

Abstract
Background
Medulloblastoma (MB) is one of the most frequent malignant brain tumors of children, and a large set of these tumors is characterized by aberrant activation of the sonic hedgehog (SHH) pathway. While some tumors initially respond to inhibition of the SHH pathway component Smoothened (SMO), tumors ultimately recur due to downstream resistance mechanisms, indicating a need for novel therapeutic options.
Methods
Here we performed a targeted small-molecule screen on a stable, SHH-dependent murine MB cell line (SMB21). Comprehensive isotype profiling of histone deacetylase (HDAC) inhibitors was performed, and effects of HDAC inhibition were evaluated in cell lines both sensitive and resistant to SMO inhibition. Lastly, distinct mouse models of SHH MB were used to demonstrate pharmacologic efficacy in vivo.
Results
A subset of the HDAC inhibitors tested significantly inhibit tumor growth of SMB21 cells by preventing SHH pathway activation. Isotype profiling of HDAC inhibitors, together with genetic approaches suggested that concerted inhibition of multiple class I HDACs is necessary to achieve pathway inhibition. Of note, class I HDAC inhibitors were also efficacious in suppressing growth of diverse SMO inhibitor‒resistant clones of SMB21 cells. Finally, we show that the novel HDAC inhibitor quisinostat targets multiple class I HDACs, is well tolerated in mouse models, and robustly inhibits growth of SHH MB cells in vivo as well as in vitro.
Conclusions
Our data provide strong evidence that quisinostat or other class I HDAC inhibitors might be therapeutically useful for patients with SHH MB, including those resistant to SMO inhibition.
Keywords: HDAC, medulloblastoma, SMO, sonic hedgehog, resistance
Key Points.
1. Sonic hedgehog medulloblastoma is sensitive to class I HDAC inhibition.
2. HDAC inhibition is efficacious in SMO inhibitor‒resistant medulloblastoma.
3. HDAC inhibitor quisinostat effectively inhibits SHH signaling and tumor growth intracranially in vivo.
Importance of the Study.
More precise treatment options for SHH subgroup MB, including those tumors resistant to SMO inhibitors, will enable improved survival with fewer adverse sequelae. Using a targeted high-throughput drug screen, we identified class I HDAC inhibitors as a major class of compounds capable of inhibiting growth of SHH-driven medulloblastoma cells. Notably, class I HDAC inhibitors, including JNJ-26481585 (quisinostat), were able to overcome multiple, well-documented resistance mechanisms that act at different nodes in the SHH pathway. Therefore, our study redefines HDAC isoform inhibitor capability for targeting SHH signaling and sets the stage for further investigation of class I HDAC inhibitors as a promising therapeutic approach for treating naïve and refractory SHH medulloblastoma.
Brain cancer is the leading cause of cancer-related death in the pediatric population, and the cerebellar tumor medulloblastoma (MB) is the most common malignant brain tumor in children. Intensive treatment has drastically improved prognosis in MB, and approximately 70% of patients can now be cured.1 Unfortunately, these treatments have profound side effects, including reduced cognitive function, hearing loss, and secondary cancers. Thus, there has been a major effort to develop novel, more targeted therapies with fewer side effects.
MB is classified into 4 subgroups that are characterized by distinct gene expression and methylation profiles, mutations, and prognoses: WNT, sonic hedgehog (SHH), group 3, and group 4.2 Tumors of the SHH subgroup exhibit mutations that lead to constitutive activity of the SHH pathway. Recently, the small-molecule inhibitors vismodegib and sonidegib targeting the SHH signaling component Smoothened (SMO) have been shown to prolong time to disease progression in patients with recurrent SHH MB.3 However, a significant proportion of SHH MBs are resistant to treatment with SMO inhibitors at time of diagnosis,4 and long-term utility of SMO inhibitors in treating MB is limited by acquired drug resistance in patients initially responding to these drugs.5,6 These findings illustrate the need for targeted therapeutics that act downstream of SMO to prevent tumor progression in SHH MB, particularly those tumors with poor prognosis due to TP53 mutations or gene amplifications of MYCN and/or GLI2.7,8
Several classes of molecular regulators have been shown to control SHH output both during normal development and SHH MB formation, including G protein-coupled receptors (GPCRs),9,10 kinases,11,12 and epigenetic modifiers.4,13,14 Given the prominent role that these protein classes play within the SHH pathway and in MB growth, we conducted a small-molecule screen of inhibitors targeting these 3 classes of regulators using recently developed, stable, TP53-mutated SHH MB cells. We find that histone deacetylase (HDAC) inhibitors specific for HDAC1 and HDAC2 reduce SHH signaling and are effective at blocking tumor growth in cells that are both responsive and resistant to SMO inhibitors. Furthermore, we demonstrate that JNJ-26481585 (quisinostat), an HDAC inhibitor with marked potency toward class I HDACs and a known safety profile in people, is efficacious in vivo and provides a promising option for treating SHH MB.
Materials and Methods
Animals
Atoh1-cre, SmoM2Fl/+ and nu/nu mice were obtained from The Jackson Laboratory and Charles River Laboratories, respectively.
Tumor Cell Culture
SMB21, SMB55, and SMB56 cell lines were derived from spontaneous MB tumors in Ptch+/− mice; generation and characterization of these cells as well as loss of function mutants from SMB21 cells were previously described.15,16 All cells were cultured as neurospheres in ultra-low attachment culture flasks with Gibco Dulbecco’s modified Eagle’s medium (DMEM)/F12 (2% B27, 1% penicillin/streptomycin).
High-Throughput Small-Molecule Screening
SMB21 cells were seeded in duplicates in 96-well plates (1 × 104 cells per well) by automated cell seeding. Twenty-four hours after seeding, 50 µL of media containing either dimethyl sulfoxide (DMSO) or a small molecule were added to the screening plates. Control compounds were included on every screening plate along with 80 test compounds. After 72 hours, cell viability was measured using CellTiter 96 Aqueous One Solution (Promega), and calculated as a percentage of control (DMSO-treated) cells. Compounds reducing cell viability by more than 50% compared with the DMSO control were considered screen hits.
