

Key Points
The IRF1/STAT1 pathway is significantly downregulated in belinostat-resistant cells.
Belinostat-resistant cells are hypersensitive to oncolytic reovirus therapy.
Abstract
Despite the promising antilymphoma activity of histone deacetylase (HDAC) inhibitors as a drug class, resistance is a significant clinical issue. Elucidating the molecular mechanisms driving HDAC inhibitor resistance and/or the specific targets that are altered in drug-resistant cells may facilitate the development of strategies that overcome drug resistance and are more effective for refractory patients. We generated novel T-cell lymphoma (TCL) cell line models of acquired resistance to the HDAC inhibitor belinostat to identify potential effective therapies. Belinostat-resistant cells displayed significant cross-resistance to other HDAC inhibitors including romidepsin, panobinostat, and vorinostat. Consistent with a lack of sensitivity to HDAC inhibitors, the resistant cells failed to induce increased acetylated histones. Drug-resistant cells featured significantly decreased expression of the key antiviral mediators IRF1 and STAT1. On the basis of these findings, we investigated the efficacy of the clinical formulation of reovirus (Reolysin) in parental and drug-resistant models. Our investigation revealed that HDAC inhibitor–resistant cells displayed enhanced vulnerability to reovirus replication and cell death in both in vitro and in vivo models compared with their parental counterparts. Importantly, Reolysin also significantly increased the antilymphoma activity of belinostat in HDAC inhibitor–resistant cells. Our data demonstrate that Reolysin alone or in combination with belinostat is a novel therapeutic strategy to treat TCL patients who develop resistance to HDAC inhibitors.
Visual Abstract
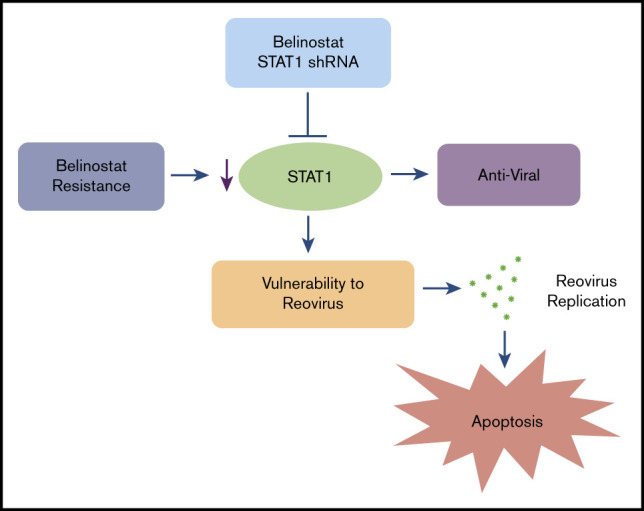
Introduction
Aberrant gene expression plays a pivotal role during the development and progression of many forms of cancer, including T-cell lymphoma (TCL).1 The acetylation status of histones is an important determinant of gene expression and is controlled by 2 opposing classes of enzymes: histone acetyl transferases and histone deacetylases (HDACs). The deacetylation of histones is associated with repression of key tumor suppressor genes and has been linked to HDAC overexpression in multiple forms of cancer including lymphomas.1-3 Based on these findings, several HDAC inhibitors have been approved for therapy of cutaneous T-cell lymphoma and peripheral T-cell lymphoma (PTCL), including belinostat, vorinostat, and romidepsin.4-7
Despite the promising antilymphoma activity of HDAC inhibitors as a drug class, resistance is a significant clinical issue.8,9 Several resistance mechanisms have been identified in preclinical models, including increased expression of multidrug resistance gene 1 (MDR1, ABCB1) and its product P-glycoprotein,9-11 high levels of antiapoptotic proteins (ie, BCL-2, BCL-XL, MCL-1),12-14 alterations in HDAC expression,15,16 and activation of various prosurvival signaling pathways (ie, MAPK, PI3K, NF-κB).17-19 In addition to these mechanisms, tumor cell plasticity enables cancer cells to resist targeted therapies, which is often mediated by epigenetic and transcriptional reprogramming.20,21 However, the development of drug resistance may simultaneously result in specific vulnerabilities that can be exploited therapeutically. Elucidating the molecular mechanisms that drive HDAC inhibitor resistance and/or the specific targets that are altered in drug-resistant cells may facilitate the development of strategies that overcome drug resistance and are effective for refractory patients.
To better understand HDAC inhibitor resistance, we generated 2 TCL models with significant resistance to belinostat. Drug-resistant cells featured dramatically decreased IRF1 and STAT1 levels, key drivers of the antiviral response, suggesting that the resistant cells may be hypersensitive to oncolytic viral therapy. Reovirus is a double-stranded RNA virus that is classified as an orphan virus because of the lack of an association with any known human disease.22,23 Importantly, we and other investigators have shown that reovirus selectively infects and kills malignant cells without harming normal cells, which makes it appealing for therapeutic development.24-26 With the goal of identifying a novel therapeutic approach for HDAC inhibitor–resistant cells, we investigated the efficacy of the proprietary clinical formulation of oncolytic reovirus (Reolysin [Pelareorep]) in parental and HDAC inhibitor–resistant models. Our investigation revealed that belinostat-resistant cells exhibited enhanced reovirus replication and were hypersensitive to Reolysin-induced cell death compared with their parental counterparts. In addition, Reolysin treatment was able to enhance the antilymphoma activity of belinostat in both parental and drug-resistant models. Administration of Reolysin and belinostat to mice bearing TCL xenografts was very well tolerated and significantly antagonized disease progression. Analysis of tumor specimens from mice treated with Reolysin and belinostat demonstrated significant in vivo reovirus accumulation, decreased tumor cell proliferation, and increased apoptosis. Our collective findings identify a vulnerability to oncolytic viral therapy for patients that develop resistance to HDAC inhibitors. This study also establishes the foundation for further investigation of oncolytic reovirus in the relapsed/refractory TCL patient population or subgroups that have defective STAT1 signaling. We are currently planning an early-phase clinical trial to further test the safety and benefit of this new approach.
Materials and methods
Cells and cell culture
Karpas-299 cells were obtained from the European Collection of Authenticated Cell Cultures (Salisbury, United Kingdom). HuT-78, HuT-102, and MJ cells were purchased from American Type Culture Collection (Manassas, VA). TCL cells were cultured with medium supplemented with 10% fetal bovine serum (FBS) at 37°C with 5% CO2. Belinostat-resistant HuT-78 and Karpas-299 cells were created by repeated exposure of cells to increasing concentrations of belinostat. Both resistant cell lines were continuously cultured in 3 μM belinostat to maintain drug resistance. Before conducting all experiments in this study, cells were removed from belinostat-containing media for 1 week. Cell lines were authenticated by using short tandem repeat DNA profiling techniques.
Chemicals and reagents
Belinostat, romidepsin, panobinostat, and vorinostat were purchased from AdooQ Bioscience (Irvine, CA). Ruxolitinib was obtained from SelleckChem (Houston, TX). Reolysin was kindly provided by Oncolytics Biotech, Inc. (Calgary, AB, Canada). Propidium iodide (PI) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma (St Louis, MO).
Quantification of drug-induced cytotoxicity
Cell viability was assessed by MTT assay as previously described.27 The proapoptotic effects of belinostat and Reolysin were quantified by PI staining and fluorescence-activated cell sorting (PI-FACS) analysis of sub-G0/G1 DNA and quantification of active caspase-3–positive cells by flow cytometry using a commercial kit (BD Biosciences, San Jose, CA).
Immunoblotting and antibodies
Immunoblotting was performed as previously described.28 Antibodies were obtained from the following sources: anti-Ac-H2A, anti-Ac-H2B, anti-Ac-HH3, anti-Ac-HH4, anti-cleaved caspase-3, anti-p-STAT1, anti-STAT1, anti-IRF1, anti-HDAC3, and anti-β-actin from Cell Signaling (Danvers, MA); anti-JAM-A was purchased from Abcam (Cambridge, MA); anti-proliferating cell nuclear antigen (PCNA) from Dako (Glostrup, Denmark); goat anti-rabbit horseradish peroxidase (HRP)–conjugated secondary antibody from The Jackson Laboratories (West Grove, PA); rat anti-mouse immunoglobulin G2a (IgG2a)-HRP from Serotec (Raleigh, NC); and sheep anti-mouse-HRP and donkey anti-rabbit-HRP from Amersham (Pittsburgh, PA).
