

Key Point
TP53 mutations were associated with poor prognosis in NCI high-risk patients but not in SR patients in pediatric B-ALL.
Abstract
Recent genetic studies using high-throughput sequencing have disclosed genetic alterations in B-cell precursor acute lymphoblastic leukemia (B-ALL). However, their effects on clinical outcomes have not been fully investigated. To address this, we comprehensively examined genetic alterations and their prognostic impact in a large series of pediatric B-ALL cases. We performed targeted capture sequencing in a total of 1003 pediatric patients with B-ALL from 2 Japanese cohorts. Transcriptome sequencing (n = 116) and/or array-based gene expression analysis (n = 120) were also performed in 203 (84%) of 243 patients who were not categorized into any disease subgroup by panel sequencing or routine reverse transcription polymerase chain reaction analysis for major fusions in B-ALL. Our panel sequencing identified novel recurrent mutations in 2 genes (CCND3 and CIC), and both had positive correlations with ETV6-RUNX1 and hypodiploid ALL, respectively. In addition, positive correlations were also newly reported between TCF3-PBX1 ALL with PHF6 mutations. In multivariate Cox proportional hazards regression models for overall survival, TP53 mutation/deletion, hypodiploid, and MEF2D fusions were selected in both cohorts. For TP53 mutations, the negative effect on overall survival was confirmed in an independent external cohort (n = 466). TP53 mutation was frequently found in IGH-DUX4 (5 of 57 [9%]) ALL, with 4 cases having 17p LOH and negatively affecting overall survival therein, whereas TP53 mutation was not associated with poor outcomes among NCI (National Cancer Institute) standard risk (SR) patients. A conventional treatment approach might be enough, and further treatment intensification might not be necessary, for patients with TP53 mutations if they are categorized into NCI SR.
Visual Abstract

Introduction
B-cell precursor acute lymphoblastic leukemia (B-ALL) accounts for 85% of pediatric ALL and has been categorized into several molecular subgroups according to heterogeneous genomic alterations such as hyperploidy, hypoploidy, ETV6-RUNX1, TCF3-PBX1, BCR-ABL1, KMT2A-rearrangements (KMT2Ar), and Philadelphia (Ph)-like profile.1,2 Genetic studies using high-throughput sequencing have disclosed landscapes of gene alterations in major subgroups of B-ALL.3-7 However, the impact of gene mutations and other genetic lesions on clinical outcomes have been investigated in a relatively small number of patients for a limited number of driver mutations and copy number lesions.8,9 Particularly, genetic profiles and their clinical relevance have been less intensively investigated in rare subtypes of B-ALL, including TCF3-PBX1–positive ALL, ZNF384, and MEF2D fusion-positive ALL, which account for ∼7%, 4%, and 2% of pediatric B-ALL, respectively.10-13 Moreover, different clinical backgrounds of patients frequently complicate the accurate determination of the effects of genetic alterations; for example, generally predicting a poor clinical outcome, IKZF1 deletions, and Ph-like signatures may not be associated with a shorter overall survival among patients who belong to the National Cancer Institute (NCI) standard risk (SR) category.9,14 To elucidate precise impacts of genetic alterations, it is instrumental to investigate a large cohort of patients for a comprehensive registry of genetic lesions in B-ALL. In the current study, we elucidated the landscape of driver mutations among 1003 pediatric patients with B-ALL and investigated the significance of driver alterations with regard to the clinical outcomes.
Patients and methods
Patients and risk stratification in treatment protocols
A total of 1003 patients with B-ALL diagnosed between April 2002 and January 2013 were included in this study; 568 and 435 patients were treated according to Japan Association Childhood Leukemia Study Group (JACLS) ALL-02 or Tokyo Children’s Cancer Study Group (TCCSG) L04-16 prospective clinical trials, respectively (supplemental Tables 1 and 2; supplemental Figure 1).15,16 Compared with the clinical background of the entire cohort, sequenced samples, where adequate materials were available for sequencing, had higher white blood cell (WBC) numbers at diagnosis and enriched in the NCI high-risk category. The diagnosis of B-ALL was based on cell morphology and immunophenotype in bone marrow aspirates or peripheral blood at diagnosis. On the basis of conventional karyotyping and reverse transcription polymerase chain reaction assays for known gene fusions routinely performed for all patients at the beginning of enrollment in the therapy protocol, 184 hyperdiploid, 6 hypodiploid, 236 ETV6-RUNX1, 95 TCF3-PBX1, and 34 KMT2Ar (17 KMT2A-AF4, 10 KMT2A-ENL, and 7 KMT2A-AF9) cases had been diagnosed. BCR-ABL1–positive and infant cases had been excluded from the analysis. Clinical characteristics of the patients are summarized in Table 1. For risk stratification in treatment protocols, patients were provisionally stratified into 3 risk groups based on the patient’s age, initial WBC count, and major gene fusions and aneuploidy, including KMT2Ar, TCF3-PBX1, and hypodiploidy. These patients were then reclassified into 4 final risk groups according to the treatment response to initial 7-day prednisolone monotherapy.16
Table 1.
Characteristics of the patients and univariate associations with overall survival
| n (%) | Hazard ratio for event (95% CI) | P | |
|---|---|---|---|
| Sample | |||
| Sample size, cases | 1003 | ||
| Follow up, median (range), y | 5.8 (0-18) | ||
| Events | |||
| Induction failure | 19 (1.9) | ||
| Relapse | 149 (15) | ||
| Death from any cause | 95 (9.5) | ||
| Treatment groups* | |||
| SR | 303 (30) | ||
| Intermediate risk | 451 (45) | ||
| High risk | 120 (12) | ||
| Extended high risk | 65 (6.5) | ||
| Discontinuation of the protocol treatment | 26 (2.6) | ||
| Age, y | <.001 | ||
| Median (range) | 5 (1-18.5) | ||
| 1-10 | 813 (81) | 1 | |
| >10 | 190 (19) | 2.6 (1.7-4.0) | |
| Sex | .3 | ||
| Female | 466 (46) | 1 | |
| Male | 537 (54) | 1.1 (0.8-1.7) | |
| Leukocyte count in peripheral blood* | <.001 | ||
| <50 × 109/L | 819 (82) | 1 | |
| ≥50 × 109/L | 183 (18) | 2.7 (1.8-4.1) | |
| NCI risk group | <.001 | ||
| SR | 671 (67) | 1 | |
| High risk | 332 (33) | 2.8 (1.9-4.2) | |
Information about final treatment risk and leukocyte count at diagnosis is missing in 38 patients and 1 patient, respectively.
