

Abstract
Background
Some patients with cancer treated with anticancer monoclonal antibodies (mAbs) develop antidrug antibodies (ADAs) that recognize and bind the therapeutic antibody. This response may neutralize the therapeutic mAb, interfere with mAb effector function or cause toxicities. We investigated the potential influence of ADA to modify the tumor-binding capability of a tumor-reactive ‘immunocytokine’ (IC), namely, a fusion protein (hu14.18-IL2) consisting of a humanized, tumor-reactive, anti-GD2 mAb genetically linked to interleukin 2. We characterize the role of treatment delivery of IC (intravenous vs intratumoral) on the impact of ADA on therapeutic outcome following IC treatments in an established antimelanoma (MEL) regimen involving radiotherapy (RT) +IC.
Methods
C57BL/6 mice were injected with human IgG or the hu14.18-IL2 IC to develop a mouse anti-human antibody (MAHA) response (MAHA+). In vitro assays were performed to assess ADA binding to IC using sera from MAHA+ and MAHA− mice. In vivo experiments assessed the levels of IC bound to tumor in MAHA+ and MAHA− mice, and the influence of IC route of delivery on its ability to bind to B78 (GD2+) MEL tumors.
Results
MAHA is inducible in C57BL/6 mice. In vitro assays show that MAHA is capable of inhibiting the binding of IC to GD2 antigen on B78 cells, resulting in impaired ADCC mediated by IC. When B78-bearing mice are injected intravenously with IC, less IC binds to B78-MEL tumors in MAHA+ mice than in MAHA− mice. In contrast, when IC is injected intratumorally in tumor-bearing mice, the presence of MAHA does not detectibly impact IC binding to the tumor. Combination therapy with RT+IT-IC showed improved tumor regression compared with RT alone in MAHA+ mice. If given intratumorally, IC could be safely readministered in tumor-bearing MAHA+ mice, while intravenous injections of IC in MAHA+ mice caused severe toxicity. Histamine levels were elevated in MAHA+ mice compared with MAHA− mice after reintroduction of IC.
Conclusions
Intratumoral injection may be a means of overcoming ADA neutralization of therapeutic activity of tumor-reactive mAbs or ICs and may reduce systemic toxicity, which could have significant translational relevance.
Keywords: antibodies, neoplasm, immunity, humoral, immunotherapy, melanoma, translational medical research
Background
Tumor-reactive monoclonal antibodies (mAbs) are a prominent class of anticancer therapeutics that have been developed to recognize clinically relevant antigens selectively expressed on tumor cells. Many mAbs are capable of inducing antibody-dependent cellular-mediated cytotoxicity (ADCC). In ADCC, a mAb binds to its tumor antigen, and the Fragment crystallizable (Fc) portion remains free to interact with Fc receptor (FcR)-expressing innate immune cells, such as natural killer cells, promoting tumor cell lysis.1 2 Immunocytokines (ICs) are genetically engineered fusions of a mAb and cytokine. These molecules can target tumors as they stimulate immune cells, enhancing the ability of ADCC effectors to mediate tumor cell lysis.3 4 One factor limiting the use of mAb and IC therapies is their intrinsic immunogenicity. Immunogenicity is defined as the ability of a substance to provoke an immune response.5 The higher the immunogenicity of any drug, the more likely the treated patient will develop antidrug antibodies (ADAs). ADAs are endogenous antibodies that target therapeutic ‘drugs’, including mAbs. The strength of the endogenous ADA response is multifactorial and impacted by patient characteristics, molecule design, molecule aggregation, timing, dosage, and route of delivery.6–10 If an ADA response is robust, it can interfere with the pharmacokinetics of the drug it recognizes and/or block its desired functions; this is described as drug ‘neutralization’. We previously demonstrated the induction of a neutralizing ADA response following intravenously administered immunocytokine (IV-IC) in mice11 and in some human patients treated with IV-IC.2 Though certain subsets of ADA may enhance therapeutic function through cross-linking at the cell surface or binding and prolongation of systemic exposure,12 13 we will focus this paper on neutralizing ADA.
We hypothesize that the level of ADA is dependent on both the drug administered and the route of administration. Currently, little is known about the immunogenicity of intratumoral delivered immunocytokine (IT-IC) and whether ADA specific for the IC influences the antitumor efficacy of IT-IC. In this study, we have used a mouse model to study the production and function of ADA, as we have observed in human patients,2 in order to test strategies that may mitigate the neutralizing effects of ADA. Due to cross-species reactivity, we have used a syngeneic murine model to reliably generate a strong production of ADA on exposure to humanized mAbs and ICs. Our model should replicate the situation for patients at the far end of the spectrum of strong ADA production who might be most impacted by ADA-associated drug neutralization. We use the term mouse anti-human antibodies (MAHAs) to refer to ADA produced by mice against humanized mAb or IC. Furthermore, we have already developed a murine therapeutic approach combining local radiotherapy (RT) with IT-IC.14 This regimen induces potent destruction of GD2+ tumors, such as B78 melanoma (MEL), in mice.14 This established murine anti-MEL regimen allows for the study of the induction of anti-IC antibodies and enables us to compare the antitumor binding and antitumor efficacy of intratumoralversus intravenous administered IC in mice that are MAHA+ or MAHA−. Herein, we report our investigations comparing the influence of intratumoral versus intravenous injection on MAHA-mediated neutralization and toxicity of IC.
Materials and methods
Study design
GD2+ B78 murine MEL cells were cultured in vitro in RPMI supplemented with 10% fetal bovine serum (FBS) and 1% penicillin streptomycin antibiotics at 37°C and 5% CO2. M21 human MEL cells (GD2+) were cultured as previously published.2 4 B78 and M21 cells were obtained as a gift from Dr Ralph Reisfeld (Scripps Research Institute, La Jolla, California, USA).
We obtained female C57BL/6 mice 5–7 weeks old from Taconic Laboratories. All mice were handled according to University of Wisconsin-Madison Research Animal Resource Center (RARC) guidelines and housed in university approved facilities. Mouse blood was collected in accordance with RARC guidelines via submandibular vein bleed. Serum was obtained using serum collection tubes followed by centrifugation and was stored at −80°C until use.