RNA isolation and expression arrays
Total RNAs were isolated from belinostat-resistant and parental Karpas-299 and HuT-78 cells using the RNeasy Plus Mini Kit (QIAGEN, Rockville, MD) and treated with a TURBO DNA-free Kit (Applied Biosystems, Foster City, CA). In all, 300 ng of total RNA per sample was amplified and hybridized to GeneChip Human Gene 1.0 ST arrays (Affymetrix, Inc., Santa Clara, CA) according to the manufacturer’s instructions. Affymetrix CEL files were imported into Partek Genomics Suite 6.4 (Partek Inc, St Louis, MO) using the default Partek normalization parameters and the robust multiarray average analysis adjusted for probe sequence and guanine and cytosine robust multiarray average content. Data normalization was performed across all arrays using quantile normalization. Finally, significantly expressed genes between parental and drug-resistant cells were searched in the DAVID database system to identify the most affected pathways as previously described.29
Quantitative real-time PCR
Complementary DNA (cDNA) from belinostat-resistant and parental Karpas-299 and HuT-78 cells was used for relative quantification by real-time polymerase chain reaction (PCR) analyses. First-strand cDNA synthesis was performed with 1 μg RNA in a 20-μL reaction mixture using the high-capacity cDNA Reverse Transcription Kit (Applied Biosystems). STAT1 and IRF1 transcripts were amplified using commercially available TaqMan gene expression assays (Applied Biosystems). Relative gene expression was calculated with the 2ΔΔCt method.30 β-Actin was used as a housekeeping gene.
Lentiviral shRNA gene silencing
Karpas-299 and HuT-78 cells were infected with lentivirus encoding a short hairpin RNA (shRNA) sequence specific for STAT1 or a nontargeted control (Origene, Rockville, MD) according to the manufacturer’s instructions. We tested 3 different STAT1 constructs, all of which displayed similar levels of silencing. Thus, STAT1 shRNA #1 (Origene) was used for all experiments. Infected cells were selected with green fluorescent protein expression using flow cytometry. STAT1 knockdown was confirmed by immunoblotting. An shRNA pool (Santa Cruz Biotech, Santa Cruz, CA) was used to silence HDAC3 in the HuT-78R and Karpas-299R cells. The knockdown efficiency in HDAC3 levels was determined by immunoblotting.
Transmission electron microscopy
Karpas-299 and HuT-78 cells were treated with 45 plaque-forming units (PFUs) of Reolysin per cell and 90 PFUs of Reolysin per cell, respectively, for 48 hours and were processed for electron microscopy as previously described.31 Xenograft tumor samples were collected at the end of the animal study and processed as previously described.31 The number of viral particles per cell was quantified by using ImageJ software (National Institutes of Health, Bethesda, MD).
ChIP assay
The ab-500 chromatin immunoprecipitation (ChIP) Kit (Abcam) was used according to the manufacturer’s instructions. Briefly, chromatin from 1 × 106 HuT-78 and Karpas-299 parental and belinostat-resistant cells was used for each immunoprecipitation. To shear DNA fragments ranging from 200 to 500 bp, we used the Diagenode SA Picoruptor (Denville, NJ) for 13 cycles with 30 seconds on and 60 seconds off. After sonication, sheared chromatin was diluted as per protocol and subjected to immunoprecipitation with antibodies against IRF1, STAT1, and normal rabbit IgG from Cell Signaling. Histone H3 was used as a positive control (Abcam). After immunoprecipitation, DNA was extracted and purified. The chromatin immunoprecipitates for the indicated antibodies were analyzed by using PCR with the following primers: IRF1 promoter32; forward: 5-CTTCGCCGCTAGCTCTACAACAG-3; reverse: 5-GCTCCGGGTGGCCTCGGTTCG-3; STAT1 promoter; SIB_forward: 5-CACCTAACGTGCTGTGCGTAG-3; SIB_reverse: 5-TAAGCCCTTCCATCTTTGAACATAGAAACA-3.
In vivo evaluation of belinostat and Reolysin combination
Human Karpas-299 parental and belinostat-resistant (2.0 × 107) cells were mixed 1:1 in Hanks’ balanced salt solution and Matrigel (Corning, NY) and implanted into 6-week-old female NOD-SCID mice (The Jackson Laboratories). When tumors reached a volume of ∼300 mm3, mice were randomly assigned to experimental groups (n = 10) and treatment was initiated. Mice were treated with 50 mg/kg belinostat intraperitoneally once per day for 5 days per week, 5.0 × 106 TCID50 Reolysin intratumorally once per week, or the combination. Tumor volume and animal weight were measured twice per week as previously described.33 Mice were euthanized at the end of treatment, and tumors were excised for further analysis. Paraffin-embedded tumor sections from each treatment cohort were used to stain for PCNA and cleaved caspase-3 as previously described.27
Statistical analyses
Power calculations were performed to determine the appropriate sample size for all experiments included in this study. Ten mice per group were used in the animal studies on the basis of statistical considerations. Statistical significance between samples was determined by using the Student t test or the 2-way analysis of variance multiple comparison test as appropriate. Differences were considered significant in all experiments at P < .05 with 2-sided comparisons.
Results
Belinostat-resistant TCL cells display cross-resistance to other HDAC inhibitors and exhibit reduced histone acetylation after belinostat treatment
TCL cell lines commonly used in preclinical studies all display high sensitivity to HDAC inhibition (supplemental Figure 1A-B). The lack of available TCL models with low intrinsic sensitivity to HDAC inhibitors has made it challenging to identify promising agents or strategies for TCL patients who are relapsed/refractory to this drug class in the preclinical setting. To specifically address this issue and to identify potential precision actionable targets in HDAC inhibitor–resistant cells, we generated 2 novel TCL cell line models with acquired HDAC inhibitor resistance through repeated exposure of parental HuT-78 and Karpas-299 cells to belinostat (Figure 1A). Initial characterization of these new models showed that resistance was associated with reduced induction of apoptosis (Figures 1B-C) and a lack of accumulation of acetylated histones (Figure 1D). Notably, belinostat-resistant cells displayed significant cross-resistance to other HDAC inhibitors, including romidepsin, panobinostat, and vorinostat (supplemental Figure 2A-C). These data suggest that TCL patients for whom an HDAC inhibitor regimen fails may not benefit significantly from subsequent treatment with other drugs of this class. Further analyses revealed that both belinostat-resistant cell lines displayed significant overexpression of HDAC3 (supplemental Figure 3) compared with their parental counterparts. Importantly, knockdown of HDAC3 partially reversed belinostat resistance in both drug-resistant TCL models (supplemental Figure 4A-B). Although the development of resistance to HDAC inhibitors is clearly multifaceted, upregulation of HDAC3 significantly contributes to drug resistance in our TCL cell lines.
Figure 1.

Belinostat-resistant TCL cells do not undergo apoptosis and exhibit reduced histone acetylation after belinostat treatment. (A) Generation of novel belinostat-resistant TCL models. HuT-78 and Karpas-299 parental and belinostat-resistant cells were treated with the indicated concentrations of belinostat for 72 hours. Cell viability was measured by 3-MTT assay. Data are shown as mean ± standard deviation (SD) (n = 3). (B-C) Measurement of apoptosis in TCL parental and belinostat-resistant cells. Induction of apoptosis was measured by active caspase-3 assay (B) and propidium iodide–fluorescence-activated cell sorting (PI-FACS) (C) analysis after treatment with the indicated concentrations of belinostat for 48 hours. Data are shown as mean ± SD (n = 3). *P < .05 indicates a significant difference from parental cells treated with the same concentration of belinostat. (D) Belinostat-resistant TCL cells display reduced histone acetylation after belinostat treatment. Acetylation of histones was measured in parental and resistant cells by immunoblotting after 24 hours of belinostat treatment.
Belinostat-resistant TCL cells exhibit downregulated IRF1 and STAT1 expression
Transcriptome analysis was conducted to further characterize the belinostat-resistant cells and to identify potential targetable vulnerabilities to overcome drug resistance. Lists of genes that were significantly altered in HuT-78R and Karpas-299R compared with parental controls are provided (supplemental Tables 1 and 2). Further analysis identified genes and pathways that were commonly modulated in both belinostat-resistant cell lines (supplemental Tables 3 and 4). Genes involved in the antiviral response pathway were significantly altered, with notable reductions in STAT1 and IRF1 levels in both the HuT-78 and Karpas-299 belinostat-resistant cells despite the diverse genetic backgrounds of their parental counterparts (Figure 2A). The decrease in STAT1 and IRF1 levels was confirmed by real-time quantitative (q)PCR (Figure 2B-C) and immunoblotting (Figure 2D). Furthermore, ChIP analyses confirmed that STAT1 and IRF1 transcriptional activity were repressed in the resistant cells (Figure 2E). IRF1 binding was reduced in HuT-78R and Karpas-299R vs parental cells by 70% and 94%, respectively. STAT1 binding was also decreased by 88% in HuT-78R and 67% in Karpas-299R cells vs parental controls. Collectively, these results establish a link between STAT1 pathway repression and belinostat resistance.
