

ABSTRACT
Introduction
The spike (S) of SARS coronavirus 2 (SARS-CoV-2) engages angiotensin-converting enzyme 2 (ACE2) on a host cell to trigger viral-cell membrane fusion and infection. The extracellular region of ACE2 can be administered as a soluble decoy to compete for binding sites on the receptor-binding domain (RBD) of S, but it has only moderate affinity and efficacy. The RBD, which is targeted by neutralizing antibodies, may also change and adapt through mutation as SARS-CoV-2 becomes endemic, posing challenges for therapeutic and vaccine development.
Areas Covered
Deep mutagenesis is a Big Data approach to characterizing sequence variants. A deep mutational scan of ACE2 expressed on human cells identified mutations that increase S affinity and guided the engineering of a potent and broad soluble receptor decoy. A deep mutational scan of the RBD displayed on the surface of yeast has revealed residues tolerant of mutational changes that may act as a source for drug resistance and antigenic drift.
Expert Opinion
Deep mutagenesis requires a selection of diverse sequence variants; an in vitro evolution experiment that is tracked with next-generation sequencing. The choice of expression system, diversity of the variant library and selection strategy have important consequences for data quality and interpretation.
KEYWORDS: Deep mutational scan, SARS coronavirus 2, ACE2, decoy receptor, mutational landscape
1. INTRODUCTION
Investigations of protein mutations have classically been approached by precision targeting, in which a small number of mutations are deliberately introduced and tested individually. This requires preconceived ideas or hypotheses on which residues and what changes to those residues might be relevant. When the important residues in a protein sequence are unknown, screens and selections can be used instead, in which a library of random mutations is in some way sorted to enrich for a small number of mutants with the intended phenotype. Both experiments are limited in the scale of information they provide. Deep mutagenesis or deep mutational scanning take advantage of next-generation sequencing to bring experimental protein mutagenesis to the realm of Big Data [1]. A screen or selection of a diverse library of variants is tracked by next-generation sequencing to observe how the population’s genetic makeup changes. Mutations with enhanced function are enriched, while deleterious mutations are depleted; the enrichment ratio comparing frequencies in the selected population with the naive library thus acts as a proxy for relative phenotype. Now, the relative effects of thousands of mutations can be assessed simultaneously in a single experiment and a comprehensive mutational landscape can be calculated from experimental data.
Deep mutagenesis has been developed by multiple groups over the past decade [2–13] and has proven especially invaluable to meet three goals: assisting protein engineering, understanding mutational tolerance within a protein sequence, and predicting which mutations might be associated with adverse disease outcomes, especially in the context of cancer or drug resistance. Two recent and prominent studies of SARS coronavirus 2 (SARS-CoV-2) have used deep mutagenesis to address each of these problems [14,15]. This Special Report summarizes the two studies with a focus on experimental details and caveats that will be unfamiliar to those outside the deep mutational scanning community.
2. CONCLUSION
Two deep mutagenesis studies have determined how thousands of mutations within the SARS-CoV-2 spike or the virus’ human receptor affect their binding. The data have proven invaluable for engineering high affinity decoy receptors that are under preclinical development as a COVID-19 therapy, and have revealed the scope of mutational tolerance within the spike that may have bearing on genetic drift as the virus becomes endemic and changes over time. While these two studies focused on expression and binding between the viral spike and its receptor, the underlying selection strategies used in deep mutational scans are increasingly tied to more complex phenotypes, such as selections for structural stability based on protease-sensitivity [16], using competing ligands to engineer specificity into proteins including viral receptors [17–19], and selections based on catalytic or biological activity [20–23]. Undoubtedly there are more questions related to SARS-CoV-2 biology and the biochemistry of its encoded proteins that will be solved using deep mutagenesis as the scientific community rises to this historical moment.
