 is abundant in skeletal and cardiac muscles and has been reported to interact with multiple proteins involved in cell scaffolding and signaling pathways, and is thought to serve essential roles in sarcomere assembly, cell growth anddifferentiation; however, its precise molecular functions remain undefined.
In humans, FHL1 gene mutations cause a spectrum of skeletal muscle and/or cardiac muscle diseases including X-linked scapuloperoneal myopathy (X-SM; 7, 8), reducing body myopathy (RBM; 9–12), X-linked myopathy with postural muscle atrophy (13–15), rigid spine syndrome (16), X-linked Emery–Dreifuss muscular dystrophy (X-EDMD; 17–19) and isolated hypertrophic cardiomyopathy (20,21). While manifesting overlapping features such as scapula-axio-peroneal weakness, these disorders differ in onset, severity, affected organs and histopathology (22,23). For example, RBM typically begins in infancy, while X-SM starts after the second decade of life. Some but not all patients with FHL1-related myopathy develop cardiomyopathy, and conversely, other patients have cardiomyopathy without skeletal myopathy. Reducing bodies are pathological hallmarks of RBM (24), but are sparse in X-SM and have not been observed in X-EDMD (17).
In 2008, we identified a missense mutation (c.365G > C, p.W122S) in FHL1 in a large Italian–American family with X-SM (7). All patients were hemizygous (males) or heterozygous (Het; females) for the mutation and developed slowly progressive myopathy after the second decade of life. Female patients were less severely affected than male patients. Of the 15 affected individuals, three had symptomatic cardiomyopathies (I1, II10 and III31) including two who developed acute heart failure (I1 and III31; 7, 25). Autopsy of one individual (III31) revealed biventricular hypertrophy with a healed and an acute infarct of a left papillary muscle. Two other affected individuals had asymptomatic moderately hypertrophic (III24) and mildly dilated (IV6) cardiomyopathy on post-mortem examination . The mutation, located in the second LIM-domain, does not directly affect the zinc-binding cysteine/histidine residues, but is adjacent to a critical histidine residue. The tryptophan residue is highly conserved across species and molecular modeling predicts that the residue is essential for LIM-domain structural stability.
To elucidate the role of FHL1 and the pathomechanism underlying X-SM, we previously generated a knock-in mouse model carrying the same mutation (c.365 G > C, p.W122S) as the X-SM family we reported. Mutant hemizygous male mice (Fhl1p.W122S/y) developed late-onset slowly progressive myopathy, which is similar to human disease (26). Cardiac involvement has been frequently reported in human patients with FHL1 mutations, but the mutant male mice did not show any cardiac abnormality. Skeletal and cardiac muscles from the mutant male mice did not show abnormal aggregation or mislocalization of mutant protein, and molecular analysis showed that the levels of Fhl1 mRNA and protein were comparable between mutant and wild-type (WT) males at the onset of the weakness; therefore, we concluded that the myopathy in our mouse model was due to altered protein function. However, the specific function of Fhl1 that was modified in mutant animals was not identified.
In contrast to the mutant male mice, a small number of Het mutant female mice (Fhl1p.W122S/+) did not manifest overt skeletal or cardiac abnormalities in our prior study (26). However, Het FHL1 p.W122S mutant women are also affected with X-SM, albeit milder than male patients. Therefore, we have studied a larger number of Het (Fhl1p.W122S/+) as well as novel homozygous (Homo; Fhl1p.W122S/p.W122S) mice to more fully characterize Fhl1 mutant female animals.
Results
Body weight and body composition
Because loss of body weight is indicative of muscle atrophy, we measured body mass of female mice at the following four different stages: 1–2, 3–5, 7–10 and >12 months. There were no differences in body weight until >12 months, when Homo mutant had 10.3% lower weight than heterozygotes (P < 0.05; Fig. 1A). However, we did not observe differences in body weight between WT and Homo mutant female mice at >12 months. Body composition analysis by nuclear magnetic resonance imaging (MRI) revealed no differences at any age (Fig. 1B).
Figure 1.
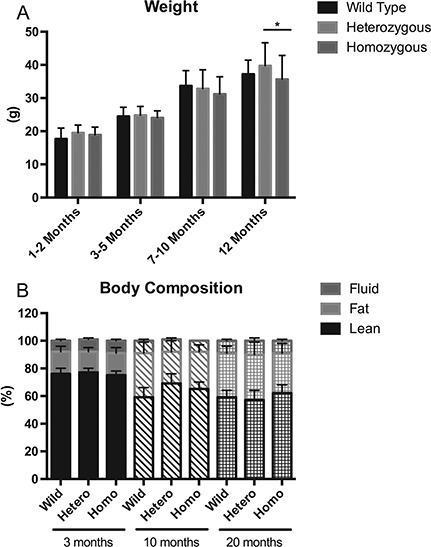
Body weight and body composition. (A) Body weight in WT, Het and Homo female mice (N ≥ 5 in all groups) were measured and analyzed at four different age groups. Body weight is expressed in grams. *P < 0.05. (B) Body composition measured by MRI is expressed as percents lean, fat and fluid components of total body composition.
Skeletal muscle phenotype
Maximal strength was measured by a grip test meter with a bar for forelimbs or a grid for four limbs. With the bar, both Het and Homo female mice showed mild forelimb weakness at ages 3–10 months (3–5 months: WT 120.8 ± 15.3 g, mean ± standard deviation (SD); heterozygotes 103.8 ± 16.5 g [P < 0.01], homozygotes 108.8 ± 18.7 g [P < 0.05]; 7–10 months: WT 111.0 ± 12.7 g, heterozygotes: 94.6 ± 11.8 g [P < 0.01], homozygotes: 94.9 ± 10.1 g [P < 0.01]); however, at age >12 months, weakness was observed only in Homo and not Het mutants (Fig. 2A). In contrast, muscle weakness was not detected with the four-limb grip test using the grid (Fig. 2B). Rotarod was also performed to assess overall exercise capacity and balance, but no difference was observed at any age (Fig. 2C). Serum creatine kinase (CK), measured to assess muscle damage, showed no difference in any age group (Fig. 2D).
Figure 2.
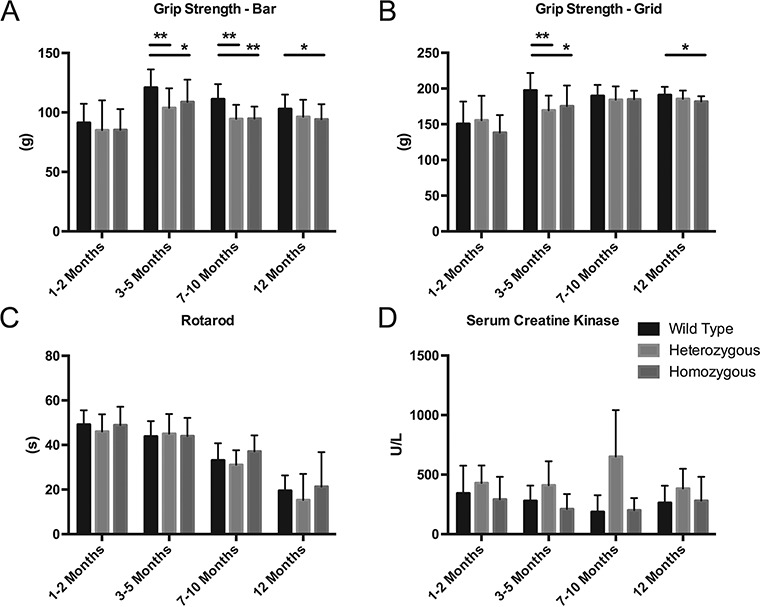
Motor functions and serum CK level in female mice (WT female mice: N = 5; heterozygotes: N = 5; homozygotes: N = 9). Maximal strength was measured using a grip strength meter with either bar (A) or grid (B), expressed in grams. *P < 0.05, **P < 0.01. Overall motor capacity was assessed by time on an accelerating rotarod (C), measured in seconds until animals fell. Serum CK level was measured to evaluate muscle damage (D). All data were stratified into four different age groups: 1–2 months (1 and 2 months), 3–5 months (3, 4 and 5 months), 7–10 months (8 and 10 months) and >12 months (12, 16 and 20 months). Data points represent the mean and bars represent SD.
