

Key Points
OM-CMML and overt CMML show a similar clinical, morphological, cytogenetic, molecular, and immunophenotypic profile.
The results support the consideration of OM-CMML as a distinctive subtype of CMML.
Abstract
Oligomonocytic chronic myelomonocytic leukemia (OM-CMML) is defined as those myelodysplastic syndromes (MDSs) or myelodysplastic/myeloproliferative neoplasms, unclassifiable with relative monocytosis (≥10% monocytes) and a monocyte count of 0.5 to <1 × 109/L. These patients show clinical and genomic features similar to those of overt chronic myelomonocytic leukemia (CMML), although most of them are currently categorized as MDS, according to the World Health Organization 2017 classification. We analyzed the clinicopathologic features of 40 patients with OM-CMML with well-annotated immunophenotypic and molecular data and compared them to those of 56 patients with overt CMML. We found similar clinical, morphological, and cytogenetic features. In addition, OM-CMML mirrored the well-known complex molecular profile of CMML, except for the presence of a lower percentage of RAS pathway mutations. In this regard, of the different genes assessed, only CBL was found to be mutated at a significantly lower frequency. Likewise, the OM-CMML immunophenotypic profile, assessed by the presence of >94% classical monocytes (MO1s) and CD56 and/or CD2 positivity in peripheral blood monocytes, was similar to overt CMML. The MO1 percentage >94% method showed high accuracy for predicting CMML diagnosis (sensitivity, 90.7%; specificity, 92.2%), even when considering OM-CMML as a subtype of CMML (sensitivity, 84.9%; specificity, 92.1%) in our series of 233 patients (39 OM-CMML, 54 CMML, 23 MDS, and 15 myeloproliferative neoplasms with monocytosis and 102 reactive monocytosis). These results support the consideration of OM-CMML as a distinctive subtype of CMML.
Visual Abstract

Introduction
Based on the World Health Organization (WHO) 2017 classification, chronic myelomonocytic leukemia (CMML) diagnosis requires the presence of persistent peripheral blood monocytosis ≥1 × 109/L, with monocytes accounting for ≥10% of the leukocytes.1,2 Although most CMML cases display dysmyelopoiesis, it may not be present. In the absence of dysplasia, a diagnosis of CMML can still be made by the demonstration of clonality by an acquired clonal cytogenetic or molecular abnormality. If no clonal marker can be found and dysplasia is not present, the diagnosis of CMML may also be established if the monocytosis persists for at least 3 months and all causes of reactive monocytosis have been excluded.1,3 In this context, a wide spectrum of neoplastic, infectious, or inflammatory conditions should be ruled out before the diagnosis of CMML is established. Nevertheless, an autoimmune or neoplastic disease may appear concomitantly, and its presence does not exclude a diagnosis of CMML. Next-generation sequencing (NGS) has emerged as the best tool for establishing diagnostic certainty, because it allows for the demonstration of clonality in most cases of CMML, but this technology is not accessible worldwide. Approximately 90% of patients with CMML display mutations of the TET2, SRSF2, and/or ASXL1 gene.4-7 By contrast, an accessible method such as flow cytometry (FC) analysis of peripheral blood (PB) monocyte subsets has attracted interest as a means of diagnosing CMML, because an increase in the classical monocyte (MO1) fraction to >94% shows high sensitivity and specificity for predicting CMML diagnosis.8
Recently, Geyer et al defined oligomonocytic chronic myelomonocytic leukemia (OM-CMML) as cases of myelodysplastic syndrome (MDSs) or MDS/myeloproliferative neoplasm (MPN), unclassifiable, with relative monocytosis (≥10% monocytes) and a total monocyte count of 0.5 to <1 × 109/L.9 According to the WHO 2017 classification, most of these patients are currently classified within the different categories of MDS. The researchers demonstrated that these cases share clinical and genomic features with overt CMML. To the best of our knowledge, there are no FC data about the distribution of the PB monocyte subset in OM-CMML. Selimoglu-Buet et al indicated that the accumulation of MO1 >94% defined a subgroup of patients with MDS that frequently evolved into CMML.10 Although some of those patients met OM-CMML criteria, there are no series that explore this aspect in a homogeneous group of patients with OM-CMML.
The purpose of this study was to provide a comprehensive comparison between a large series of well-annotated patients with OM-CMML or CMML, with particularly novel data concerning the immunophenotypic and molecular characteristics of OM-CMML. In addition, we assessed the accuracy of the MO1 percentage >94% method of predicting CMML and OM-CMML diagnosis in a large series (n = 233) of patients.
Methods
Patients
We prospectively studied 236 patients and assessed the PB distribution of monocyte subsets by FC in 233 of them. This assessment has been part of the diagnostic routine in our laboratory since 2016. The patients were either initially diagnosed or followed up during this period. All diagnostics were established according to WHO 2017 criteria.
Of the 236 patients, 56 were diagnosed with CMML (16 with “proliferative type” CMML [p-CMML] and 40 with “dysplastic type” [d-CMML]), and 40 met OM-CMML diagnostic criteria. According to the WHO 2017 MDS classification, patients with OM-CMML were classified into the following categories: 1 MDS with single-lineage dysplasia (MDS-SLD), 16 MDS with multilineage dysplasia (MDS-MLD), 4 MDS with ring sideroblasts and single-lineage dysplasia (MDS-RS-SLD), 9 MDS with ring sideroblasts and multilineage dysplasia (MDS-RS-MLD), 9 MDS with excess blasts-1 (MDS-EB-1), and 1 MDS with excess blasts-2 (MDS-EB-2). In addition, we identified 23 patients with MDS who did not meet OM-CMML diagnostic criteria (1 MDS-SLD, 11 MDS-MLD, 3 MDS-RS-SLD, 6 MDS-RS-MLD, 1 MDS-EB-1, and 1 MDS with isolated deletion of 5q), 15 had Ph-negative MPNs with ≥1 × 109/L monocytes (7 essential thrombocytosis, 6 polycythemia vera, and 2 primary myelofibrosis), and 102 patients had absolute monocytosis of reactive origin. The study was conducted according to the biomedical research guidelines of the Declaration of Helsinki.
NGS
Molecular characterization by targeted NGS was performed on DNA extracted from total PB or bone marrow (BM). Targeted amplicon libraries (QIAseq Custom DNA Panels; Qiagen, Hilden, Germany) were prepared from a custom panel covering the full exonic regions of 25 genes associated with myeloid malignancies (ASXL1, CALR, CBL, CSF3R, DNMT3A, ETV6, EZH2, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NRAS, PRPF8, RUNX1, SETBP1, SF3B1, SH2B3, SRSF2, STAG2, TET2, TP53, U2AF1, and ZRSR2).11 Libraries were sequenced with MiSeq or NextSeq (Illumina, San Diego, CA) with a 2000× minimum coverage. The variant allele frequencies (VAF; proportion of mutated reads out of the total NGS reads) for each mutation were recorded. The NGS methodology is described in further detail in the supplemental Material.