3. EXPERT OPINION
3.1. Engineered, high affinity decoy receptors for SARS-CoV-2
While much attention has been given to isolating monoclonal antibodies with tight affinity for the SARS-CoV-2 spike (S) glycoprotein [24–30], an alternative is to use the entry receptor as a soluble decoy to neutralize infection. S is a class I viral fusion protein that is proteolytically processed into two subunits, S1 and S2, that are non-covalently associated and decorate the coronavirus envelope [31–33]. S recognizes angiotensin-converting enzyme 2 (ACE2) on host cells to initiate attachment and fusion of the viral and plasma membranes [33–38]. Soluble ACE2 (sACE2) blocks receptor-binding sites on S [15,37,39–42] and while escape mutations in S rapidly emerge in tissue culture in the presence of monoclonal antibodies [43], in principle the virus has limited mechanisms to escape a soluble decoy receptor without simultaneously losing affinity for the natural receptor. The decoy receptor might also have a virucidal effect by inducing conformational changes and S1 shedding, such that virus particles are inactivated even if sACE2 dissociates. However, monoclonal antibodies have superior affinity and neutralization efficacy.
To improve the therapeutic potential of decoy receptors, my group used deep mutagenesis to find mutations in ACE2 that enhance affinity [15]. A library of over 2,000 single amino acid substitutions in ACE2 was constructed, focused on diversification of residues at the structurally defined interface with the receptor-binding domain (RBD) of S [44,45] and also within the ACE2 catalytic cleft. The library was expressed in a human cell line, with a c-myc epitope tag fused to the extracellular N-terminus of ACE2 for detection of surface expressed protein. Other than the presence of the epitope tag, ACE2 expressed in this experimental selection system matches native ACE2 in the human body. The culture expressing the ACE2 library was then selected by fluorescence activated cell sorting (FACS) to collect cells expressing ACE2 variants with tight affinity for fluorescently labeled RBD from S of SARS-CoV-2 (Figure 1A).
Figure 1.
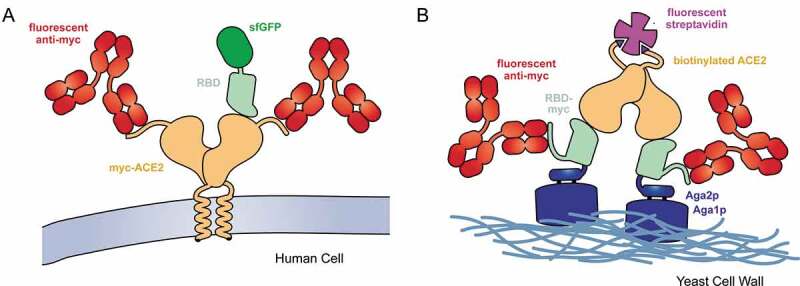
Selection strategies for deep mutational scans of the RBD•ACE2 complex
(A) A library of ACE2 variants was expressed in human cells. Full-length ACE2 (tan) was tagged with a c-myc epitope at its extracellular N-terminus for detection of surface expression with a fluorescent antibody (red). SARS-CoV-2 RBD (pale green) genetically fused with superfolder green fluorescent protein (sfGFP; dark green) was incubated with the cell culture. FACS was used to collect fluorescent cells expressing ACE2 with bound RBD-sfGFP. (B) The isolated RBD of SARS-CoV-2 (pale green) was fused at its N-terminus to Aga2p (blue) and at its C-terminus to a c-myc epitope tag. A saturation mutagenesis library of the RBD was expressed on the yeast surface. Following induction of RBD expression, the yeast were incubated with dimeric, biotinylated sACE2 (tan). Bound ACE2 was detected with fluorescent streptavidin (purple) and surface expressed RBD was detected with a fluorescent antibody (red).