Nocturnal activity of mice at age 20 months was decreased in Homo mutant female mice (n = 5) compared to WT female mice (n = 4) (WT female mice 59 931 ± 21 733 counts [mean ± SD]; Homo female mice 36 897 ± 18 638 counts, P < 0.01).
Cardiac function
To screen for cardiac phenotypes, we performed transthoracic echocardiography on female mutant mice at ages 10 and 20 months. At 10 months, there was no difference in systolic diameter), diastolic diameter (Dd), fractional shortening (FS), left ventricular mass (LVM) index and relative wall thickness (RWT). However, at 20 months, both Het and Homo mutant female mice showed increased Ds (WT: 2.74 ± 0.22 mm [mean ± SD]; heterozygotes: 3.13 ± 0.11 mm, P < 0.01; homozygotes: 3.08 ± 0.37 mm, P < 0.05) and lower FS (WT: 31.1 ± 4.4% [mean ± SD]; Het: 22.7 ± 2.5%, P < 0.01; homozygous: 22.4 ± 6.9%, P < 0.01) (Table 1). There was no difference in Dd, LVM index and RWT at 20 months. No arrhythmias were observed during the echocardiogram.
Table 1.
Cardiac function
| 10 months | 20 months | |||||
|---|---|---|---|---|---|---|
| WT | Het | Homo | WT | Het | Homo | |
| n = 4 | n = 7 | n = 5 | n = 8 | n = 5 | n = 8 | |
| HR (bpm) | 497.8 ± 2.99 | 504 ± 3.21 | 504.2 ± 1.79 | 502.9 ± 6.15 | 497.4 ± 10.55 | 504.9 ± 2.95 |
| Dd (mm) | 3.96 ± 0.23 | 3.73 ± 0.4 | 3.74 ± 0.13 | 3.98 ± 0.26 | 4.05 ± 0.1 | 3.96 ± 0.22 |
| Ds (mm) | 2.95 ± 0.37 | 2.54 ± 0.47 | 2.59 ± 0.37 | 2.74 ± 0.22 | 3.13 ± 0.11 | 3.08 ± 0.37 |
| FS (%) | 25.6 ± 6.5 | 32.3 ± 6.4 | 30.7 ± 7.4 | 31.1 ± 4.4 | 22.7 ± 2.5 | 22.4 ± 6.9 |
| LVM index | 2.72 ± 0.19 | 2.79 ± 0.29 | 2.64 ± 0.26 | 2.68 ± 0.53 | 2.19 ± 0.22 | 2.5 ± 0.27 |
| RWT | 0.38 ± 0.038 | 0.42 ± 0.086 | 0.42 ± 0.033 | 0.38 ± 0.022 | 0.35 ± 0.016 | 0.38 ± 0.036 |
Echocardiogram was performed at 10 and 20 months in WT (N = 4 at 10 months; 8 at 20 months), Het (N = 7 at 10 months; 5 at 20 months) and Homo female (N = 5 at 10 months; 8 at 20 months) mice. Cardiac function was assessed by Dd (mm), Ds (mm) and FS (%). *P < 0.05, **P < 0.01. Two assessments of cardiac hypertrophy, LVM index (mg/g) and RWT (mm/mm), were also calculated. Data are shown as mean ± SD.
Histological analysis
We performed histological analyses of deltoid and heart muscle of 10- and 20-month-old mice. Although mild fiber-size variation was observed especially in aged mice, we did not observe any histological alterations in deltoid muscle of mutant female mice, at 10 months (Fig. 3A–I) and at 20 months (data not shown). Modified Gomori Trichrome stain and menadinone-nitro blue tetrazolium (NBT) stain did not show any structural abnormalities. In skeletal muscle of Homo female mice compared to WT and Het female mice, anti-Fhl1 immunohistochemical staining revealed decreased signal intensity (Fig. 3J–L). In addition, numerous extraordinary enlarged rectangular nuclei were observed in heart tissue of Het and Homo female mice (Fig. 4A–C), which were also present in human cardiac muscle from the patients. Sections of the ventricles showed mild peri-vascular fibrosis and enlarge myocytes with enlarged rectangular nuclei (Fig. 4D); examples of normal and enlarged rectangular nuclei in human heart tissue are shown in Supplementary Material, Figure A. Ultrastructural analysis of the heart of 20-month-old Homo female mice did not show sarcomeric alterations (Fig. 4E–H).
Figure 3.
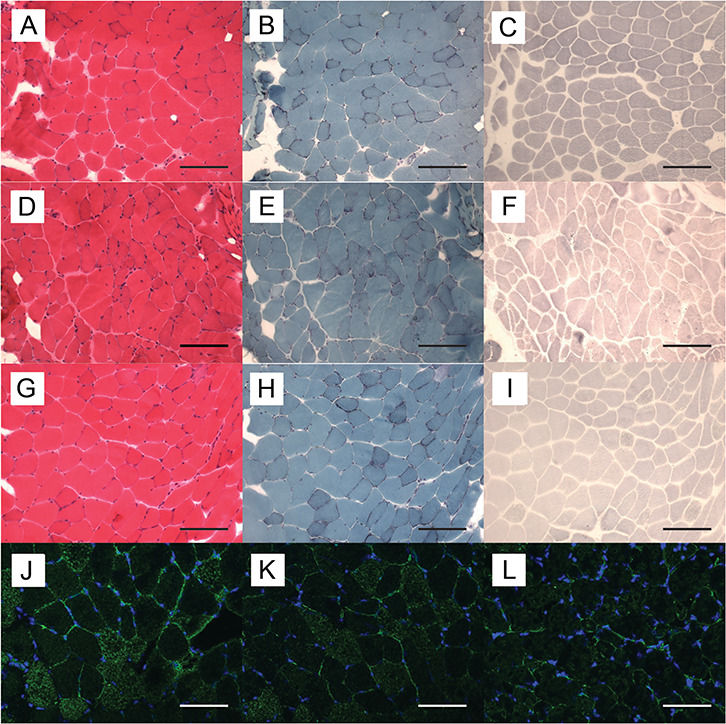
Histological and immunohistochemical findings in deltoid muscle of female mice. Representative serial cryosections of deltoid muscle of a WT (A–C), Het (D–F) and Homo female mice (G–I) at age 10 months. (A, D and G) hematoxylin-eosin stain; (B, E and H) modified Gomori-Trichrome stain; (C, F and I) menadione-NBT stain. Bar = 75 μm. There were no differences in staining intensities or structural abnormalities. Frozen sections were stained with anti-Fhl1 antibody (green) and DAPI (blue). Representative immunohistochemistry of deltoid muscle of a WT (J), a Het and (K) and a Homo female mouse (L) at age 20 months. Bar = 75 μm. Homo female mouse showed decreased Fhl1 signal intensity compared to WT and Het female mice.