Figure 2.
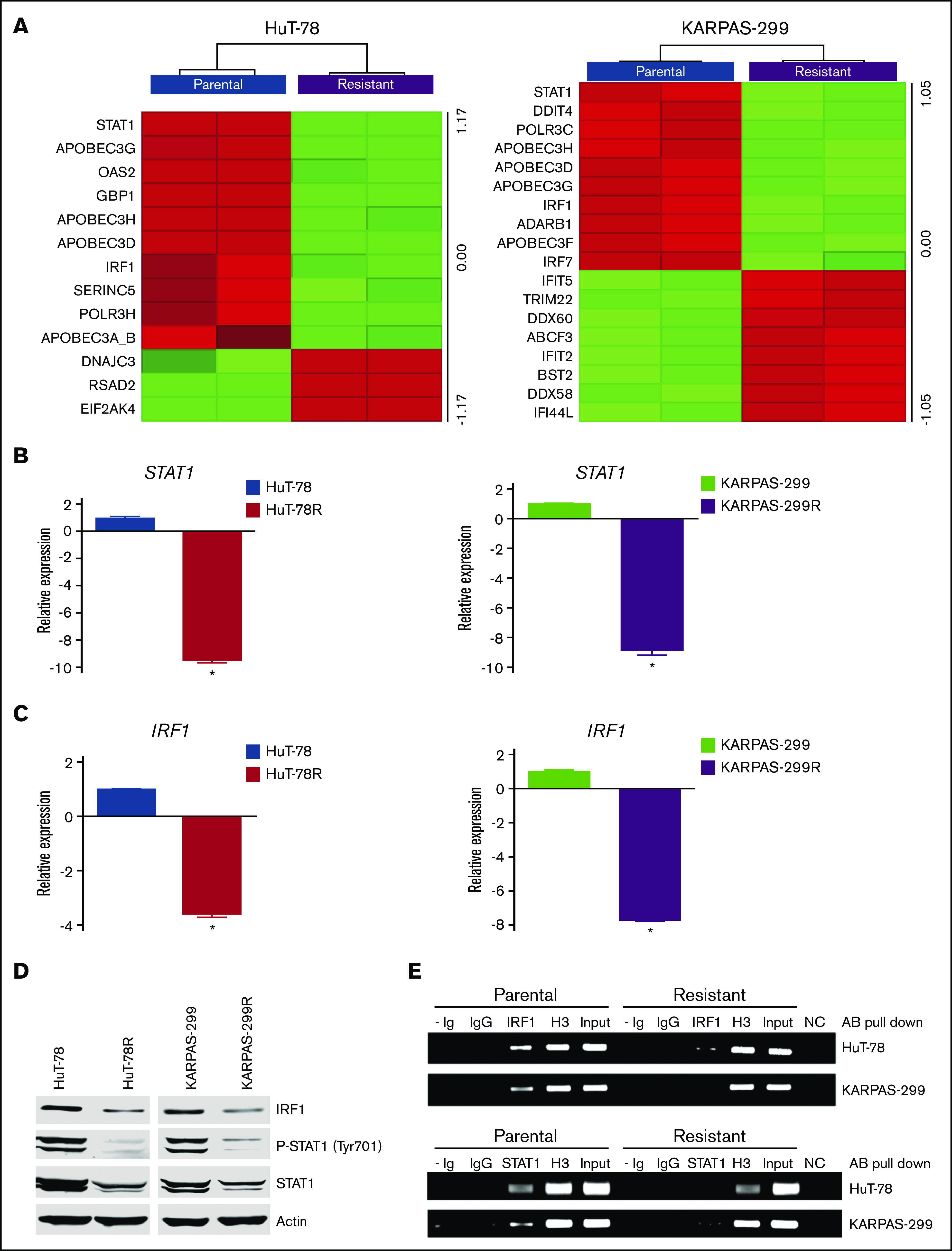
Belinostat-resistant TCL cells exhibit downregulated IRF1 and STAT1 expression. (A) Transcriptome analysis of HuT-78 and Karpas-299 parental and resistant cell lines reveal significant alterations in antiviral genes, including reductions in STAT1 and IRF1. Gene expression changes were determined by using Affymetrix expression arrays. Virus-related genes with significant induction/repression are illustrated in the heat maps. (B-C) Real-time qPCR analysis of STAT1 and IRF1 levels. STAT1 and IRF1 expression was measured by real-time qPCR in parental and belinostat-resistant TCL cells. Data are shown as mean ± SD (n = 3). *P < .05 indicates a significant difference compared with parental cells. (D) STAT1 and IRF1 protein expression is decreased in belinostat-resistant TCL cells. IRF1, phospho-STAT1, and STAT1 protein expression was determined by immunoblotting. (E) Belinostat-resistant cells show reduced IRF1 and STAT1 promoter site binding. ChIP assays were used to quantify the binding of IRF1 and STAT1 to their respective promoters. Absence of Ig (- Ig) served as a negative control, and normal rabbit IgG was used as an isotype control. Histone H3 (H3) served as a positive control, and chromatin input was used as a loading control. NC represents a negative control for the PCR reaction.
Belinostat-resistant TCL cells are hypersensitive to oncolytic reovirus replication and Reolysin-mediated cell death
Because STAT1 is a critical mediator of antiviral response, we hypothesized that belinostat-resistant cells may be particularly vulnerable to oncolytic viral therapy. In agreement with this hypothesis, belinostat-resistant TCL cells were hypersensitive to reovirus replication compared with their parental counterparts (Figure 3A). Quantification of viral load in TCL cells revealed that the belinostat-resistant cells were dramatically more vulnerable to reovirus infection (Figure 3B). Reovirus particles were approximately threefold higher in Karpas-299–resistant cells and eightfold higher in HuT-78–resistant cells compared with their parental counterparts. Consistent with increased viral replication in the resistant cells, oncolytic reovirus treatment significantly decreased cell viability (Figure 3C) and induced apoptosis (Figure 3D) more effectively in the belinostat-resistant cells. Upregulation of the reovirus receptor junctional adhesion molecule-A (JAM-A) has previously been identified as a key mediator of sensitivity to reovirus infection and cell death.34,35 To determine whether JAM-A expression was altered in parental vs resistant cells, we measured its expression by immunoblotting. JAM-A levels were similar in expression in both parental and belinostat-resistant cells, suggesting that differences in receptor expression were not leading to enhanced reovirus susceptibility in our models (supplemental Figure 5).
Figure 3.