Informed consent was obtained from the patients’ guardians according to the Declaration of Helsinki. This study was approved by the institutional review boards at National Hospital Organization Nagoya Medical Center and Kyoto University.
Targeted gene panel sequencing
Genomic DNA was obtained from peripheral blood or bone marrow at diagnosis and subjected to hybridization-based capture by using a SureSelect custom kit (Agilent Technologies), followed by high-throughput sequencing on an Illumina HiSeq 2500 platform. The targeted gene panel included 110 known or putative driver genes in B-ALL and/or target genes of activation-induced cytidine deaminase (supplemental Table 3).17 In addition, 1484 single-nucleotide polymorphism probes were designed across the entire genome to detect genomic copy numbers, together with an additional 662 probes, which cover the IGH enhancer locus to capture IGH-involving rearrangements. In sequencing-based copy number analysis, lesions affecting >95% of the entire chromosome were defined as whole chromosome changes. GenomonPipeline18 was used for mutation calling with stringent criteria, in which all missense single-nucleotide variants (SNVs) with a variant allele frequency of 0.4 to 0.6 were eliminated as germline variants; the exception was the pathogenic SNVs registered in the Catalogue of Somatic Mutations in Cancer as somatic mutations in hematopoietic and lymphoid neoplasms. Methods of mutation calling, curation of the oncogenic variants, and detection of structural variations are detailed in the supplemental Appendix. Copy number alterations were evaluated by using our in-house pipeline “CNACS.”19 CNACS is a UNIX-based program for sequencing-based copy-number analysis, which is available at (https://github.com/papaemmelab/toil_cnacs).
Transcriptome sequencing and expression array
Gene expression profile was analyzed for 203 patients using Microarray GeneChip Human Genome U133 Plus 2.0 (n = 120) and/or RNA-sequencing (n = 116). RNA samples were extracted from diagnostic specimens using RNeasy Mini Kit. For microarray analysis, GeneChip Operating Software 1.2 (Affymetrix) and GeneSpring GX 13.1 software (Agilent Technologies) were used. Construction of an RNA-sequencing library was performed by using an NEBNext Ultra RNA Library Prep Kit for Illumina according to the manufacturer’s instructions. Mapped reads were counted for each gene using our in-house GenomonExpression pipeline. Gene expression levels were normalized by using the R package “DESeq2” and were subjected to clustering analysis to detect Ph-like and ETV6-RUNX1–like ALL. Fusion transcripts were detected by Genomon version 2.3.4. Methods of detection of Ph-like and ETV6-RUNX1–like ALL and identification of fusion transcripts are detailed in the supplemental Appendix.
Statistical analysis
Associations of genetic alterations with B-ALL subgroups and clinical factors were analyzed by using the Fisher’s exact test or the Wilcoxon rank sum test. Multiple testing was adjusted by calculating q values using Benjamini-Hochberg’s method, in which q < 0.1 was considered statistically significant. Event-free and overall survivals were estimated by using the Kaplan-Meier method. An event was defined as either a failure to achieve remission, a relapse after remission, or any cause of death. Univariate analyses were performed by using the Cox proportional hazards regression model, in which P < .05 was considered statistically significant. Using these variables with NCI risk criteria and adjustment to treatment intensity, multivariate analyses were conducted based on the Cox proportional hazards model, and optimal combination of covariates was selected with the least absolute shrinkage and selection operator using R package “glmnet” for overall survival. All analyses were performed by using R software (www.r-project.org).
Results
Panel sequencing of 1003 B-ALL SNVs
In total, we detected 1300 SNVs (median, 1 per patient; range, 0-8) and 398 indels (median, 0 per patient; range, 0-5), which frequently affected transcription factors (ETV6, PAX5, and IKZF1), epigenetic regulators (ATF7IP, SETD2, KMT2D, and CREBBP), cell cycle regulators (CDKN2A/B, BTG1, and RB1), RAS pathway genes (KRAS, NRAS, and PTPN11), and FLT3 (Figure 1A; supplemental Figure 2; supplemental Tables 4 and 5). Novel recurrent mutations were identified in 2 genes, CCND3 (n = 13) and CIC (n = 5) (Figure 1B). CCND3 were mutated in 13 cases, 8 of which were positive for ETV6-RUNX1. Although recurrent CCND3 mutations have recently been reported in KMT2Ar AML as well as non-Hodgkin lymphomas, none of the cases with KMT2Ar ALL carried these mutations.20,21 As is the case with mutations in lymphomas and KMT2Ar AML, CCND3 mutations exhibited a prominent mutational hotspot at R271. Except for 1 missense mutation, all CCND3 mutations caused a truncating protein lacking the PEST domain, which is known to be involved in casein-mediated degradation of this cyclin.22 Thus, the consequence of these mutations would be an elevated expression of this cyclin. CIC encodes a transcriptional repressor containing a Sox-like high-mobility group box, which was mutated in 4 of 9 patients with hypodiploid ALL (Fisher’s exact test, P = 1.5 × 10−8), showing multiple discrete mutations in 3 cases (converging mutations); this finding suggests strong positive selection of CIC-mutated cells, as seen in lower grade gliomas.23
Figure 1.