Figure 4.
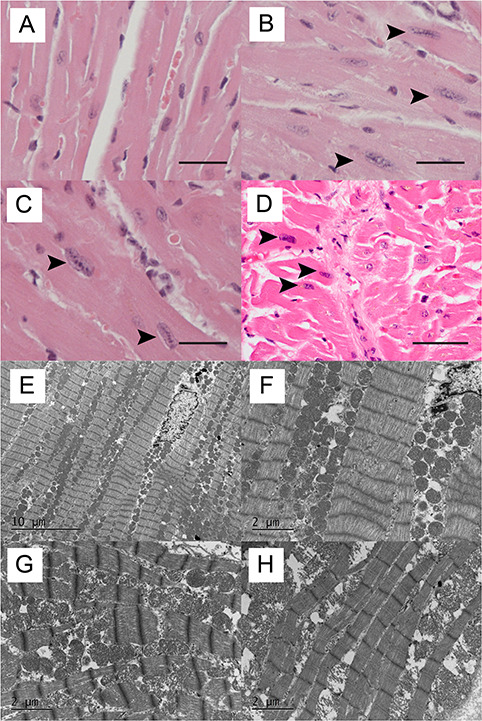
Histological findings in hearts of female mice and human patients. Representative formalin-fixed, paraffin-embedded section of heart of a WT (A), Het (B), Homo mutant female mouse (C) at 20 months and a human X-SM patient (D). Numerous enlarged rectangular nuclei (arrows) were observed in cardiac muscle of Het and Homo female mice as well as a human X-SM patient. Bar = 37.5 μm. Transmission electron micrographs of heart in 20-month-old WT (E, F), Het and (G) Homo female mice (H). No significant structural alterations, including sarcomere abnormalities, are seen.
Proteomic analysis
To investigate the molecular changes in the heart of Homo mutants at 20 months, we performed quantitative proteomic profiling (WT N = 4 versus homozygotes N = 4). Of the 8527 proteins detected by liquid chromatography tandem mass spectrometry (LC-MS/MS), 2178 were identified and quantified by spectra counting. Volcano plot analysis revealed that levels of only 27 proteins were significantly altered in Homo mutant relative to WT mice (Fig. 5A). As depicted in the Venn diagram, 24 were significantly up-regulated and only three were down-regulated (Fig. 5B). Similarly, proteomic analysis with filtering conditions of false-discovery rate (FDR) of 30% (q < 0.3) and an adjusted P < 0.01 identified 42 proteins that are dysregulated in the heart tissue of homozygotes (Fig. 5C). The list included proteins involved in extracellular matrix assembly (PGM5, ASPN, PGS2 and LAMA4) and an integrin-actin network (ACTN4, ARPC1A, CAP1 and MYLK3). Ingenuity Pathway Analysis (IPA) predicted dysregulation of multiple signaling cascades with the most prominent alterations of integrin signaling and actin cytoskeleton signaling in heart tissue of homozygotes (Fig. 5D).
Figure 5.
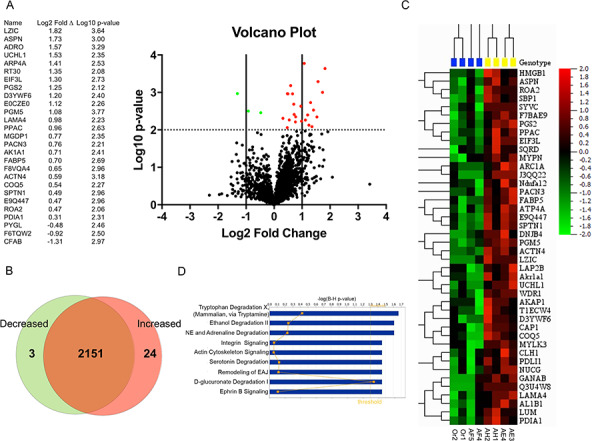
(A) Volcano plot showing the distribution of the fold change (log2) versus the P-values (log10) for 2178 proteins identified in all samples. Significant (P < 0.01) up-regulated proteins are shown as red circles and significant down-regulated proteins are shown as green circles. (B) Venn diagram of identified proteins showing shared proteins between WT and Homo mutant female mice. (C) Heatmap representing proteomic analysis with filtering condition of FDR 30% (q < 0.3) and adjusted P < 0.01. (D) IPA of differentially regulated proteins. Logarithmic transformation of Benjamini–Hochberg adjusted P-values [log (B-H P-value)]; NE = noradrenaline; EAJ = epithelial adherens junctions.
Western blot
Fhl1 protein in deltoid and heart muscles was measured by western blot (Fig. 6A–B). In deltoid, levels of Fhl1 protein were comparable in heterozygotes, Homo mutant and WT female mice at both ages 10 and 20 months. However, in heart muscle, Homo mutant females showed trends toward decreased Fhl1 at both ages relative to age-matched WT females (10 months 56.3 ± 11.4% P = 0.4 and 20 months 59.1 ± 7.2% of WT P = 0.2). Heterozygotes at age 20 months also revealed trends toward lower amounts of Fhl1 protein (75.1 ± 16.6% of WT P = 0.4).
Figure 6.
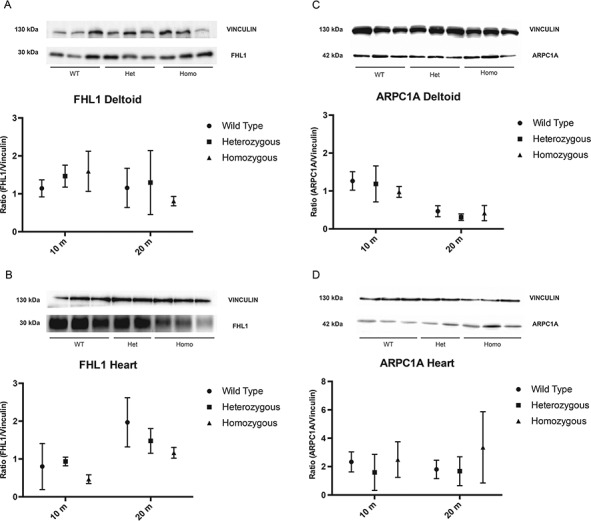
Fhl1 and Arpc1a quantitation by Immunoblot analysis. (A) Representative immunoblot of Fhl1 and vinculin in deltoid muscle of WT, Het and Homo mutant female mice at 20 months of age. The known molecular masses (in kDa) of proteins are indicated in the left margin of the gel. (B) Immunoblot analysis of Fhl1 in heart of 10- and 20-month-old WT, Het and Homo female mice. (C) Representative immunoblot of Arpc1a and vinculin in deltoid muscle of WT, Het and Homo female mice at 20 months of age. The known molecular masses (in kDa) of proteins are indicated in the left margin of the gel. (D) Immunoblot analysis of Arpc1a in heart of 10- and 20-month-old WT, Het and Homo female mice. Data are expressed as ratio (Arpc1a /Vinculin). Data points represent the mean of at least three animals. Bars represent SDs.