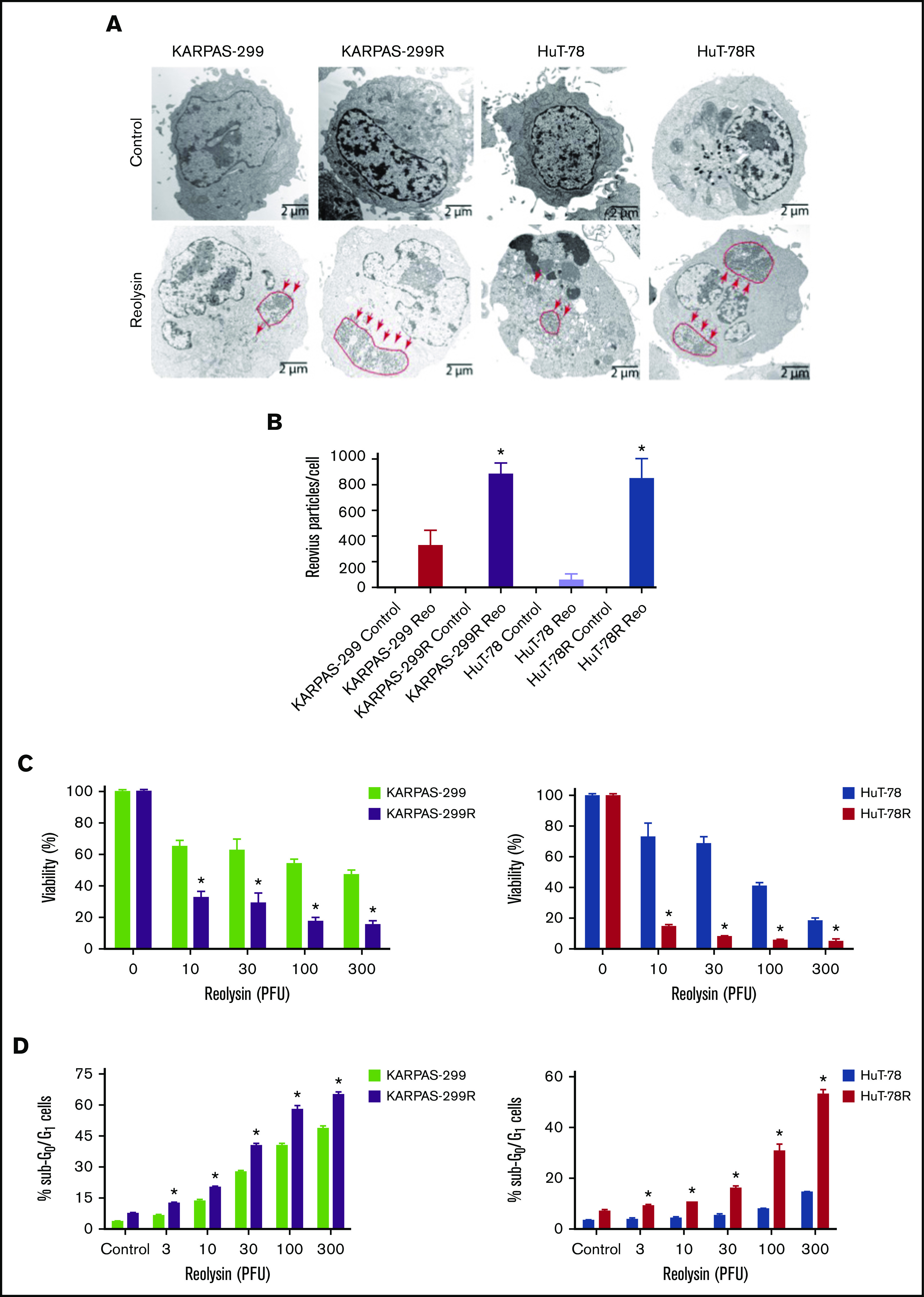
Belinostat-resistant TCL cells are hypersensitive to oncolytic reovirus replication and Reolysin-mediated cell death. (A) Reovirus viral load is significantly greater in belinostat-resistant TCL cells. Karpas-299 and HuT-78 cells were treated for 48 hours with 45 and 90 PFUs of Reolysin per cell, respectively. Reovirus accumulation was visualized by electron microscopy (arrows indicate reovirus accumulation). (B) Quantification of reovirus in parental and belinostat-resistant TCL cells. The number of reovirus particles per cell was quantified using ImageJ software. Data are shown as mean ± SD (n = 5). *P < .05 indicates a significant difference compared with parental cells treated with Reolysin. (C) Belinostat-resistant cells are hypersensitive to Reolysin-mediated cell death. Belinostat-resistant and parental cells were treated with the indicated amounts of Reolysin, and cell viability was determined by MTT assay after 72 hours of treatment. Data are shown as mean ± SD (n = 3). *P < .05 indicates a significant difference compared with parental cells treated with the same concentration of Reolysin. (D) Belinostat-resistant cells display increased sensitivity to Reolysin-mediated apoptosis. TCL cells were treated with Reolysin for 48 hours, and apoptosis was quantified by PI-FACS analysis in parental and belinostat-resistant Karpas-299 and HuT-78 cells. Data are shown as mean ± SD (n = 3). *P < .05 indicates a significant difference compared with parental cells.
Targeting STAT1 enhances the antilymphoma activity of oncolytic reovirus
To confirm the mechanistic relationship between STAT1 levels and Reolysin efficacy, STAT1 was silenced in the parental Karpas-299 and HuT-78 TCL cells using lentiviral shRNA (Figure 4A). As expected, knockdown of STAT1 significantly enhanced the anticancer activity of Reolysin in the parental TCL cell lines (Figure 4B). Consistent with the enhanced anticancer activity of Reolysin in TCL cells with knockdown of STAT1, we also observed significantly increased reovirus replication by transmission electron microscopy in cells with diminished STAT1 expression (Figure 4C). We next evaluated whether pharmacologic inhibition of JAK/STAT signaling with the JAK inhibitor ruxolitinib could also augment Reolysin-mediated cell death. Treatment with ruxolitinib significantly enhanced the efficacy of Reolysin in both TCL models (Figure 4D). Collectively, our data demonstrate that TCL cells with low STAT1 expression are hypersensitive to oncolytic reovirus therapy.
Figure 4.
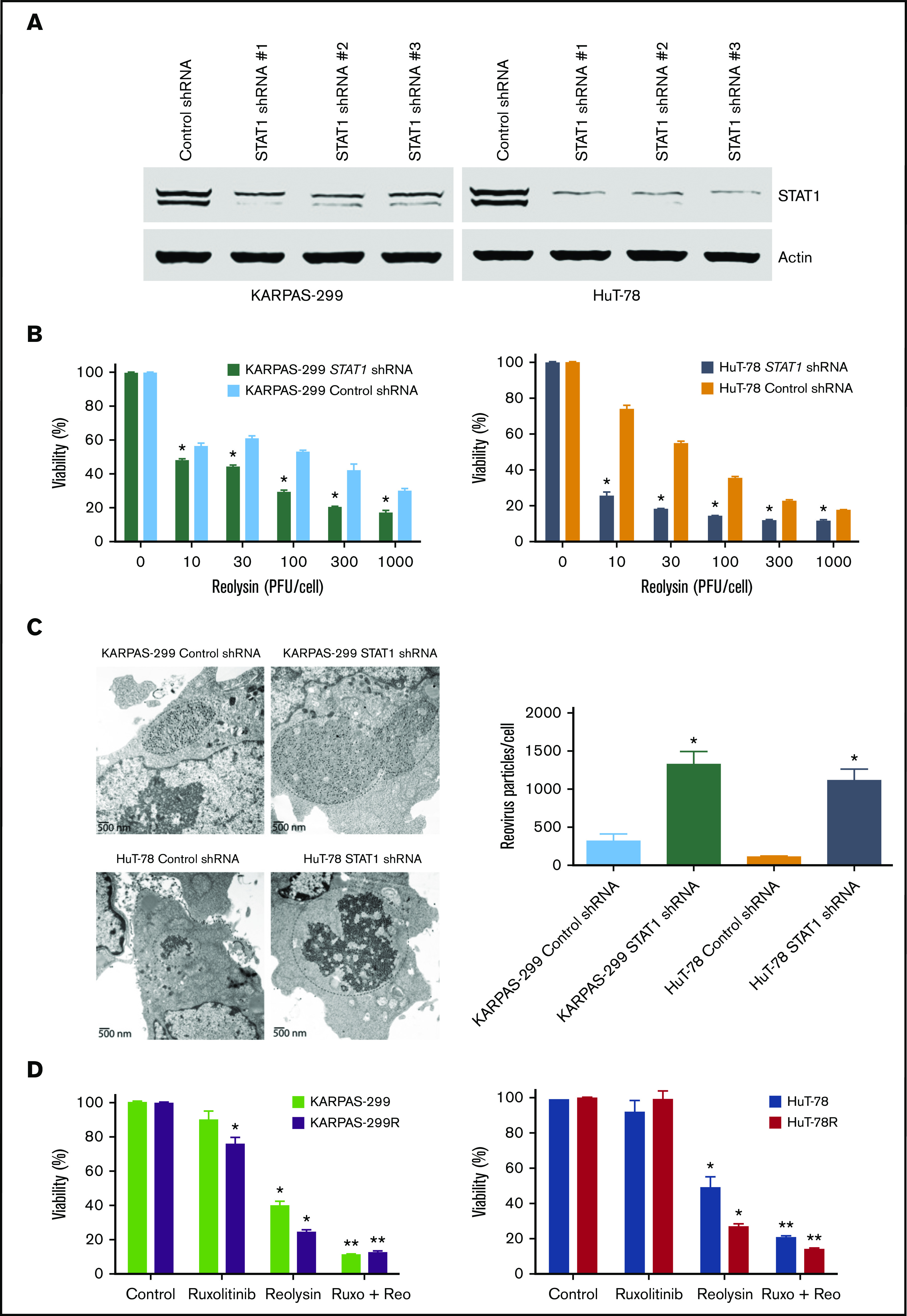
Targeting STAT1 enhances the antilymphoma activity of oncolytic reovirus. (A) Silencing STAT1 expression in TCL cell lines. STAT1 was knocked down in parental Karpas-299 and HuT-78 cells using 3 different lentiviral shRNA constructs, and efficiency was assessed by immunoblotting. STAT1 shRNA #1 was used for all experiments. (B) Reolysin exhibits greater antilymphoma activity in TCL cells with diminished STAT1 levels. TCL cells infected with Control and STAT1 shRNA were treated with the indicated concentrations of Reolysin for 72 hours, and cell viability was determined by MTT assay. Data are shown as mean ± SD (n = 3). *P < .05 indicates a significant difference compared with Control shRNA cells treated with the same concentration of Reolysin. (C) Reovirus viral load is significantly greater in TCL cells with STAT1 knockdown. Karpas-299 and HuT-78 cells were treated for 48 hours with 45 and 90 PFUs Reolysin per cell, respectively. Reovirus accumulation was visualized by electron microscopy. Circles indicate reovirus accumulation. Quantification of reovirus in Control and STAT1 shRNA TCL cells. The number of reovirus particles per cell was quantified using ImageJ software. Data are shown as mean ± SD (n = 5). *P < .05 indicates a significant difference compared with Control shRNA cells treated with Reolysin. (D) Treatment with the JAK inhibitor ruxolitinib sensitizes TCL cells to Reolysin treatment. Parental and belinostat-resistant Karpas-299 and HuT-78 cells were treated with 45 and 90 PFUs Reolysin per cell, respectively, 5 μM ruxolitinib, or the combination for 72 hours, and cell viability was measured by MTT assay. Data are shown as mean ± SD (n = 3). *P < .05 indicates a significant difference compared with Control or **P < .05 for treatment with either ruxolitinib or Reolysin as a single agent.