IC and mAb
MAHA was generated by intradermal (ID) injection of 15 µg hu14.18-IL2 IC (by Apeiron Biologics of Vienna Austria) diluted in commercial-grade phosphate-buffered saline (PBS) for five consecutive daily doses, as previously published.11 For certain treatment groups, soluble interleukin (IL)-2 was added to hu14.18K322A (St. Jude Children’s Research Hospital, Memphis, Tennessee, USA14) at equivalent dosages to that present in comparable doses of IC. Murine IgG2a anti-GD2 mAb, 14G2a (Dr Ralph Reisfeld, Scripps Research Institute), and rituximab (commercial grade from the UW Pharmacy) were also used.
Detection of IC bound to tumor in vivo
A cohort of C57BL/6 mice in each experiment was immunized as stated previously to induce a MAHA+ response. After confirmation of MAHA+ status using ELISA, all mice were engrafted with 2×106 B78-MEL cell ID on the right flank. Once tumors reached the target volume (200 mm3), mice were randomized and treated with 50 µg IC by intratumoral or intravenous injections. Mice exhibiting dyspnea due to suspected anaphylaxis were placed in an oxygen chamber until 1 hour, at which time all mice were sacrificed by CO2. Blood was extracted via cardiac bleed, and tumors were harvested using a surgical blade and dissociated to extract live cells. The disaggregated tumor cell preparation was FcR blocked with anti-mouse CD16/CD32 Fc block (Tonbo Biosciences #70–0161 U500) and stained with anti-mouse CD45-FITC (Tonbo Biosciences #35–0451 U100), anti-Human IgG-AF647 (Life Technologies #A21445), and anti-human IL-2-PE (Biolegend Mq1-17H12). 4′,6-diamidino-2-phenylindole (DAPI) was added for live cell identification, and samples were analyzed using a ThermoFisher Attune NxT flow cytometer. Tumor-bound ICs were identified as CD45−, huIgG+ or huIL2+ populations. Fluorescence minus one controls were used.
Binding inhibition flow cytometry
Mouse anti-GD2 mAb 14G2a-PE (Biolegend #357304) was added to either MAHA+ or naive mouse serum (1–10 μL) to a total volume of 50 µL of 2% FBS flow cytometry buffer. The light-protected mixture was incubated for 30 min at 4°C. The serum-14G2a mixture was added to GD2+ M21 cells aliquoted to 96-well plates and incubated for 45 min at 4°C, and then analyzed by flow cytometer, as described earlier.
In situ vaccine treatment efficacy
C57BL/6 mice were immunized to induce a MAHA+ response (half of the cohort), and B78-MEL flank tumors were implanted, as previously mentioned. Once the tumors reached a volume of 80–120 mm3 for RT+IT-IC groups, all tumors were treated with 12 Gy external beam RT via an XRAD 320 (Precision X-ray, North Bradford, Connecticut, USA) and custom lead shielding, designated day 1 (D1) of the treatment. MAHA+ and MAHA− mice were treated with RT alone, RT+IT-IC, or RT+IV-IC injection over D6–D10, as published.14 Tumor volume was tracked biweekly using digital calipers and mice were sacrificed if any measure of the tumor exceeded 20 mm or if recommended by veterinary staff. Serum for MAHA detection was collected at three timepoints: a pretreatment time point (D−7 to D0), D22, and after rendering of disease-free status.
ELISAs
MAHA detection
Reagents were added 100 µL/well in 96-well plates, coated overnight at 4°C with 0.1 µg/mL of hu14.18K322A hIgG1 (diluted in PBS buffer, pH 7.4). All wash steps were performed three times with 100 mM Tris 0.05%–Tween 20 (pH 7.4). Plates were blocked for 3 hours using PBS–5% milk, followed by a wash. Serum samples were diluted 1:250 in PBS–0.5% bovine serum albumin (BSA) and added in triplicate for overnight incubation at 4°C. Plates were washed; goat anti-mouse IgG alkaline phosphatase (Sigma ImmunoChemicals #A-4656) was added; and plates were incubated light protected for 2 hours at 20°C. Detection of anti-mouse IgG was emphasized due to the role of IgG in inducing memory and mediating effector functions via FcRs.15 Plates were washed; 100 µL of p-nitrophenylphosphate (PNPP, alkaline phosphatase substrate, S-0942, Sigma) in diethanolamine buffer was added; and plates were incubated light protected for 1 hour at 20°C. The plates were read at 450 nm with a 570 nm reference filter. The standard curve reagent used for this assay was obtained by performing these measurements on serial dilutions of mouse anti-human IgG (BD Pharmingen #555784) in concentrations between 5 and 250 ng/mL in twofold increments.
IC detection
Plates were coated overnight with 2 µg/mL 1A7 mAb at 4°C. The 1A7 mAb is specific for the idiotypical (Id) determinant of the 14.18 anti-GD2 antibody.2 16 The IC detection ELISA was performed similarly to the MAHA detection ELISA, with the following adjustments: serum samples were diluted 1:50 in PBS–0.5% BSA and added to 1A7-coated plates overnight at 4°C. Then 0.035 µg/mL biotinylated anti-human IL-2 (R&D Systems #BAF202) was added, and plates were incubated for 3 hours at 20°C light protected. Extravidin alkaline phosphatase was prepared (Sigma, #14M4781V) and added for 1 hour at 20°C prior to the addition of PNPP in diethanolamine buffer. The standard curve reagent used for this assay was obtained by performing these measurements on serial dilutions of hu14.18-IL2 in concentrations between 0.78 and 50 ng/mL in twofold increments.
Binding inhibition
This method assesses the ability of MAHA to inhibit the specific binding of the IC as previously published.2 Briefly, hu14.18-IL2 was diluted to 12.5 ng/mL and combined with 1 µL of serum from MAHA+ or MAHA− mice for 30 min. This mixture was then added to wells in duplicate, and the assay was conducted as previously published2 using biotinylated goat–anti-human-IL-2 antibody (R&D Systems #BAF202) followed by ExtrAvidien-PE (Sigma). The percent binding inhibition was calculated as previously published.2
Histamine detection
Sera from MAHA+ or MAHA− mice that were subsequently injected with IC by either intravenous or intratumoral injections were tested for histamine production (run in duplicate, n=8 mice per group; HIS ELISA Kit, K4163-100, BioVision). Sera were diluted 1:10 in the sample dilution buffer following the manufacturer’s instructions; optical density was read (SpectraMax). Histamine concentrations were interpolated using standard curves provided in each kit.