Generation of the HDAC1/2 Selective Inhibitor DLS-3
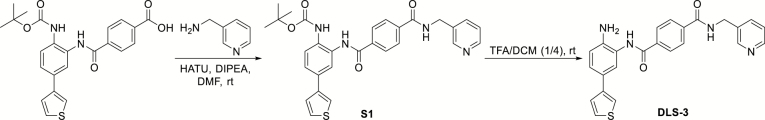
Schematic detailing the synthesis of the HDAC1/2 selective inhibitor DLS-3. For details of the reaction, consult the Supplementary Materials.
Cell Survival Assays with Pharmacologic Inhibition
Cell lines (SMB21 or SMB21-derived mutants) were seeded at a density of 1 × 104 cells in 96-well plates. Drugs were added 24 hours after seeding, and viability was measured 72 hours later. Survival curves were modeled using nonlinear regression with a sigmoid dose response for calculation of half-maximal inhibitory concentration (IC50). HDAC inhibitor OJI-1 was a gift from Aaron Beeler and previously described17; pandacostat was a gift from Jay Bradner18; WT161 was a gift from Ken Anderson19; and Merck 60 was obtained from Merck Pharmaceuticals. Sonidegib (LDE225) and JNJ-26481585 were purchased from Selleck Chemicals.
Plasmids
Specifics for all plasmids used in this study can be seen in the Supplementary Materials.
Lentiviral Production
Lentivirus was generated using human embryonic kidney 293T cells maintained in DMEM (10% heat inactivated fetal bovine serum, 1% penicillin/streptomycin). After transfection, media were collected sequentially over the next 3 days. Viral supernatant was spun at 1100 rpm to remove cell debris and then concentrated via filter centrifugation at 2000 g.
Analysis of Cell Cycle Distribution and Apoptosis
For cell cycle analysis, tumor cells were treated with quisinostat (hereafter called JNJ) for 2 h, fixed in 100% ethanol, and subsequently stained with propidium iodide/RNase staining solution (Cell Signaling). The cells were analyzed in a BD LSR Fortessa. For analysis of apoptosis, cells were treated with JNJ for indicated periods and subsequently analyzed using the Abcam annexin V‒fluorescein isothiocyanate apoptosis detection kit according to the manufacturer’s protocol. All flow cytometry analyses were performed with FlowJo.
HDAC Isoform Inhibition Assay
Fluorogenic HDAC isoform inhibition assay for all indicated HDAC inhibitors was conducted as previously described.20 Recombinant HDAC protein was obtained from BPS Bioscience.
Cell Proliferation Assay
SMB21 cells were infected with short hairpin (sh)RNA lentiviruses and selected with puromycin 48 hours post-transduction. Subsequently, cells were counted and plated on 96-well plates. Remaining cells were collected for cell lysates after 7 days of puromycin selection. Readings for MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] were taken 24 hours after plating and then every 48 hours thereafter up to day 6. Cell proliferation was normalized to MTS signal on day 0.
Immunoblots
Briefly, cells were lysed and resolved by sodium dodecyl sulfate gel electrophoresis on 4–12% Bis-Tris gels. Proteins were transferred to polyvinylidene difluoride membranes and probed with the indicated primary antibodies. Horseradish peroxidase–linked secondary antibodies were used for visualization. Blots were quantified using ImageJ.
RNA-Sequencing
RNA-sequencing was performed on 2 Illumina NextSeq 500 runs with single-end 75 bp reads. Alignment and quantification were performed using the STAR aligner version 2.5.1b.21 Principal component analysis and differential gene expression analysis were performed using the R/Bioconductor package DESeq2.22 For additional information, consult the Supplementary Materials.
Data Availability
All sequencing data generated in this study are available under Gene Expression Omnibus accession number GSE129512.
Pharmacokinetic Analyses and In Vivo Efficacy
For pharmacokinetic studies, wildtype mice received either 5 or 10 daily doses 5 milligram per kilogram body weight (mpk) JNJ or vehicle control. Drug penetration into the brain was evaluated using a mass spectrometry–based imaging approach (consult Supplementary Materials for details). For tolerability studies, nu/nu mice were treated once daily with a single dose of 10 mpk JNJ or a combination dose of 60 mpk LDE225 with escalating dose of JNJ. For efficacy studies, tumor cells were injected subcutaneously into the right flank of nu/nu mice, and mice were separated into treatment groups when tumor reached 150 mm3. Mice were euthanized when tumors exceeded 2000 mm3. For JNJ-26481585 treatment experiments with Atoh1-cre::SmoM2Fl/+ mice, all animals received the drug (8 mg/kg) or corresponding vehicle daily starting at postnatal day P20. Animals were monitored for clinical symptoms.
Statistical Analysis
Statistical analyses were performed with one-way or two-way ANOVA with Bonferroni correction for multiple comparisons. Significance threshold for P-value was 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). If not stated otherwise, all data are displayed as mean ± SD. All statistical analyses were performed using Microsoft Excel or GraphPad Prism 7.
Study Approval
All experimental procedures were done in accordance with National Institutes of Health guidelines and approved by the Dana-Farber Cancer Institutional Animal Care and Use Committee. For further details, consult the Supplementary Materials.
