

SUMMARY
Several cytosolic pattern-recognition receptors (PRRs) form multiprotein complexes called canonical inflammasomes in response to intracellular danger signals. Canonical inflammasomes recruit and activate caspase-1 (CASP1), which in turn cleaves and activates inflammatory cytokines and gasdermin D (GSDMD), inducing pyroptotic cell death. Inhibitors of the dipeptidyl peptidases DPP8 and DPP9 (DPP8/9) activate both the human NLRP1 and CARD8 inflammasomes. NLRP1 and CARD8 have different N-terminal regions but have similar C-terminal regions that undergo autoproteolysis to generate two non-covalently associated fragments. Here, we show that DPP8/9 inhibition activates a proteasomal degradation pathway that targets disordered and misfolded proteins for destruction. CARD8’s N terminus contains a disordered region of ~160 amino acids that is recognized and destroyed by this degradation pathway, thereby freeing its C-terminal fragment to activate CASP1 and induce pyroptosis. Thus, CARD8 serves as an alarm to signal the activation of a degradation pathway for disordered and misfolded proteins.
Graphical Abstract
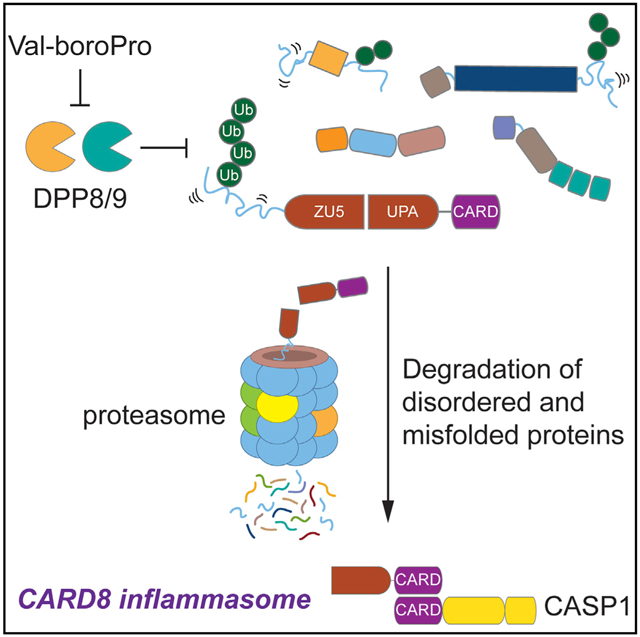
In Brief
Inflammasomes are multiprotein complexes that detect intracellular danger signals and stimulate powerful immune responses. DPP8/9 inhibitors activate the CARD8 inflammasome through an unknown mechanism. Here, Chui et al. show that DPP8/9 inhibitors induce the degradation of many disordered and misfolded proteins. CARD8 has an N-terminal disordered region that is degraded upon DPP8/9 inhibition, triggering inflammasome formation.
INTRODUCTION
Inflammasomes are multiprotein complexes that are rapidly assembled in response to intracellular pathogen- or danger-associated signals (Broz and Dixit, 2016; Lamkanfi and Dixit, 2014). Typically, inflammasome formation involves a pattern-recognition receptor (PRR) protein sensing a specific pathogen-associated signal, oligomerizing with the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD), and recruiting pro-caspase-1 (CASP1). Pro-CASP1 then undergoes proximity-induced autoprocessing on this structure to generate a fully active enzyme, which subsequently cleaves and activates inflammatory cytokines (interleukin [IL]-1β and IL-18) and gasdermin D (GSDMD) (Kayagaki et al., 2015; Shi et al., 2015). The N-terminal fragment of GSDMD forms pores in the cell membrane, releasing the activated cytokines and inducing pyroptotic cell death.