ADCC
To evaluate ADCC induced by hu14.18-IL2 and the potential impact of MAHA+ serum on this response, M21 (GD2+) human MEL cells and freshly purified human volunteer peripheral blood mononuclear cells (PBMCs) as ADCC effector cells were analyzed using a 51Cr release cytotoxicity assay as previously described.17 Diluted IC (PBS buffer) was incubated with MAHA+ or naive serum prior to addition to effector cells and 51Cr-labeled M21 cells. Controls included effectors alone and IC alone, each added to the 51Cr-labeled M21 cells. M21 cells incubated with medium alone or with cetrimide detergent represented spontaneous and maximum release, respectively. All samples were analyzed in triplicate in at least two replicate experiments (n=20/group). ADCC calculations were as follows:
Lytic units were defined as the number of effector cells per 106 PBMC, which resulted in 20% lysis of 5×103 target cells.17
Binding inhibition flow cytometry
Mouse anti-GD2 mAb 14G2a-PE (Biolegend #357304) was added to either MAHA+ or naive mouse serum (1–10 μL) to a total volume of 50 µL of 2% FBS flow cytometry buffer. The light-protected mixture was incubated for 30 min at 4°C. The serum-14G2a mixture was added to GD2+ M21 cells aliquoted to 96-well plates and incubated for 45 min at 4°C, and then analyzed by flow cytometer, as described previously. Percent binding inhibition by flow cytometry was calculated similarly to that done for the ELISA binding inhibition assay.2
Statistics
MAHA ELISA results were analyzed using one-way analysis of variance (ANOVA) or mixed effects analysis. Binding inhibition assays and ADCC results were analyzed using an unpaired, non-parametric Mann-Whitney calculation. Wilcoxon rank-sum tests were used to compare serum MAHA levels after IC reintroduction and differences in IC bound to tumor detected by flow cytometry between matched treatment groups. Mixed effects analysis was used to determine differences in tumor growth over time between treatment groups. Histamine ELISA analysis were calculated using Brown-Forsythe and Welch one-way ANOVA test. Survival was analyzed using the Kaplan-Meier method and a log-rank (Mantel-Cox) test.
Results
MAHA is induced in tumor-bearing mice following therapy with IT-IC
To determine the ability of IT-IC to provoke a MAHA response in C57BL/6 mice bearing B78-MEL flank tumors treated with 12 Gy RT and IT-IC,14 serum was collected from mice treated with five daily IT-IC injections of 50 µg of hu14.18-IL2. Serological MAHA levels were elevated in tumor-bearing mice after IT-IC administration relative to naive controls (figure 1A). We next sought to develop a model in which MAHA was produced in non-tumor-bearing mice in order to obtain serial serum samples collected longitudinally for study of MAHA without the need to terminate analyses due to increasing tumor burden and animal sacrifice.
Figure 1.

MAHA can safely be induced in a murine model. (A) MAHA is measurable in tumor-bearing mice after intratumoral injection of IC. Serum was collected in tumor-bearing mice before and after treatment with IC. Mean MAHA levels measured by ELISA from four individual mice performed in triplicate, at D0 (just before initiating IT-IC), D6 and D22 are shown from a representative experiment (of three replicate experiments). (B) MAHA response induced by intraperitoneal injection of 40 µg mAb on D1–D3 compared with PBS control, measured 29 days after initial immunization. Mean and SE are shown; a one-way analysis of variance was used to assess significance. A MAHA detection ELISA was performed using serum collected after treatment with 14G2a (mouse mAb which recognizes GD2), hu14.18K322A (K322A, a humanized mAb identical to the antibody portion of IC), or rituximab (humanized mAb with a similar Fc region to IC), none of which were significantly different from the background signal obtained from mice receiving intraperitoneal PBS. (C) MAHA induced by daily ID injection versus intravenous injection of 15 µg of IC on D1–D5 was compared on days 1–36 (p=0.012, n=7 per group). (D) MAHA responses induced by daily ID injection of 15 µg of IC, hu14.18K322A, or hu14.18K322A+45,000 IU of IL-2 (an equivalent dose to the IL-2 present on 15 µg IC), on D1–D5 were compared (n=7). *P<0.05. Fc, ID, intradermal; IC, immunocytokine; IL-2, interleukin 2; IT-IC, intratumoral delivered immunocytokine; MAHA, mouse anti-human antibody; ns, not significant; PBS, phosphate-buffered saline; RT, radiotherapy.
To evaluate induction of a MAHA response in non-tumor-bearing mice, mice were injected with mAb, or underwent mAb-based therapies, as described in the Materials and methods section. The antibodies that were chosen to analyze induction of a MAHA response were the following, and were chosen in order to test whether mAb construct impacted immunogenicity and subsequent MAHA production: 14G2a (mouse anti-GD2 IgG2a mAb), hu14.18K322A (humanized mAb identical to the antibody portion of IC but has a single amino acid substitution in the Fc component), or rituximab (chimeric anti-huCD20 (not expressed on B78s) mAb with a similar Fc region to IC). All of these mAbs share varying degrees of similarity in their Fab or Fc regions to the IC. IC or mAbs were injected intraperitoneally, intravenously, or ID (not intratumorally, as these mice had no tumors) in order to determine whether the route of injection impacted MAHA production.
When mice were injected with 14.G2a, hu14.18K322A, or rituximab mAb alone by intraperitoneal injection, a detectible MAHA response was not induced (p>0.05, figure 1B). When IC was injected ID or intravenously, a MAHA response was observed relative to naive serum samples taken from the same mice prior to IC injection. ID injection induced higher MAHA levels than intravenous injection (p=0.012, figure 1C). Additionally, ID-IC induced higher MAHA levels than hu14.18K322A, and hu14.18K322A+IL-2 at equivalent doses (figure 1D). ID-mAb alone (hu14.18K322A) did not reliably induce an elevated MAHA response from baseline levels in all mice, reported out to 36 days after the initial injection (4/7 mice had observable change from baseline levels; figure 1D, individual curves shown in online supplemental figure 1). In this model, the IC is more immunogenic than mAb and ID injection is more immunogenic than intravenous injection.