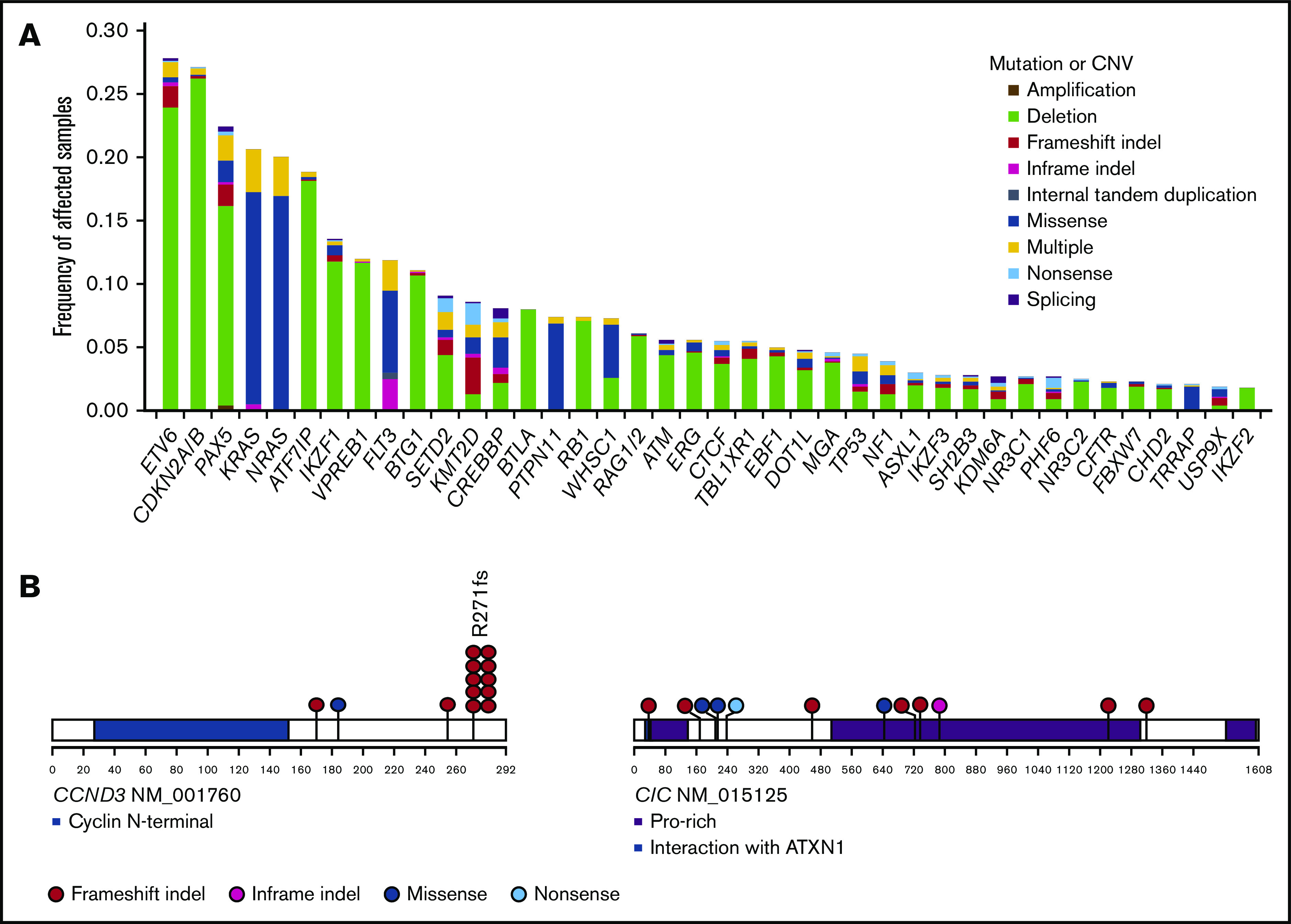
Frequently altered genes and novel driver genes. (A) Frequencies of recurrent mutations and copy number variations (CNVs) identified by targeted capture sequencing. Types of mutations and CNVs are indicated by color. Multiple: more than 1 mutation or mutation accompanied by CNV. (B) Positions and types of somatic mutations in novel driver genes.
Some of mutational targets, including ETV6, CDKN2A/B, PAX5, IKZF1, and ERG, were more commonly affected by focal deletions, which were sensitively detected by sequencing-based copy number analysis (Figure 1A; supplemental Figure 3). In addition to these focal events, whole chromosomal events were frequently observed; according to sequencing-based analysis, we found 307 hyperdiploid and 9 hypodiploid (7 were near-haploid and 2 were low-hypodiploid) cases, as well as 13 iAMP21-positive cases (supplemental Appendix). In accordance with a previous report,24 a substantial number of cases with hyperdiploid ALL were overlooked in conventional metaphase karyotyping; in the latter analysis, only 57% (n = 174 of 307) of all hyperdiploid cases were detected (supplemental Figure 4A). In particular, hyperdiploid cases with lower peripheral WBC counts at diagnosis tended to escape more from metaphase karyotyping (supplemental Figure 4B). Notably, patients with hyperdiploid ALL detected by sequencing-based analysis alone exhibited an overall survival almost identical to that of other hyperdiploid cases detected by metaphase karyotyping (supplemental Figure 4C). We found 9 cases apparently showing hypodiploid on the basis of extensive LOH in the majority of chromosome; 2 of these cases exhibited a hyperdiploid karyotype, and 2 other cases had a normal karyotype (supplemental Figure 5). Breakpoints of IGH-involving translocations were successfully captured in 71 patients, which most frequently affected DUX4 (n = 57 [5.7%]), followed by EPOR (n = 7 [0.6%]), CRLF2 (n = 4 [0.4%]), BCL2 (n = 2 [0.2%]), and MYC (n = 1 [0.1%]) (supplemental Table 4).