We also assessed levels of Arpc1A protein, a subunit of a downstream molecule of the integrin signaling pathway (ISP), in deltoid and heart of 10- and 20-month-old female mice (Fig 6C–D and Supplementary Figure B). In 10-month-old Homo mutant mice, Arpc1a levels in deltoid muscles showed trends toward decreases compared to WT animals (WT: 100 ± 14.4; Homo mutants: 77.14 ± 11.3, P = 0.2) while hearts of 10-month-old mutant and WT mice showed similar Arpc1a protein levels. In 20-month-old mutant mice, levels of Arpc1a were normal in deltoid while Homo mutants revealed trends toward increased levels in heart compared with WT (Homo mutants: 226.1 ± 141.8, WT: 100 ± 37.1; P = 0.2).
We also measured protein levels of FHL1 and ARPC1A, the human homologue of Arpc1A, in autoptic heart and psoas from two previously reported patients with FHL1 mutations [P1 and P2 are IV18 and IV6 in (7,25)], four control subjects matched by age and sex, and two patients diagnosed with amyotrophic lateral sclerosis (ALS), as disease controls (Fig. 7). Both patients had declining respiratory functions and pneumonias, but no symptomatic cardiomyopathy; P1 died at age 49 years-old and P2 died at age 42 years old. As shown in Figure 7, FHL1 and ARPC1A levels were drastically reduced in both heart and psoas muscle (FHL1; 100 ± 22.1 versus 2.7 ± 1.8 [heart], versus 79.2 ± 35.7 [psoas] (P < 0.001) and 100 ± 71.9 versus 1.4 ± 1.3 (P < 0.05), ARPC1A; 100 ± 17.5 versus 6.4 ± 3.0 and 50.8 ± 22.7 (P < 0.001) and 100 ± 10.6 versus 33.5 ± 15.9 (P < 0.05)).
Figure 7.
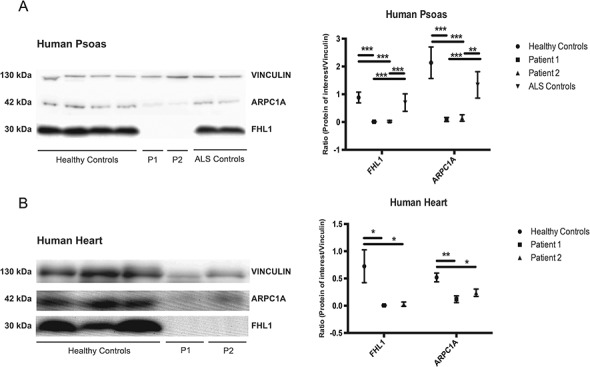
FHL1 and ARPC1A quantitation by immunoblot analysis of human psoas and heart muscles. (A) Representative immunoblot of vinculin, ARPC1A and FHL1 in psoas muscle of Controls, Patient 1 (P1), Patient 2 (P2). (B) Immunoblot analysis of vinculin, ARPC1A and FHL1 in heart of Controls, P1 and P2. The molecular masses (in kDa) of proteins are indicated on the left side of the blots. Ratios of FLH1 and ARPC1A signal normalized to vinculin are shown in graphs. Data are expressed as ratios (FHL1/Vinculin and ARPC1A/Vinculin). Bars represent SDs. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
In humans, FHL1 mutations cause a spectrum of skeletal myopathy, cardiomyopathy, or both (7–19). We previously observed that hemizygous Fhl1 p.W122S male mice develop late-onset myopathy without cardiac dysfunction (26). In this study, we have characterized Homo female mice and observed that, unlike male mutant mice, female mutant mice do not develop skeletal myopathy but rather cardiomyopathy.
In both Het and Homo mutant female mice at age 20 months, echocardiograms revealed systolic dysfunction. Histological examination of heart tissue revealed numerous extraordinarily enlarged rectangular nuclei in female mutant mice, and similar abnormal nuclei were also found in post-mortem heart tissue of a human patient with X-SM. At age 20 months, female mutant mice showed comparable strength and short-term exercise capacity, but decreased overall nocturnal activity, which may be a manifestation of cardiac dysfunction. FHL1 mutations in patients, including individuals with X-SM (25), are often associated with hypertrophic cardiomyopathy and/or arrhythmia (27,28); however, in our female mutant mice, we did not observe cardiac hypertrophy by echocardiogram, cardiac myofiber hypertrophy by histological examination, or arrhythmias.
In contrast to mutant males, mutant female mice did not show differences in the amounts of Fhl1 in deltoid muscle at any age, but Fhl1 revealed trends toward decreases in cardiac muscle of Homo mutant female mice at 10 and 20 months, and in heart of Het female mice at 20 months. The decreased Fhl1 protein in cardiac muscle appeared to correlate with the heart phenotype and decreased nocturnal activity in female mutant mice. However, several aspects of our findings were puzzling: 1) while Fhl1 protein was decreased in hearts of 10-month-old Homo mutant female mice, cardiac dysfunction was not evident until 20 months; 2) reduction of Fhl1 protein was more marked in Homo mutants than heterozygotes, but both mutants showed similar levels of cardiac dysfunction and 3) in our previous study, hemizygous mutant male mice did not develop cardiac dysfunction despite manifesting more pronounced reductions of Fhl1 protein in heart than mutant female mice. These observations indicate that loss of Fhl1 protein is not the primary cause of the cardiomyopathy. In our previous study, we also noted that skeletal muscle weakness preceded reduction of Fhl1 in mutant male mice, therefore, we postulated that mutant Fhl1 causes functional abnormalities that contribute the muscle phenotype more than decreases of Fhl1 protein levels.
Evidence of mutant Fhl1 functional defects were revealed by our proteomics analysis of cardiac muscle that demonstrated upregulation of proteins involved in extracellular matrix assembly and integrin-actin network in Homo mutant female mice. The IPA indicated dysregulation of ISP and actin cytoskeleton pathway. Dysregulation of ISP was further supported by western blot analysis. Arpc1a in mice and ARPC1A in human is a subunit of a downstream molecule of ISP (29). Arpc1a and ARPC1A was selected for this analysis, because those molecules are the furthest downstream of ISP. Western blot analysis of Arpc1a showed trends toward increased levels in heart of Homo mutant mice at 20 months. We postulate that altered levels of Arpc1a/ARPC1A are compensatory responses to ISP dysregulation triggered by mutant Fhl1 protein dysfunction. Western blot analysis of human psoas and cardiac muscle revealed significantly reduced ARPC1A as well as in X-SM patients, confirming dysregulation of ISP in X-SM.