Flow cytometry analysis of monocyte subsets in peripheral blood
Multiparametric FC analysis of monocyte subsets was performed on whole PB collected on EDTA. Based on Euroflow Consortium recommendations we followed the stain-lyse-wash procedure with FACS Lysing Solution (BD Biosciences, San Jose, CA).12 Cell surface staining of 2 × 106 cells was performed, and at least 500 000 total events were acquired per tube (FACS Canto II; BD Biosciences). Analysis was performed with Infinicyt, version 1.7 (Cytognos SL, Salamanca, Spain). The strategy of analysis and the 5-tube experimental panel are described in the supplemental Data and supplemental Figure 1.
Statistical analysis
Categorical variables are described by frequencies and percentages and continuous variables as means, medians, and ranges. For categorical data, comparisons of proportions were evaluated by χ2 test, χ2 test with Yates continuity correction, or Fisher’s exact test, as appropriate. For continuous variables, comparisons were assessed by nonparametric Mann-Whitney U test. No adjustments were made to P-values for multiple tests. We assessed the Spearman rank correlation or the Φ coefficient to evaluate the strength of association between 2 variables. The area under the receiver operating characteristic (ROC) curve (AUC) of the percentage of MO1s and MO3s was calculated to assess its accuracy for predicting CMML diagnosis. We used the Youden index (J = sensitivity + specificity − 1) for evaluating the balance between sensitivity and specificity. Survival curves were constructed by the Kaplan-Meier method, using the interval from the date of diagnosis to the date of last contact or death and compared by log-rank test. Differences were considered statistically significant when P < .05 in a 2-tailed test. The code used in R v3.6.2 to create the figures is displayed in supplemental Data 2.
Results
OM-CMML and overt CMML show similar clinical, morphological, and cytogenetic features
The clinical findings for the 40 patients with OM-CMML and the 56 patients with CMML are compared in Table 1. As shown, we observed no significant differences in age, sex, platelet count, BM dysgranulopoiesis, BM dysthrombopoiesis, percentage of BM blasts, percentage of abnormal karyotypes, distribution of the Spanish cytogenetic risk groups,13 and proportion of patients showing blasts in PB. Patients with OM-CMML showed lower absolute leukocyte and monocyte count, a predictable finding, given the definition of OM-CMML. Moreover, they showed a lower percentage of PB and BM monocytes and BM promonocytes.
Table 1.
Comparison of clinical findings between patients with OM-CMML and CMML
| Characteristic | OM-CMML (n = 40) | CMML (n = 56) | d-CMML (n = 40) | p-CMML (n = 16) | P (OM-CMML vs CMML) | P (OM-CMML vs d-CMML) | P (OM-CMML vs p-CMML) | P (d-CMML vs p-CMML) |
|---|---|---|---|---|---|---|---|---|
| Age, median (range), y | 77.5 (44-91) | 76.5 (60-94) | 76.5 (60-88) | 77 (61-94) | .997 | .95 | .913 | .835 |
| Male sex, n (%) | 27 (68) | 29 (52) | 21 (52.5) | 8 (50) | .124 | .171 | .222 | .866 |
| Hemoglobin, median (range), g/L | 11.35 (7.3-14.6) | 12.3 (5.4-15.7) | 12.55 (8.6-15.7) | 11.85 (5.4-14.6) | .019 | .005 | .650 | .198 |
| WBC count, median (range), ×109/L | 4.58 (2.49-7.02) | 9.03 (2.81-34.82) | 7.95 (2.81-12.62) | 17.11 (13.18-34.82) | < .001 | < .001 | < .001 | < .001 |
| Neutrophil count, median (range), ×109/L | 2.15 (0.38-4.42) | 4.86 (0.47-19.62) | 3.35 (0.47-7.02) | 8.77 (4.67-19.62) | < .001 | < .001 | < .001 | < .001 |
| Platelet count, median (range), ×109/L | 138.5 (36-318) | 138.5 (15-559) | 137 (33-559) | 140 (15-294) | .565 | .462 | .986 | .618 |
| Monocyte count, median (range), ×109/L | 0.71 (0.53-0.96) | 1.96 (1-13.33) | 1.67 (1-3.01) | 5.02 (2.69-13.33) | <.001 | <.001 | <.001 | <.001 |
| PB monocyte %, median (range) | 17 (10-26) | 24.85 (13-60) | 23.85 (13-42) | 28.7 (20-60) | <.001 | <.001 | <.001 | .002 |
| BM monocyte %, median (range) | 5 (1-13) | 10 (2-27) | 9 (2-16) | 12 (2-27) | <.001 | <.001 | .001 | .175 |
| BM promonocyte %, median (range) | 1 (0-6) | 3 (0-14) | 2 (0-7) | 4 (1-14) | <.001 | <.001 | <.001 | .012 |
| BM blast %, median (range) | 3 (0-8) | 3 (1-15) | 3 (1-9) | 4 (1-15) | .838 | .798 | .338 | .252 |
| Dyserythropoiesis, median (range), % | 31 (0-80) | 23 (0-90) | 22 (0-90) | 24 (3-77) | .04 | .026 | .411 | .252 |
| ≥10%, % | 74 | 42.7 | 71.4 | 80 | .027 | .03 | .329 | .728 |
| Dysgranulopoiesis, median (range), % | 46 (0-100) | 44 (0-92) | 40 (0-92) | 55 (9-78) | .917 | .636 | .530 | .233 |
| ≥10%, % | 87.2 | 88.5 | 86.5 | 93.3 | 1 | .929 | 1 | .659 |
| Dysthrombopoiesis, median (range), % | 13 (0-64) | 8 (0-69) | 7 (0-69) | 16 (0-54) | .15 | .063 | .794 | .142 |
| ≥10%, % | 65.7 | 46.7 | 42.9 | 60 | .089 | .055 | .726 | .476 |
| Karyotype, abnormal/total cases (%) | 10/38 (26) | 9/50 (18) | 7/36 (19.4) | 2/14 (14.3) | .498 | .482 | .475 | .99 |
| Spanish cytogenetic risk group, n (%) | .384 | .559 | .546 | .809 | ||||
| Low | 33 (87) | 45 (90) | 32 (88.9) | 13 (92.9) | ||||
| Intermediate | 3 (8) | 1 (2) | 1 (2.8) | 0 (0) | ||||
| High | 2 (5) | 4 (8) | 3 (8.3) | 1 (7.1) | ||||
| Presence PB blasts, n (%) | 2/40 (5) | 8/50 (16) | 3/36 (8.3) | 5/15 (33.3) | .176 | .663 | .013 | .039 |
| 2017 WHO classification, n (%) | .013 | .121 | .002 | .023 | ||||
| CMML-0 | 22 (55) | 13 (25) | 12 (32.4) | 1 (6.7) | .003 | .078 | .002 | .078 |
| CMML-1 | 14 (35) | 29 (56) | 21 (56.8) | 8 (53.3) | .048 | .055 | .354 | .822 |
| CMML-2 | 4 (10) | 10 (19) | 4 (10.8) | 6 (40) | .256 | .99 | .018 | .024 |
| Patients with an associated autoimmune disease, n (%)* | 4 (10) | 5 (9) | 4 (10) | 1 (6.3) | .99 | .99 | .99 | .99 |
| Number of patients with mutations (%) | 40 (100) | 52/53 (98) | 36/37 (97.3) | 16 (100) | .99 | .481 | .99 | .99 |
| Number of mutated genes, median (range) | 2 (1-8) | 3 (0-5) | 2 (0-5) | 4 (2-5) | .407 | .46 | .001 | <.001 |
| Number of mutations, median (range) | 3 (1-9) | 3 (0-9) | 3 (0-9) | 4.5 (2-7) | .134 | .859 | .002 | .003 |
Bold P values are statistically significant.