Correlation between genetic alteration and disease subgroup
On the basis of panel sequencing, 760 of 1003 B-ALL cases were classified into 10 nonoverlapping subgroups, either of hyperdiploid (n = 307 [31%]) or hypodiploid (n = 9 [1%]) karyotypes; ETV6-RUNX1 (n = 236 [23%]), TCF3-PBX1 (n = 95 [10%]), and IGH-DUX4 (n = 57 [6%]) fusions; PAX5 p.Pro80Arg (n = 4 [0.4%]) and IKZF1 p.Asn159Tyr (n = 3 [0.3%]) mutations; or KMT2Ar (n = 34 [3%]), iAMP21 (n = 13 [1%]), and BCL2/MYC (n = 2 [0.2%]) (supplemental Figure 6). In accordance with a previous report,1 PAX5 p.P80R and IKZF1 N159Y (novel subgroup-defining SNVs) were rare in our pediatric cohort. Of the remaining 243 cases, high-quality RNA was available in 203 (84%) for gene expression analysis to identify cases with Ph-like or ETV6-RUNX1–like phenotype and other fusions, using transcriptome sequencing (n = 116) and/or array-based gene expression analysis (n = 120) (supplemental Figure 7). Twenty-one patients were classified as Ph-like ALL. Transcriptome sequencing also revealed ETV6-RUNX1–like profile (n = 6) and ZNF384 (n = 15), MEF2D (n = 12), and NUTM1 (n = 4) fusions, including novel fusion partners (MEF2D-PYGO2 and NUTM1-ATAD5) (supplemental Table 6).
Altogether, we classified 880 B-ALL cases into 15 discrete subtypes; genetic features of these subtypes were further investigated, focusing on 50 recurrent alterations that were detected in at least 10 patients (supplemental Table 7). We first investigated a significant enrichment of these genetic lesions within each B-ALL subtype. Deletions in hypodiploid ALL were excluded from the analysis, because almost all driver genes were deleted in this subtype. In total, we found 62 significant enrichment events, q < 0.1 (Figure 2). Most common driver changes were significantly enriched in one or more disease subgroups. Although most of these correlations had previously been reported, we newly identified a significant enrichment of PHF6 and PAX5 mutations in TCF3-PBX1 ALL and an SETD2 deletion in iAMP21 ALL. In addition, IKZF1 deletions were significantly enriched in the IGH-DUX4, Ph-like, iAMP21, and ETV6-RUNX1–like subgroups. Recently, IKZF1plus has been reported to be a very poor prognostic factor.25 We therefore further analyzed the corelationship of the IKZF1plus pattern with genetic subgroups. Among 81 cases with IKZF1 deletion, 31 cases fulfilled the IKZF1plus definition, and Ph-like ALL was enriched in IKZF1plus (Fisher’s exact test, P = .002) (supplemental Appendix; supplemental Table 8).
Figure 2.
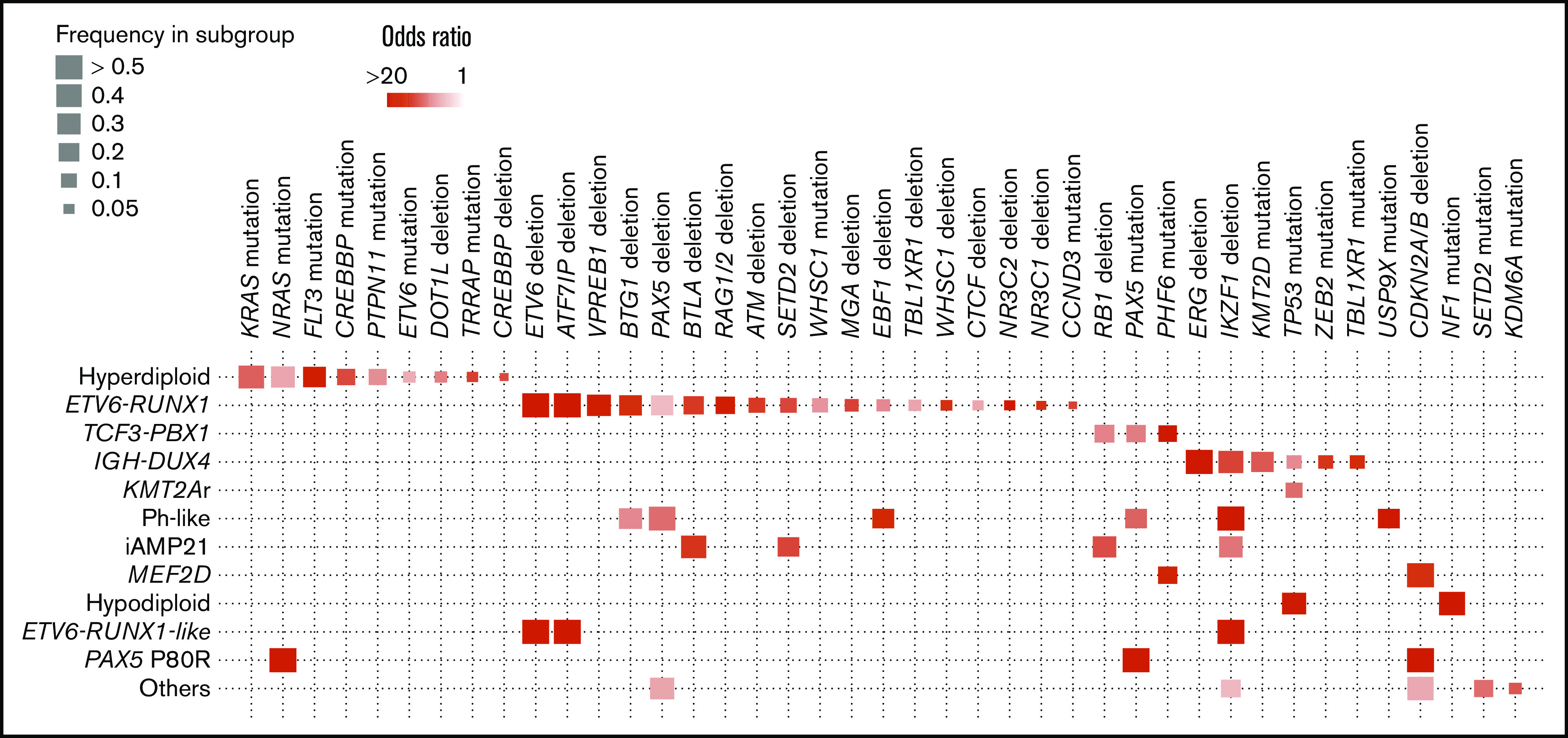
Positive correlations between genetic alterations with disease subgroups. Statistically significant pairwise relationships (q < 0.1) for cooccurrence between a genetic alteration and a disease subgroup. Color gradient indicates odds ratio, and size of boxes show frequency of affected samples in each subgroup.