Belinostat decreases IRF1 and STAT1 expression and significantly enhances the antilymphoma activity of Reolysin
The striking downregulation of IRF1 and STAT1 in our models of acquired belinostat resistance suggested that belinostat treatment triggers IRF1/STAT1 repression. To determine whether acute belinostat treatment was sufficient to decrease IRF1 and STAT1 levels, parental TCL cells were treated with belinostat for 24 hours, and IRF1 and STAT1 expression was determined by immunoblotting. STAT1 and IRF1 levels were significantly decreased after short-term belinostat treatment (Figure 5A). This indicated that combination therapy with belinostat and Reolysin may be effective in both the newly diagnosed HDAC inhibitor–sensitive and relapsed/refractory setting. We next evaluated whether the combination of Reolysin and belinostat would exhibit enhanced anticancer activity in both parental and resistant TCL cell lines. Co-treatment with both agents resulted in significantly decreased cell viability in parental and belinostat-resistant cell lines (Figure 5B). In addition, the combination also promoted increased apoptosis compared with either single-agent treatment (Figure 5C). We further investigated the efficacy, safety, and tolerability of this combination treatment in parental and belinostat-resistant xenograft models. Mice were administered vehicle control, belinostat (50 mg/kg intraperitoneally once per day), Reolysin (5.0 × 106 TCID50 intratumorally once per week), or the combination. Disease burden and animal weight were measured twice per week. As expected, belinostat did not significantly reduce tumor burden in the belinostat-resistant xenograft model, but it possessed significant activity against parental tumors (Figure 6A). Reolysin displayed dramatic antilymphoma activity in both models with greater activity observed in the belinostat-resistant tumors (Figure 6A). Importantly, the combination of belinostat and Reolysin significantly reduced tumor burden in both parental and belinostat-resistant models (Figure 6A). In addition, all treatments were very well tolerated; we did not observe any adverse effects on the mice or significant differences in animal weight (Figure 6B).
Figure 5.
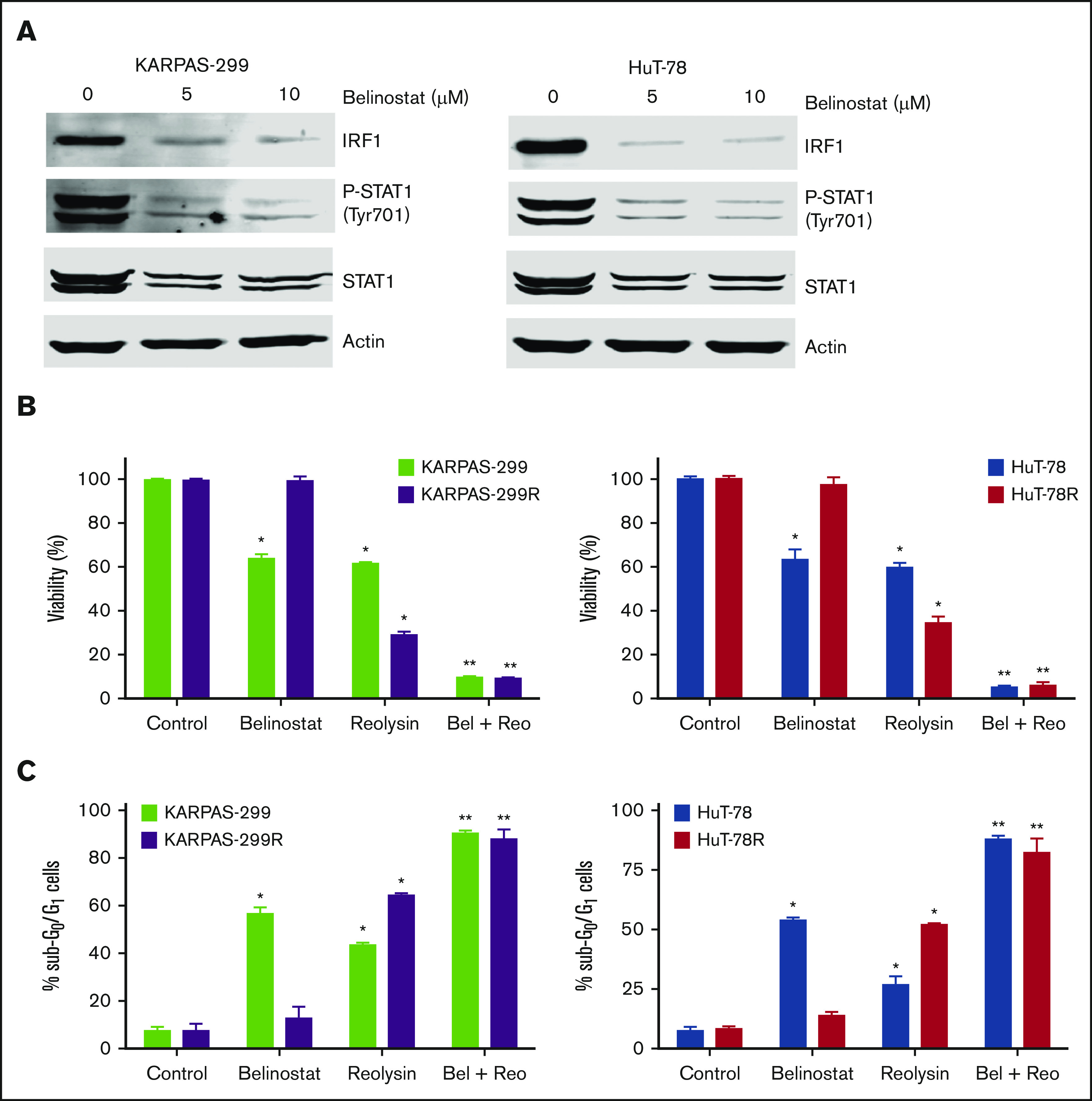
Belinostat decreases STAT1 and IRF1 expression and significantly enhances the antilymphoma activity of Reolysin. (A) Belinostat treatment decreases STAT1 and IRF1 expression. Parental TCL cells were treated with 5 and 10 μM belinostat for 24 hours. STAT1 and IRF1 expression were determined by immunoblotting. (B-C) Reolysin enhances the activity of belinostat in both parental and belinostat-resistant TCL cells. Cells were treated with 0.25 μM belinostat and 45 or 90 PFUs Reolysin per cell, respectively, for Karpas-299 and HuT-78 and the combination for 72 hours (B) or 48 hours (C). Cell viability was determined by MTT assay and apoptosis was measured by PI-FACS analysis. Data are shown as mean ± SD (n = 3). *P < .05 indicates a significant difference compared with Control or **P < .05 compared with either monotherapy.
Figure 6.