WBC, white blood cell; Spanish cytogenetic risk group: low, normal, and isolated Y chromosome loss; intermediate, other abnormalities except those mentioned in low- and high-risk categories; and high, +8, abnormalities of chromosome 7 and complex karyotype (≥3 abnormalities).
Four patients with OM-CMML presented with a concomitant autoimmune disease: 1, antiphospholipid syndrome; 2, systemic scleroses; and 1, immune thrombocytopenic purpura. Five patients with CMML presented with a concomitant autoimmune disease: 1, systemic sclerosis; 1, dermatomyositis; 1, ankylosing spondylitis HLA-B27+; 1, antiphospholipid syndrome; 1, immune thrombocytopenic purpura.
Patients with OM-CMML were also more anemic and showed more evident dyserythropoiesis. In this sense, we observed a higher proportion of OM-CMML showing SF3B1 mutation and ≥5% ring sideroblasts (28% vs 12%; P = .056). Patients with OM-CMML or CMML who displayed this feature showed a significantly lower hemoglobin level and a higher median percentage of dyserythropoiesis (Hb, median: 11 vs 12 g/dL; P = .010; dyserythropoiesis: median, 60% vs 22%; P < .001, SF3B1 mutated vs unmutated).
OM-CMML and overt CMML show a similar mutational profile
Molecular characterization by NGS was performed in all patients with OM-CMML and in 53 of 56 patients with CMML. As depicted in Table 1, there were no significant differences in the proportion of patients showing at least 1 mutation (40 of 40 vs 52 of 53; P = .99; OM-CMML vs CMML) in the median number of mutated genes per patient (2 vs 3; P = .407, OM-CMML vs CMML) or in the median number of mutations per patient (3 vs 3; P = .134, OM-CMML vs CMML).
The mutation patterns of OM-CMML and CMML are depicted in Figure 1. The genes mutated at a frequency >10% in patients with OM-CMML were TET2 (72%), SRSF2 (30%), SF3B1 (27.5%), ZRSR2 (20%), ASXL1 (17.5%), DNMT3A (15%), and RUNX1 (12.5%). In patients with CMML, the genes mutated at a frequency >10% were TET2 (75%), ASXL1 (28.3%), SRSF2 (26.4%), CBL (20.8%), SF3B1 (17%), NRAS (11.3%), and KRAS (11.3%). The VAFs were similar between both groups in all genes, except for DNMT3A (supplemental Table 1). In line with the literature, the 3 most frequently mutated genes in CMML group were TET2, ASXL1, and SRSF2.4-6,14 Remarkably, no significant difference was observed in the proportion of patients showing concurrent TET2 and SRSF2 mutations, the gene signature of CMML15 (27.5% vs 22.6%, P = .591, OM-CMML vs CMML). Only 1 gene mutated at a significantly different frequency: CBL (2.5% vs 20.8%; P = .011, OM-CMML vs CMML; Figure 2A). Notably, we found no gene mutated at a significantly different proportion when comparing OM-CMML and d-CMML (Table 2). As expected, CMML showed a higher percentage of mutations in RAS pathway genes (mutations in CBL, NRAS, and/or KRAS) than did OM-CMML, given that these genes have been associated with proliferative features16,17 (37.7% vs 5%; P < .001). Although d-CMML showed a significantly higher percentage of RAS-pathway mutations than OM-CMML (27% vs 5%; P = .011), this difference was especially evident in p-CMML (62.5% vs 5%; P < .001), in which genes associated with proliferation were present at higher frequencies16-18: CBL (2.5% vs 31.3%; P = .006), NRAS and/or KRAS (2.5% vs 31.3%; P = .006), and ASXL114 (17.5% vs 62.5%; P = .003) (Table 2). These mutations were also more frequent in p-CMML than in d-CMML (ASXL1: 62.5% vs 13.5, P = .001; RAS-pathway: 62.5% vs 27%, P = .014) (Table 2). It is also worth noting that the proliferative condition associated with the presence of ASXL1 mutations in our series could be explained in part by the presence of concomitant RAS-pathway mutations. In this sense, we found a positive correlation between mutations in the RAS pathway and ASXL1 (Φ coefficient, 0.23; P = .029). It would be interesting to explore this association in larger series of patients with CMML, because ASXL1 mutation is a well-established independent adverse prognostic factor in CMML,5,6,14 but it also seems to be partially interrelated with RAS mutations and p-CMML type, 2 other well-accepted independent adverse prognostic factors in this disease.6,19
Figure 1.
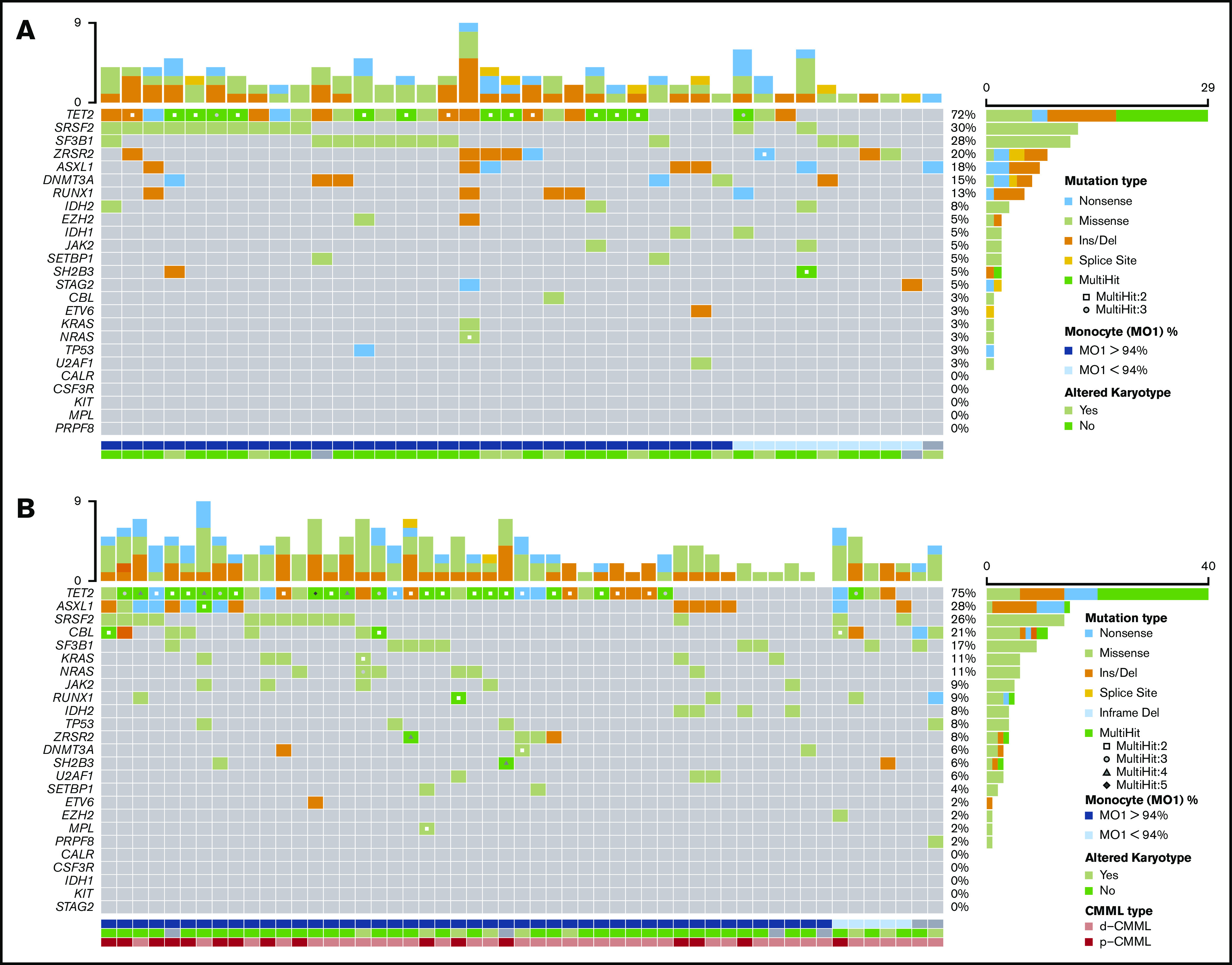