jitc-2020-001262supp001.pdf (50.8KB, pdf)
MAHA+ sera inhibit the in vitro binding of hu14.18-IL2 to GD2 or GD2-like targets
To test the ability of MAHA+ serum to inhibit the binding of the IC to GD2 in vitro we used a 1A7 binding inhibition ELISA. 1A7 is a mouse IgG1 anti-idiotype mAb; its Fab region recognizes the idiotype of the IC, the hu14.18K322A and the 14G2a mAbs. As such, the antigen-binding portions of these anti-GD2 agents (IC, hu14.18K322A and 14G2a) recognize the 1A7 mAb as if it resembles GD2, as previously published.16 17 For this ELISA, MAHA− pretreatment serum was matched with MAHA+ serum from each mouse to determine baseline interference of serum components to the binding of IC to 1A7. When MAHA+ serum was added to IC prior to addition to the 1A7 plate, significant inhibition of IC binding to the plate was observed compared with MAHA− serum (p=0.008, figure 2A). To determine the capability of MAHA+ serum to inhibit IC binding to GD2 in a cellular model, human M21-MEL cells (highly GD2+) were incubated with a mixture of 14G2a-PE mAb+ MAHA+ or MAHA− sera. 14G2a is a mouse IgG2a (which should not be recognized as foreign by C57BL/6 mice) anti-GD2 mAb, and is identical to the Id region of the IC. Thus, any ADA recognizing 14G2a in the serum from MAHA+ mice would be anticipated to be recognizing the Id region of 14G2a. When 14G2a-PE was added to M21 cells in the presence of MAHA+ serum, nearly complete inhibition of 14G2a-PE binding to M21 cells was observed relative to MAHA− control serum (figure 2B). This indicates the MAHA+ sera contains anti-Id neutralizing antibody, that is binding to the Id of the IC and preventing its interaction with the GD2 on the M21 cells.
Figure 2.

Serum containing MAHA inhibits IC in vitro (A) using a binding inhibition detection ELISA; serum from MAHA+ mice or MAHA− naive serum was combined with IC prior to addition to the 1A7 coated plates. Hu14.18-IL2 bound to 1A7 was detected and the percent binding inhibition was calculated for each mouse (p=0.008, n=5 mice per group in each experiment; two replicate experiments were performed, one replicate shown); each dot represents one individual sample, with group mean and SE of the mean shown. Non-parametric t-tests were performed. (B) Human M21 melanoma cells were incubated with a mixture of MAHA+ or MAHA− serum and 14G2a-PE antibody. The Median Fluorescence Intensity (MFI) of 14G2a-PE in the presence of MAHA+ or MAHA− serum was detected by flow cytometry; the binding histograms for a single representative mouse are shown. (C) The cell-mediated cytotoxicity of IC, in an ADCC assay, in the presence of MAHA+ versus MAHA− serum, was determined (p<0.0001). IC concentration of 20 ng/mL was chosen after testing multiple concentrations of IC without serum (not shown). Two separate human effectors were used. Results from one of two replicates are shown with an effector to target ratio of 40:1; total n=40. Welch’s t-test was used to compare groups. **P<0.01, ****P<0.0001. ADCC, antibody-dependent cellular-mediated cytotoxicity; IC, immunocytokine; MAHA, mouse anti-human antibody.
To determine if the anti-Id antibody observed could influence ADCC activity, MAHA+ or MAHA− sera were incubated with a known concentration of IC. These sera/IC samples were then added with effector cells (human PBMCs) to 51Cr-labeled M21 cells. Relative to MAHA− serum, ADCC was significantly reduced in the presence of MAHA+ serum (p<0.0001, figure 2C). Average lytic units (online supplemental figure 2) for IC in the presence of MAHA− serum was 85.74 (95% CI 77.28 to 94.20), and this average was reduced to −0.37 lytic units (95% CI −2.94 to 2.20) in the presence of MAHA+ serum. Collectively, these findings indicate reduced IC function in the presence of MAHA in vitro.
jitc-2020-001262supp002.pdf (23.9KB, pdf)
MAHA does not detectibly dampen delivery of IT-IC to the tumor
MAHA+ and naive (non-immunized MAHA−) mice were engrafted with B78-MEL cells in the right flank and randomized to receive IC (50 µg), either intravenously or intratumorally. Serum samples obtained 1 day before starting IC treatment and 1 hour after IC treatment were assessed for MAHA levels in the MAHA+ cohort (IC treatment was delivered either intravenously or intratumorally). MAHA+ sera showed a significant reduction in detectable MAHA from samples obtained 1 hour after the IC treatment (p=0.004, figure 3). This result suggests that IC in the serum is adsorbing the pre-existing MAHA, leading to a decrease in detectible serum MAHA.
Figure 3.
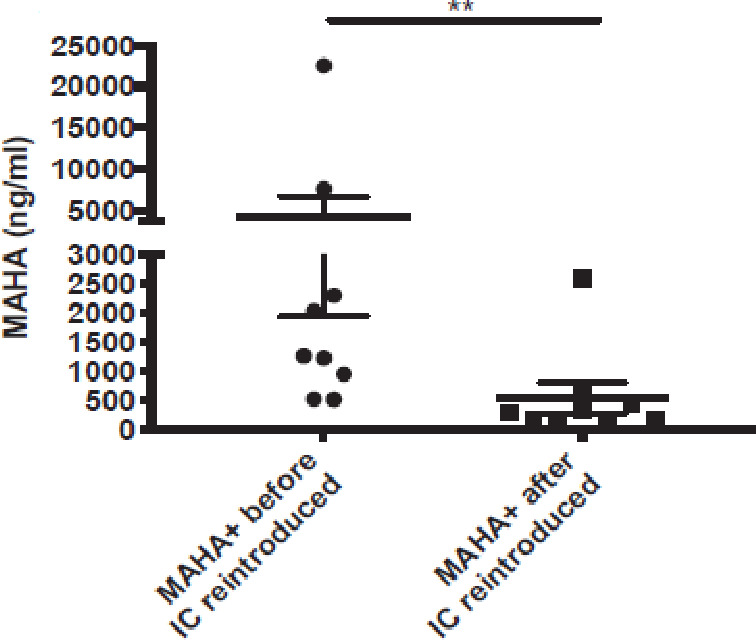
Treatment of MAHA+ mice with IC causes an immediate decrease in detectible MAHA in serum. C57BL/6 mice were immunized with IC as described previously to induce MAHA+ and then engrafted with B78 melanoma. Tumor-bearing MAHA+ mice were treated with a single dose of 50 µg IC half by it and half by intravenous injection. Serum was obtained from all mice 1 day prior to the IC treatment and 1 hour after the IC treatment. these serum samples were then assayed for MAHA levels by ELISA, and the pretreatment samples were compared with all post-treatment samples. The serum obtained just after IC administration showed a significant decrease in detectible MAHA level (p=0.004). Statistics were conducted using Mann-Whitney t-test. **P<0.01. IC, immunocytokine; MAHA, mouse anti-human antibody.