Integrin, a cell adhesion molecule, also mediates bidirectional signaling between extracellular matrix and intracellular space (30). Activation of integrin by extracellular stimulus results in activation of many intracellular pathways, which, in turn, affects multiple cellular functions including: migration, spreading, survival, proliferation and differentiation (31). The ISP involves an incompletely characterized set of intracellular molecules including integrin-linked kinase, Particularly Interesting Cys-His-rich protein (PINCH), and parvin that form complexes that modulate transcription and cell–cell adhesion (32). It is noteworthy that, like FHL1, PINCH is a LIM-domain only adaptor molecule (33), which raises the possibility that FHL1 may be a structural component of the ISP. Molecular and animal model studies have indicated that integrin and its associated complexes play critical roles in cardiac function (34–37). In support of this concept, a mutation in ILK gene was reported in a human patient with dilated cardiomyopathy (38). Thus, our results suggest that the p.W122S Fhl1 mutation disrupts the integrin-actin signaling pathway leading to cardiac dysfunction in our mouse model. The abnormally enlarged rectangular nuclei in mutant mice and a human X-SM patient is a striking feature and potentially a clue to the pathomechanism of X-SM because integrins have been reported to play important roles in internal nuclear structure and nuclear alteration (39). We postulate that dysregulation of ISP may contribute to the aberrant nuclear shape, but further studies are necessary to determine the precise significance of the aberrant nuclei.
In addition to our knock-in mouse model, there are two published animal models of FHL1 related diseases: a Fhl1-null mouse model and a fhl1a-knockdown zebrafish model. Fhl1-null male mice developed more severe muscle weakness and atrophy than our hemizygous male mice but showed no cardiac phenotype (40). Interestingly, Fhl1-null mice showed blunted response to transverse aortic constriction indicating that absence of WT Fhl1 protein may prevent cardiac hypertrophy (41). This observation supports our hypothesis that Fhl1 mutations cause dysfunction, which is responsible for the cardiomyopathy. We did not observe menadione-NBT positive aggregates (reducing bodies) in skeletal muscle of hemizygous male mice (26) or cardiac muscle of Het and Homo female mice, indicating that intracellular Fhl1 aggregates are not the cause of skeletal or cardiac muscle dysfunction in our mouse model. In a morpholino-modified antisense oligonucleotide fhl1a knockdown zebrafish model, both skeletal and cardiac muscle were affected (42). In skeletal muscle, myofibrillar disarray and dysmorphic mitochondria, but no reducing bodies, were observed. Heart manifested arrhythmias as well as reduced contractility. Those phenotypes were rescued by expression of WT human FHL1, but not human FHL1 with pathogenic mutations. The mutant human FHL1 did not show dominant negative effect indicating that the phenotypes in the fhl1a-morphant zebrafish were due to loss of fhl1a function. These findings reinforce our hypothesis that loss-of-function of FHL1 protein can be a cause of FHL1-related disease. It is interesting that the zebrafish model developed arrhthymias, which were not observed in our animal model. The reason for the absence of arrhythmia in our mouse model is unclear, but may be species-dependent.
In our previous study, we did not identify skeletal or cardiac muscle phenotypes in Het female mice; however, in this more detailed study, Het female mice showed cardiac dysfunction comparable to Homo female mice. We excluded skewed X-chromosome inactivation in all Het female mice (Supplementary Material, Figure C). Thus, our positive findings can be attributed to our deeper phenotyping of Het female mice.
An important unresolved issue is why mutant male mice developed only a skeletal muscle phenotype, whereas mutant female mice with the same mutation manifested a cardiac phenotype. Further analyses are needed to address this conundrum, but in human patients with FHL1 mutations, dissociation between skeletal and cardiac phenotype has been seen frequently. In X-SM and RBM, due to FHL1 mutation that typically affect the second LIM-domain, the skeletal muscle phenotype is predominant while cardiac abnormalities are rare (12,25). In contrast, other FHL1 mutations cause isolated hereditary hypertrophic cardiomyopathy without skeletal myopathy (20,21). It is noteworthy that some patients, who carry FHL1 frameshift or splice-site mutations that totally diminish expression of one or more isoforms of FHL1 mRNA, manifest more severe cardiomyopathy than myopathy supporting the hypothesis that severe loss of specific FHL1 function(s) is more detrimental to heart than skeletal muscle (28,43).
In conclusion, Het and Homo female mutant mice harboring the missense mutation p.W122S in Fhl1 gene developed late-onset cardiac dysfunction without skeletal myopathy. Histological analysis revealed numerous extraordinarily enlarged rectangular nuclei in heart of mutant female mice, and similar shaped nuclei were also observed in autopsy heart tissue of an X-SM patient with the same mutation in the FHL1 gene. This is the first animal model of FHL1 related disease, manifesting cardiac dysfunction. The amount of Fhl1 protein did not correlate well with cardiac function and there was no aggregation of Fhl1 protein, which supports the idea that loss of protein function is critical for the cardiac muscle dysfunction. Proteomic analysis suggest that dysregulation of integrin-actin pathway contributes to cardiac dysfunction in our mouse model. Decreased levels of FHL1 and ARPC1A in heart and skeletal muscle of X-SM patients further implicates alterations of the integrin-actin pathway in human FHL1-associated myopathies.
Materials and Methods
Studies in mice
Generation of mutant Fhl1 female mice
Generation of Fhl1 c.365G > C mutation (p.W122S) knock-in mice was previously described (26). Fhl1p.W122S/+ female mice were obtained by mating Het female mice and either WT or mutant male mice. Fhl1p.W122S/p.W122S female mice were obtained by mating mutant male mice and either Het or Homo female mice. Fecundity and viability were similar in Het and Homo female mice and WT female mice (Supplementary table, Table S1).
Animal care
All experiments were performed according to a protocol approved by the Institutional Animal Care and Use Committee at Columbia University Medical Center and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were housed and bred according to international standard conditions, with a 12 h light and 12 h dark cycle. Het and Homo mutant female mice were studied and compared with age-matched WT female mice.
Mice were sacrificed at four different ages (1–2 months, 3–5 months, 7–10 months and 12–20 months) using rapid asphyxiation with carbon dioxide followed by cervical dislocation. Deltoid, triceps and heart muscles were dissected and either frozen in liquid isopentane, pre-cooled to near freezing with dry ice or fixed in 10% neutral buffered formalin and embedded in paraffin using standard procedures. All the experiments were performed in at least five mice per group, unless otherwise specified.
Nuclear MRI
Body composition was analyzed in 3-month-old, 10-month-old and 20-month-old mice. Lean, fat and fluid percentages of body mass were assessed by nuclear MRI using a mini-Spec nuclear magnetic resonance (NMR) analyzer (Bruker, Woodlands, TX).
Grip strength test
Measurement of the maximal muscle strength of forelimbs and combined forelimbs and hind limbs was performed using a grip strength meter following manufacturer instructions (Bioseb BP 32025, 13845 Vitrolles, Cedex, France). Briefly, animals were allowed to grab the grid/bar of the tester and then pulled gradually by the tail until grip was released. The force applied at the moment of release was recorded as the maximal grip strength. Mice were not trained. Grip strength was measured three times for each animal.
Rotarod test
Whole body mobility and coordination were assessed with an accelerating rotarod performance test (Economex Rotarod, Columbus Instruments, Columbus, OH). We measured latency to fall off a 3.5 cm diameter rod rotating initially at 4 rpm constant speed for 15 s, followed by a continuous acceleration of 1 rpm/s for a maximum of 60 s. After 60 s without falling, the test was ended. Four mice were tested simultaneously, each mouse being separated from the others by a 30 cm wide and 60 cm high opaque plexiglass wall. Three consecutive trials for each animal were evaluated after a training period.