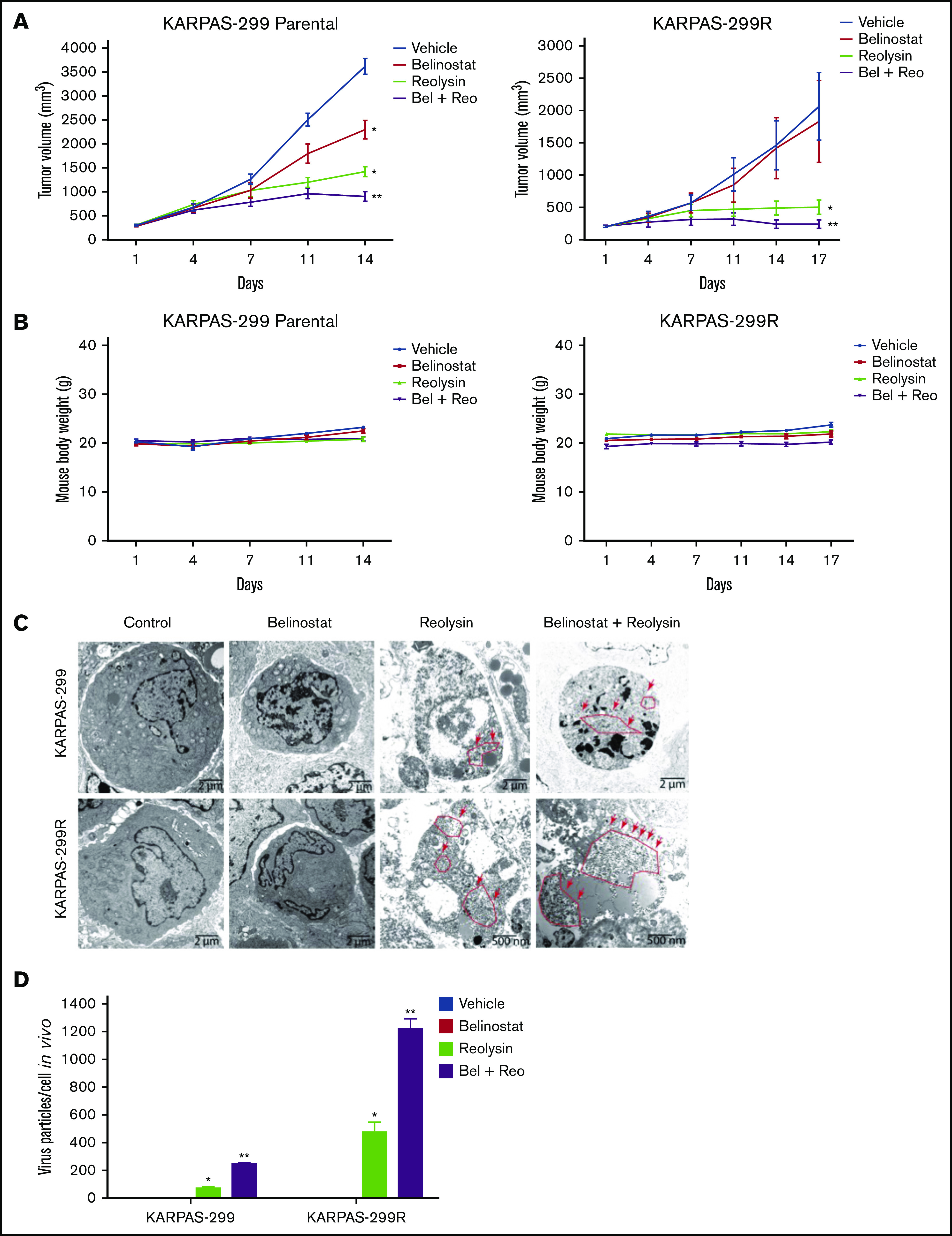
The combination of Reolysin and belinostat results in significantly enhanced antilymphoma activity and reovirus replication in parental and belinostat-resistant Karpas-299 tumors. (A) Reolysin enhances the antilymphoma activity of belinostat in vivo in both Karpas-299 parental (left) and belinostat-resistant tumors (right). Karpas-299 parental and belinostat-resistant tumors were established in NOD-SCID mice. Mice were treated with 5.0 × 106 TCID50 Reolysin once per week intratumorally, 50 mg/kg belinostat once per day intraperitoneally, or the combination of both agents. Tumor volume was measured twice per week. Data are shown as mean ± SEM (n = 10). *P < .05 indicates a significant difference compared with vehicle control or **P < .05 compared with either belinostat or Reolysin single agent treatment. (B) The combination of Reolysin and belinostat is well tolerated in mice. Parental and belinostat-resistant Karpas-299 tumors were established in immunodeficient NOD-SCID mice. Mice were then treated with 5.0 × 106 TCID50 Reolysin, 50 mg/kg belinostat, and the combination. Mouse body weight was measured twice per week. Data are shown as mean ± SD (n = 10). No significant animal weight loss was observed in any of the groups. (C) Reovirus accumulation is significantly greater in belinostat-resistant tumors and when given in combination with belinostat. Tumors were harvested at the end of treatment, and reovirus replication was visualized by electron microscopy. Arrows denote reovirus particles. (D) Quantification of reovirus particles inside parental and belinostat-resistant Karpas-299 tumors. Reovirus accumulation was determined by counting viral particles using ImageJ software. Data are shown as mean ± SD (n = 3). *P < .05 indicates a significant difference from vehicle or belinostat or **P < .05 for treatment with Reolysin monotherapy.
Combined Reolysin and belinostat results in significantly enhanced viral replication, decreased tumor cell proliferation, and augmented apoptosis in Karpas-299 tumors
To further evaluate the antilymphoma effects of Reolysin and belinostat in vivo, we conducted pharmacodynamic analyses by using specimens obtained from all treatment groups at the study end point. We first evaluated in vivo reovirus replication by transmission electron microscopy. Consistent with our in vitro findings, oncolytic reovirus accumulation was significantly more pronounced in belinostat-resistant tumors (Figure 6C). Importantly, treatment with belinostat further enhanced reovirus tumor load in both parental and belinostat-resistant tumors (Figure 6D). The increase in reovirus accumulation was also associated with inhibition of tumor cell proliferation (Figure 7A) and induction in apoptosis (Figure 7B) as measured by PCNA and cleaved caspase-3 immunohistochemistry, respectively.
Figure 7.
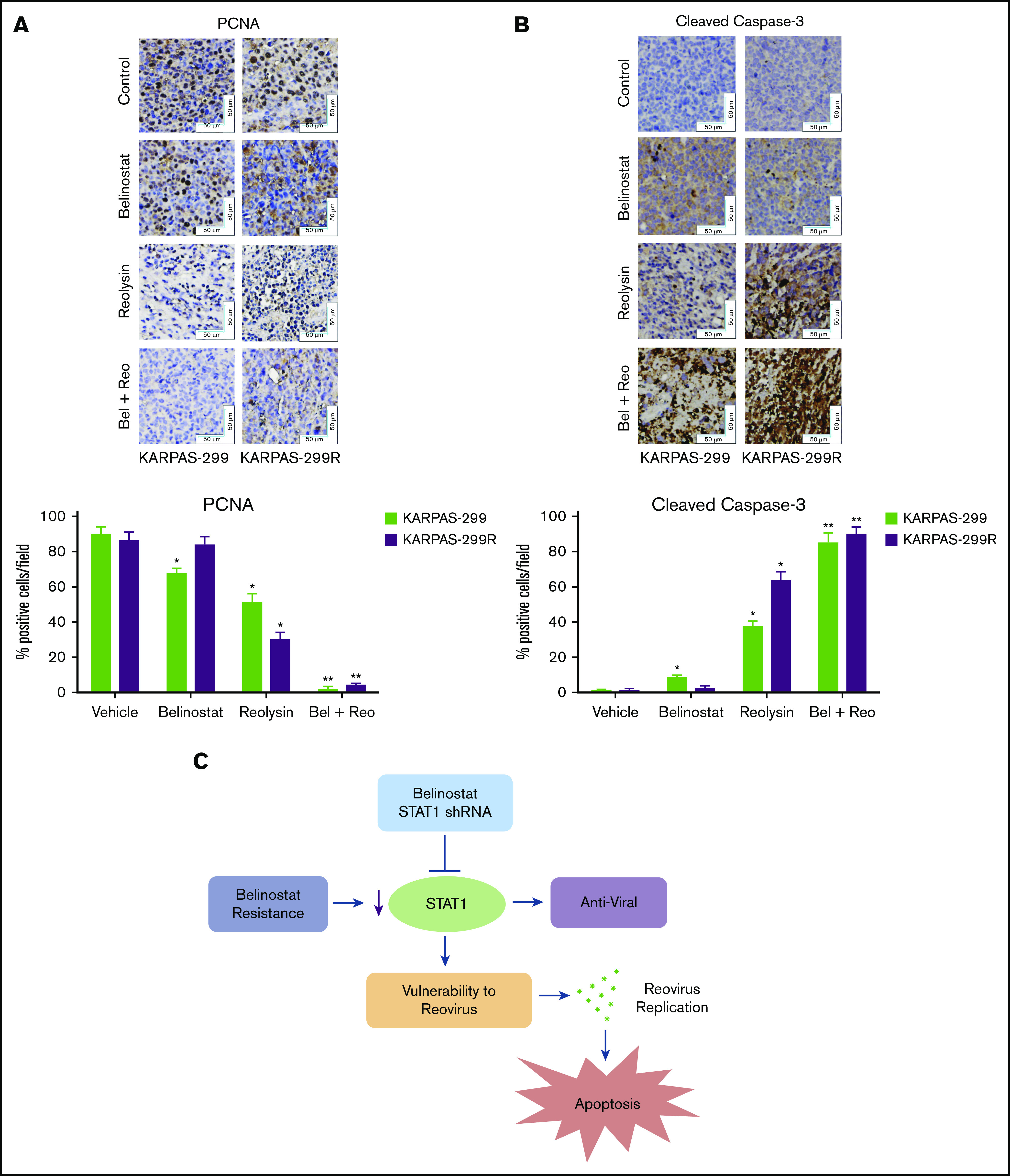
The combination of Reolysin and belinostat results in significantly decreased tumor cell proliferation and augmented apoptosis in parental and belinostat-resistant Karpas-299 tumors. (A-B) Immunohistochemical analysis demonstrates that administration of Reolysin and belinostat effectively decreases PCNA-positive cells (A) and induces significant levels of caspase-3–positive cells (B). Percent PCNA and active caspase-3–positive cells were determined by manual counting. Data are shown as mean ± SD (n = 3 random fields). *P < .05 indicates a significant difference from vehicle or **P < .05 for treatment with either belinostat or Reolysin as a single agent. Scale bars represent 50 μm. (C) Belinostat-induced reduction in STAT1 expression increases TCL cell vulnerability to oncolytic reovirus-mediated cell death. The development of belinostat resistance, acute treatment with belinostat, or STAT1 shRNA infection decreases the expression of STAT1. Oncolytic reovirus replicates significantly more effectively in belinostat-resistant cells and when administered in combination with belinostat resulting in enhanced antilymphoma activity.