Serum IC levels from these mice were also evaluated. Importantly, MAHA+ mice showed lower serum IC levels than the MAHA− mice, when the IC was injected intratumorally (p=0.02) and when the IC was injected intravenously (p=0.11), though the intravenous comparison did not reach statistical significance likely due to small sample size (online supplemental figure 3). These findings (figure 3 and online supplemental figure 3) indicate the presence of MAHA in the serum at the time of IC treatment can neutralize the IC in the serum, interfering with its detection by ELISA.
jitc-2020-001262supp003.pdf (25.9KB, pdf)
Tumors from these mice were disaggregated and analyzed by flow cytometry for the presence of tumor-bound IC using anti-huIgG and anti-huIL2 detection antibodies. When the IC was given intravenously, the MFI value for IC detected on tumor cells was significantly lower for MAHA+ mice than for MAHA− mice (p=0.02). This may indicate MAHA can neutralize IC in the vascular compartment, mirroring the IC levels shown in online supplemental figure 3, thereby limiting the amount of IC delivered to and bound by tumor cells. Importantly, and in contrast, when IC was administered intratumorally, the MFI value for the IC detected on tumor cells was not different between MAHA+ and MAHA− mice (p=0.49, figure 4). This result is consistent with the hypothesis that the presence of MAHA in the circulation is not interfering with the ability of IC to bind to tumor cells when the IC is injected intratumorally rather than intravenously.
Figure 4.
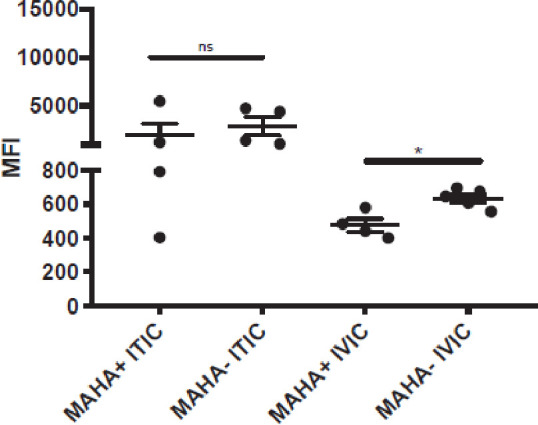
IC delivery to the tumor is impacted by MAHA when given intravenously, but not it C57BL/6 mice were immunized with IC as described previously to become MAHA+. MAHA+ and naive (non-immunized MAHA) mice were engrafted with B78 melanoma cells in the right flank. After tumor detection, MAHA+ and MAHA− mice were randomized into treatment groups. Tumor sizes were comparable across all groups (approximately 200 mm3). Different groups received IV-IC or IT-IC. IC treatment was 50 µg for a single dose. Mice were all sacrificed at the same time point (1 hour after IC administration), at which point tumors were collected for analysis. Tumors were disaggregated and analyzed as described in the Materials and methods section by flow cytometry for the presence of tumor-bound hu-IgG+ and hu-IL2+ populations (anti-hu-IL2 data are shown here; data using hu-IgG+ showed a similar distribution for all groups and are not shown), which were considered reflective of the binding of IC to the B78 tumor. The Median Fluorescence Intensity (MFI) of anti-hu-IL2+, live CD45− populations is reported. *P<0.05. IC, immunocytokine; IT-IC, intratumoral delivered immunocytokine; IV-IC, intravenously administered immunocytokine; MAHA, mouse anti-human antibody; ns, not significant,
IV-IC treatment (but not IT-IC) results in acute toxicity in MAHA+ but not MAHA− mice
These observations led to testing whether MAHA might impact antitumor efficacy of RT+IC anti-MEL treatment. We hypothesized that RT+IT-IC might remain efficacious in MAHA+ mice, based on our observation that tumor-bound IC was not detectibly reduced in MAHA+ intratumorally treated mice (figure 4). C57BL/6 mice were engrafted with B78-MEL tumors as described previously. Tumor growth prior to treatment was not significantly different between the MAHA+ and MAHA− animals (p>0.05, figure 5A). MAHA+ and MAHA− mice were treated with RT alone, RT+IT-IC, or RT+IV-IC. Treatment consisted of RT on D1 and either IT-IC or IV-IC on D6–D10. RT treatment alone was used as a control for tumor growth in the absence of immunotherapy. Within minutes after IC treatment on D6 (the first day of IC treatment), 12 of 13 MAHA+ RT+IV-IC treated mice displayed toxicity for greater than 15 min (dyspnea, hunched posturing, and lethargy) and were euthanized. One of 13 mice displayed signs of illness but eventually recovered to baseline health and was able to receive all five doses of IC. The MAHA− RT+IV IC and MAHA+ RT+IT-IC groups in this study tolerated all 5 days of IC treatment without any signs of severe toxicity as assessed by two blinded investigators. A small proportion of MAHA+ RT+IT-IC mice showed signs of lethargy after IC injection but quickly recovered to baseline health within 15 min of injection. Thus, MAHA+ mice receiving IV-IC had significantly reduced overall survival compared with the MAHA− RT+IV IC group (p<0.0001). These observations were seen in repeated experiments (summarized in table 1), with only 1/32 MAHA+ RT+IT-IC mouse displaying the acute toxicity seen in all but 1 MAHA+ mice treated with RT+IV-IC.
Figure 5.

Intratumoral treatment remains efficacious even in the presence of MAHA. C57BL/6 mice were engrafted with 2 million B78 melanoma cells in the right flank to induce tumor growth. (A) Tumor growth prior to treatment of MAHA+ and MAHA− mice on day 38 after engraftment (p>0.05). Mice were then treated with RT, or RT+IT-IC, or RT+IV-IC. RT is given on day 1, and IC is given on days 6–10. (B) average tumor size for RT and RT+IT IC mice is shown. during the first injection of IC, 12/13 mice in the MAHA+ RT+IV-IC group displayed substantial toxicity requiring euthanasia. as such, survival was significantly different between the MAHA+ RT+IV-IC and all other groups (p<0.0001). Consequentially, it was not possible to evaluate the in vivo antitumor efficacy of IV-IC in MAHA+ mice or compare it to the efficacy of IV-IC in MAHA− mice (and thus these data are not shown). The mean tumor sizes of MAHA+ RT+IT-IC mice are significantly less than those of MAHA+ RT-treated mice (p=0<0.001), indicating additional antitumor efficacy with the addition of IT-IC to RT treatment. MAHA− RT+IT-IC mean tumor volumes are significantly less than those of MAHA− RT groups, consistent with historical controls (p<0.001). One of three replicate experiments is shown; the number of mice per group was 9–16 in total, as represented in the figure. ***P<0.001. IT-IC, intratumoral delivered immunocytokine; IV-IC, intravenously administered immunocytokine; MAHA, mouse anti-human antibody; ns, not significant; RT, radiotherapy.