NLRP1 (nucleotide-binding domain leucine-rich repeat pyrin domain-containing 1) and CARD8 (caspase activation and recruitment domain-containing protein 8) are related inflammasome-forming PRRs. NLRP1 has an N-terminal pyrin domain (PYD), followed by a nucleotide-binding (NACHT) domain, leucine-rich repeat (LRR), function-to-find domain (FIIND, which has ZU5 and UPA-like subdomains), and CARDs (Figure S1A) (Finger et al., 2012). CARD8 only has a predicted N-terminal ~160-amino-acid-long unstructured region followed by a FIIND and a CARD (Figure 1A; Figure S1B). Both NLRP1 and CARD8 undergo post-translational autoproteolysis near the C-terminal end of their ZU5 subdomains, generating two non-covalently associated polypeptide chains (D’Osualdo et al., 2011; Frew et al., 2012). The serine proteases DPP8 and DPP9 (DPP8/9) bind directly to the FIINDs of both CARD8 and NLRP1 (Griswold et al., 2019; Zhong et al., 2018). Small-molecule DPP8/9 inhibitors, including Val-boroPro (VbP), induce the proteasome-mediated degradation of the N-terminal fragments, releasing the C-terminal UPA-CARD domains to form inflammasomes (Ball et al., 2019; Chui et al., 2019; Johnson et al., 2018). However, the molecular mechanisms that control NLRP1 and CARD8 N-terminal degradation, and whether they are similar or different, are unknown.
Figure 1. The N-Terminal Disordered Region of CARD8 Is Required for Inflammasome Activation.

(A) Diagram of human CARD8 (above) and its predicted disorder (below) as determined by the Sequence-Based Prediction of Disordered Residues for Proteins (SPOT-Disorder) program (Hanson et al., 2017).
(B, C, E, and F) HEK293T cells stably expressing human CASP1 and GSDMD were transiently transfected with the indicated constructs (0.02 μg). After 16–20 h, samples were treated with VbP (10 μM) for 6 h before lysates were evaluated by immunoblotting (B and E) and cell viability was evaluated by lactate dehydrogenase (LDH) release (C and F).
(D) HEK293T cells were transiently transfected with the indicated constructs (2 μg). After 48 h, samples were treated with VbP (10 μM) for 6 h before lysates were immunoprecipitated by anti-FLAG and evaluated by immunoblotting. Immunoblot depicts input whole-cell lysate (WCL) and captured proteins (immunoprecipitation [IP]: FLAG). Residues that were mutated to create start sites are indicated. Data are means ± SEM of three biological replicates.
***p < 0.001 and **p < 0.01 by two-sided Student’s t test compared with mock. Data are representative of three or more independent experiments. FL, full length; CL, cleaved GSDMD; ZUC, ZU5-UPA-CARD domains of CARD8. Related to Figure S1.
RESULTS
The Unstructured N-Terminal Region of CARD8 Is Required
Here, we wanted to study the mechanism of CARD8 activation. The ectopic expression of CARD8, CASP1, and GSDMD renders HEK293T cells sensitive to DPP8/9 inhibitor-induced pyroptosis (Johnson et al., 2018). To investigate the function of the putative disordered region of CARD8, we transiently transfected constructs encoding either full-length CARD8 (isoform 5, the longest isoform) or the ZU5-UPA-CARD (ZUC) domains of CARD8 (starting at residue 162) into HEK293T cells stably expressing CASP1 and GSDMD. Both proteins expressed at similar levels and underwent autoproteolysis to similar extents (Figure 1B). However, VbP induced GSDMD cleavage and lactate dehydrogenase (LDH) release only in cells expressing full-length CARD8 (Figures 1B and 1C), indicating that the predicted disordered region of CARD8 plays a functional role in VbP-induced inflammasome activation. As expected, similar results were observed with the highly selective DPP8/9 inhibitor 8j (Figures S1C and S1D). Notably, removal of the disordered region did not affect DPP9 binding to CARD8 FIIND (Figure 1D). We found that only ~15 amino acids N-terminal to the ZU5 domain were required for VbP-dependent CARD8 inflammasome activation (Figures 1E and 1F). Consistent with previous data (Johnson et al., 2018), transfection of the C-terminal UPA-CARD region induced VbP-independent GSDMD cleavage and cell death.