For the artificial selection to be successful, cells must express a single protein variant from a single sequence variant, thereby providing a tight physical link between the phenotype of ACE2 expressed at the plasma membrane and a single sequence within the cell. Getting human cells in culture to acquire and express a single coding variant is no trivial feat, as transfection methods typically introduce many plasmid copies. Different methods to solve this technical challenge have included the use of episomal plasmids that randomly partition to daughter cells during division until progeny harbor a single coding variant over time [4], the use of engineered integration sites in the genome [9,46,47], or the use of viral vectors at low multiplicities-of-infection [48,49]. My group used carrier DNA to sufficiently dilute the ACE2 plasmid library such that each cell typically acquired no more than a single coding variant [11]. An episomal plasmid is used for the library so that extrachromosomal replication within the cell enhances expression of the protein under investigation. (The carrier DNA, itself a modified episomal plasmid, further assists in this process [50].) The disadvantage to this simple solution for linking a single genotype to phenotype is that the coding sequence is so diluted with carrier DNA, most cells in the culture do not express ACE2 and FACS time is wasted on sorting a large number of negative cells. This has important consequences on the data, as time spent sampling negative cells is time not spent sampling cells expressing the protein under investigation, and consequently variants in the library may be under-sampled giving poor data accuracy. Under-sampling becomes exceptionally concerning as the library size increases, and for this reason the library was limited to single amino acid substitutions at just 117 positions in ACE2.
Following FACS selection of the human culture to enrich a cell population with high binding activity for SARS-CoV-2 protein S, RNA transcripts were isolated and Illumina sequenced. An enrichment ratio is calculated for each mutation by dividing its frequency in the sorted cell transcripts by its frequency in the naive plasmid library [51]. Illumina sequencing did not cover the full length of ACE2 and instead the cDNA was sequenced as a series of fragments that together provided full coverage of the diversified regions. One assumes during analysis that there are no additional mutations outside a sequenced fragment, a reasonable assumption when a mutation is found because the library was constructed to have only one amino acid substitution per plasmid. However, the assumption breaks down when no mutations are observed in the sequenced fragment, as one cannot know whether there was a mutation elsewhere outside the sequenced region. As a consequence, the wild type sequence is not directly observed and is instead only estimated. There are strategies using the introduction and analysis of silent mutations that can resolve this issue [52]. Overall, there was close agreement between the mutation enrichment ratios from two independent replicates of the FACS experiments, indicating that the ACE2 library was well sampled and there was high confidence in the data [15].
The enrichment ratios calculated for each variant in the sorted ACE2 library provide a mutational landscape that defines the relative phenotypes of thousands of ACE2 mutations for binding to SARS-CoV-2 S [15]. The data in this experiment are qualitative and it is unclear how a log2 enrichment ratio of, say, −2 or +3 translates to an exact change in a biophysical parameter such as KD. Furthermore, mutations can impact not only binding affinity for the RBD of S but also ACE2 surface expression. To filter out the contribution of mutations to expression, two populations of cells were collected by FACS. In addition to collecting cells that express ACE2 and tightly bind RBD, cells were simultaneously collected in the same experiment that express ACE2 but have weak RBD binding. ACE2 mutants that were not expressed at the cell surface would be depleted from both sorted populations, which was apparent from tracking the depletion of nonsense mutations. In this way, information was collected on how ACE2 mutations impact expression and RBD binding from a single FACS experiment.
The deep mutational scan of ACE2 revealed that mutations can indeed be found to enhance binding toward SARS-CoV-2 RBD (Figure 2), suitable for engineering high affinity soluble decoy receptors [15]. Mutations were found at the binding interface where they enhance specific atomic contacts, and were also found distally in the second shell and beyond where they may impact ACE2 conformation, folding and dynamics. A soluble ACE2 variant that combines three mutations, called sACE22.v2.4, was found to be highly expressed, is a stable monodisperse dimer, binds SARS-CoV-2 S with picomolar affinity and potently neutralizes infection of a susceptible cell line by authentic virus. Its properties rival affinity-matured monoclonal antibodies under commercial development for therapy and prophylaxis. Despite only affinity toward SARS-CoV-2 being considered during the engineering process, sACE22.v2.4 also potently neutralizes authentic SARS-CoV-1, and we speculate that it will have broad activity against betacoronaviruses that use ACE2 as an entry receptor. In unpublished work that has yet to be peer reviewed, we have found sACE22.v2.4 broadly and tightly binds bat coronaviruses that may be a source for future pandemics, supporting the concept of receptor-based decoys as antiviral biologics with exceptional breadth.
Figure 2.