Mutational profile in patients with OM-CMML and CMML. Mutations were identified by NGS in 40 patients with OM-CMML (A) and in 53 patients with CMML (B). Results of the sequencing of the 25 genes are shown in the plot, where each column represents a patient and each row represents a gene. The number of mutations identified per patient is represented as columns in the top row. Genes are ordered from the most to the least frequently mutated, and frequencies for each gene are displayed (right), as well as the mutation type (nonsense, missense, insertion/deletion, splice site, or multihit). Patients with more than 1 mutation in the same gene are represented as shown in the key (2, 3, 4, or 5 mutations in the same gene). The immunophenotypic profile, assessed by the presence of MO1s upper 94%, is shown (bottom; MO1 >94%, blue, MO1 ≤94%, light blue, nonanalyzed, gray). Cytogenetic results are also displayed (bottom row; altered karyotype, lime green; normal karyotype, light green; nonanalyzed, gray). CMML types are also shown (bottom row: d-CMML, light red; p-CMML, red).
Figure 2.

Distribution of mutated genes in CMML and OM-CMML. (A) Frequencies of the 25 genes analyzed by NGS in the CMML and OM-CMML groups. Genes are ordered from the most to the least frequently mutated, combining the CMML and OM-CMML cases. CBL was the only gene mutated at a significantly different frequency in the groups (2.5% vs 20.8%; OM-CMML vs CMML; P = .011). (B) The plot represents all the mutations identified in the TET2 gene classified by the type of alteration, with insertions or deletions of nucleotides (orange) being the most frequent mutations identified. Nonsense mutations, producing a stop codon in the sequence (light blue), and missense mutations, producing a change in 1 amino acid (lime green), were the second most commonly identified. The least common alterations in our cohort were splice site mutations (yellow). No significant differences were observed in the distribution of mutations in TET2 when both disease groups were compared.
Table 2.
Distribution of somatic mutations in patients with OM-CMML, d-CMML, or p-CMML
| OM-CMML, n = 40, % | d-CMML, n = 37, % | p-CMML, n = 16, % | P (OM-CMML vs d-CMML) | P (d-CMML vs p-CMML) | P (OM-CMML vs p-CMML) | |
|---|---|---|---|---|---|---|
| ASXL1 | 17.5 | 13.5 | 62.5 | .757 | .001 | .003 |
| CALR | — | — | — | — | — | — |
| CBL | 2.5 | 16.2 | 31.3 | .051 | .275 | .006 |
| CSF3R | — | — | — | — | — | — |
| DNMT3A | 15 | 5.4 | 6.3 | .266 | .99 | .660 |
| ETV6 | 2.5 | 2.7 | — | .99 | .99 | .99 |
| EZH2 | 5 | — | 6.3 | .494 | .302 | .99 |
| IDH1 | 5 | — | — | .494 | — | .99 |
| IDH2 | 7.5 | 2.7 | 18.8 | .616 | .077 | .338 |
| JAK2 | 5 | 10.8 | 6.3 | .419 | .99 | .99 |
| KIT | — | — | — | — | — | — |
| KRAS | 2.5 | 10.8 | 12.5 | .189 | .99 | .193 |
| MPL | — | — | 6.3 | — | .302 | .286 |
| NRAS | 2.5 | 8.1 | 18.8 | .346 | .351 | .066 |
| RUNX1 | 12.5 | 10.8 | 6.3 | .99 | .99 | .662 |
| PRPF8 | — | 2.7 | — | .481 | .99 | — |
| SETBP1 | 5 | 2.7 | 6.3 | .99 | .517 | .99 |
| SF3B1 | 27.5 | 16.2 | 18.8 | .279 | .99 | .734 |
| SH2B3 | 5 | 2.7 | 12.5 | .99 | .213 | .570 |
| SRSF2 | 30 | 18.9 | 43.8 | .299 | .123 | .362 |
| STAG2 | 5 | — | — | .494 | — | .99 |
| TET2 | 72 | 73 | 81.3 | .99 | .731 | .734 |
| TP53 | 2.5 | 8.1 | 6.3 | .346 | .99 | .494 |
| U2AF1 | 2.5 | 2.7 | 12.5 | .99 | .213 | .193 |
| ZRSR2 | 20 | 10.8 | — | .352 | .303 | .089 |
| NRAS and/or KRAS | 2.5* | 16.2 | 31.3 | .051 | .275 | .006 |
| RAS pathway | 5 | 27 | 62.5 | .011 | .014 | <.001 |
Bold P values are statistically significant.
NRAS and/or KRAS, mutations in both genes or one of them; RAS pathway, mutations in CBL, NRAS, and/or KRAS genes.
One patient showed concurrent NRAS and KRAS mutations (Figure 1A).
As previously reported, mutations in the hydroxymethylation pathway (mutations in IDH1, IDH2, and/or TET2) are almost mutually exclusive in acute myeloid leukemia20 and CMML.21 In our CMML series, we found no concomitant mutations in TET2 and IDH1 or IDH2, but surprisingly, 3 patients with OM-CMML showed simultaneous mutations in these genes, 2 with TET2 and IDH2 mutations and 1 with TET2 and IDH1 mutations (Figures 1A and 3). The impairment of the hydroxymethylation pathway was present in the majority of these patients (78%, 83%; OM-CMML, CMML) and, remarkably, in all p-CMML cases in our series. Moreover, a high proportion of patients with OM-CMML or CMML showed more than 1 TET2 mutation (37.5% vs 52.8%, P = .142, OM-CMML vs CMML) and the distribution of the different TET2 subtype of mutations was almost identical in a comparison of both groups (Figure 2B). These findings suggest that the impairment of this pathway could be the pathophysiological hallmark of these entities.
Figure 3.

Co-occurrence or mutual exclusivity of genes in both OM-CMML and CMML. The plot shows, for the whole group of OM-CMML and CMML, all genes found to be altered in our cohort ordered by the number of mutations identified. In those genes that were frequently found to be comutated in the same patient, the interactions are depicted in lime green. In genes that were observed to be mutually exclusive and thus not frequently altered in the same patient, the interactions are depicted in brown. *P < .05; •P < .1.