Clinical effects of genetic alterations
We examined the effects of 50 genetic lesions and 8 subtypes detected in at least 10 cases on survival in the JACLS and TCCSG cohorts. As in the correlation analysis, all deletions in hypodiploid ALL were also excluded in the survival analysis. In univariate analyses, 10 variables were significantly associated with shorter overall survival in each cohort (supplemental Table 9). We next performed multivariate Cox proportional hazard models in each cohort using these 10 variables and factors relevant to treatment stratification: NCI category, prednisone poor response, hypodiploidy, TCF3-PBX1, and KMT2Ar. After adjustment to treatment intensity, TP53 mutation, TP53 deletion, hypodiploidy, and MEF2D fusions were selected in the model in both the JACLS and the TCCSG cohort (Figure 3; Table 2; supplemental Table 10). To confirm the finding, we analyzed whole-exome sequencing data of 466 patients with B-ALL publicly available from the TARGET (Therapeutically Applicable Research to Generate Effective Treatments) project (AALL0331 and AALL0232), in which paired germline control samples were sequenced in all cases. Clinical backgrounds were almost the same as those in the Japanese cohorts, suggesting that the TARGET cohort was suitable for verification (supplemental Table 11). In this external data set, TP53 mutation was associated with shorter overall survival (log-rank test, P < .001); however, TP53 deletion was not associated with shorter event-free or overall survival in the TARGET cohort (supplemental Figure 8; supplemental Table 12).
Figure 3.
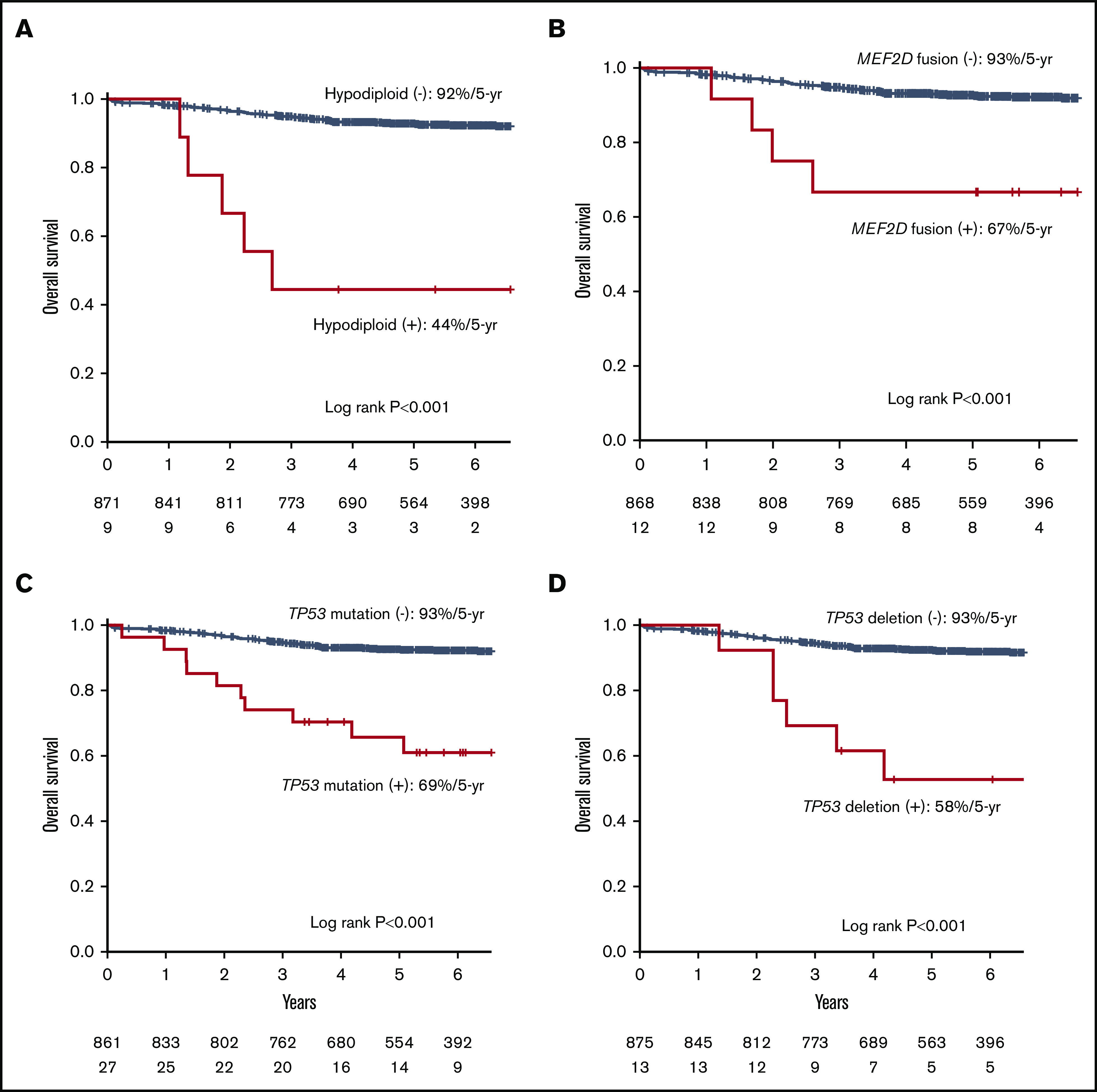
Kaplan-Meier curves for hypodiploid, MEF2D fusion, TP53 mutation, and TP53 deletion in the Japanese cohort. Kaplan-Meier curves of overall survival for individuals in the Japanese cohort with or without hypodiploid (A), MEF2D fusion (B), TP53 mutation (C), and TP53 deletion (D).