Activity test
Spontaneous activity was measured using an activity monitor (Automex II, Columbus Instruments, Columbus, OH). Mice were placed individually in cages, and each cage was put on the activity monitor, isolated from light and sound. Each mouse was acclimated on the activity monitor for two days and nocturnal spontaneous activity was measured for 15 h daily for four days.
Plasma CK activity
Blood (50–100 μl) was collected in heparinized tubes. Plasma was obtained following centrifugation of the blood at 10 000 g for 5 min at room temperature. We used the CK Liquid-ultraviolet (UV) Test NAC (Stanbio, Boerne, TX) and followed the manufacturer’s protocol for determination of serum CK activity. Briefly, CK working reagent was prepared and warmed at 37°C. Samples were added to the reagent and mixed gently. Absorbance was read at 2, 3 and 4 min. The average absorbance per minute multiplied by 3376 to determine results in U/L.
Cardiac function measurement
Echocardiogram was performed in 10- and 20-month-old animals using Vevo 770 imaging system (VisualSonics, Toronto, Canada) equipped with a 30 MHz transducer. Mice were sedated with isoflurane in O2 and placed on a heating pad (37°C) attached to an electrocardiographic monitor. After the mouse chest was shaved, the transducer was gently placed on the hairless region for recordings. The depth of anesthesia was adjusted to maintain a heart rate of approximately 500 bpm. Dd and Ds (mm), FSs (%), LVM (mg) and posterior wall thickness (mm) were measured in 2D mode and M-mode three times for each mouse. LVM index was calculated by dividing LVM by body weight. RWT was calculated from posterior wall thickness and Dd (2× posterior wall thickness/Dd).
Histology
To evaluate structural alterations, 5 μm thick sections of formalin-fixed, paraffin-embedded skeletal and heart muscle were stained with hematoxylin and eosin (H&E) and Masson trichrome following standard protocols. Approximately 8 μm thick frozen sections of the dissected deltoid muscle and heart were cut in a cryostat and stained with H&E, modified Gomori trichrome and menadione-NBT stains following standard protocols. Human samples were flash frozen in isopentane chilled with liquid nitrogen and processed for routine histology including H&E, nicotinamide dehydrogenase tetrazolium reductase, succinate dehydrogenase, modified Gomori trichrome and menadione-NBT stains.
Transmission electron microscopy
Cardiac tissue was fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer. After dehydration, tissue was embedded in Lx-112 (Ladd Research Industries, Inc., Williston, VT). Thin sections were cut on the MT-7000 ultramicrotome at 60 nm thick. The sections were stained with uranyl acetate and lead citrate examined under a JEOL JEM-1200 EXII electron microscope. Images were captured with an ORCA-HR digital camera (Hamamatsu Photonics, Hamamatsu, Japan) recorded with an AMT Image Capture Engine.
Immunofluorescence
Fhl1 localization in skeletal muscle was detected by immunofluorescence stain applied to 8 μm thick frozen sections fixed in ice-cold acetone for 10 min, rinsed and incubated overnight with blocking solution (PBS with 0.1% Triton X-100 and 3% BSA) at 4°C. The slides were subsequently incubated overnight at 4°C with rabbit polyclonal anti-FHL1 antibody, C-terminal region (1:100 dilution; ARP34378 T100, Aviva Systems Biology, San Diego, CA) and then with Alexa Fluor®-conjugated secondary antibody 488 goat anti-rabbit (1:1000 dilution; A11034, Molecular Probes by Life Technologies, Carlsbad, CA) at room temperature for 1 h, with DAPI for nuclear staining. Sections were imaged using an SP5 Leica confocal microscope (Leica Microsystems, Wetzlar, Germany).
Proteomics analysis
Frozen tissue samples were cut in half and homogenized to generate tissue lysates. Cell lysate (10 μg) were digested by trypsin and 2 μg aliquots were analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS). Thermo Orbitrap Fusion Tribrid Mass Spectrometer (ThermoFisher, Waltham, MA) was used for MS/MS analysis.
Western blot
Mouse deltoid and cardiac muscle were dissected and frozen in the liquid phase of isopentane, pre-cooled near its freezing point with dry ice. Human psoas muscles were obtained from human autopsy samples of X-SM patients, control subjects and patients with ALS. Human cardiac muscle of X-SM patients and control subjects were also obtained. Muscle tissue samples were homogenized in lysis buffer containing 50 mm Tris–HCl, 150 mm NaCl, 1 mm ethylenediaminetetraacetic acid (EDTA) and 1% nonyl phenoxypolyethoxylethanol (NP)-40, and supernatants were collected after centrifugation. Whole tissue extracts (25 μg) were electrophoresed in an sodium docecyl sulfate (SDS)-12% polyacrylamide gel electrophoresis (PAGE) system and transferred to Immunoblot polyvinylidene fluoride (PVDF) membranes (Immobilon-P transfer membrane, Millipore, Burlington, MA). Non-specific binding was blocked for 1 h at room temperature in 5% non-fat milk. Incubation was carried out at 4°C overnight with the following primary antibodies: goat anti-FHL1 polyclonal antibody (1:1000 dilution; AHP2070, AbD Serotec, Kidlington, UK); goat anti-ARPC1A polyclonal antibody (1:1000 dilution; SAB2502033, SIGMA-ALDRICH, Saint Louis, MO); mouse anti-vinculin antibody (1:5000 dilution; ab18058, Abcam, Cambridge, MA) and mouse anti β-tubulin antibody (1:1000 dilution; clone AA2, T8328, SIGMA-ALDRICH). Protein–antibody interaction was detected by peroxidase-conjugated mouse antibody, peroxidase-conjugated rabbit antibody or peroxidase-conjugated goat antibody using ECL Prime Western Blotting Detection Reagents® (GE Healthcare, Buckinghamshire, UK). Images of the membranes were taken by G:BOX Chemi IR6® (SYNGENE, Cambridge, UK). Quantification of bands intensities was carried out using NIH imageJ 1.50i software.
Restriction fragment length polymorphism analyses
Total RNA was extracted from frozen muscle using RNeasy Fibrous Tissue Mini Kit® (Qiagen Sciences, Germantown, MD). Complementary DNA (cDNA) was synthesized from the extracted total RNA using SuperScript® VILO™ cDNA Synthesis Kit (Invitrogen, Grand Island, NY). Fhl1 cDNA sequence spanning the mutation site was amplified with a pair of primers (Forward: 5′- GCGCTGATGCCAAGGAGGTGCATTATAAG-3′; Reverse: 5′- TTGCAGTTGCTGCAGGTGAAGCAGTCTTTATTC-3′). The reverse primer was designed to introduce a single-base substitution of c.367 C > A, which creates a restriction site of Taqα I in mutant cDNA only. The amplified nucleotides comprised by 265 base pairs were digested by Taqα I at 6°C for 10 min, and polymerase chain reaction (PCR) product from mutant cDNA was cleaved into two fragments of 222 and 41 base pairs. The digested products were separated on 5% acrylamide gel, incubated in Tris-Borate-EDTA buffer with 0.1% Gel Red™ Nucleic Acid Gel stain (Biotium, Hayward, CA) for 1 h, and was visualized under UV light. Images were analyzed by ImageJ (National Institute of Health, Bethesda, MD), and the intensities of 265 base-pairs (from WT cDNA) and 222 base pairs (from mutant cDNA) bands were measured. Ratios of WT and mutant RNA were calculated.