Results
Small-Molecule Screen Identifies HDAC Inhibitors as Potential SHH MB Therapeutics
The development of novel therapies for MB has been limited by the lack of stable, SHH pathway–dependent MB cell lines amenable to large scale screens. Here we performed a drug screen using an SHH-dependent cell line (SMB21) derived from Ptch1−/− MB,16 employing a chemical library representing 960 small molecules that are predominantly inhibitors of GPCRs, kinases, or epigenetic modifiers, and including sonidegib and 2 selected inhibitors of MEK (mitogen extracellular-signal-regulated kinase) as positive and negative controls, respectively.16 We assessed tumor cell viability in response to 3 days of treatment at 2 different concentrations for each compound (Fig. 1A, Supplementary Fig. 1A and B). In the initial screen, 105 of 960 molecules reduced the number of viable cells by at least 50% at a concentration of 1 µM (Supplementary Fig. 1C and D, Supplementary Table 1), including several small molecules previously reported to inhibit SHH signaling, such as JQ1, JK184, and sonidegib.23–25 Within those 105 effective compounds, 9 HDAC inhibitors dramatically reduced cell survival at tested doses (Supplementary Fig. 1E), representing the largest group of related compounds in our list of hits. However, our compound library included a total of 67 HDAC inhibitors, suggesting specificity among the HDAC inhibitors. HDAC inhibitors have been previously proposed as potential therapies for SHH MB,26,27 but it is not yet understood whether HDAC inhibition is effective for tumors resistant to SMO inhibitors, and whether there is selectivity for specific HDACs among the inhibitors that impact SHH MB.
Fig. 1.

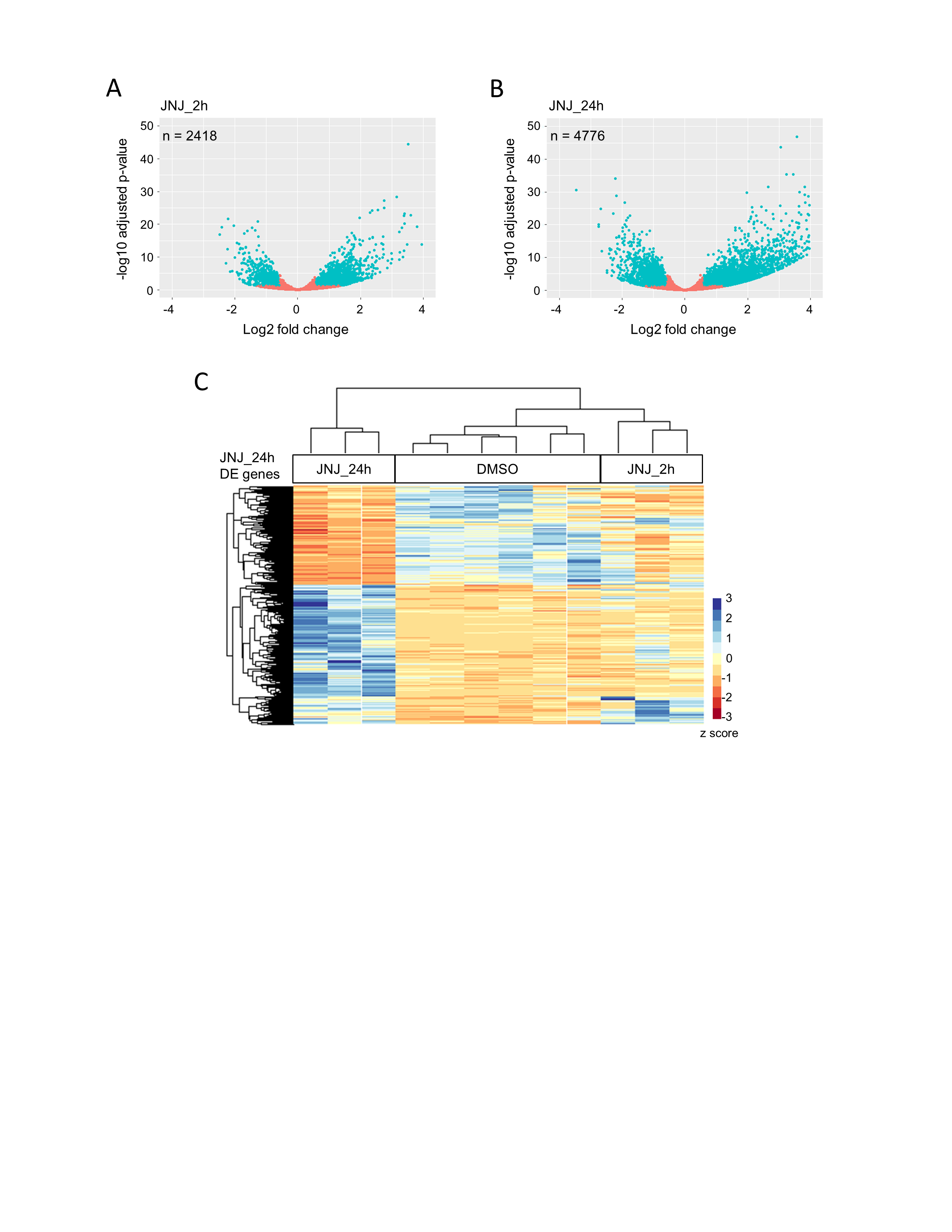
A small-molecule screen identifies a subset of HDAC inhibitors that reduce SHH MB cell survival. (A) Screen schematic: 960 compounds were screened in a 2-dose primary screen, followed by a 5-dose secondary screen using SMB21 and SMB21-GLI2ΔN cells. (B) Results from 8 HDAC inhibitors tested in a 5-dose secondary screen in SMB21 and SMB21-GLI2ΔN cells (n = 2 biological repeats). (C) Dose-dependent response in SMB21, SMB55, and SMB56 cells after 24 h of JNJ treatment (n = 3). (D) Dose-dependent response in SMB21 cells after 24 h of treatment with JNJ, vincristine, and JQ1 (n = 3). (E) Cell cycle analysis as determined by propidium iodide (PI) staining for SMB21 cells treated with JNJ (20 nM) or DMSO control for 2 h (n = 3). (F) Analysis of apoptosis as determined by annexin-V/PI staining for SMB21 cells treated with JNJ (20 nM) for 2 h or 24 h and corresponding DMSO controls (n = 3).