Table 2.
Selected variables by using the least absolute shrinkage and selection operator in a Cox proportional hazards multiple regression model
| JACLS | TCCSG | ||
|---|---|---|---|
| Selected variables | Hazard ratio | Selected variables | Hazard ratio |
| TP53 deletion | 14 | TP53 deletion | 3.8 |
| Hypodiploid | 8.4 | Hypodiploid | 14 |
| MEF2D | 4.4 | MEF2D | 5.3 |
| TP53 mutation | 1.3 | TP53 mutation | 2.6 |
| IKZF1 deletion | 1.1 | Prednisone poor response | 1.5 |
| IGH-DUX4 | 1.7 | JAK2 mutation | 2.4 |
| NCI HR | 1.7 | Ph-like | 2.7 |
| TCF3-PBX1 | 2.2 | KMT2Ar | 3.0 |
| PAX5 mutation | 2.5 | ||
| iAMP21 | 2.7 | ||
In the total 1003 cases, 36 TP53 mutations were identified in 30 patients: 22 missense and 2 nonsense mutations, as well as 7 frameshift and 5 in-frame indels (supplemental Tables 13-15). Among them, 11 cases (37%) were accompanied by 17p LOH. As for pathogenicity, 21 of 22 missense mutations are believed to result in complete (n = 19) or partial (n = 2) loss of transcriptional activity in the International Agency for Research on Cancer TP53 database26; the remaining mutation (p.Y236N) was predicted to be damaging by PolyPhen2, SIFT,27 and M-CAP.28 The size of the TP53-mutated clones exhibited a substantial difference among patients (0.03-1, with a median of 0.3), and from the view of this distribution, most of the TP53 mutations were considered to be somatic (supplemental Figure 9). The clone size of TP53 mutations did not significantly correlate with poor prognosis or high NCI risk (supplemental Figure 10).
In terms of disease subtype, TP53 mutations were enriched in hypodiploid (3 of 9 [33%]), KMT2Ar (4 of 34 [12%]), and IGH-DUX4 (5 of 57 [9%]) (Figures 2 and 4A). Of note, TP53 mutations predicted a dismal prognosis even in IGH-DUX4 fusion-positive ALL, which has otherwise been associated with favorable clinical outcomes (Figure 4B). In IGH-DUX4 fusion-positive ALL, TP53 mutations were frequently accompanied by 17p LOH (n = 4 of 5); only 7 of the other 25 TP53 mutated cases harbored 17p LOH (Fisher’s exact test, P = .047). In NCI-SR patients, the presence of a TP53 mutation did not predict poor prognosis (log-rank test, P = .9) (Figure 4C), and the same was true in the TARGET cohort (log-rank test, P = .6).
Figure 4.
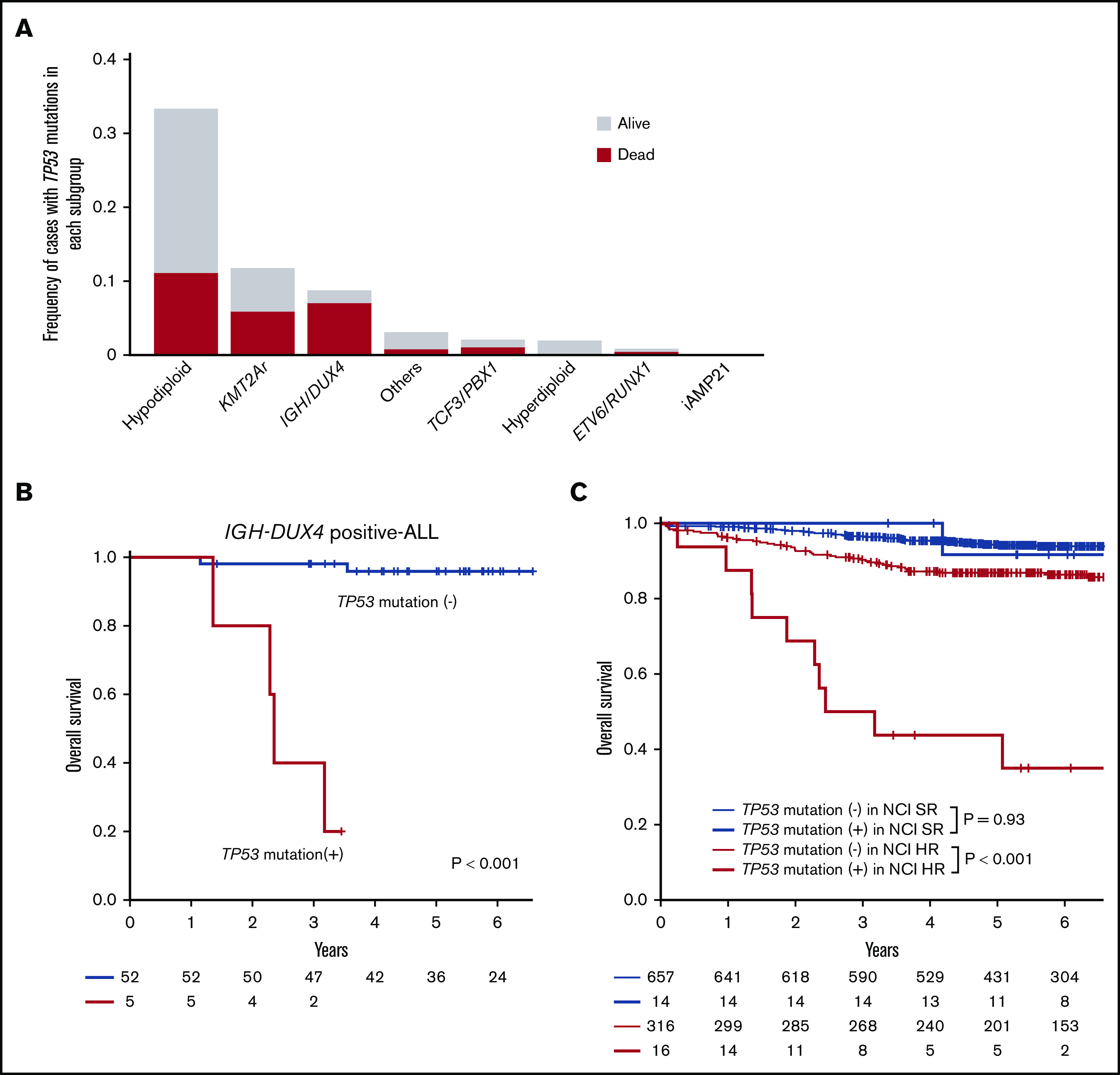
Prevalence and prognostic impact of TP53 mutations. (A) Frequencies of TP53 mutations in each subgroup. Outcomes are indicated by colors. (B) Kaplan-Meier curves of overall survival for individuals with and without TP53 mutations in IGH-DUX4 positive-ALL. (C) Kaplan-Meier curves of overall survival among patients with or without TP53 mutations stratified with NCI criteria.
Discussion
During the past decade, a complete registry of driver alterations in pediatric B-ALL have been clarified by using advanced sequencing technologies. However, exact frequencies and combinations of these mutations, as well as their effects on clinical outcomes, have not been fully elucidated in a larger cohort of patients, including rare subtypes of B-ALL. Enrolling >1000 cases, our study represents one of the largest genetic analyses on pediatric B-ALL, through which we have revealed a complete landscape of major genetic alterations in this common pediatric malignancy and their clinical effects.
Through the analysis, different subtypes of B-ALL were shown to be characterized by a unique pattern of driver alterations, including previously unknown correlations between ETV6-RUNX1 ALL with CCND3 mutations, hypodiploid ALL with CIC mutations, and TCF3-PBX1 ALL with PHF6 and PAX5 mutations. The unique enrichment of mutations suggests a discrete pathophysiology of different B-ALL subtypes, which might promote our understanding of the molecular pathogenesis of B-ALL. Our results also support the usefulness of clinical application of next-generation sequencing for accurate diagnosis in pediatric B-ALL; it allows for detection of copy number change, focal deletion, and SNVs in the single platform.