Discussion
Frequently used front-line therapies for PTCL include cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (EPOCH), and cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with high-dose methotrexate and cytarabine (hyper-CVAD).6 Unfortunately, relapse is common after treatment, and options for the relapsed/refractory patient population are limited.36 HDAC inhibitors such as belinostat and romidepsin have demonstrated significant promise for the treatment of relapsed/refractory PTCL, but the development of drug resistance continues to be a major obstacle.6 Resistance to HDAC inhibitors is a complex phenomenon, and multiple mechanisms of drug resistance have been previously described.1 Interestingly, the development of belinostat resistance in our drug-resistant cell lines was associated with increased expression of HDAC3. We plan to evaluate whether alterations in HDAC3 contribute to belinostat resistance in clinical samples in a future investigation. Given the poor prognosis of patients with PTCL, new therapeutic approaches are desperately needed. Although drug resistance limits the efficacy of targeted agents, new vulnerabilities may be produced that can be therapeutically targeted. Here we demonstrate that TCL cells with acquired resistance to belinostat exhibit significantly decreased IRF1 and STAT1 expression. Dysregulation of STAT1 signaling has been associated with both prodeath and prosurvival processes, depending on the tumor type and specific conditions.37 However, the role of STAT1 in interferon signaling and stimulation of an antipathogen (viral and microbial) response is well established.38 Indeed, STAT1 knockout mice are especially susceptible to viral infections and tumor formation.39,40 Because belinostat-resistant TCL cells display significant downregulation of STAT1, we hypothesized that the resistant cells may be hypersensitive to oncolytic viral therapy.
Reolysin is a clinical formulation of oncolytic reovirus that has been established to be safe and well tolerated in multiple clinical studies.41,42 Previous investigations have demonstrated that its selective replication properties are attributable to an activated RAS pathway.24,43 Our parental and belinostat-resistant TCL cell line models exhibit constitutive RAS activity because HuT-78 cells possess mutant NRAS (Q61K), and Karpas-299 cells carry the NPM-ALK fusion protein, which leads to downstream activation of the RAS pathway.44,45 Another significant mediator of reovirus replication is increased expression of the reovirus cell surface receptor JAM-A.35 Interestingly, JAM-A levels were previously reported to be upregulated after treatment with an HDAC inhibitor leading to sensitization to reovirus therapy.46 However, we did not observe a significant difference in JAM-A expression levels between parental and belinostat-resistant TCL cells. This may be a result of differences in the cancer cell types that were evaluated or that JAM-A expression is not upregulated during the development of acquired resistance to belinostat. These results led us to further investigate the role of STAT1 in susceptibility to reovirus-mediated cell death in TCL models.
Some TCLs have been reported to display defective interferon signaling, including the failure to induce various antiviral proteins.47 Interestingly, this process was selected for following repeated exposure to belinostat and the development of resistance. The inability of these cells to mount an effective antiviral response renders them vulnerable to oncolytic reovirus therapy. Our results demonstrate that Reolysin may be an effective agent against belinostat-resistant TCL and select patients with defective STAT1 signaling. It will be important to investigate whether STAT1 is also decreased in TCL patients that receive belinostat treatment or develop resistance to HDAC inhibitors. We are currently evaluating this phenomenon in clinical specimens. Indeed, STAT1 expression has been reported to be an important host defense mechanism that can reduce the effectiveness of oncolytic virus therapy.48 Consistent with this idea, treatment with a JAK inhibitor significantly enhanced the antilymphoma activity of Reolysin in parental TCL cell lines. These results also indicate that combination treatment with Reolysin and JAK inhibitors may be effective in TCL cells with high basal STAT1 signaling.
Although Reolysin demonstrated dramatic efficacy as a monotherapy, particularly in the belinostat-resistant TCL models, it is best used clinically in combination with conventional chemotherapy.25,49,50 This is a result of the ability of standard chemotherapy to blunt the effect of antireovirus neutralizing antibodies, enhance viral replication, and augment the proapoptotic anticancer response. In addition, Reolysin is extremely well tolerated and does not exacerbate dose-limiting toxicities of other agents. Interestingly, Reolysin was also effective at enhancing the activity of belinostat in both parental and belinostat-resistant TCL models. Our collective findings establish that the evolution of HDAC inhibitor resistance in TCL is linked to the development of an immunosuppressed phenotype characterized by STAT1 repression in a manner that renders them hypervulnerable to oncolytic virus therapy (Figure 7C). Furthermore, because acute treatment with belinostat in sensitive cells also effectively reduces STAT1 expression, our findings provide the scientific rationale for clinical testing of the combination of Reolysin and belinostat for patients with TCL who are relapsed/refractory to frontline therapy irrespective of previous HDAC inhibitor exposure.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by grants from the National Institutes of Health, National Cancer Institute (NCI) (R01CA190789 [S.T.N.] and R01CA172443 [J.S.C.]), and by an award from the NCI Cancer Center Support Grant developmental funds of the University of Arizona (P30CA023074).
Authorship
Contribution: S.I. performed research and contributed to data analysis and manuscript preparation; C.M.E. performed research and contributed to data analysis; D.O.P. contributed to data analysis; J.S.C. and S.T.N. designed and directed the study and contributed to data analysis and manuscript preparation; and all authors approved the manuscript before submission.
Conflict-of-interest disclosure: D.O.P. received funding from Merck and Spectrum, honoraria from Genentech, and consultant fees from Morphosys. The remaining authors declare no competing financial interests.
Correspondence: Steffan T. Nawrocki, Department of Medicine, Translational Medical Oncology, University of Arizona Cancer Center, 1515 N Campbell Ave, PO Box 245024, Tucson, AZ 85724; e-mail: snawrocki@arizona.edu.