A Lysine Is Not Required in Unstructured Region
We reasoned that one or more lysine residues in the predicted disordered region preceding CARD8’s ZU5 domain might be required for the ubiquitination and degradation of the N-terminal fragment. The truncated construct starting at residue 147 that still responded to VbP (Figures 1E and 1F) has only a single lysine (K157) before the start of the ZU5 subdomain (Figure 1A). We mutated this lysine to an arginine in this construct (K147M-537 [K157R]) and found that the resulting protein was still sensitive to VbP (Figures 1E and 1F). To further investigate where N-terminal ubiquitination was occurring after VbP treatment, we mutated all 10 lysines N-terminal to the CARD8 autoproteolysis site (CARD8 K10R), including K175 and K272 within the ZU5 domain. As expected, CARD8 K10R was insensitive to VbP (Figures S1E and S1F), although this mutant protein did not express as highly as the wild-type (WT) protein. Regardless, we found that reintroducing a single lysine either in the disordered N terminus (K3) or well into the ZU5 domain (K272) (Figure 1A) largely restored CARD8’s ability to induce pyroptosis while still being expressed at a low level similar to the K10R protein (Figures S1E and S1F). Collectively, these data show that a lysine is required before the autoproteolysis site but that the lysine does not have to be located in the unstructured N-terminal region for activation. Thus, the putative disordered region is not required simply to present a lysine for ubiquitination and N-terminal degradation. However, although the overall amount of CARD8 degradation is typically slight and ubiquitination is not observed by gel (Figures 1B and 1E), the requirement of at least one lysine strongly indicates that the N terminus is indeed being directly ubiquitinated.
Disorder Is Necessary for CARD8 Activation
We next wanted to determine whether the identity of the N-terminal amino acids was important. This region, unlike the ZUC region, is not evolutionarily conserved (Figure S1G), suggesting the exact residues are perhaps not critical. Moreover, CARD8 has at least five isoforms generated by alternative mRNA splicing, three of which (UniProt isoforms 1, 4, and 5) contain ZUC but have different N-terminal regions (Figure S1G) (Bagnall et al., 2008; D’Osualdo et al., 2011). We found that all three of these CARD8 isoforms mediated VbP-induced pyroptosis (Figures S1H and S1I), although isoform 1 induced the least cell death. Isoform 1 underwent considerably less autoproteolysis than isoforms 4 and 5 (Figure S1H). In addition to having a shorter N terminus than isoforms 4 and 5, isoform 1 lacks an alanine in its ZU5 subdomain (residue 153 in isoform 1; Figure S1G, yellow box). We made a mutant of isoform 1 with an alanine inserted after Q152 (Q152QA) and found that this mutation restored the ability of isoform 1 to undergo autoproteolysis and mediate VbP-induced cell death at levels comparable to isoforms 4 and 5 (Figures S1H and S1I). Thus, this single amino acid deletion, not its dissimilar N terminus, is responsible for the activity difference of isoform 1. Overall, the lack of N-terminal conservation across species and the similar activities of the different splice isoforms in humans indicate that a specific N-terminal sequence is likely not critical for CARD8 function.
We next fused a 9-amino-acid-long hemagglutinin (HA) tag either directly to CARDZUC or separated by flexible 2×, 4×, or 6× GSSG (Gly-Ser-Ser-Gly) linkers. We found that the latter constructs, which have longer unstructured N-terminal regions, mediated VbP-induced pyroptosis (Figures 2A and 2B; Figure S2A). The HA-GSSGn× tags do not contain lysine residues, confirming that a lysine is not needed before the ZU5 domain. In contrast, the fusion of well-folded green fluorescent protein (GFP) or glutathione S-transferase (GST) tags to the CARD8ZUC N terminus did not restore CARD8’s pyroptosis-inducing activity (Figures 2C and 2D; Figures S2B-S2D). GFP and GST tags are separated from CARDZUC by a 13-amino-acid-long linker spanning the attB Gateway recombination site, but this linker region is not predicted to be disordered (Figure S2B). However, appending a disordered 14-residue V5 tag on the N terminus of the GFP-CARD8ZUC construct restored sensitivity to DPP8/9 inhibition (Figures 2C and 2D; Figures S1C, S1D, and S2B). Collectively, these data suggest that some N-terminal disorder is critical for CARD8’s ability to sense DPP8/9 inhibition.
Figure 2. Unrelated Disordered Sequences Create Functional CARD8 Chimeras.

(A–D, G, and H) HEK293T cells stably expressing human CASP1 and GSDMD were transiently transfected with plasmids encoding full-length CARD8, CARD8ZUC, or variously N-terminally tagged CARD8ZUC (0.02 μg). In (G) and (H), GFP11 was co-transfected at the indicated plasmid ratios. After 16–20 h, samples were treated with VbP (10 μM) for 6 h before lysates were evaluated by immunoblotting (B, D, and H) and cell viability was evaluated by LDH release (A, C, and G). Cropped images in (D) are from the same membrane.