Table 1.
Intratumoral injection reduces toxicity observed when IC is injected intravenously in MAHA+ mice
| Treatment | Toxicity requiring euthanasia |
| MAHA+ IT-IC | 1/32 |
| MAHA− IT-IC | 0/32 |
| MAHA+ IVIC | 31/32 |
| MAHA− IVIC | 0/32 |
Acute toxicity observed meeting endpoints for euthanasia was stratified by intervention, in multiple experiments, n=32/group. Toxicity was determined by dyspnea, lethargy, hunched posturing, and unresponsiveness to tactile stimuli on days that IC was administered. All animals were assessed by a second blinded observer familiar with objective endpoints determined for this study to confirm the need for euthanasia.
Based on the nature of the toxic symptoms displayed by MAHA+ RT+IV-IC treated mice, we suspected a compromise of pulmonary function and systemic circulation, likely due to anaphylaxis. Thus, we used ELISA to quantify histamine in the serum, a key mediator of anaphylactoid responses in mice.18 Serum histamine levels taken 1 hour after IC injection were elevated in the MAHA+ IT-IC group and the MAHA+ IV-IC group compared with MAHA− mice (p=0.01 and p=0.0088, respectively). There was no significant difference in the histamine levels in MAHA+ IT-IC versus MAHA+ IV-IC mice (figure 6, p=0.5384), suggesting histamine alone does not account for the difference in toxicity observed between these groups.
Figure 6.

MAHA+ mice have increased histamine after IC injection blood was collected by cardiac bleed 1 hour after injection of IC. Serum was isolated and histamine levels were determined by ELISA (n=8/group). No significant difference was detected between MAHA+ IT-IC and IV-IC groups (p=0.54); However, MAHA+ IT-IC and MAHA+ IV-IC histamine levels were significantly elevated compared with MAHA− mice after IC injection (p=0.012 and p=0.0088, respectively). Welch analysis of variance was used to compare groups. *P<0.05, **P<0.01. IC, immunocytokine; IT-IC, intratumoral delivered immunocytokine; IV-IC, intravenously administered immunocytokine; OD, Optical Density; MAHA, mouse anti-human antibody; ns, not significant.
Antitumor efficacy of IT-IC is retained in MAHA+ mice
In MAHA− mice, the addition of IT-IC to RT provides a significant benefit in tumor growth inhibition (p<0.001, figure 5B). Similarly, the addition of IT-IC to RT in MAHA+ mice provides a significant benefit in tumor growth inhibition (p<0.001, figure 5B). The MAHA+ mice treated with IT-IC showed tumor growth inhibition that was not inferior to that seen in the MAHA− mice (figure 5B). Additionally, complete tumor resolution, without tumor regrowth, was observed in 4/9 MAHA+ RT+IT-IC mice as well as in 4/9 MAHA− RT+IT-IC mice. In summary, when treated with RT+IT-IC, MAHA+ mice still achieved improved antitumor efficacy as compared with MAHA+ RT-treated groups. This corresponds with our flow cytometric analysis showing similar levels of IC binding to the tumor, regardless of MAHA status following IT-IC, suggesting MAHA+ does not abrogate the effects of IT-IC in mice treated with RT+IT-IC. MAHA− mice treated with RT had significantly greater mean tumor sizes compared with MAHA− RT+IT-IC treated mice (p<0.001), consistent with historical controls.14
Discussion
We have established a murine model to investigate the influence of the MAHA response seen in mice receiving mAbs or ICs to determine the impact of ADA on therapeutic efficacy and toxicity. In tumor-bearing mice that receive IT-IC, we confirm the generation of MAHA as a by-product of therapy. We show both in vitro and in vivo that MAHA can neutralize the binding of IC to GD2, and verify a reduction in IC-dependent effector function (ADCC) in the presence of MAHA. A reduction in IC binding to the tumor was observed in MAHA+ mice for IV-IC but not for IT-IC. This finding predicted a maintenance of beneficial antitumor effects of RT+IT-IC therapy in MAHA+ mice compared with MAHA+ RT alone, consistent with previous MAHA− experiments.14 We confirmed this prediction using the in vivo conditions here. In addition, intratumoral delivery of IC in MAHA+ mice was well tolerated, while intravenous delivery of IC in MAHA+ resulted in substantial toxicity. These findings demonstrate the potential importance of therapeutic delivery route. Specifically, as studied here, intratumoral injection allows superior efficacy and tolerance to IC over intravenous delivery, in mice with substantial pre-existing ADA against hu14.18-IL2 IC. Furthermore, these studies include analyses in non-tumor-bearing mice and confirm in this setting that ID injection of immune activating agents is more immunogenic than intravenous.6 7 We additionally confirm that the presence of IL-2, an immune activating cytokine, increases the immunogenicity of mAbs.2 To expand on the findings described here, investigations are currently under way to detect ADA using use surface plasmon resonance, a highly sensitive and label-free modality for detecting ADA in the serum.
One mechanism of accelerated clearance of mAb-based therapeutics can be attributed to the formation of ADA–mAb complexes, which are rapidly cleared by the reticuloendothelial system.19 Phagocytic cellular mediators of the reticuloendothelial system predominate in the blood; by using IV-IC in MAHA+ mice, IC is in an environment optimal for clearance. The reduction in detectable serum levels of MAHA from pre-IC to post-IC reintroduction in MAHA+ mice (figure 3) is comparable to that seen for serum samples from human patients that develop a human anti-human antibody (HAHA) response following IV-IC and then show a transient decrease in HAHA when they are retreated with IV-IC.2 These data, taken together with the reduction in detectable IC in the serum in MAHA+ mice (online supplemental figure 3), suggest a process which may be consuming MAHA and IC, such as the formation of immune complexes. This inhibits the detection of MAHA or IC, either through increased clearance of complexes by means of phagocytosis or by blocking the portions of the MAHA or IC which would be recognized by ELISA reagents. This reduction of detectible IC in the serum also translates to a decrease in the amount of functional IC able to bind to the tumor itself. As expected, based on the detectable serum levels of IC in MAHA+ serum, less IV-IC is able to reach the tumor and be detected on tumor cells in MAHA+ mice than in MAHA− mice. However, the amount of IC bound to tumor cells following IT-IC was not detectibly different for MAHA+ versus MAHA− mice (figure 4). This could possibly reflect a lower concentration of MAHA in the tumor interstitial fluid than in the plasma, as well as a far greater concentration of IC in the interstitial space after IT-IC than after IV-IC.20
Given the immediacy of the toxicity observed directly following the administration of IV-IC in MAHA+ mice, we suspect exposure to a large bolus of IV-IC in the presence of MAHA+ leads to acute systemic toxicity. One key pathway mediated by increased platelet-activating factor levels,21 22 and modest histamine elevation is the IgG-mediated anaphylaxis pathway.22 This pathway has similar manifestations to classic anaphylaxis but is mediated by high levels of antigen-specific IgG, which activate neutrophils, basophils, and monocytes23–25 rather than by IgE and mast cells. These responses are classically provoked by large, parenteral doses of antigen in subjects who have known antibodies against that antigen. In this experiment, parenteral intravenous is reintroduced in an anti-IC-rich environment, and anaphylactic-like symptoms result within minutes.