Finally, in line with published data, mutations in the assessed splicing factors in our series (SF3B1, SRSF2, ZRSR2, U2AF1, and PRPF8) were almost mutually exclusive.22-26 In the OM-CMML group, we observed 1 patient with concomitant SRSF2 and SF3B1 mutations and another with simultaneous SRSF2 and ZRSR2 mutations (Figures 1A, and 3). Only 1 patient with CMML showed simultaneous SF3B1 and ZRSR2 mutations (Figures 1B and3).
Graphic representations of the mutations are depicted in supplemental Figure 2, and the full list of variants identified is shown in supplemental Data 3.
The increase in the fraction of MO1 >94% is shown as the approach with the highest accuracy for predicting CMML diagnosis
The repartition of monocyte subsets in PB was assessed in 233 patients (39 OM-CMML, 54 CMML, 23 MDS that did not meet OM-CMML diagnostic criteria, 15 MPN with ≥1 × 109/L monocytes, and 102 with reactive monocytosis). The percentage of MO1s in these groups of patients is shown in Figure 4. As Selimoglu-Buet et al and other later studies have shown, the increase in MO1 fraction >94% is a very sensitive and specific predictor of CMML diagnosis.8,27,28 We explored the sensitivity and specificity of this method in our series. Because the minimum diagnostic criterion for considering the diagnosis of CMML is the presence of at least 1 × 109/L monocytes in PB, we first analyzed the 171 patients in our cohort with ≥1 × 109/L monocytes (54 CMML and 15 MPN with monocytosis and 102 reactive monocytosis). The presence of MO1 percentage >94% predicted the diagnosis of CMML with a high sensitivity (90.7%) and specificity (92.2%). Because another group proposed MO1 percentage >95% as the best cutoff for predicting CMML diagnosis,29 we assessed according to that criterion in our series. MO1 percentage >95% showed a lower sensitivity (83.3%) and a slightly better specificity (95.7%), and the balance between sensitivity and specificity calculated by the Youden index (J = 79) was worse than the 94% cutoff (J = 82.9). The AUC of the percentage of MO1 in our series was 0.941 (Figure 5), in line with the previous literature.8,27,28 Other authors have proposed the reduction of the percentage of MO3 as the best predictor for CMML diagnosis.30 In our series, the MO1 population showed a better AUC than did the MO3 population (0.933). Moreover, we found the cutoff in the percentage of MO3s under 3.18% to have the best predictive capacity in our series, but it performed worse than did the MO1 >94% cutoff (sensitivity, 92.6%; specificity, 83.8%; J = 76.4).
Figure 4.
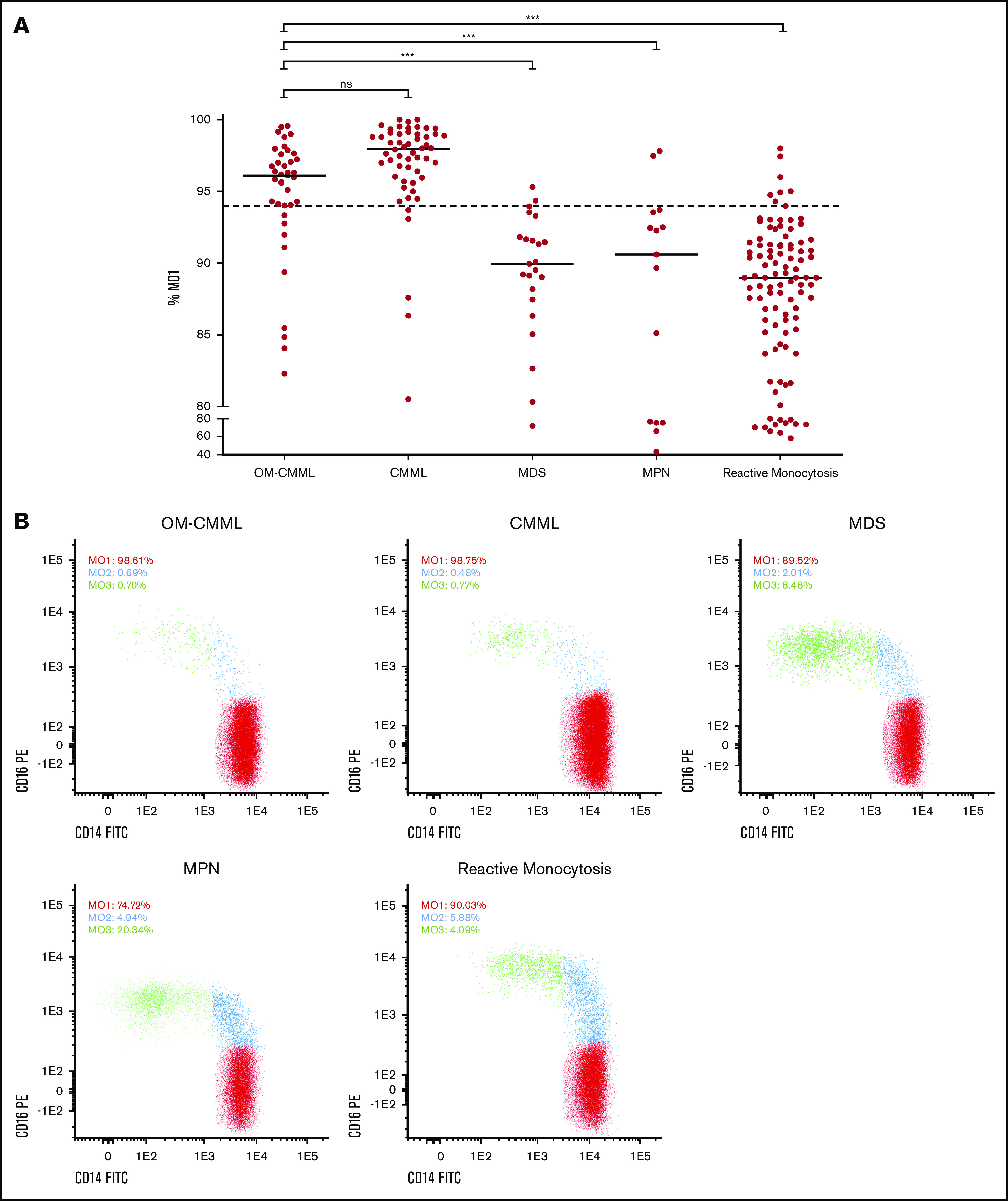
Percentage of MO1s in the 233 cases grouped by disease. (A) Flow cytometry results are shown as the percentage of MO1 identified for the 5 groups analyzed (39 OM-CMML, 54 CMML, and 23 MDS that did not meet OM-CMML diagnostic criteria; 15 MPN with ≥1 × 109/L monocytes, and 102 reactive monocytosis). Each dot represents a patient result for the MO1 test, and lines represent the median percentage for each disease group. The dotted line indicates the 94% cutoff of the test. ***P < .001. (B) The distribution of monocyte subsets is shown for 5 examples, each one from a different disease group. The percentage of the 3 monocyte subsets (MO1, MO2, and MO3) out of the total monocytes is displayed for each example. ns, not significant.