(E) HEK293T cells were transiently transfected with the indicated constructs (2 μg). After 16–20 h, cells were harvested, lysed, and incubated with varying concentrations of trypsin (20, 2, and 0.2 ng/μL) or proteinase K (PROK) (4, 0.8, 0.16, and 0.032 ng/μL) before immunoblotting.
(F) HEK293T cells were transiently transfected with the indicated CARD8ZUC-containing constructs (0.02 μg), together with a red fluorescent protein (RFP)-encoding filler plasmid. GFP11 was co-transfected at the indicated plasmid ratios. After 16–20 h, cells were imaged by fluorescence microscopy. A RFP-positive signal was used to show total cell area. Representative images are shown. Data are means ± SEM of three biological replicates.
***p < 0.001 and **p < 0.01 by two-sided Student’s t test compared with mock. Data are representative of three or more independent experiments. Related to Figures S1 and S2.
To investigate whether the disordered sequence can be internal (i.e., not on the N terminus), we appended GFP or GST (including the 13-amino-acid attB linker) to the N terminus of full-length CARD8 and the functional truncated CARD8 proteins (Figures S2B-S2E). We found that these constructs all retained their ability to induce GSDMD cleavage and cell death. Thus, the disordered sequence does not have to be on the extreme N terminus of the protein.
These preceding experiments demonstrated that appending amino acid sequences with no predicted secondary structure could restore function to the isolated CARD8ZUC. We next asked whether a misfolded N-terminal domain, which is able to adopt some secondary structure but is not entirely globular, could similarly restore CARD8 function. Split GFP, in which the last β strand of GFP (GFP11) is physically separated from the first 10 β strands (GFP1–GFP10), fluoresces when the two fragments associate to form GFP in cells (Feng et al., 2017; Kamiyama et al., 2016). GFP1–GFP10 are reported to be prone to misfolding in the absence of GFP11 (Lee et al., 2016). We found that limited proteolysis of GFP1–GFP10 with trypsin or proteinase K resulted in more rapid protein digestion than full-length GFP (Figure 2E), consistent with a less globular structure. We next fused GFP1–GFP10 to the N terminus (including the same 13-amino-acid-long linker as for full-length GFP earlier) of the CARD8ZUC, and observed that this construct does not fluoresce (Figure 2F) but does induce pyroptosis in response to VbP (Figures 2G and 2H). However, complementation of GFP11, which resulted in fluorescence consistent with formation of a folded, globular structure in cells (Figure 2F), inactivated this chimera (Figures 2G and 2H). Similarly, fusion of the partially folded protein α-synuclein (Breydo et al., 2012) to the N terminus of the CARDZUC generated a VbP-activatable protein (Figure S2F). Overall, these results suggest that a disordered and/or misfolded region in the N-terminal fragment is critical for CARD8 inflammasome activation.
DPP8/9 Inhibition Alters Proteostasis
Because VbP-induced protein degradation was independent of sequence identity, we predicted that proteins in addition to CARD8, in particular those with high levels of intrinsic disorder and/or misfolding, would be degraded. To test this hypothesis, we treated HEK293T cells with DMSO or VbP before evaluating protein levels by quantitative mass spectrometry and gene expression by RNA sequencing (RNA-seq). Because we have consistently observed more CARD8 degradation in previous studies with longer (>12 h) incubation times (Griswold et al., 2019; Johnson et al., 2018), we chose 48 h of VbP treatment for this experiment. We used HEK293T cells lacking inflammasome components so that VbP-induced cell death would not confound our results. Consistent with our hypothesis, we observed a shift in protein abundance across the entire proteome, with the levels of many proteins slightly reduced in VbP-treated cells (Figures 3A and 3B; Table S1). In contrast, we observed no significant changes in gene expression (Figures S3A and S3B; Table S2). These data indicate that VbP induces a small yet global change in proteostasis that does not measurably affect transcription.
Figure 3. VbP Induces the Degradation of Many Proteins.

(A–C) HEK293T cells were treated with VbP (10 μM, 48 h) before protein abundance was determined by quantitative tandem mass spectrometry (MS) (A and B) or immunoblotting (C). In (C), bortezomib (1 μM) or MLN4924 (1 μM) was added 6 h before harvesting.
(D) CASP1−/− OCI-AML2 and MV-4-11 cells were treated with VbP (10 μM) for 24 h before protein levels were evaluated by immunoblotting. Bortezomib (1 μM) was added 6 h before harvesting.