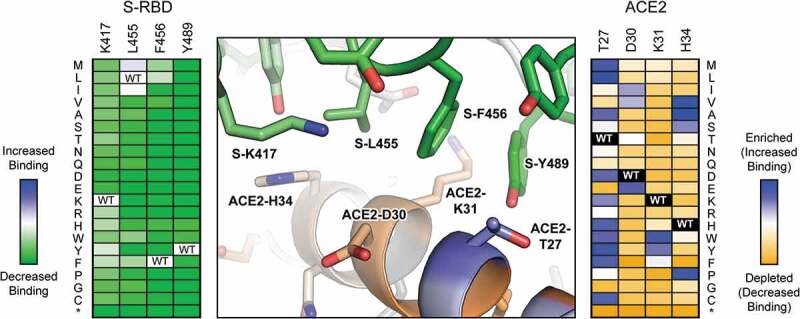
Substitutions at the RBD ACE2 interface have different outcomes on binding
As determined by yeast display, the effects of mutations in the RBD of SARS-COV-2 protein S on receptor affinity are plotted in the heat map at left, with dark green indicating the mutations are deleterious and pale colors indicating the mutations are neutral. The effects of mutations in human cell-expressed ACE2 on binding to soluble RBD are plotted in the heat map at right, with depleted mutations in orange, neutral mutations in white and enriched mutations in blue. Positional scores are mapped to the atomic structure of RBD-bound ACE2 (PDB 6M17) at center. Conserved ACE2 residues for RBD binding are orange, while ACE2 residues that are hot spots for mutations with increased affinity are blue. RBD residues conserved for ACE2 binding are green. Most RBD mutations in this region of the interface are deleterious, whereas numerous mutations were found in ACE2 that increased affinity.
3.2. Sequence constraints on the RBD of SARS-CoV-2 S for binding ACE2
In Starr et al, deep mutagenesis was applied to the SARS-CoV-2 spike to assess mutational tolerance for expression and ACE2 interactions [14]. Instead of investigating the entire trimeric S protein expressed on a cellular or viral membrane, the isolated RBD was fused to the yeast mating factor Aga2p and displayed on the yeast surface [53] (Figure 1B). This is an artificial display platform that removes the RBD from its native context. N-Glycosylation in yeast is also of high-mannose type and lacks the complex, terminally sialylated glycans produced by human cells [54], which can be important when binding interactions are glycan-dependent as is seen for some antibodies targeting viral spikes [55]. However, this display platform harnesses yeast genetics to confer tremendous advantages for in vitro selection and evolution. Using yeast display, large diverse libraries can be readily sorted by FACS to provide high-quality data. Separate selections were completed at a range of different sACE2 concentrations to simulate a titration experiment, from which the data could be converted to quantitative changes in apparent KD on the yeast surface (Figure 2). As a surrogate for how RBD mutations may impact expression of the viral spike, the effects of mutations on RBD surface display were also assessed in a standalone FACS selection. Quality control pathways for protein secretion in yeast can be forgiving of misfolded protein sequences [16] and there are residues of the RBD that would ordinarily be buried in the context of the full S protein; it therefore remains to be seen how closely the yeast display data will correlate with equivalent experiments in more physiologically relevant expression systems. Nonetheless, the predicted effects by yeast display of some mutations were validated using full length S expressed in human cells and packaged in pseudovirus [14].
The library encoding nearly 4,000 single amino acid substitutions in the SARS-CoV-2 RBD was PacBio sequenced, providing long reads that match untranslated nucleotide barcodes to a specific protein variant. Following FACS-based selection, only the barcodes are read to determine how favorable sequence variants are enriched or deleterious sequence variants are depleted. This resolves issues with Illumina sequencing failing to cover the full cDNA length, and because multiple barcodes are associated with any given protein variant, there are additional internal checks for data quality and consistency.
Despite the limitations of a yeast display platform, the deep mutational scan of the isolated RBD provides a high quality and useful data set from which several important conclusions were drawn. First, the ACE2 binding surface of SARS-CoV-2 RBD tolerates surprisingly high sequence diversity, even though it is a critical site for function [14]. High diversity is also seen in the ACE2-binding sites of S proteins from SARS-related bat coronaviruses, but this matches corresponding diversity in ACE2 from ecologically diverse bat species [56] and does not necessarily mean that the RBD tolerates mutations for binding ACE2 from a single species. The deep mutational scan addresses this uncertainty and is further supported by evidence showing that diverse RBD sequences from bat coronaviruses are all competent for binding human ACE2 with varying affinities [38].