Figure 5.

ROC AUC curves of the percentage of MO1s in our series. (A) ROC curve analysis of diagnostic sensitivity and specificity of the MO1 percentage in 171 patients with ≥1 × 109/L PB monocytes (54 CMML, 15 MPN with monocytosis, and 102 with reactive monocytosis). (B) ROC curve analysis of diagnostic sensitivity and specificity of the MO1 percentage in PB monocytes of 233 patients (93 recoded CMML, including 39 OM-CMML and 54 overt CMML; 23 MDS not meeting OM-CMML diagnostic criteria; 15 MPN with monocytosis; and 102 with reactive monocytosis).
OM-CMML and overt CMML show similar immunophenotypic features
The comparison between OM-CMML and CMML showed that the MO1 percentage was significantly lower in OM-CMML, but it is noteworthy that the median and mean MO1 percentages in OM-CMML were above the 94% cutoff (median, 96.11 vs 97.96; mean, 94.76 vs 96.93; P = .001, OM-CMML vs CMML). Moreover, the proportion of patients with MO1 percentage >94% was not significantly different when OM-CMML was compared with CMML (76.9% vs 90.7%; P = .122; Figure 4). Although probably achieved in the context of a type II error, this result is impressive because, as previously mentioned, the specificity of the MO1 percentage >94% test is ∼90% to 95% and, therefore, only a 5% to 10% false-positive rate should be expected. However, in the group of patients with OM-CMML, a 76.9% false-positive rate was observed, because these patients had a current diagnosis of MDS according to the 2017 WHO recommendation. Likewise, no differences were observed in the percentage of patients showing CD56 positivity in monocytes (61.5% vs 63%; P = .889, OM-CMML vs CMML) or in the percentage of them showing CD2 (28.2% vs 35.2%; P = .477, OM-CMML vs CMML; supplemental Table 2). On the contrary, we found significant differences between patients with OM-CMML and those with MDS who did not meet OM-CMML diagnostic criteria in MO1 percentage (median: 96.11 vs 89.95; mean: 94.76 vs 89.01; P < .001, OM-CMML vs MDS), the proportion of patients with MO1 percentage >94% (76.9% vs 8.7%; P < .001, OM-CMML vs MDS), and the percentage of patients showing CD56 (61.5% vs 8.7%; P < .001, OM-CMML vs MDS) or CD2 (28.2% vs 0; P = .005, OM-CMML vs MDS; supplemental Table 2). No patient with OM-CMML, CMML, or MDS showed CD7 positivity. It is remarkable that we found no significant difference in the distribution of patients with OM-CMML and the comparator group of patients with MDS (P = .433), among the MDS categories stipulated by the WHO 2017 classification. Therefore, the differences detected between the OM-CMML and MDS groups are not attributable to their primary WHO 2017 classification.
Interestingly, a significantly higher proportion of patients with OM-CMML with TET2 mutations had a MO1 percentage >94% (89.7% vs 40%, P = .004). Notably, patients with OM-CMML with TET2 mutations demonstrated this feature in a percentage similar to overt CMML (89.7% vs 90.7%; P = .99). This mutation was the only one of the assessed mutations that enabled division of the OM-CMML series into 2 groups, which showed a significant difference in the proportion of patients with MO1 percentage >94% (supplemental Table 3).
As published by Cargo et al,31 CD56 positivity in monocytes correlated positively with TET2 mutation in our series, both as a binary value (Φ coefficient, 0.45; P < .001) or as a continuous variable (ρ Spearman, 0.4; P < .001). Likewise, the median expression of CD56 in monocytes was significantly higher in the patients with TET2 mutations (34% vs 3%; P < .001), and the proportion of patients showing CD56 positivity was also higher in the TET2-mutated group (75% vs 24%; P < .001).
Given the similarities observed between patients with OM-CMML or CMML, we placed them together in a single category (93 recoded CMML: 39 OM-CMML and 54 overt CMML) and assessed the strength of the MO1 >94% method in all 233 patients of our series (93 recoded CMML, 23 MDS, 15 MPN with monocytosis, and 102 with reactive monocytosis). The presence of MO1 percentage >94% predicted the diagnosis of these patients with high sensitivity (84.9%) and specificity (92.1%; J = 77). The AUC of the percentage of MO1 was 0.908, and the best MO1 cutoff was >94% (Figure 5). Because a similar proportion of patients with OM-CMML or CMML showed CD56 and CD2 positivity and these findings were rarely seen in the other groups of patients analyzed (supplemental Table 2), we tried to improve the performance of the method by using a combined approach: the presence of MO1 percentage >94% and CD56 positivity and/or CD2 positivity. The presence of at least 1 of these features presented a better sensitivity (94.6%) with a slightly lower specificity (87.8%), and the balance between sensitivity and specificity was clearly better (J = 82.4). The sensitivity of this approach when evaluating patients with OM-CMML was 89.7%, whereas the sensitivity in patients with CMML increased to 98.1%. Thus, given its sensitivity, this combined assay may be of high utility as a screening test in this context.
As previously reported by Tarfi et al,32 we observed a significantly higher false-negative rate of the MO1 >94% test in those patients with a concomitant autoimmune disease (44.4% vs 11.9%, patients with OM-CMML or CMML, analyzed together, with and without an associated autoimmune disease; P = .027).
Finally, we compared the percentage of plasmacytoid dendritic cells (pDCs) in PB from total leukocytes among the OM-CMML, d-CMML, and p-CMML groups (median, 0.05%, 0.04%, and 0.015%, respectively). We observed that p-CMML had a significantly lower percentage of pDCs than OM-CMML (P = .022). Likewise, we observed a trend when comparing OM-CMML with CMML as a whole group (median, 0.05% vs 0.02%; P = .067). In this regard, progression from low- to high-risk categories or even leukemic transformation in MDS patients has been associated with a progressive decrease in pDCs.33,34 These data enable us to infer that the transition of one stage to another may be partially favored by the progressive decline of pDCs, which would lead to a decrease in immune surveillance.
Patients with OM-CMML that evolved to CMML showed inferior survival
At a median follow-up of 31.1 months, 18% of patients with OM-CMML that evolved to CMML showed a median time to evolution of 34.3 months. The overall survival (OS) and cumulative incidence of evolution to CMML at 3 years of the 40 patients with OM-CMML were 85.9% and 15.7%, respectively. Seven patients with OM-CMML that evolved to CMML had a significantly shorter OS than did those in whom it did not evolve (median OS: not reached vs 64.62 months; P = .026). Patients in whom the disease evolved showed no significant differences regarding immunophenotypic or molecular patterns. In this regard, we did not find any variable showing a significant influence in predicting time to CMML (number of mutations, number of mutated genes, RAS-pathway mutations, number of TET2 mutations, truncating vs nontruncating type TET2 mutations, and molecular CMML-specific prognostic scoring system). Notably, 4 of 7 patients with OM-CMML that evolved to CMML died, showing a very short median OS from the moment of progression (3.42 months; 95% CI, 0.6-6.2). One patient progressed to acute myeloid leukemia and the other 3 patients died of severe infections. Although this finding deserves to be taken into consideration, larger series of patients are needed before generating warnings in this area.