Data are representative of three or more independent experiments. Related to Tables S1 and S2 and Figure S3.
Although VbP induced only a slight change in the abundance of many proteins, several were significantly depleted, including D-2-hydroxyglutarate dehydrogenase (D2HGDH), myotubularin-related protein 1 (MTMR1), and activating transcription factor 3 (ATF3) (Figures 3B and 3C, red dots). These HEK293T cells do not express CARD8; therefore, CARD8 was not evaluated in this assay. The depleted proteins are not linked in any known biological pathway or complex. We confirmed that these proteins, but not control proteins that did not change in abundance, were indeed depleted by immunoblotting (Figure 3C). We observed VbP-induced depletion of these proteins, as well as CARD8, in CASP1−/− (to prevent cell death) OCI-AML2 and MV4;11 monocytes (Figure 3D), demonstrating that this degradation pathway is not restricted to HEK293T cells. Consistent with proteasome-mediated degradation, protein depletion was blocked by the proteasome inhibitor bortezomib (Figures 3C and 3D; Figures S3C and S3D). Although the identity of the E3 ligase that mediates this degradation has not yet been established, the NEDD8-activating enzyme inhibitor MLN4924 (Soucy et al., 2009) similarly blocked VbP-induced protein degradation and cell death (Figure 3C; Figures S3C-S3H), indicating a cullin E3 ligase is likely responsible.
Disorder Controls Depletion of Proteins
We predicted that intrinsically disordered regions were likely common features among the proteins most depleted upon VbP treatment. MTMR1, D2HGDH, and other significantly depleted proteins have predicted disordered regions (Figure 4A; Figure S4A). Consistent with these computational assessments, we observed that limited proteolysis of full-length MTMR1, D2HGDH, and CARD8 with trypsin or proteinase K resulted in rapid protein digestion (Figure 4B). In contrast, truncated proteins lacking the predicted disordered N-terminal regions were more stable under protease treatment (Figure 4B). This analysis also revealed that the globular CARD8 C terminus was similarly more stable against limited proteolysis than full-length CARD8. We next ectopically expressed full-length or N-terminally truncated MTMR1, D2HGDH, and CARD8 in HEK293T cells. We discovered that VbP only induced the degradation of the full-length proteins (Figure 4C), demonstrating that the predicted disordered regions are required for the degradation of all three proteins. Consistent with these data, we found that VbP and compound 8j induced LDH release and GSDMD cleavage in HEK293T cells expressing a MTMR1M1-Q94-CARD8ZUC fusion construct, together with CASP1 and GSDMD (Figures 4D and 4E; Figures S1C and S1D).
Figure 4. Disorder Is the Key Feature of Degraded Proteins.

(A) Diagrams of human MTMR1 and D2HGDH (above) and corresponding predicted disorder plots (below) (Hanson et al., 2017).
(B) HEK293T cells were transiently transfected with plasmids encoding either the full-length or N-terminally truncated, disorder free (ΔN-term), indicated constructs (2 μg). After 16–20 h, cells were harvested, lysed, and incubated with varying concentrations of trypsin (20, 2, and 0.2 ng/μL) or PROK (4, 0.8, 0.16, and 0.032 ng/μL) before immunoblotting.
(C) HEK293T cells were transiently transfected with plasmids encoding the full-length or N-terminally truncated indicated constructs (0.1 μg). After 16–20 h, cells were treated with VbP (10 μM) for 18 h before protein levels were evaluated by immunoblotting.
(D and E) HEK293T cells stably expressing human CASP1 and GSDMD were transiently transfected with plasmids encoding the indicated constructs (0.02 μg). After 16–20 h, samples were treated with VbP (10 μM) for 6 h before cell viability was evaluated by LDH release (D) and lysates were evaluated by immunoblotting (E).
(F and G) THP-1 CARD8−/− monocytes stably expressing the indicated constructs were treated with compound 8j (20 μM) or VbP (10 μM). After 24 h, cell viability was evaluated by LDH release (F) and lysates were evaluated by immunoblotting (G).
(H) Schematic of the proposed CARD8 activation mechanism, which involves the degradation of its disordered N-terminus.
Related to Figure S4.