Second, mutations were found in the RBD that enhance binding to ACE2, yet there does not appear to be positive selective pressure for these variants in the human population [14]. SARS-CoV-2 affinity for ACE2 is therefore ‘good enough,’ with no additional fitness benefit for higher affinity. It is worth noting that classical SARS-CoV-1 is also a highly infectious and virulent pathogen, despite having weaker ACE2 affinity [36,57]. The rapid spread of SARS-CoV-2 probably has more to do with asymptomatic and presymptomatic transmission than enhanced receptor binding.
Third, mutations were found within the epitopes for monoclonal antibodies but maintain high ACE2 binding, and it is likely that SARS-CoV-2 can easily mutate to escape neutralization without losing infectivity [14]. This agrees with selection experiments of pseudovirus expressing SARS-CoV-2 S variants, in which escape mutants in the viral spike rapidly emerge to neutralizing antibodies in a single passage [43]. This has profound implications for antibody therapy, where the standard has become combinations of non-competing monoclonals in a cocktail to prevent rapid resistance. It is currently unknown whether an engineered soluble decoy receptor, such as sACE22.v2.4, will similarly be susceptible to the emergence of viral spike variants that can discriminate between the engineered decoy and the native receptor. We hypothesize that engineered decoys will be broadly active against SARS-CoV-2 variants and this remains an active area of investigation.
Article highlights
In deep mutagenesis, the relative phenotypes of thousands of mutations in a protein sequence are determined in a single experiment.
The experimental mutational landscape of ACE2 for binding the RBD of SARS-CoV-2 provides a blueprint for engineering high affinity decoy receptors.
A deep mutational scan of the SARS-CoV-2 RBD reveals considerable opportunity for genetic drift without loss of receptor affinity.
Different expression systems for selecting ACE2 or spike variants have inherent advantages and disadvantages.
There are opportunities for deep mutagenesis to provide biochemical insights on other SARS-CoV-2 proteins.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••)
- 1.Fowler DM, Fields S.. Deep mutational scanning: a new style of protein science. Nat Methods. 2014;11(8):801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fowler DM, Araya CL, Fleishman SJ, et al. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7(9):741–746. DOI: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Araya CL, Fowler DM, Chen W, et al. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proceedings of the National Academy of Sciences. 2012;109(42):16858–16863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forsyth CM, Juan V, Akamatsu Y, et al. Deep mutational scanning of an antibody against epidermal growth factor receptor using mammalian cell display and massively parallel pyrosequencing. MAbs. 2013;5(4):523–532. DOI: 10.4161/mabs.24979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu Z, Juan V, Ivanov A, et al. Affinity and cross-reactivity engineering of CTLA4-Ig to modulate T cell costimulation. The Journal of Immunology. 2012;189(9):4470–4477. DOI: 10.4049/jimmunol.1201813. [DOI] [PubMed] [Google Scholar]
- 6.Harris DT, Singh NK, Cai Q, et al. An engineered switch in T cell receptor specificity leads to an unusual but functional binding geometry. Structure. 2016;24(7):1142–1154. DOI: 10.1016/j.str.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Procko E, Hedman R, Hamilton K, et al. Computational design of a protein-based enzyme inhibitor. Journal of Molecular Biology. 2013;425(18):3563–3575. DOI: 10.1016/j.jmb.2013.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haddox HK, Dingens AS, Bloom JD. Experimental estimation of the effects of all amino-acid mutations to HIV’s envelope protein on viral replication in cell culture. PLoS Pathog. 2016;12(12):e1006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matreyek KA, Stephany JJ, Fowler DM. A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res. 2017;45(11):e102–e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matreyek KA, Starita LM, Stephany JJ, et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet. 2018;50(6):874–882. DOI: 10.1038/s41588-018-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heredia JD, Park J, Brubaker RJ, et al. Mapping interaction sites on human chemokine receptors by deep mutational scanning. J Immunol. 2018;200(11):ji1800343–3839. DOI: 10.4049/jimmunol.1800343. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study established a simple and effective method for linking phenotype to a single genotype in transfected human cells. This technical accomplishment is necessary for selection and deep mutational scanning in human cells.