Discussion
We analyzed the clinicopathologic features of the largest series of patients with OM-CMML reported to date, with extensively studied clinical, morphological, cytogenetic, molecular, and immunophenotypic data. In addition, we compared the features of these patients to those of a large series of patients with CMML, with data concerning immunophenotypic characteristics of OM-CMML being especially novel. In this sense, we compared the utility of the MO1 >94% test between these 2 groups of patients and collected a large series of patients with MPN with absolute monocytosis and reactive monocytosis and a subset of patients with MDS who did not fulfill OM-CMML diagnostic criteria. Notably, we report one of the largest published series to assess the MO1 >94% criterion, in either the total number of patients assessed (n = 233) or in the number of patients with CMML analyzed (n = 54). The increase of MO1 >94% provided high sensitivity (90.7%) and specificity (92.2%) for CMML diagnosis in our series. Although the 94% threshold was initially validated by 2 studies,27,28 some controversial results have recently appeared in the literature. Picot et al29 detected the 95% cutoff as the one with the best sensitivity and specificity, and later, Hudson et al30 found that the MO3 percentage <1.13% was the best predictor of a diagnosis of CMML. Although valuable, these studies were based on a small number of patients with CMML (15 in Picot et al and 16 in Hudson et al). In addition, the different series in the literature assessing the performance of the MO1 >94% criterion are not well studied from a molecular point of view.8,10,27-30 In contrast, molecular characterization by targeted NGS was performed in all patients with OM-CMML and in 53 of 56 patients with CMML in our series. The lack of molecular data could diminish the accuracy of the results of the MO1 >94% test because, as previously stated, some uncertainty may exist when establishing a CMML diagnosis in some cases (eg, absence of dysmyelopoiesis, absence of clonality assessed by cytogenetics, and coexistence of autoimmune or neoplastic diseases). In our series, the best cutoff in MO1 percentage was >94% and the MO1 population showed the best predictive capacity for the diagnosis of CMML, validating the results of the French group.8,27
Focusing on the comparison between OM-CMML and CMML, we found no significant differences in the proportion of patients with MO1 percentage >94% or in those who showed CD56 or CD2 positivity in monocytes. Based on this, we tried to improve the performance of the MO1 >94% method by using a combined approach: the presence of a percentage of MO1s >94% and CD56 and/or CD2 positivity in monocytes. This method afforded better sensitivity (94.6%) with slightly lower specificity (87.8%) than the MO1 >94% cutoff, and the balance between sensitivity and specificity was clearly superior. Thus, given its high sensitivity, this combined assay emerged as an excellent screening test in this context. Interestingly, as previously reported by Tarfi et al,32 we observed a significantly higher false-negative rate of the MO1 >94% test in those patients with a concomitant autoimmune disease. They showed that a decrease in the 6-sulfo lac-nac (slan)+ MO3 monocytes below 1.7% is characteristic of CMML and persists in those exhibiting an associated inflammatory state.32 Therefore, in future studies, it would be interesting to dispose of the anti-slan antibody, to further improve the precision of the method.
OM-CMML and overt CMML showed similar clinical, morphological, and cytogenetic features, with the exception that patients with OM-CMML showed lower hemoglobin levels and more evident dyserythropoiesis. This finding could be partially explained by a higher proportion of patients with OM-CMML showing SF3B1 mutation and ≥5% ring sideroblasts in BM. In our series, patients with OM-CMML or CMML displaying this feature showed a significantly lower hemoglobin level and a higher median percentage of dyserythropoiesis.
OM-CMML and overt CMML show a similar mutational profile. We found no significant difference in the proportion of patients showing concurrent TET2 and SRSF2 mutations, the well-accepted gene signature of CMML.15 As previously shown, the impairment of the hydroxymethylation pathway (mutations in IDH1, IDH2, and/or TET2 genes) is present in most of these patients. Moreover, in line with previous data,35 a high proportion of patients with OM-CMML or CMML showed multiple TET2 mutations. Interestingly, patients with OM-CMML who had TET2 mutations had MO1 percentage >94% in a rate similar to those with overt CMML. Moreover, as previously reported by Cargo et al,31 CD56 positivity in monocytes was significantly associated with TET2 mutation in our series. These findings suggest that the impairment of this pathway could be the pathophysiological hallmark of these entities. Hydroxymethylation has been recognized as a physiological passive DNA demethylation process.36-38 Therefore, it is expected that patients with OM-CMML or CMML will present aberrant DNA hypermethylation states mediated by an ineffective production of 5-hydroxymethylcytosines.20,39,40 In future studies, it would be interesting to explore the implication of DNA methylation, and especially 5-hydroxymethylcytosine levels, in prognosis, disease progression, and prediction of response to hypomethylating agents,21 in this group of patients.
The only gene mutated at a significantly lower frequency when comparing OM-CMML with CMML was CBL. This fining was expected, because mutations in genes of the RAS pathway (CBL, NRAS, KRAS, NF1, and PTPN11) are well-known secondary events in CMML and have been associated with proliferative features.16,17 This finding is in line with that of Geyer et al,9 who found a significantly lower proportion of patients with OM-CMML with CBL mutations (0% vs 28%; OM-CMML vs CMML). It is also remarkable that a significantly higher proportion of patients with p-CMML carried ASXL1 mutations when compared to those with OM-CMML and d-CMML. This result agrees with the published data showing that patients with CMML harboring ASXL1 mutations have more prominent leukocytosis than the group not displaying this mutation.14
Finally, in our series, 18% of patients had OM-CMML that evolved to CMML. This observation supports considering OM-CMML as an early stage of d-CMML.9 If true, it would allow for the establishment of a continuum of OM-CMML, d-CMML, and p-CMML. In this sense, as previously mentioned, we inferred that second genetic hits, such as the acquisition of RAS-pathway mutations, could promote the transition from one stage to another.16 In addition, immune dysregulation, together with a progressive decrease in immune surveillance, could play a pivotal role in the progression of the disease. In this regard, as previously reported by other researchers,33,34 we found a decline in pDCs when comparing OM-CMML with CMML, and this was especially evident when OM-CMML was compared to p-CMML. Interestingly, in ours series, the patients with OM-CMML that evolved to CMML showed a significantly shorter overall survival than did those in whom it did not evolve.
In summary, OM-CMML and overt CMML show similar clinical, morphological, cytogenetic, molecular, and immunophenotypic features. In addition, the MO1 percentage >94% method showed a high accuracy for predicting CMML and OM-CMML diagnosis in our series. The results reinforce the consideration of OM-CMML as a distinctive subtype of CMML.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This study was supported in part by grants from Instituto de Salud Carlos III (ISCIII) and Spanish Ministry of Health grants FIS PI16/0153, FIS PI19/0005, and FEDER 2017SGR205, 2017SGR437, and PT17/0015/0011; Beca Gilead 2016; and Xarxa de Banc de Tumors de Catalunya.
Footnotes
Presented at the 62nd annual meeting of the American Society of Hematology, Orlando, FL, 7-10 December 2019 and at the 24th Congress of the European Hematology Association, Amsterdam, The Netherlands, 13-16 June 2019.
Original data are available by e-mail request to the corresponding author, Xavier Calvo (e-mail: xcalvo@parcdesalutmar.cat).