Data analysis
Data are expressed as mean ± SD of at least five animals per group, unless otherwise specified. Statistical analyses were performed using the unpaired Student’s t-test. P ≤ 0.05 were considered significant. Proteome Discoverer software package (ThermoFisher) was used to search the acquired MS/MS data against a mouse protein database downloaded from the UniProt website (December 2014). Spectra counts (# of MS/MS) were used for quantitative analysis. Global normalization of spectra counts was applied to all samples. Qlucore Omics Explorer software was used to perform statistical analysis. Volcano plot analysis was performed using Prism 7 for Mac (GraphPad Software Inc., San Diego, CA). Power calculations for FS and Ds in 20-month-old female mice were assessed using a two-sample t-test. At alpha level of 0.05, comparison of FS of heterozygotes (N = 5) and homozygotes (N = 8) to wild female mice (N = 8) gave 98.5 and 88.7% power, and comparison of Ds of heterozygotes and homozygotes to wild female mice gave 97.9 and 68.4% power.
Supplementary Material
Acknowledgements
We are grateful to Hasan Orhan Akman, PhD, for his assistance with the acquisition of blood samples from mice; to Beatriz Garcia-Diaz, PhD, for her assistance with the acquisition of tissue samples from mice; and to Saba Tadesse, BS, for her assistance with the preparation of experiments.
Conflict of Interest statement. None declared.
Funding
This work was supported by the Muscular Dystrophy Association (MDA) grant number 115567 and by the Caffarelli Family Study Research (CSFR) Foundation, Inc.
References
- 1. Lee S.M., Tsui S.K., Chan K.K., Garcia-Barcelo M., Waye M.M., Fung K.P., Liew C.C. and Lee C.Y. (1998) Chromosomal mapping, tissue distribution and cDNA sequence of four-and-a-half LIM domain protein 1 (FHL1). Gene, 216, 163–170. [DOI] [PubMed] [Google Scholar]
- 2. Greene W.K., Baker E., Rabbitts T.H. and Kees U.R. (1999) Genomic structure, tissue expression and chromosomal location of the LIM-only gene, SLIM1. Gene, 232, 203–207. [DOI] [PubMed] [Google Scholar]
- 3. Lee S.M., Li H.Y., Ng E.K., Or S.M., Chan K.K., Kotaka M., Chim S.S., Tsui S.K., Waye M.M., Fung K.P. et al. (1999) Characterization of a brain-specific nuclear LIM domain protein (FHL1B) which is an alternatively spliced variant of FHL1. Gene, 237, 253–263. [DOI] [PubMed] [Google Scholar]
- 4. Morgan M.J., Madgwick A.J., Charleston B., Pell J.M. and Loughna P.T. (1995) The developmental regulation of a novel muscle LIM-protein. Biochem. Biophys. Res. Commun., 212, 840–846. [DOI] [PubMed] [Google Scholar]
- 5. Kadrmas J.L. and Beckerle M.C. (2004) The LIM domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell. Biol., 5, 920–931. [DOI] [PubMed] [Google Scholar]
- 6. Shathasivam T., Kislinger T. and Gramolini A.O. (2010) Genes, proteins and complexes: the multifaceted nature of FHL family proteins in diverse tissues. J. Cell. Mol. Med., 14, 2702–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quinzii C.M., Vu T.H., Min K.C., Tanji K., Barral S., Grewal R.P., Kattah A., Camano P., Otaegui D., Kunimatsu T. et al. (2008) X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am. J. Hum. Genet., 82, 208–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen D.H., Raskind W.H., Parson W.W., Sonnen J.A., Vu T., Zheng Y., Matsushita M., Wolff J., Lipe H. and Bird T.D. (2010) A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: phenotypic spectrum and structural study of FHL1 mutations. J. Neurol. Sci., 296, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schessl J., Zou Y., McGrath M.J., Cowling B.S., Maiti B., Chin S.S., Sewry C., Battini R., Hu Y., Cottle D.L. et al. (2008) Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J. Clin. Invest., 118, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shalaby S., Hayashi Y.K., Nonaka I., Noguchi S. and Nishino I. (2009) Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology, 72, 375–376. [DOI] [PubMed] [Google Scholar]
- 11. Schessl J., Taratuto A.L., Sewry C., Battini R., Chin S.S., Maiti B., Dubrovsky A.L., Erro M.G., Espada G., Robertella M. et al. (2009) Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain, 132, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schreckenbach T., Henn W., Kress W., Roos A., Maschke M., Feiden W., Dillmann U., Schulz J.B., Weis J. and Claeys K.G. (2013) Novel FHL1 mutation in a family with reducing body myopathy. Muscle Nerve, 47, 127–134. [DOI] [PubMed] [Google Scholar]
- 13. Windpassinger C., Schoser B., Straub V., Hochmeister S., Noor A., Lohberger B., Farra N., Petek E., Schwarzbraun T., Ofner L. et al. (2008) An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am. J. Hum. Genet., 82, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schoser B., Goebel H.H., Janisch I., Quasthoff S., Rother J., Bergmann M., Muller-Felber W. and Windpassinger C. (2009) Consequences of mutations within the C terminus of the FHL1 gene. Neurology, 73, 543–551. [DOI] [PubMed] [Google Scholar]
- 15. Binder J.S., Weidemann F., Schoser B., Niemann M., Machann W., Beer M., Plank G., Schmidt A., Bisping E., Poparic I. et al. (2012) Spongious hypertrophic cardiomyopathy in patients with mutations in the four-and-a-half LIM domain 1 gene. Circ. Cardiovasc. Genet., 5, 490–502. [DOI] [PubMed] [Google Scholar]
- 16. Shalaby S., Hayashi Y.K., Goto K., Ogawa M., Nonaka I., Noguchi S. and Nishino I. (2008) Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1). Neuromuscul. Disord., 18, 959–961. [DOI] [PubMed] [Google Scholar]
- 17. Gueneau L., Bertrand A.T., Jais J.P., Salih M.A., Stojkovic T., Wehnert M., Hoeltzenbein M., Spuler S., Saitoh S., Verschueren A. et al. (2009) Mutations of the FHL1 gene cause Emery–Dreifuss muscular dystrophy. Am. J. Hum. Genet., 85, 338–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tiffin H.R., Jenkins Z.A., Gray M.J., Cameron-Christie S.R., Eaton J., Aftimos S., Markie D. and Robertson S.P. (2013) Dysregulation of FHL1 spliceforms due to an indel mutation produces an Emery-Dreifuss muscular dystrophy plus phenotype. Neurogenetics, 14, 113–121. [DOI] [PubMed] [Google Scholar]
- 19. Pen A.E., Nyegaard M., Fang M., Jiang H., Christensen R., Molgaard H., Andersen H., Ulhoi B.P., Ostergaard J.R., Vaeth S. et al. (2015) A novel single nucleotide splice site mutation in FHL1 confirms an Emery-Dreifuss plus phenotype with pulmonary artery hypoplasia and facial dysmorphology. Eur. J. Med. Genet., 58, 222–229. [DOI] [PubMed] [Google Scholar]