To confirm the screen hits, we performed a 5-dose secondary screen for 43 compounds identified in the primary screen, excluding a total of 62 compounds for potential nonspecific toxicity based on our own experience.28 To test their potential for treating SMO inhibitor–resistant disease, we performed this screen on SMB21 cells as well as SMB21 cells overexpressing a truncated, constitutively active form of human GLI2 (GLI2ΔN), rendering those cells SHH pathway–dependent but resistant to SMO inhibition.29,30 Using a stringent cutoff of greater than 80% cell viability reduction, 27 out of 43 (63%) and 21 out of 43 (49%) compounds were effective on SMB21 and SMB21-GLI2ΔN cells, respectively (Supplementary Table 2). Specifically, all 8 HDAC inhibitors identified as hits in the primary screen significantly reduced SMB21 cell survival (Fig. 1B), while 6 out of 8 HDAC inhibitors also inhibited survival of SMB21 cells expressing GLI2ΔN.
Given its potency and previous safety profile in phase I and II testing,31–34 we focused further studies on the HDAC inhibitor JNJ-26481585 (quisinostat). JNJ significantly inhibited viability of several murine cell lines derived from Ptch1−/− MB16 at nanomolar concentrations (Fig. 1C). Furthermore, JNJ exhibited similar potency in inhibiting cell survival as did vincristine, a broadly cytotoxic compound used in the clinic for treatment of MB (Fig. 1D), while it is significantly more potent than the bromodomain inhibitor JQ1, which has been discussed as a targeted therapeutic avenue for MB.35 Strikingly, JNJ significantly increased the fraction of G0/G1 cells and decreased the number of cells in S phase as early as 2 hours after treatment start (Fig. 1E). While we did not detect an increase of apoptosis at this early stage of HDAC inhibition, JNJ treatment led to a significant increase in cell death after 24 hours (Fig. 1F). Together, these data suggest that a subset of HDAC inhibitors may provide an effective and highly potent therapeutic approach for treating both SMO inhibitor–sensitive as well as SMO inhibitor–resistant SHH MB.
The HDAC Inhibitor JNJ-26481585 Inhibits SHH Pathway Activity
As expected, JNJ treatment of SHH MB cells leads to an increase in histone acetylation (Fig. 2A). To determine the specificity of the effect of JNJ on SHH-dependent cells, we investigated the effect of JNJ on SMB21 cells expressing a constitutively active human HRAS (G12V), as these cells depend on RAS/mitogen-activated protein kinase signaling rather than on active SHH signaling.16 In contrast to parental SMB21 cells as well as SMO inhibitor‒resistant SMB21 derivatives with loss of Sufu (Sufu knockout [KO]) or loss of function mutations in the ciliary component Ofd1 (Ofd1 KO),15,16 SMB21-HRAS cells showed decreased response to JNJ (Fig. 2B), providing evidence that JNJ specifically inhibits growth of SMB21 cells by inhibiting the SHH pathway.
Fig. 2.

JNJ reduces survival of SHH MB cell lines by inhibiting SHH pathway activity. (A) Western blot analyses of H3 and H4 histone acetylation marks in SMB21 cells treated with JNJ for 24 h. (B) Dose-dependent response of parental SMB21, SMO inhibitor–resistant derivatives of SMB21 cells (Sufu KO and Ofd1 KO), and HRAS-driven G12V SMB21 cells treated with JNJ for 24 h (n = 3; P-values depict difference between SMB21 and SMB21-HRAS cells). (C) Principal component analysis of global gene expression profiles from JNJ-treated SMB21 cells (20 nM) at 2 h and 24 h and corresponding DMSO controls. (D) Unsupervised hierarchical clustering of genes differentially depleted in SMB21 cells treated with DMSO or JNJ for 2 h or 24 h (Euclidean distance, average linkage). (E) mRNA levels for genes associated with SHH signaling after treatment with JNJ. P-values are derived from Wald test statistics in the DESeq2 package. (F) Gene set enrichment plots for a gene set associated with active SHH signaling in cerebellar granule neuron precursors (normalized enrichment score NES: 2 h = 1.44, 24 h = 2.37). (G) Unsupervised hierarchical clustering of single sample GSEA scores for selected gene sets. (H) Western blot analyses of GLI1 protein levels in SMB21 cells treated with either 1 μM LDE-225 or indicated concentrations of JNJ for 24 h. (I) Quantification of western blots shown in H (n = 3).
We performed RNA sequencing of SMB21 tumor cells to investigate effects of JNJ treatment on global gene expression. We identified 2418 and 4776 genes that were differentially expressed at 2 hours or 24 hours of JNJ, respectively, with a high degree of consistency across biologic replicates (Fig. 2C and D, Supplementary Table 3, Supplementary Fig. 2). Strikingly, gene ontology (GO) analysis revealed that early changes primarily involve gene networks associated with acetyltransferase activity, while the top GO terms repressed at late stages were associated with DNA replication, cell cycle progression, and mitosis. In line with previous results,36 JNJ treatment significantly and rapidly induced expression of the tumor suppressor gene Foxo1 (Fig. 2E).36 In contrast, expression of the transcription factor Atoh1 and several of its direct targets such as Pax6 and Barhl137 were significantly downregulated at early stages of JNJ treatment (Fig. 2E). It has been shown previously that deletion of Atoh1 disrupts SHH signaling and prevents the formation of SHH MBs,38 consistent with data that several direct targets of Atoh1 such as Mycn, D-type cyclins, and primary cilia components15 are involved in SHH output. In line with the inhibition of major downstream targets of the pathway, the SHH effector Gli1 was also downregulated at late stages of JNJ treatment.
To test whether a disruption of the SHH pathway was responsible for the effects seen after JNJ treatment, we performed gene set enrichment analysis (GSEA). The top gene set that is downregulated in response to JNJ treatment was one indicative of active SHH signaling in cerebellar granule neuron precursors (GCNPs), suggesting that JNJ inhibits SHH signaling in a rapid and sustained fashion (Fig. 2F). Further single sample GSEA analyses revealed that even 2 h of treatment with JNJ caused a significant reduction of a gene set implicated in SHH pathway activation in GCNPs (P = 0.044, GCNP_SHH_UP_EARLY.V1_UP) (Fig. 2G). The ability of JNJ to inhibit the SHH pathway was corroborated by analysis at the protein level, as GLI1 protein levels were decreased in SMB21 cells following 24 h of treatment with JNJ at nanomolar concentrations (Fig. 2H and I). Taken together, these data indicate that the HDAC inhibitor JNJ rapidly attenuates the SHH pathway transcriptional network in MB cells, leading to a dramatic decrease in tumor cell viability.