References
- 1.Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008;269(1):7-17. [DOI] [PubMed] [Google Scholar]
- 2.Wilson AJ, Byun DS, Popova N, et al. . Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281(19):13548-13558. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Z, Yamashita H, Toyama T, et al. . HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res. 2004;10(20):6962-6968. [DOI] [PubMed] [Google Scholar]
- 4.Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs. 2007;16(7):1111-1120. [DOI] [PubMed] [Google Scholar]
- 5.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12(10):1247-1252. [DOI] [PubMed] [Google Scholar]
- 6.Lee HZ, Kwitkowski VE, Del Valle PL, et al. . FDA approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res. 2015;21(12):2666-2670. [DOI] [PubMed] [Google Scholar]
- 7.Grant C, Rahman F, Piekarz R, et al. . Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev Anticancer Ther. 2010;10(7):997-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JH, Choy ML, Marks PA. Mechanisms of resistance to histone deacetylase inhibitors. Adv Cancer Res. 2012;116:39-86. [DOI] [PubMed] [Google Scholar]
- 9.Robey RW, Chakraborty AR, Basseville A, et al. . Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol Pharm. 2011;8(6):2021-2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frommel TO, Coon JS, Tsuruo T, Roninson IB. Variable effects of sodium butyrate on the expression and function of the MDR1 (P-glycoprotein) gene in colon carcinoma cell lines. Int J Cancer. 1993;55(2):297-302. [DOI] [PubMed] [Google Scholar]
- 11.Mickley LA, Bates SE, Richert ND, et al. . Modulation of the expression of a multidrug resistance gene (mdr-1/P-glycoprotein) by differentiating agents. J Biol Chem. 1989;264(30):18031-18040. [PubMed] [Google Scholar]
- 12.Vrana JA, Decker RH, Johnson CR, et al. . Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. 1999;18(50):7016-7025. [DOI] [PubMed] [Google Scholar]
- 13.Shao W, Growney JD, Feng Y, et al. . Activity of deacetylase inhibitor panobinostat (LBH589) in cutaneous T-cell lymphoma models: Defining molecular mechanisms of resistance. Int J Cancer. 2010;127(9):2199-2208. [DOI] [PubMed] [Google Scholar]
- 14.Inoue S, Walewska R, Dyer MJ, Cohen GM. Downregulation of Mcl-1 potentiates HDACi-mediated apoptosis in leukemic cells. Leukemia. 2008;22(4):819-825. [DOI] [PubMed] [Google Scholar]
- 15.Fiskus W, Rao R, Fernandez P, et al. . Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood. 2008;112(7):2896-2905. [DOI] [PubMed] [Google Scholar]
- 16.Bandyopadhyay D, Mishra A, Medrano EE. Overexpression of histone deacetylase 1 confers resistance to sodium butyrate-mediated apoptosis in melanoma cells through a p53-mediated pathway. Cancer Res. 2004;64(21):7706-7710. [DOI] [PubMed] [Google Scholar]
- 17.Wozniak MB, Villuendas R, Bischoff JR, et al. . Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica. 2010;95(4):613-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rundall BK, Denlinger CE, Jones DR. Combined histone deacetylase and NF-κB inhibition sensitizes non-small cell lung cancer to cell death. Surgery. 2004;136(2):416-425. [DOI] [PubMed] [Google Scholar]
- 19.Ozaki K, Minoda A, Kishikawa F, Kohno M. Blockade of the ERK pathway markedly sensitizes tumor cells to HDAC inhibitor-induced cell death. Biochem Biophys Res Commun. 2006;339(4):1171-1177. [DOI] [PubMed] [Google Scholar]
- 20.Knoechel B, Roderick JE, Williamson KE, et al. . An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46(4):364-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramirez M, Rajaram S, Steininger RJ, et al. . Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun. 2016;7(1):10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hingorani P, Zhang W, Lin J, Liu L, Guha C, Kolb EA. Systemic administration of reovirus (Reolysin) inhibits growth of human sarcoma xenografts. Cancer. 2011;117(8):1764-1774. [DOI] [PubMed] [Google Scholar]
- 23.Zhao X, Chester C, Rajasekaran N, He Z, Kohrt HE. Strategic combinations: The future of oncolytic virotherapy with reovirus. Mol Cancer Ther. 2016;15(5):767-773. [DOI] [PubMed] [Google Scholar]
- 24.Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282(5392):1332-1334. [DOI] [PubMed] [Google Scholar]
- 25.Kelly KR, Espitia CM, Zhao W, et al. . Oncolytic reovirus sensitizes multiple myeloma cells to anti-PD-L1 therapy. Leukemia. 2018;32(1):230-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly KR, Espitia CM, Mahalingam D, et al. . Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene. 2012;31(25):3023-3038. [DOI] [PubMed] [Google Scholar]
- 27.Carew JS, Espitia CM, Zhao W, et al. . Rational cotargeting of HDAC6 and BET proteins yields synergistic antimyeloma activity. Blood Adv. 2019;3(8):1318-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Islam S, Vick E, Huber B, et al. . Co-targeting aurora kinase with PD-L1 and PI3K abrogates immune checkpoint mediated proliferation in peripheral T-cell lymphoma: a novel therapeutic strategy. Oncotarget. 2017;8(59):100326-100338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Islam S, Paek AL, Hammer M, et al. . Drug-induced aneuploidy and polyploidy is a mechanism of disease relapse in MYC/BCL2-addicted diffuse large B-cell lymphoma. Oncotarget. 2018;9(89):35875-35890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carew JS, Espitia CM, Zhao W, et al. . Disruption of autophagic degradation with ROC-325 antagonizes renal cell carcinoma pathogenesis. Clin Cancer Res. 2017;23(11):2869-2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carew JS, Espitia CM, Zhao W, Mita MM, Mita AC, Nawrocki ST. Oncolytic reovirus inhibits angiogenesis through induction of CXCL10/IP-10 and abrogation of HIF activity in soft tissue sarcomas. Oncotarget. 2017;8(49):86769-86783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartman SE, Bertone P, Nath AK, et al. . Global changes in STAT target selection and transcription regulation upon interferon treatments. Genes Dev. 2005;19(24):2953-2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Islam S, Qi W, Morales C, et al. . Disruption of aneuploidy and senescence induced by aurora inhibition promotes intrinsic apoptosis in double hit or double expressor diffuse large B-cell lymphomas. Mol Cancer Ther. 2017;16(10):2083-2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barton ES, Forrest JC, Connolly JL, et al. . Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104(3):441-451. [DOI] [PubMed] [Google Scholar]
- 35.Kelly KR, Espitia CM, Zhao W, et al. . Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget. 2015;6(38):41275-41289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011;117(25):6756-6767. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Liu Z. STAT1 in cancer: friend or foe? Discov Med. 2017;24(130):19-29. [PubMed] [Google Scholar]
- 38.Kim HS, Lee MS. STAT1 as a key modulator of cell death. Cell Signal. 2007;19(3):454-465. [DOI] [PubMed] [Google Scholar]
- 39.Lesinski GB, Anghelina M, Zimmerer J, et al. . The antitumor effects of IFN-α are abrogated in a STAT1-deficient mouse. J Clin Invest. 2003;112(2):170-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan SR, Vermi W, Luo J, et al. . STAT1-deficient mice spontaneously develop estrogen receptor α-positive luminal mammary carcinomas. Breast Cancer Res. 2012;14(1):R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vidal L, Pandha HS, Yap TA, et al. . A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14(21):7127-7137. [DOI] [PubMed] [Google Scholar]
- 42.Forsyth P, Roldán G, George D, et al. . A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16(3):627-632. [DOI] [PubMed] [Google Scholar]
- 43.Shmulevitz M, Pan LZ, Garant K, Pan D, Lee PW. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Res. 2010;70(12):4912-4921. [DOI] [PubMed] [Google Scholar]
- 44.Kiessling MK, Oberholzer PA, Mondal C, et al. . High-throughput mutation profiling of CTCL samples reveals KRAS and NRAS mutations sensitizing tumors toward inhibition of the RAS/RAF/MEK signaling cascade. Blood. 2011;117(8):2433-2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ceccon M, Merlo MEB, Mologni L, et al. . Excess of NPM-ALK oncogenic signaling promotes cellular apoptosis and drug dependency. Oncogene. 2016;35(29):3854-3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stiff A, Caserta E, Sborov DW, et al. . Histone deacetylase inhibitors enhance the therapeutic potential of reovirus in multiple myeloma. Mol Cancer Ther. 2016;15(5):830-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun WH, Pabon C, Alsayed Y, et al. . Interferon-alpha resistance in a cutaneous T-cell lymphoma cell line is associated with lack of STAT1 expression. Blood. 1998;91(2):570-576. [PubMed] [Google Scholar]
- 48.Goody RJ, Beckham JD, Rubtsova K, Tyler KL. JAK-STAT signaling pathways are activated in the brain following reovirus infection. J Neurovirol. 2007;13(4):373-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carew JS, Espitia CM, Zhao W, et al. . Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013;4(7):e728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Comins C, Spicer J, Protheroe A, et al. . REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010;16(22):5564-5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.