- 20. Friedrich F.W., Wilding B.R., Reischmann S., Crocini C., Lang P., Charron P., Muller O.J., McGrath M.J., Vollert I., Hansen A. et al. (2012) Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum. Mol. Genet., 21, 3237–3254. [DOI] [PubMed] [Google Scholar]
- 21. Hartmannova H., Kubanek M., Sramko M., Piherova L., Noskova L., Hodanova K., Stranecky V., Pristoupilova A., Sovova J., Marek T. et al. (2013) Isolated X-linked hypertrophic cardiomyopathy caused by a novel mutation of the four-and-a-half LIM domain 1 gene. Circ. Cardiovasc. Genet., 6, 543–551. [DOI] [PubMed] [Google Scholar]
- 22. Cowling B.S., Cottle D.L., Wilding B.R., D'Arcy C.E., Mitchell C.A. and McGrath M.J. (2011) Four and a half LIM protein 1 gene mutations cause four distinct human myopathies: a comprehensive review of the clinical, histological and pathological features. Neuromuscul. Disord., 21, 237–251. [DOI] [PubMed] [Google Scholar]
- 23. Malfatti E., Olive M., Taratuto A.L., Richard P., Brochier G., Bitoun M., Gueneau L., Laforet P., Stojkovic T., Maisonobe T. et al. (2013) Skeletal muscle biopsy analysis in reducing body myopathy and other FHL1-related disorders. J. Neuropathol. Exp. Neurol., 72, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brooke M.H. and Neville H.E. (1972) Reducing body myopathy. Neurology, 22, 829–840. [DOI] [PubMed] [Google Scholar]
- 25. Wilhelmsen K.C., Blake D.M., Lynch T., Mabutas J., De Vera M., Neystat M., Bernstein M., Hirano M., Gilliam T.C., Murphy P.L. et al. (1996) Chromosome 12-linked autosomal dominant scapuloperoneal muscular dystrophy. Ann. Neurol., 39, 507–520. [DOI] [PubMed] [Google Scholar]
- 26. Emmanuele V., Kubota A., Garcia-Diaz B., Garone C., Akman H.O., Sanchez-Gutierrez D., Escudero L.M., Kariya S., Homma S., Tanji K. et al. (2015) Fhl1 W122S causes loss of protein function and late-onset mild myopathy. Hum. Mol. Genet., 24, 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roman I.S., Navarro M., Martinez F., Albert L., Polo L., Guardiola J., Garcia-Molina E., Munoz-Esparza C., Lopez-Ayala J.M., Molina M.S. et al. (2016) Unclassifiable arrhythmic cardiomyopathy associated with Emery–Dreifuss caused by a mutation in FHL1. Clin. Genet., 90, 171–176. [DOI] [PubMed] [Google Scholar]
- 28. Gallego-Delgado M., Gonzalez-Lopez E., Garcia-Guereta L., Ortega-Molina M., Gonzalez-Vioque E., Cobo-Marcos M., Alonso-Pulpon L. and Garcia-Pavia P. (2015) Adverse clinical course and poor prognosis of hypertrophic cardiomyopathy due to mutations in FHL1. Int. J. Cardiol., 191, 194–197. [DOI] [PubMed] [Google Scholar]
- 29. Blystone S.D. (2004) Integrating an integrin: a direct route to actin. Biochim. Biophys. Acta., 1692, 47–54. [DOI] [PubMed] [Google Scholar]
- 30. Hynes R.O. (2002) Integrins: bidirectional allosteric signaling machines. Cell, 110, 673–687. [DOI] [PubMed] [Google Scholar]
- 31. Legate K.R., Wickstrom S.A. and Fassler R. (2009) Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev., 23, 397–418. [DOI] [PubMed] [Google Scholar]
- 32. Legate K.R., Montanez E., Kudlacek O. and Fassler R. (2006) ILK, PINCH and parvin: the tIPP of integrin signalling. Nat. Rev. Mol. Cell Biol., 7, 20–31. [DOI] [PubMed] [Google Scholar]
- 33. Kovalevich J., Tracy B. and Langford D. (2011) PINCH: more than just an adaptor protein in cellular response. J. Cell. Physiol., 226, 940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen H., Huang X.N., Yan W., Chen K., Guo L., Tummalapali L., Dedhar S., St-Arnaud R., Wu C. and Sepulveda J.L. (2005) Role of the integrin-linked kinase/PINCH1/alpha-parvin complex in cardiac myocyte hypertrophy. Lab. Invest., 85, 1342–1356. [DOI] [PubMed] [Google Scholar]
- 35. Bendig G., Grimmler M., Huttner I.G., Wessels G., Dahme T., Just S., Trano N., Katus H.A., Fishman M.C. and Rottbauer W. (2006) Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev., 20, 2361–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. White D.E., Coutu P., Shi Y.F., Tardif J.C., Nattel S., St Arnaud R., Dedhar S. and Muller W.J. (2006) Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes Dev., 20, 2355–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meder B., Huttner I.G., Sedaghat-Hamedani F., Just S., Dahme T., Frese K.S., Vogel B., Kohler D., Kloos W., Rudloff J. et al. (2011) PINCH proteins regulate cardiac contractility by modulating integrin-linked kinase-protein kinase B signaling. Mol. Cell. Biol., 31, 3424–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knoll R., Postel R., Wang J., Kratzner R., Hennecke G., Vacaru A.M., Vakeel P., Schubert C., Murthy K., Rana B.K. et al. (2007) Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation, 116, 515–525. [DOI] [PubMed] [Google Scholar]
- 39. Madrazo E., Conde A.C. and Redondo-Munoz J. (2017) Inside the cell: integrins as new governors of nuclear alterations? Cancers (Basel), 9, E82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Domenighetti A.A., Chu P.H., Wu T., Sheikh F., Gokhin D.S., Guo L.T., Cui Z., Peter A.K., Christodoulou D.C., Parfenov M.G. et al. (2014) Loss of FHL1 induces an age-dependent skeletal muscle myopathy associated with myofibrillar and intermyofibrillar disorganization in mice. Hum. Mol. Genet., 23, 209–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sheikh F., Raskin A., Chu P.H., Lange S., Domenighetti A.A., Zheng M., Liang X., Zhang T., Yajima T., Gu Y. et al. (2008) An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J. Clin. Invest., 118, 3870–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kessler M., Kieltsch A., Kayvanpour E., Katus H.A., Schoser B., Schessl J., Just S. and Rottbauer W. (2018) A zebrafish model for FHL1-opathy reveals loss-of-function effects of human FHL1 mutations. Neuromuscul. Disord., 28, 521–531. [DOI] [PubMed] [Google Scholar]
- 43. Xue Y., Schoser B., Rao A.R., Quadrelli R., Vaglio A., Rupp V., Beichler C., Nelson S.F., Schapacher-Tilp G., Windpassinger C. et al. (2016) Exome sequencing identified a splice site mutation in FHL1 that causes Uruguay syndrome, an X-linked disorder with skeletal muscle hypertrophy and premature cardiac death. Circ. Cardiovasc. Genet., 9, 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.