Inhibitors Targeting Class I HDACs Reduce Viability of SHH Pathway–Dependent MB Cells
Isoform specificity profiling indicates that JNJ is a pan-HDAC inhibitor with marked potency toward HDAC1.33 RNA sequencing analysis of SMB21 cells (Fig. 3A) and expression analysis of a cohort of 223 human SHH MBs39 (Supplementary Fig. 3) revealed that class I HDACs are consistently expressed at higher levels compared with other HDAC isoforms. Thus, variable efficacy of HDAC inhibitors in our screen may be due to differing inhibition of the class I HDACs. To investigate this, we obtained and tested isoform specificity of several known and novel HDAC inhibitors. We found that DLS-3 is highly selective for HDAC1/2; Merck 60 is an HDAC1/2/3 selective inhibitor; WT161 is HDAC6 selective; while OJI-1 is an HDAC8 inhibitor and pandacostat is a broad pan-HDAC inhibitor (Fig. 3B). These results were validated by including the pan-HDAC inhibitor SAHA and class I HDACi MS275 as controls for HDAC activity (Supplementary Fig. 4). In agreement with previous results,33 JNJ is highly potent in inhibiting both classes I and II HDACs, displaying much higher potency than all other tested HDAC inhibitors in this study. The class I specific inhibitors DLS-3 and Merck 60, like JNJ, reduced SMB21 cell survival below 50%, while neither OJI-1 nor pandacostat reduced cell survival (Fig. 3C). Of note, the selective HDAC6 inhibitor WT161 also significantly decreased survival of tumor cells. Moreover, Merck 60, which exhibits the highest potency for inhibition of HDAC1/2, also demonstrated the lowest IC50 for SMB21 cells of the novel compounds tested (Fig. 3D), providing further correlative evidence for a relationship between HDAC1/2 inhibition and antitumor potency. In line with our previous results, both class I HDAC-specific inhibitors DLS-3 and Merck 60 as well as the HDAC6-specific WT161 decreased GLI1 protein in SMB21 cells (Fig. 3E and F). As HDAC6 alters tubulin acetylation, this effect may reflect changes in microtubule stability within the primary cilia implicated in SHH signaling. However, although WT161 has highest potency against HDAC6, it has IC50 values for HDAC1/2/3 that are similar to DLS-3, suggesting that the effects on tumor cell proliferation might also reflect inhibition of class I HDACs. Together our data indicate that the HDAC inhibitors selective for class I HDACs or for HDAC6 can be useful in treating SHH MBs.
Fig. 3.

HDAC1/2 isoform specific inhibitors reduce SHH MB cell survival. (A) Reads per kilobase of transcript per million mapped reads (RPKM) of HDAC isoforms in SMB21 cells. (B) HDAC selectivity profiling for various HDAC inhibitors (n = 2). (C) Single dose screen for SMB21 cells treated with isoform specific HDAC inhibitors for 24 h (n = 3). All drugs except JNJ dosed at 10 μM, JNJ positive control dosed at 1 μM. (D) Dose response curve of SMB21 cells treated with DLS-3, Merck 60, and WT161 for 24 h (n = 3). The table shows IC50 values for all 3 compounds as tested in SMB21 cells, and corresponding IC50 values for HDAC1, HDAC2, and HDAC3 derived from the in vitro enzymatic assay. (E) Western blot analyses of GLI1 protein levels in SMB21 cells treated with indicated HDAC inhibitors for 24 h. DMSO (vehicle) and LDE225 were included as controls. (F) Quantification of western blots shown in E (n = 3).
Individual Knockdown of Hdac1 or Hdac2 Only Partially Phenocopies Pharmacologic Class I HDAC Inhibition
We used genetic approaches to reduce expression of Hdac1 and Hdac2 to understand the contributions of these 2 closely related class I enzymes to the growth of MB cells. While knockdown of each HDAC by shRNA approaches individually was confirmed by western blot analysis, we detected a compensatory increase of the HDAC2 protein after Hdac1 knockdown and a tendency for higher HDAC1 protein levels after Hdac2 knockdown (Fig. 4A and B), a mechanism that has already been described for Hdac1 and Hdac2.40,41 Since we did not detect any compensatory increase of the corresponding paralog at the RNA level (Fig. 4C), we assume that this compensatory effect is mediated at the protein level. Reduced expression of either Hdac1 or Hdac2 using selective shRNAs clearly decreases cell viability relative to a luciferase shRNA control by 56% and 42%, respectively (Fig. 4D). However, unlike treatment with JNJ or SMO knockdown, reducing expression of either Hdac1 or Hdac2 alone was not effective at reducing viability or GLI1 protein levels (Fig. 4E and F). In contrast, a simultaneous knockdown for Hdac1 and Hdac2 (Fig. 4G and H) reduced cell viability of SMB21 cells on average by 73% relative to control cells (Fig. 4I), and was therefore dramatically more potent than knockdown of either one of the HDAC isoforms alone. Furthermore, while the knockdown of either Hdac1 or Hdac2 alone altered expression of some SHH target genes, we found that simultaneous knockdown of both genes was more efficacious in downregulating those genes (Supplementary Fig. 5). This was especially true for the main SHH effector Gli1, which was only affected by knockdown of both enzymes. These data provide evidence that, while expression of both Hdac1 and Hdac2 is essential for growth of SHH MB cells, targeting either of these enzymes individually is not sufficient to inhibit SHH signaling. Compensatory changes and overlapping functions of these 2 paralog enzymes could account for these findings.
Fig. 4.