We next wanted to confirm that CARD8 similarly required a disordered N terminus in human monocytes. Thus, we re-expressed full-length CARD8, CARD8ZUC, and various N-terminal CARD8ZUC fusions in THP-1 CARD8−/− monocytes before treating these cells with DPP8/9 inhibitors. As expected, VbP and 8j induced GSDMD cleavage and LDH release in cells expressing CARD8 proteins with disordered N termini (CARD8FL, V5-GFP-CARD8ZUC, MTMR1M1-Q94-CARD8ZUC, and D2HGDHM1-R51-CARD8ZUC) (Figures 4F and 4G). In contrast, VbP and 8j did not trigger cell death in cells expressing the CARD8 constructs lacking disordered N termini (CARD8ZUC or GFP-CARD8ZUC) (Figures 4F and 4G). Overall, these data demonstrate that DPP8/9 inhibition stimulates a pathway that degrades proteins with disordered and/or misfolded regions and that this pathway activates the CARD8 inflammasome.
DISCUSSION
NLRP1 was the first protein discovered to form an inflammasome (Martinon et al., 2002), but the pathogen-associated structure or activity that it detects has not been identified. Because NLRP1 is a highly polymorphic protein even across inbred rodent strains (Boyden and Dietrich, 2006; Newman et al., 2010), it has been suggested that the different alleles may sense entirely different signals (Sandstrom et al., 2019). However, we recently reported that DPP8/9 inhibitors are universal activators of all functional NLRP1 and CARD8 alleles in humans and rodents (Gai et al., 2019). Moreover, the intracellular parasite Toxoplasma gondii has been shown to activate several NLRP1 alleles in rodents (Cirelli et al., 2014; Ewald et al., 2014). Intriguingly, the relative sensitivities of NLRP1 alleles to DPP8/9 inhibitors closely mirrors their relative sensitivities to T. gondii (Gai et al., 2019). Based on these data, we speculate that DPP8/9 inhibitors and T. gondii induce the same cellular perturbation and that all NLRP1 and CARD8 alleles evolved to detect this perturbation (Taabazuing et al., 2020).
Here, we have demonstrated that DPP8/9 inhibition activates a proteasomal degradation pathway that selectively destroys proteins with disordered and/or misfolded structures (Figure 4H). It is tempting to speculate that this degradation pathway is activated in response to the unknown cellular perturbation to restore cellular homeostasis. For example, it is possible that the perturbation is the depletion of a key nutrient and that increased protein degradation helps to replenish this nutrient. Because disordered and/or misfolded proteins are more likely to be non-functional than well-folded proteins, their degradation has the potential to restore homeostasis with minimal detrimental impact on other biological processes. This degradation pathway exists in many cell types, including HEK293T cells, not just in immune cells. However, in immune cells that express inflammasome components, CARD8 uses its disordered N terminus to detect the activation of this pathway and to mount a rapid immune response.
NLRP1, unlike CARD8, has several well-folded N-terminal domains, although a disordered region is predicted to exist between the PYD and the NACHT domains (Figure S1A). We hypothesize that the N terminus of NLRP1 may sense the same danger-associated signal using its disordered region but that the folded domains provide additional regulation or sense an alternative stimulus. We expect that these results reported herein will provide the foundation for future studies to characterize the features that control the degradation of the NLRP1 N terminus and to identify the elusive cellular perturbation that activates both CARD8 and NLRP1 inflammasomes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel Bachovchin (bachovcd@mskcc.org).
Materials Availability
All cell lines and plasmids generated in this study are available upon request to the lead contact.
Data and Code Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez-Riverol et al., 2019) with the dataset identifier PXD015978. The RNA-Seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE153744 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153744).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and cell culture information is listed below. HEK293T cells, THP-1 cells, and RAW 264.7 cells were purchased from ATCC. OCI-AML2 and MV-4-11 cells were purchased from DSMZ. HEK293T and RAW 264.7 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with L-glutamine and 10% fetal bovine serum (FBS). THP-1 and OCI-AML2 cells were grown in Roswell Park Memorial Institute (RPMI) medium 1640 with L-glutamine and 10% FBS. All cells were grown at 37°C in a 5% CO2 atmosphere incubator. Cell lines were regularly tested for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza).