The relative absence of these toxic responses in IT-IC-treated mice is likely multifactorial. When IC is injected intratumorally, the IC is released slowly to the blood relative to when IC is delivered as an intravenous bolus (online supplemental figure 3). As a result, serum levels of IC after intratumoral administration would be expected to reach much lower peak serum levels than with intravenous bolus, comparable to that seen with ID or subcutaneous administration.26 27 In addition, as the IC is slowly released from the treated tumor ‘depot’ into circulation, MAHA may encounter it at the site of vascular entry and neutralize it, resulting in lower levels of IC entering systemic circulation (online supplemental figure 3). This could prevent a strong systemic anaphylactoid response to IC. Furthermore, compensatory mechanisms against characteristic anaphylactoid physiology could have time to counteract the mechanisms of developing toxicity, potentially reducing the strength of the anaphylactic response.
It is difficult to correlate preclinical immunogenicity data to human responses due to the species differences of the treatment molecules and resultant increased foreignness of the drug construct, producing augmented ADA responses. However, HAHAs have been characterized clinically, showing that though cross-species reactivity is important in ADA generation, it is not the only means of immune activation.2 28 The significance of these antibodies in humans requires further study; when meaningful titers are achieved, clinical manifestations of ADA include loss of efficacy, neutralization, and antibody-mediated adverse effects,6 all of which have been observed in the experiments presented in this report. Importantly, in this study, these consequences are all attenuated when the therapeutic antibody is delivered intratumorally.
This finding may have clinical significance, especially for delayed salvage therapies (therapies that are offered once it has been determined that the initial treatment has failed). ADA may have an impact on salvage therapies; therefore, it is important to consider possible mediating factors, such as route of injection selected for salvage therapy. In a metastatic model, it would be intuitive to choose intravenous delivery due to the propensity to deliver antitumor therapy to all distant tumor sites. However, in the case of in situ vaccination,29–32 such as our RT+IC treatment,14 33 in addition to local destruction mediated by ADCC, we seek to turn on a systemic antitumor adaptive immune response propagated by local therapy. Thus, by ensuring adequate delivery of IC to the tumor, even in a single location, through intratumoral delivery, we may be more likely to induce an adaptive systemic response. This has already been observed in a small cohort of patients with metastatic MEL with PD1 therapy resistance, for whom the addition of intratumoral IL-2 resulted in improved systemic tumor control.34 For therapies that use tumor-directed agents like IC or mAbs in regimens designed to locally induce a subsequent systemic immune response, the ability of intratumoral administration to reliably deliver IC to the tumor, even in the face of a potentially neutralizing ADA found in serum, may prove useful.
Conclusions
A further understanding of endogenous antibodies and their interactions with the tumor, the host response, and therapeutic interventions will be helpful in augmenting the immunotherapeutic treatments of certain cancers. With this knowledge, treatment doses, schedules, and even drug design may be optimized. This study provides insight regarding how the route of administration influences IC delivery to the tumor, as well as tumor regression and toxicity in the setting of potent ADA. In the conditions studied here, intratumoral administration provided superior results, compared with intravenous delivery, for all these parameters in MAHA+mice. The potential screening for endogenous ADA in previously treated patients as prognostic markers to aid in choosing the route of drug administration could prevent adverse effects to a treatment that may have a low probability of success if delivered by a suboptimal route. Further cautious testing of these concepts in the clinical setting is warranted.
Acknowledgments
The UWCCC Flow Cytometry Laboratory is supported by the University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520.
Footnotes
Correction notice: This article has been corrected since it was published online. The author name ‘Jacquelyn A Hank’ was incorrectly spelt as ‘Jacqueline A Hank'.
Funding: Claire Baniel is a Howard Hughes Medical Institute medical research fellow. This research was supported by Hyundai Hope on Wheels grant; Midwest Athletes Against Childhood Cancer; University of Wisconsin Carbone Cancer Center; The Crawdaddy Foundation; Children’s Neuroblastoma Cancer Foundation; Stand Up 2 Cancer; the St. Baldrick’s Foundation; Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. This research was also supported in part by public health service grants from the National Cancer Institute (grant numbers U54 CA232568, CA014520, CA166105, and CA197078) and from the National Institutes of Health and the Department of Health and Human Services (grant number TL1 TR002375). Its content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available upon reasonable request from the corresponding author.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
References
- 1.Yang RK, Sondel PM. Anti-Gd2 strategy in the treatment of neuroblastoma. Drugs Future 2010;35:665. 10.1358/dof.2010.035.08.1513490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hank JA, Gan J, Ryu H, et al. Immunogenicity of the hu14.18-IL2 immunocytokine molecule in adults with melanoma and children with neuroblastoma. Clin Cancer Res 2009;15:5923–30. 10.1158/1078-0432.CCR-08-2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sondel PM, Gillies SD. Current and potential uses of immunocytokines as cancer immunotherapy. Antibodies 2012;1:149–71. 10.3390/antib1020149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol 2016;40:96–102. 10.1016/j.coi.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy K. Janeway’s immunobiology. New York: Garland Science, 2012. [Google Scholar]
- 6.Schellekens H. Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transplant 2005;20 Suppl 6:vi3–9. 10.1093/ndt/gfh1092 [DOI] [PubMed] [Google Scholar]