Knockdown of HDAC1/2 decreases SHH MB cell proliferation. (A) Western blot analyses of HDAC1 and HDAC2 protein levels in SMB21 cells after shRNA-guided knockdown of Hdac1 and Hdac2. shRNAs against luciferase (Luc) and Smoothened (SMO) were included as controls. (B) Quantification of western blot analyses shown in A (n = 4). (C) mRNA levels of Hdac1 and Hdac2 in shHDAC-treated SMB21 cells as determined by RNA sequencing (n = 3). P-values are derived from Wald test statistics in the DESeq2 package. (D) MTS assays for SMB21 cells transduced with indicated shRNA constructs (n = 4). (E) Western blot analyses of GLI1 protein levels in SMB21 cells transduced with indicated shRNA constructs. (F) Quantification of western blots shown in E (n = 3). (G) Western blot analyses of HDAC1 and HDAC2 protein levels in SMB21 cells simultaneously transduced with shRNA constructs for Hdac1 and Hdac2. (H) Quantification of western blots shown in G (n = 3). (I) MTS assays for SMB21 cells with a simultaneous knockdown of Hdac1 and Hdac2 (n = 3).
Pharmacologic HDAC Inhibition via JNJ Is Effective Against SMO Inhibitor–Resistant SHH MB Cells
The major targeted treatments for SHH subgroup MB are the SMO inhibitors, vismodegib and sonidegib. However, primary and secondary resistance mechanisms preclude a durable response in the clinic.4–6 We therefore tested the ability of JNJ to treat several distinct models of SHH MB cells that are resistant to SMO inhibitors, including SMB21-GLI2ΔN, Sufu KO, and Ofd1 KO cells. Of note, Trp53 is mutated in the SMB21 cells,16 and so these already represent a good model of aggressive tumors. Treatment with JNJ effectively reduced viability of GLI2ΔN-expressing cells (as predicted based on the secondary screen results), while LDE225 was not able to inhibit proliferation of these cells at any concentration tested (Fig. 5A). In fact, JNJ displayed IC50 values in the nanomolar range for all SMO inhibitor–resistant cell types tested (Fig. 5B), suggesting that JNJ acts downstream of these SHH pathway components. Furthermore, as observed for parental SMB21 cells, JNJ was similarly potent at inhibiting cell survival of SMB21-GLI2ΔN cells as was vincristine, while both of these compounds were more potent than the bromodomain inhibitor JQ1 (Fig. 5C). Notably, JNJ significantly reduced GLI1 protein levels in GLI2ΔN-expressing as well as in parental SMB21 tumor cells (Fig. 5D and E). While the class I HDAC selective inhibitors DLS-3 and Merck 60 as well as the HDAC6 inhibitor WT161 also somewhat affected GLI1 levels, LDE225, pandacostat, and OJI-1 did not reduce GLI1 protein expression in this resistant line. Thus, JNJ potently reduces viability in both SMO inhibitor–resistant and –sensitive SHH MB cells through abrogating SHH pathway activity.
Fig 5.

JNJ treatment decreases survival and reduces SHH signaling of SMO inhibitor–resistant mutants. (A) Dose response curve for SMO inhibitor–resistant SMB21-GLI2ΔN cells treated with LDE225 and JNJ for 24 h (n = 3). (B) Dose response curves for parental SMB21 cells and multiple SMO inhibitor–resistant derivatives of SMB21 cells treated with JNJ for 24 h (n = 3). This experiment was conducted in parallel with JNJ testing on HRAS cells in Fig. 2B, and the same survival curves for control SMB21 cells are shown in these figures. (C) Dose-dependent response in SMB21-GLI2ΔN cells after 24 h of treatment with JNJ, vincristine, and JQ1 (n = 3). (D) Western blot analyses of GLI1 protein levels in SMB21-GLI2ΔN cells treated with indicated HDAC inhibitors for 24 h. (E) Quantification of western blots shown in D (n = 3).
JNJ Is Well Tolerated and Effective in Mouse Models of SHH MB
To determine whether JNJ might be useful as a clinical treatment for SHH subgroup MB either alone or in combination with a SMO inhibitor, we carried out in vivo testing. First, we used a mass spectrometry–based in situ derivatization protocol to indirectly assess JNJ penetration in vivo into the brain after intraperitoneal application of 5 mg/kg of the drug. While the drug derivative was below limit of detection, segmentation by bisecting K-means of the multivariate spectral information highlighted marked differences between the brain chemistry of vehicle- and JNJ-treated mice, suggesting that JNJ is able to cross the blood–brain barrier (Supplementary Fig. 6). In subsequent tolerability studies, we found that mice treated with up to 8 mg/kg of JNJ in combination with sonidegib (10 mg/kg) exhibited acceptable levels of weight loss (less than 10%) (Supplementary Fig. 7). We then examined the efficacy of JNJ treatment in vivo, using an allograft model of SHH MB cells treated daily with vehicle, JNJ, sonidegib, or a combination of the two (Fig. 6A). Tumor growth was observed in all control mice, and 7 out of 10 had to be sacrificed due to tumor burden before the conclusion of the 50-day treatment period (Fig. 6B and C). Treatment with JNJ, or with sonidegib alone dramatically reduced tumor growth, and all mice in these groups survived beyond the 50-day endpoint. A combination of sonidegib and JNJ reduced tumor growth in 9 out of 10 animals treated. One mouse in this group had a complete response, with no measurable tumor from day 14 to day 50 of treatment. Overall no significant differences were observed among the 3 treatment groups, indicating that JNJ efficacy is comparable to that of a SMO inhibitor, and that JNJ could potentially be used in combination with sonidegib to target SHH MB.
Fig. 6.