Except for the germline TP53 mutations and somatic TP53 mutations at relapse,29,30 the clinical significance of TP53 mutations at diagnosis has not fully been investigated in pediatric ALL. In our cohort, TP53 mutations were more commonly found in hypodiploid, KMT2Ar, and IGH-DUX4 ALL. Particularly, even in IGH-DUX4 ALL, which generally predicts a favorable prognosis, most cases with TP53 mutations exhibited a dismal outcome in this cohort. This finding needs to be validated in an external cohort, but these results may suggest the need for novel therapeutic approaches in these patients. Of particular note, however, is that the negative effects of TP53 mutations were not observed in NCI-SR patients, suggesting that for these individuals, the presence of TP53 mutations may not necessarily predict a poor prognosis, and they therefore could be successfully treated with conventional protocols with no further intensification.
In addition, the negative prognostic impact of TP53 deletion was not validated in the TARGET cohort. Most of the TP53 deletions (n = 23 of 26) were consequences of deletion involving >50% of chromosome 17p. In the United Kingdom cohort, 17p abnormality was associated with poor outcome, but only for patients without one of the good-risk abnormalities of ETV6-RUNX1 and high hyperdiploidy.31 In the TARGET cohort, TP53 deletion was not associated with poor outcome even after excluding these good-risk subgroups (data not shown). Although TP53 mutations were frequently (37%) accompanied by 17pLOH, the functional relevance of exclusive TP53 deletions remains unclear.
In conclusion, we comprehensively elucidated genetic alterations and their clinical significance in pediatric B-ALL. Our study should provide an essential guide for diagnosis, prediction of prognosis, and optimal therapy using a genomics-based approach in pediatric patients with B-ALL.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Tomomi Ishida, Mayumi Kibe, Miho Yamada, Mika Fuyama, and Kanako Okada for excellent technical assistance. They also thank all the staffs in the Organization for Supporting Clinical Research data center for data management and the physicians who registered patients in the JACLS ALL-02 or TCCSG L04-16 clinical trials. The authors also express their deep gratitude to those patients who have provided consent to use their biological materials and clinical data.
This work was supported by Grant-in-Aid for Practical Research for Innovative Cancer Control (16ck0106066h0003, 17ck0106253h0001, and 18ck0106253h0002) (M. Sanada), the Program for an Integrated Database of Clinical and Genomic Information (17kk0205005h0002 [K.H.] and 18kk0205005s0203 [S.O.]), and the Program for a Cancer Research and Therapeutic Evolution (18cm0106501h0003JP) (S.O.) from the Japan Agency for Medical Research and Development, Scientific Research from the Japan Society for the Promotion of Science (17K10136) (M. Sanada), and the Takeda Science Foundation (M. Sanada). It was also supported by Grants for Clinical Cancer Research from the Ministry of Health, Labour and Welfare of Japan: H14-Koka(Gan)-031, H15-Koka(Gan)-024, H16-GanRinsho-004, H17-GanRinsho-004, H20-GanRinsho-Ippan-017, and H23-GanRinsho-Ippan-014 (K.H.). This research used computational resources of the K computer provided by the RIKEN Advanced Institute for Computational Science through the HPCI System Research project (hp150232) (S.O.).