- 12.Olson CA, Wu NC, Sun R. A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain. Current Biology. 2014;24(22):2643–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu NC, Young AP, Al-Mawsawi LQ, et al. High-throughput profiling of influenza A virus hemagglutinin gene at single-nucleotide resolution. Sci Rep. 2014;4:4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starr TN, Greaney AJ, Hilton SK, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–1310.e20. DOI: 10.1016/j.cell.2020.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The isolated RBD of SARS-CoV-2 was displayed on yeast and deep mutationally scanned to understand the mutational landscape for yeast surface expression (a surrogate for folding) and ACE2 binding. The data reveal substantial sequence diversity is tolerated on the RBD surface, including within the ACE2 binding site.
- 15.Chan KK, Dorosky D, Sharma P, et al. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science. 2020;4:eabc0870. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Chan et al. deep mutationally scanned ACE2 expressed on a human cell membrane to identify substitutions that enhance binding to S of SARS-CoV-2. This guided the engineering of high affinity and potently neutralizing decoy receptors.
- 16.Rocklin GJ, Chidyausiku TM, Goreshnik I, et al. Global analysis of protein folding using massively parallel design, synthesis, and testing. Science. 2017;357(6347):168–175. DOI: 10.1126/science.aan0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Procko E, Berguig GY, Shen BW, et al. A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell. 2014;157(7):1644–1656. DOI: 10.1016/j.cell.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berger S, Procko E, Margineantu D, et al. Computationally designed high specificity inhibitors delineate the roles of BCL2 family proteins in cancer. Elife. 2016;5:1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J, Gill KS, Aghajani AA, et al. Engineered receptors for human cytomegalovirus that are orthogonal to normal human biology. PLoS Pathog. 2020;16(6):e1008647. DOI: 10.1371/journal.ppat.1008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wrenbeck EE, Azouz LR, Whitehead TA. Single-mutation fitness landscapes for an enzyme on multiple substrates reveal specificity is globally encoded. Nat Commun. 2017;8(1):15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Firnberg E, Labonte JW, Gray JJ, et al. A comprehensive, high-resolution map of a gene’s fitness landscape. Molecular Biology and Evolution. 2014;31(6):1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klesmith JR, Bacik J-P, Michalczyk R, et al. Comprehensive sequence-flux mapping of a levoglucosan utilization pathway in E. coli. ACS Synthetic Biology. 2015;4(11):1235–1243. [DOI] [PubMed] [Google Scholar]
- 23.McShan AC, Devlin CA, Overall SA, et al. Molecular determinants of chaperone interactions on MHC-I for folding and antigen repertoire selection. Proceedings of the National Academy of Sciences. 2019;116(51):25602–25613. DOI: 10.1073/pnas.1915562116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinto D, Park Y-J, Beltramello M, et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583(7815):290–295. DOI: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- 25.Hansen J, Baum A, Pascal KE, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020. eabd0827 10.1126/science.abd0827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brouwer PJM, Caniels TG, van der Straten K, et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science. 2020;369(6504):643–650. DOI: 10.1126/science.abc5902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wec AZ, Wrapp D, Herbert AS, et al. Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science. 2020;369(6504):731–736. DOI: 10.1126/science.abc7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C, Li W, Drabek D, et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun. 2020;11(1):2251. DOI: 10.1038/s41467-020-16256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Y, Wang F, Shen C, et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science. 2020;368(6496):1274. DOI: 10.1126/science.abc2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers TF, Zhao F, Huang D, et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020. eabc7520 10.1126/science.abc7520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walls AC, Park Y-J, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tortorici MA, Veesler D. Structural insights into coronavirus entry. Adv Virus Res. 2019;105:93–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann M, Kleine-Weber H, Schroeder S, et al.. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. DOI: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. DOI: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wan Y, Shang J, Graham R, et al. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS . Coronavirus. Journal of Virology. 2020;94(7). DOI: 10.1128/JVI.00127-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. 10.1126/science.abb2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W, Moore MJ, Vasilieva N, et al., Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 426(6965): 450–454. 2003. 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study reports the original discovery of ACE2 as the entry receptor for classical SARS-CoV-1.