JNJ has in vivo efficacy in models of MB. (A) Body weight of mice treated with LDE225 (60 mg/kg), JNJ (8 mg/kg), or a combination of both. Arrow indicates day 21; from day 22 onward, JNJ dose was reduced to 4 mg/kg in combination-treated mice (n = 10 per group). (B) Tumor volumes of SMB21 allograft mice treated with LDE225, JNJ, or combination of both (n = 10 per group). (C) Kaplan–Meier analyses of SMB21 allografts treated with indicated drugs or vehicle (****log rank test). (D) Kaplan–Meier analyses of Atoh1-cre::SmoM2Fl/+ mice treated with vehicle or JNJ (8 mg/kg) starting at postnatal day P20 (n = 6 per group, ***log rank test). (E) Relative mRNA levels of SHH downstream targets Gli1 and Ptch1 in tumors isolated from Atoh1-cre::SmoM2Fl/+ mice treated with vehicle (n = 4) and JNJ (n = 3). All data are mean ± SEM, except for E, which displays mean ± SD.
To validate these findings in an endogenously arising intracranial SHH MB mouse model, we carried out further testing in Atoh1-cre::SmoM2Fl/+ mice. JNJ treatment was started at postnatal day P20, when sizable tumors have already developed, and mice were treated daily until they became symptomatic and needed to be euthanized. Kaplan–Meier survival analyses revealed that mice treated with JNJ display a significant survival benefit (P = 0.0008) compared with vehicle treated mice (Fig. 6D). In addition, tumors isolated from JNJ-treated mice had lower expression levels of SHH target genes Gli1 and Ptch1 (Fig. 6E), indicating that JNJ inhibits SHH signaling in MB cells. These findings provide critical evidence that JNJ is capable of crossing the blood–brain barrier. Therefore, JNJ represents a novel therapeutic option for the clinical treatment of SHH MB.
Discussion
We made use of a stable, SHH-dependent MB cell line to screen inhibitors of GPCRs, kinases, and epigenetic modifiers for their efficacy in treating this malignant brain tumor. Here we demonstrate that inhibition of class I HDACs by use of the novel HDAC inhibitor JNJ constitutes a promising therapeutic strategy for SHH MB, including tumors resistant to targeted therapies with SMO inhibitors. Somatic and germline mutations of TP53 are relatively common in children with SHH MB, and these children often experience recurrent disease following standard therapy.8 Of note, the model system used here for large-scale drug screening harbors a mutation in Trp53,16 indicating that JNJ or other therapeutic options identified in this study might be an appropriate strategy for those high risk patients. The comprehensive isotype selectivity profiling of multiple HDAC inhibitors presented in this study will also be broadly useful for clinical scientists beyond the MB field.
While our screen panel included 67 HDAC inhibitors, only 9 of these agents exhibited efficacy in our cell lines. Notably, all inhibitors that exhibited efficacy are reported to be potent inhibitors of HDAC1/2 isoforms, and our detailed analyses reveal that structural class and isoform specificity of HDAC inhibitors predict the ability to restrain growth of SHH MB. This accords well with the preferential expression of class I HDACs in SHH MB shown here and by others.26,42 Thus, class I HDACs are the primary mediators of tumor cell growth in SHH MB. In this regard, JNJ and other class I HDAC inhibitors, such as MGCD0103, which has recently been shown to be efficacious in SHH MB,27 represent promising options for therapeutic intervention.36,42–44
Several mechanisms by which HDAC1/2 activity contributes to tumor growth and survival have been suggested, including promotion of replication fork progression during S phase45 and G1-to-S phase transition.40 Here, we demonstrate that an early and stable effect of HDAC1/2 inhibition is a reduction in SHH pathway signaling. In our experiments, JNJ was effective against SMO inhibitor–resistant SHH MB cells with mutations at various steps in the SHH pathway, suggesting that HDACs act at late stages in the SHH pathway. Late stage events in the pathway entail epigenetic changes in gene expression, which could be affected by changes in acetylation of histones or of the transcription factors GLi1 and GLi2.46,47 HDAC inhibitors are effective in MB cells expressing truncated, constitutively active GLI2, which lacks many identified acetylation sites. Thus histone deacetylation by HDAC1 or 2 is likely to be critical in this response. We find that an HDAC6 inhibitor, WT161, also reduced SHH MB cell survival and SHH pathway activity. Conflicting data on the role of HDAC6 in SHH signaling have been reported.26,48 HDAC6 has distinct substrates from most other isoforms, and is best known for its role in microtubule deacetylation, which might impact primary cilia signaling. Even though WT161 has highest potency toward HDAC6, it also has considerable activity toward class I HDACs, which may also contribute to its efficacy.
At present, approximately one third of patients with SHH subgroup MB succumb to their disease, and this is particularly true for children with TP53-mutant tumors. Moreover, given the large proportion of MB that exhibit de novo or acquired resistance to SMO inhibitors, new therapeutics should circumvent documented mechanisms of resistance. We find that JNJ is effective in reducing expression of SHH-associated gene transcripts and limiting tumor growth in multiple cell lines resistant to SMO inhibition. Our data and previous reports indicate that JNJ is well tolerated at clinically relevant concentrations, crosses the blood–brain barrier, and is effective in multiple models of SHH MB.31,32,34 Thus, JNJ may provide an effective clinical agent for further development as a novel therapeutic option for SHH MB.
Funding
This work was supported by NIH (PO1CA142536), Alex’s Lemonade Stand Foundation Award (to R.A.S.), F31CA183145 (to E.P.), Samuel E. Lux fellowship (to E.L.M.), German Cancer Aid (Mildred Scheel postdoctoral fellowship), and Deutsche Forschungsgemeinschaft (ZUK 63 to D.J.M.).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Anthony Arvanites, Vatsal Oza, Lance S. Davidow, and Kathleen L. Pfaff for assistance with the high-throughput screen. We also acknowledge support from the Dana-Farber Experimental Therapeutics Core.