Footnotes
The data reported in this article have been deposited in the DNA Data Bank of Japan (accession number PRJDB8942).
Authorship
Contribution: K.H., S.O., and M. Sanada designed the study; Y.I.-Y., T.D., N. Kiyokawa, K.O., A.S., H. Takahashi, Y.H., S.T., J.H., Y.K., K. Kato, T. Inukai, J.T., T. Imamura, A.M., K.H., and M. Sanada collected clinical data and DNA specimens; H.U. and Y.I.-Y. performed sample preparation and sequencing; H.U., K.Y., Y. Shiozawa, Y.N., Y.I.-Y., Y. Shiraishi, K.C., H. Tanaka, N. Kiyokawa, T. Isobe, M. Seki, S.K., H.M., M.M.N., N. Kakiuchi, K. Kataoka, T.Y., D.N., and S.M. analyzed data; H.U., S.O., and M. Sanada wrote the manuscript; and all the authors reviewed and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Seishi Ogawa, Department of Pathology and Tumor Biology, Graduate School of Medicine, Kyoto University, Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan; e-mail: sogawa-tky@umin.ac.jp; or Masashi Sanada, Clinical Research Center, National Hospital Organization Nagoya Medical Center, 4-1-1, Sannomaru, Naka-ku Nagoya-shi, Aichi, 460-0001, Japan; e-mail: masashi.sanada@nnh.go.jp.
References
- 1.Gu Z, Churchman ML, Roberts KG, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51(2):296-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15):1535-1548. [DOI] [PubMed] [Google Scholar]
- 3.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Rapado I, Li Y, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46(2):116-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paulsson K, Lilljebjörn H, Biloglav A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet. 2015;47(6):672-676. [DOI] [PubMed] [Google Scholar]
- 6.Yasuda T, Tsuzuki S, Kawazu M, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults [published correction appears in Nat Genet. 2016;48(12):1591]. Nat Genet. 2016;48(5):569-574. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, McCastlain K, Yoshihara H, et al. ; St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project . Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet. 2016;48(12):1481-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malinowska-Ozdowy K, Frech C, Schönegger A, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015;29(8):1656-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen IM, Harvey RC, Mullighan CG, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. 2012;119(15):3512-3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asai D, Imamura T, Yamashita Y, et al. ; Japan Association of Childhood Leukemia Study (JACLS) & Children’s Cancer and Leukemia Study Group (CCLSG) . Outcome of TCF3-PBX1 positive pediatric acute lymphoblastic leukemia patients in Japan: a collaborative study of Japan Association of Childhood Leukemia Study (JACLS) and Children’s Cancer and Leukemia Study Group (CCLSG). Cancer Med. 2014;3(3):623-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirabayashi S, Ohki K, Nakabayashi K, et al. ; Tokyo Children’s Cancer Study Group (TCCSG) . ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica. 2017;102(1):118-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohki K, Kiyokawa N, Saito Y, et al. ; Tokyo Children’s Cancer Study Group (TCCSG) . Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica. 2019;104(1):128-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu Z, Churchman M, Roberts K, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016;7:13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts KG, Reshmi SC, Harvey RC, et al. Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: a report from the Children’s Oncology Group. Blood. 2018;132(8):815-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasegawa D, Imamura T, Yagi K, et al. Risk-adjusted therapy of acute lymphoblastic leukemia can optimize the indication of stem cell transplantation and cranial irradiation: results of Japan Association Childhood Leukemia Study Group (JACLS) Protocol ALL-02. Blood. 2016;128:3973. [Google Scholar]
- 16.Takahashi H, Kajiwara R, Kato M, et al. Treatment outcome of children with acute lymphoblastic leukemia: the Tokyo Children’s Cancer Study Group (TCCSG) Study L04-16. Int J Hematol. 2018;108(1):98-108. [DOI] [PubMed] [Google Scholar]
- 17.Swaminathan S, Klemm L, Park E, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol. 2015;16(7):766-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiraishi Y, Sato Y, Chiba K, et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41(7):e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshizato T, Nannya Y, Atsuta Y, et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood. 2017;129(17):2347-2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmitz R, Young RM, Ceribelli M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490(7418):116-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuo H, Yoshida K, Fukumura K, et al. Recurrent CCND3 mutations in MLL-rearranged acute myeloid leukemia. Blood Adv. 2018;2(21):2879-2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234(4774):364-368. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47(5):458-468. [DOI] [PubMed] [Google Scholar]
- 24.Nygaard U, Larsen J, Kristensen TD, et al. Flow cytometric DNA index, G-band karyotyping, and comparative genomic hybridization in detection of high hyperdiploidy in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2006;28(3):134-140. [DOI] [PubMed] [Google Scholar]
- 25.Stanulla M, Dagdan E, Zaliova M, et al. ; International BFM Study Group . IKZF1plus defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36(12):1240-1249. [DOI] [PubMed] [Google Scholar]
- 26.Kato S, Han SY, Liu W, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100(14):8424-8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073-1081. [DOI] [PubMed] [Google Scholar]
- 28.Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48(12):1581-1586. [DOI] [PubMed] [Google Scholar]
- 29.Qian M, Cao X, Devidas M, et al. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J Clin Oncol. 2018;36(6):591-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hof J, Krentz S, van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185-3193. [DOI] [PubMed] [Google Scholar]
- 31.Moorman AV, Ensor HM, Richards SM, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11(5):429-438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.