- 38.Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;11:1860. [DOI] [PMC free article] [PubMed] [Google Scholar]; * ACE2 is shown to be a shared entry receptor for a clade of SARS-associated betacoronaviruses, including diverse strains from bats and human virus SARS-CoV-2.
- 39.Hofmann H, Geier M, Marzi A, et al. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochemical and Biophysical Research Communications. 2004;319(4):1216–1221. DOI: 10.1016/j.bbrc.2004.05.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lei C, Qian K, Li T, et al. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat Commun. 2020;11(1):2070. DOI: 10.1038/s41467-020-16048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monteil V, Kwon H, Prado P, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 181(4): 905–913.e7. 2020. 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Soluble ACE2 effectively neutralizes SARS-CoV-2 in vitro, inspiring the repurposing of sACE2 as an investigational drug for respiratory distress to treat COVID-19 patients.
- 42.Iwanaga N, Cooper L, Rong L, et al. Novel ACE2-IgG1 fusions with improved in vitro and in vivo activity against SARS-CoV2. bioRxiv. 2020;(2020(6):15.152157. [Google Scholar]
- 43.Baum A, Fulton BO, Wloga E, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020; eabd0831. DOI: 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The SARS-CoV-2 spike is able to rapidly acquire mutations to escape neutralizing monoclonal antibodies in tissue culture, necessitating the combination of multiple non-competing monoclonals in a cocktail to prevent resistance.
- 44.Yan R, Zhang Y, Li Y, et al. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. 10.1126/science.abb2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li F. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. [DOI] [PubMed] [Google Scholar]
- 46.Matreyek KA, Stephany JJ, Chiasson MA, et al. An improved platform for functional assessment of large protein libraries in mammalian cells. Nucleic Acids Res. 2020;48(1):e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruun T-H, Grassmann V, Zimmer B, et al. Mammalian cell surface display for monoclonal antibody-based FACS selection of viral envelope proteins. MAbs. 2017;9(7):1052–1064. DOI: 10.1080/19420862.2017.1364824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steichen JM, Kulp DW, Tokatlian T, et al. HIV vaccine design to target germline precursors of glycan-dependent broadly neutralizing antibodies. Immunity. 2016;45(3):483–496. DOI: 10.1016/j.immuni.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kulp DW, Steichen JM, Pauthner M, et al. Structure-based design of native-like HIV-1 envelope trimers to silence non-neutralizing epitopes and eliminate CD4 binding. Nat Commun. 2017;8(1):1655. DOI: 10.1038/s41467-017-01549-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park J, Selvam B, Sanematsu K, et al. Structural architecture of a dimeric class C GPCR based on co-trafficking of sweet taste receptor subunits. J Biol Chem. 2019;294(13):4759–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fowler DM, Araya CL, Gerard W, et al. Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics. 2011;27(24):3430–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jardine JG, Kulp DW, Havenar-Daughton C, et al. HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science. 2016;351(6280):1458–1463. DOI: 10.1126/science.aad9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chao G, Lau WL, Hackel BJ, et al. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1(2):755–768. [DOI] [PubMed] [Google Scholar]
- 54.Wildt S, Gerngross TU. The humanization of N-glycosylation pathways in yeast. Nat Rev Microbiol. 2005;3(2):119–128. [DOI] [PubMed] [Google Scholar]
- 55.Cohen YZ, Lavine CL, Miller CA, et al. Glycan-dependent neutralizing antibodies are frequently elicited in individuals chronically infected with HIV-1 Clade B or C. AIDS Research and Human Retroviruses. 2015;31(11):1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frank HK, Enard D, Boyd SD. Exceptional diversity and selection pressure on SARS-CoV and SARS-CoV-2 host receptor in bats compared to other mammals. bioRxiv. 2020;5:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;382:1199. [DOI] [PMC free article] [PubMed] [Google Scholar]