References
- 1. Rutkowski S, von Hoff K, Emser A, et al. Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol. 2010;28(33):4961–4968. [DOI] [PubMed] [Google Scholar]
- 2. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robinson GW, Orr BA, Wu G, et al. vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33(24):2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kool M, Jones DT, Jäger N, et al. ; ICGC PedBrain Tumor Project Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25(3):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361(12):1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326(5952):572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwalbe EC, Lindsey JC, Nakjang S, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol. 2017;18(7):958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramaswamy V, Remke M, Bouffet E, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016;131(6):821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mukhopadhyay S, Rohatgi R. G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin Cell Dev Biol. 2014;33:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sasai K, Romer JT, Kimura H, Eberhart DE, Rice DS, Curran T. Medulloblastomas derived from Cxcr6 mutant mice respond to treatment with a smoothened inhibitor. Cancer Res. 2007;67(8): 3871–3877. [DOI] [PubMed] [Google Scholar]
- 11. Jacob LS, Wu X, Dodge ME, et al. Genome-wide RNAi screen reveals disease-associated genes that are common to Hedgehog and Wnt signaling. Sci Signal. 2011;4(157):ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Evangelista M, Lim TY, Lee J, et al. Kinome siRNA screen identifies regulators of ciliogenesis and hedgehog signal transduction. Sci Signal. 2008;1(39):ra7. [DOI] [PubMed] [Google Scholar]
- 13. Zhan X, Shi X, Zhang Z, Chen Y, Wu JI. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc Natl Acad Sci U S A. 2011;108(31):12758–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shi X, Zhang Z, Zhan X, et al. An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nat Commun. 2014;5:5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao X, Pak E, Ornell KJ, et al. A transposon screen identifies loss of primary cilia as a mechanism of resistance to SMO inhibitors. Cancer Discov. 2017;7(12):1436–1449. [DOI] [PubMed] [Google Scholar]
- 16. Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75(17):3623–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ingham OJ, Paranal RM, Smith WB, et al. Development of a potent and selective HDAC8 inhibitor. ACS Med Chem Lett. 2016;7(10):929–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bradner JE, West N, Grachan ML, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6(3):238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hideshima T, Mazitschek R, Qi J, et al. HDAC6 inhibitor WT161 downregulates growth factor receptors in breast cancer. Oncotarget. 2017;8(46):80109–80123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalin JH, Wu M, Gomez AV, et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat Commun. 2018;9(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12): 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee J, Wu X, Pasca di Magliano M, et al. A small-molecule antagonist of the hedgehog signaling pathway. Chembiochem. 2007;8(16): 1916–1919. [DOI] [PubMed] [Google Scholar]
- 24. Pan S, Wu X, Jiang J, et al. Discovery of NVP-LDE225, a potent and selective Smoothened antagonist. ACS Med Chem Lett. 2010;1(3):130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang Y, Gholamin S, Schubert S, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20(7):732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee SJ, Lindsey S, Graves B, Yoo S, Olson JM, Langhans SA. Sonic hedgehog-induced histone deacetylase activation is required for cerebellar granule precursor hyperplasia in medulloblastoma. PLoS One. 2013;8(8):e71455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coni S, Mancuso AB, Di Magno L, et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH medulloblastoma. Sci Rep. 2017;7:44079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang YM, Gupta SK, Kim KJ, et al. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell. 2013;12(6):713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006;20(22):3161–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roessler E, Ermilov AN, Grange DK, et al. A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum Mol Genet. 2005;14(15):2181–2188. [DOI] [PubMed] [Google Scholar]
- 31. Carol H, Gorlick R, Kolb EA, et al. Initial testing (stage 1) of the histone deacetylase inhibitor, quisinostat (JNJ-26481585), by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2014;61(2):245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Child F, Ortiz-Romero PL, Alvarez R, et al. Phase II multicentre trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB-IVA mycosis fungoides/Sézary syndrome. Br J Dermatol. 2016;175(1):80–88. [DOI] [PubMed] [Google Scholar]
- 33. Arts J, King P, Mariën A, et al. JNJ-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin Cancer Res. 2009;15(22):6841–6851. [DOI] [PubMed] [Google Scholar]
- 34. Venugopal B, Baird R, Kristeleit RS, et al. A phase I study of quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin Cancer Res. 2013;19(15): 4262–4272. [DOI] [PubMed] [Google Scholar]
- 35. Bandopadhayay P, Bergthold G, Nguyen B, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. 2014;20(4):912–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pei Y, Liu KW, Wang J, et al. HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell. 2016;29(3):311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Klisch TJ, Xi Y, Flora A, Wang L, Li W, Zoghbi HY. In vivo Atoh1 targetome reveals how a proneural transcription factor regulates cerebellar development. Proc Natl Acad Sci U S A. 2011;108(8):3288–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Flora A, Klisch TJ, Schuster G, Zoghbi HY. Deletion of Atoh1 disrupts Sonic Hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science. 2009;326(5958):1424–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31(6):737–754 e736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamaguchi T, Cubizolles F, Zhang Y, et al. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010;24(5):455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wilting RH, Yanover E, Heideman MR, et al. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29(15):2586–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ecker J, Oehme I, Mazitschek R, et al. Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol Commun. 2015;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stubbs MC, Kim W, Bariteau M, et al. Selective Inhibition of HDAC1 and HDAC2 as a potential therapeutic option for B-ALL. Clin Cancer Res. 2015;21(10):2348–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khabele D, Son DS, Parl AK, et al. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: implications for therapy. Cancer Biol Ther. 2007;6(5):795–801. [DOI] [PubMed] [Google Scholar]
- 45. Bhaskara S, Jacques V, Rusche JR, Olson EN, Cairns BR, Chandrasekharan MB. Histone deacetylases 1 and 2 maintain S-phase chromatin and DNA replication fork progression. Epigenetics Chromatin. 2013;6(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Canettieri G, Di Marcotullio L, Greco A, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12(2):132–142. [DOI] [PubMed] [Google Scholar]
- 47. Coni S, Antonucci L, D’Amico D, et al. Gli2 acetylation at lysine 757 regulates hedgehog-dependent transcriptional output by preventing its promoter occupancy. PLoS One. 2013;8(6):e65718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dhanyamraju PK, Holz PS, Finkernagel F, Fendrich V, Lauth M. Histone deacetylase 6 represents a novel drug target in the oncogenic Hedgehog signaling pathway. Mol Cancer Ther. 2015;14(3):727–739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated in this study are available under Gene Expression Omnibus accession number GSE129512.