Authorship
Contribution: X.C. designed the study; X.C., I.P., N.G.-G., L.A., M.A.-C., B.M., S.G.-A., D.R.-B., B.B., L.C., and L.F. collected and assembled data from the study patients; X.C., N.G.-G., L.A., C.F.-R., J.G., M.S., A.P., B.E., S.M., and A.F. analyzed the data; X.C., N.G.-G., L.A., and J.G. interpreted the data; X.C. wrote the final version of the manuscript; and all authors reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xavier Calvo, Paseo Marítimo, 25, 08003 Barcelona, Spain; e-mail: xcalvo@parcdesalutmar.cat.
References
- 1.Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues 4th ed, revised Lyon, France: International Agency for Research on Cancer, WHO; 2017. [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 3.Valent P, Orazi A, Savona MR, et al. Proposed diagnostic criteria for classical chronic myelomonocytic leukemia (CMML), CMML variants and pre-CMML conditions. Haematologica. 2019;104(10):1935-1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meggendorfer M, Haferlach T, Alpermann T, et al. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica. 2014;99(12):e244-e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itzykson R, Kosmider O, Renneville A, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428-2436. [DOI] [PubMed] [Google Scholar]
- 6.Elena C, Gallì A, Such E, et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood. 2016;128(10):1408-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patnaik MM, Tefferi A. Cytogenetic and molecular abnormalities in chronic myelomonocytic leukemia. Blood Cancer J. 2016;6(2):e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selimoglu-Buet D, Wagner-Ballon O, Saada V, et al. ; Francophone Myelodysplasia Group . Characteristic repartition of monocyte subsets as a diagnostic signature of chronic myelomonocytic leukemia. Blood. 2015;125(23):3618-3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geyer JT, Tam W, Liu Y-C, et al. Oligomonocytic chronic myelomonocytic leukemia (chronic myelomonocytic leukemia without absolute monocytosis) displays a similar clinicopathologic and mutational profile to classical chronic myelomonocytic leukemia. Mod Pathol. 2017;30(9):1213-1222. [DOI] [PubMed] [Google Scholar]
- 10.Selimoglu-Buet D, Badaoui B, Benayoun E, et al. ; Groupe Francophone des Myélodysplasies . Accumulation of classical monocytes defines a subgroup of MDS that frequently evolves into CMML. Blood. 2017;130(6):832-835. [DOI] [PubMed] [Google Scholar]
- 11.Palomo L, Ibáñez M, Abáigar M, et al. ; Spanish Group of MDS (GESMD) . Spanish Guidelines for the use of targeted deep sequencing in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2020;188(5):605-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalina T, Flores-Montero J, van der Velden VHJ, et al. ; EuroFlow Consortium (EU-FP6, LSHB-CT-2006-018708) . EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia. 2012;26(9):1986-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Such E, Cervera J, Costa D, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96(3):375-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patnaik MM, Itzykson R, Lasho TL, et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014;28(11):2206-2212. [DOI] [PubMed] [Google Scholar]
- 15.Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122(25):4021-4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itzykson R, Solary E. An evolutionary perspective on chronic myelomonocytic leukemia. Leukemia. 2013;27(7):1441-1450. [DOI] [PubMed] [Google Scholar]
- 17.Itzykson R, Kosmider O, Renneville A, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121(12):2186-2198. [DOI] [PubMed] [Google Scholar]
- 18.Patnaik MM, Padron E, LaBorde RR, et al. Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia. 2013;27(7):1504-1510. [DOI] [PubMed] [Google Scholar]
- 19.Such E, Germing U, Malcovati L, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood. 2013;121(15):3005-3015. [DOI] [PubMed] [Google Scholar]
- 20.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meldi K, Qin T, Buchi F, et al. Specific molecular signatures predict decitabine response in chronic myelomonocytic leukemia. J Clin Invest. 2015;125(5):1857-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bejar R. Myelodysplastic syndromes diagnosis: what is the role of molecular testing? Curr Hematol Malig Rep. 2015;10(3):282-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bejar R, Steensma DP. Recent developments in myelodysplastic syndromes. Blood. 2014;124(18):2793-2804. [DOI] [PubMed] [Google Scholar]
- 24.Papaemmanuil E, Gerstung M, Malcovati L, et al. ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium . Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616-3627, quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurtovic-Kozaric A, Przychodzen B, Singh J, et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia. 2015;29(1):126-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarfi S, Harrivel V, Dumezy F, et al. ; Groupe Francophone des Myélodysplasies (GFM) . Multicenter validation of the flow measurement of classical monocyte fraction for chronic myelomonocytic leukemia diagnosis. Blood Cancer J. 2018;8(11):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talati C, Zhang L, Shaheen G, et al. Monocyte subset analysis accurately distinguishes CMML from MDS and is associated with a favorable MDS prognosis. Blood. 2017;129(13):1881-1883. [DOI] [PubMed] [Google Scholar]
- 29.Picot T, Aanei CM, Flandrin Gresta P, et al. Evaluation by flow cytometry of mature monocyte subpopulations for the diagnosis and follow-up of chronic myelomonocytic leukemia. Front Oncol. 2018;8:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudson CA, Burack WR, Leary PC, Bennett JM. Clinical utility of classical and nonclassical monocyte percentage in the diagnosis of chronic myelomonocytic leukemia. Am J Clin Pathol. 2018;150(4):293-302. [DOI] [PubMed] [Google Scholar]
- 31.Cargo C, Cullen M, Taylor J, et al. The use of targeted sequencing and flow cytometry to identify patients with a clinically significant monocytosis. Blood. 2019;133(12):1325-1334. [DOI] [PubMed] [Google Scholar]
- 32.Tarfi S, Badaoui B, Freynet N, et al. ; Groupe Francophone des Myelodysplasies (GFM) . Disappearance of slan-positive non-classical monocytes for diagnosis of chronic myelomonocytic leukemia with associated inflammatory state. Haematologica. 2019;105(4):e147-e152. [DOI] [PubMed] [Google Scholar]
- 33.Saft L, Björklund E, Berg E, Hellström-Lindberg E, Porwit A. Bone marrow dendritic cells are reduced in patients with high-risk myelodysplastic syndromes. Leuk Res. 2013;37(3):266-273. [DOI] [PubMed] [Google Scholar]
- 34.Kerkhoff N, Bontkes HJ, Westers TM, de Gruijl TD, Kordasti S, van de Loosdrecht AA. Dendritic cells in myelodysplastic syndromes: from pathogenesis to immunotherapy. Immunotherapy. 2013;5(6):621-637. [DOI] [PubMed] [Google Scholar]
- 35.Patnaik MM, Zahid MF, Lasho TL, et al. Number and type of TET2 mutations in chronic myelomonocytic leukemia and their clinical relevance. Blood Cancer J. 2016;6(9):e472-e472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo JU, Su Y, Zhong C, Ming GL, Song H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle. 2011;10(16):2662-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Zhang G, Yi Y, et al. Decreased 5-hydroxymethylcytosine levels are associated with TET2 mutation and unfavorable overall survival in myelodysplastic syndromes. Leuk Lymphoma. 2013;54(11):2466-2473. [DOI] [PubMed] [Google Scholar]
- 40.Figueroa ME, Skrabanek L, Li Y, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood. 2009;114(16):3448-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.