- 7.Ross C, Clemmesen KM, Svenson M, et al. Immunogenicity of interferon-beta in multiple sclerosis patients: influence of preparation, dosage, dose frequency, and route of administration. Danish multiple sclerosis Study Group. Ann Neurol 2000;48:706–12. 10.1002/1531-8249(200011)48:5<706::AID-ANA3>3.0.CO;2-V [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg AS. Effects of protein aggregates: an immunologic perspective. Aaps J 2006;8:E501–7. 10.1208/aapsj080359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang WYK, Foote J. Immunogenicity of engineered antibodies. Methods 2005;36:3–10. 10.1016/j.ymeth.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 10.Fathallah AM, Bankert RB, Balu-Iyer SV. Immunogenicity of subcutaneously administered therapeutic proteins--a mechanistic perspective. Aaps J 2013;15:897–900. 10.1208/s12248-013-9510-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imboden M, Murphy KR, Rakhmilevich AL, et al. The level of MHC class I expression on murine adenocarcinoma can change the antitumor effector mechanism of immunocytokine therapy. Cancer Res 2001;61:1500–7. [PubMed] [Google Scholar]
- 12.Shan D, Ledbetter JA, Press OW. Signaling events involved in anti-CD20-induced apoptosis of malignant human B cells. Cancer Immunol Immunother 2000;48:673–83. 10.1007/s002620050016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan HTC, Hughes D, French RR, et al. CD20-induced lymphoma cell death is independent of both caspases and its redistribution into Triton X-100 insoluble membrane rafts. Cancer Res 2003;63:5480. [PubMed] [Google Scholar]
- 14.Morris ZS, Guy EI, Francis DM, et al. In situ tumor vaccination by combining local radiation and tumor-specific antibody or immunocytokine treatments. Cancer Res 2016;76:3929–41. 10.1158/0008-5472.CAN-15-2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008;8:34–47. 10.1038/nri2206 [DOI] [PubMed] [Google Scholar]
- 16.Foon KA, Sen G, Hutchins L, et al. Antibody responses in melanoma patients immunized with an anti-idiotype antibody mimicking disialoganglioside GD2. Clin Cancer Res 1998;4:1117–24. [PubMed] [Google Scholar]
- 17.Hank JA, Surfus JE, Gan J, et al. Activation of human effector cells by a tumor reactive recombinant anti-ganglioside GD2 interleukin-2 fusion protein (ch14.18-IL2). Clin Cancer Res 1996;2:1951–9. [PubMed] [Google Scholar]
- 18.Wechsler JB, Schroeder HA, Byrne AJ, et al. Anaphylactic responses to histamine in mice utilize both histamine receptors 1 and 2. Allergy 2013;68:1338–40. 10.1111/all.12227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johansson A, Erlandsson A, Eriksson D, et al. Idiotypic-anti-idiotypic complexes and their in vivo metabolism. Cancer 2002;94:1306–13. 10.1002/cncr.10301 [DOI] [PubMed] [Google Scholar]
- 20.Yang RK, Kalogriopoulos NA, Rakhmilevich AL, et al. Intratumoral hu14.18-IL-2 (IC) induces local and systemic antitumor effects that involve both activated T and NK cells as well as enhanced IC retention. J Immunol 2012;189:2656–64. 10.4049/jimmunol.1200934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terashita Z, Imura Y, Shino A, et al. A lethal role of platelet activating factor in anaphylactic shock in mice. J Pharmacol Exp Ther 1987;243:378–83. [PubMed] [Google Scholar]
- 22.Tsujimura Y, Obata K, Mukai K, et al. Basophils play a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity 2008;28:581–9. 10.1016/j.immuni.2008.02.008 [DOI] [PubMed] [Google Scholar]
- 23.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012;119:5640–9. 10.1182/blood-2012-01-380121 [DOI] [PubMed] [Google Scholar]
- 24.Finkelman FD, Khodoun MV, Strait R. Human IgE-independent systemic anaphylaxis. J Allergy Clin Immunol 2016;137:1674–80 https://doi.org/ 10.1016/j.jaci.2016.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jönsson F, Mancardi DA, Kita Y, et al. Mouse and human neutrophils induce anaphylaxis. J Clin Invest 2011;121:1484–96. 10.1172/JCI45232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lansigan F, Nakamura R, Quick DP, et al. Di-Leu16-IL2, an anti-CD20-interleukin-2 immunocytokine, is safe and active in patients with relapsed and refractory B-cell lymphoma: a report of maximum tolerated dose, optimal biologic dose, and recommended phase 2 dose. Blood 2016;128:620 10.1182/blood.V128.22.620.620 [DOI] [Google Scholar]
- 27.Danielli R, Patuzzo R, Di Giacomo AM, et al. Intralesional administration of L19-IL2/L19-TNF in stage III or stage IVM1a melanoma patients: results of a phase II study. Cancer Immunol Immunother 2015;64:999–1009. 10.1007/s00262-015-1704-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ritter G, Cohen LS, Williams C, et al. Serological analysis of human anti-human antibody responses in colon cancer patients treated with repeated doses of humanized monoclonal antibody A33. Cancer Res 2001;61:6851–9. [PubMed] [Google Scholar]
- 29.Shekarian T, Sivado E, Jallas A-C, et al. Repurposing rotavirus vaccines for intratumoral immunotherapy can overcome resistance to immune checkpoint blockade. Sci Transl Med 2019;11:eaat5025. 10.1126/scitranslmed.aat5025 [DOI] [PubMed] [Google Scholar]
- 30.Marabelle A, Tselikas L, de Baere T, et al. Intratumoral immunotherapy: using the tumor as the remedy. Ann Oncol 2017;28:xii33–43. 10.1093/annonc/mdx683 [DOI] [PubMed] [Google Scholar]
- 31.Marabelle A, Kohrt H, Caux C, et al. Intratumoral immunization: a new paradigm for cancer therapy. Clin Cancer Res 2014;20:1747–56. 10.1158/1078-0432.CCR-13-2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sagiv-Barfi I, Czerwinski DK, Levy S, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med 2018;10:eaan4488. 10.1126/scitranslmed.aan4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morris ZS, Guy EI, Werner LR, et al. Tumor-Specific Inhibition of In Situ Vaccination by Distant Untreated Tumor Sites. Cancer Immunol Res 2018;6:825–34. 10.1158/2326-6066.CIR-17-0353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rafei-Shamsabadi D, Lehr S, von Bubnoff D, et al. Successful combination therapy of systemic checkpoint inhibitors and intralesional interleukin-2 in patients with metastatic melanoma with primary therapeutic resistance to checkpoint inhibitors alone. Cancer Immunol Immunother 2019;68:1417–28. 10.1007/s00262-019-02377-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2020-001262supp001.pdf (50.8KB, pdf)
jitc-2020-001262supp002.pdf (23.9KB, pdf)
jitc-2020-001262supp003.pdf (25.9KB, pdf)