METHOD DETAILS
Antibodies and reagents
Antibodies used include: GSDMD Rabbit polyclonal Ab (Novus Biologicals, NBP2-33422), GSDMD Rabbit monoclonal Ab (Abcam, Ab209845), FLAG® M2 monoclonal Ab (Sigma, F3165), CARD8 C terminus Rabbit polyclonal Ab (Abcam, Ab24186), CARD8 N-terminal Rabbit polyclonal Ab (Abcam, Ab194585), GAPDH Rabbit monoclonal Ab (Cell Signaling Tech, 14C10), MTMR1 (Abcam, Ab 240569), D2HGDH (Abcam, Ab233516), ATF-3 (Cell Signaling Tech, D2Y5W), IGBP1 (Cell Signaling Tech, 5F6), SMAD9 (Abcam, Ab124094), ARF1 (Abcam, Ab58578), ACTR5 (Proteintech, 21505), AKAP8 (Abcam, Ab72196), GSTP1 (Cell Signaling Tech, 3F2), SET (Abcam, Ab1183), NLRP1B (Vance laboratory, 2A12), IRDye 800CW anti-rabbit (LICOR, 925-32211), IRDye 800CW anti-mouse (LI-COR, 925-32210), IRDye 680CW anti-rabbit (LI-COR, 925-68073), IRDye 680CW anti-mouse (LI-COR, 925-68072). Other reagents used include: Val-boroPro (VbP) (Okondo et al., 2017), Bortezomib (Bort; MilliporeSigma, 504314), MLN4924 (Cayman, 15217), bestatin methyl ester (Me-Bs; Sigma, 200485), sequencing grade modified trypsin (Promega, V5113), proteinase K (PROK; Invitrogen, 25530049), FuGENE HD (Promega, E2311), TMTsixplex Isobaric Label Reagents (ThermoFisher Scientific).
Cell Culture
HEK293T cells, THP-1 cells, and RAW 264.7 cells were purchased from ATCC. OCI-AML2 and MV-4-11 cells were purchased from DSMZ. HEK293T and RAW 264.7 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with L-glutamine and 10% fetal bovine serum (FBS). THP-1 and OCI-AML2 cells were grown in Roswell Park Memorial Institute (RPMI) medium 1640 with L-glutamineand 10% FBS. All cells were grown at 37°C in a 5% CO2 atmosphere incubator. Cell lines were regularly tested for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza). Stable cell lines were generated as described previously (Johnson et al., 2018).
Cloning
Plasmids for full-length and truncated CARD8 were cloned as described previously (Johnson et al., 2018) and as annotated in Figure 1A. The CARD8ZUC construct starts from L162 of human origin. DNA encoding GFP, V5-GFP, GFP(1-10), GST, or α-synuclein were inserted before the attR1 recombination site in a modified pLEX_307 vector (immediately after the EcoRV site), and DNA encoding indicated CARD8 variants were shuttled into these modified vectors using Gateway technology. The resulting proteins expressed from these modified vectors includes an attB linker (GSTSLYKKAGFAT) N-terminal to CARD8 variants. The HA-CARD8ZUC and HA-GSSGnX-CARD8ZUC (n = 2, 4, or 6) constructs were cloned via PCR using template DNA from CARD8 with primers that included 5′ flanking DNA encoding the hemagglutinin (HA) tag or the HA tag with a GSSG2X linker sequence. Plasmids encoding MTMR1 and D2HGDH were purchased from Origene (RC212024) and GenScript (OHu27566), respectively. Constructs encoding full-length MTMR1 (residues 1-665), MTMR1Δ1-94 (residues 95-665), full-length D2HGDH (residues 1-521), and D2HGDHΔ1-51 (residues 52-521) were cloned into the Gateway system. The CARD8 Kfree construct contains all 10 lysines N-terminal to the CARD8 autoproteolysis site mutated to arginine (residues 3, 4, 9, 32, 41, 55, 147, 157, 175, 272). pEGFP-GFP11-Clathrin light chain and pcDNA3.1-GFP(1-10) were gifts from Bo Huang (Addgene plasmids #70217 and #70219, respectively). With the exception of pEGFP-GFP11-Clathrin light chain, all constructs prepared for transient transfection or lentiviral infection of cells were shuttled into modified pLEX_307 vectors (Addgene) using Gateway technology (Thermo Fisher Scientific). A pLEX_307 vector containing eGFP was used for transfection controls. Point mutations were generated using the QuikChange II site-directed mutagenesis kit (Agilent, 200523) following the manufacturer’s instructions.
Transient transfections
HEK293T cells were plated in 6-well culture plates at 5.0 × 105 cells/well in DMEM. The next day, the indicated plasmids were mixed with an empty vector to a total of 2.0 μg DNA in 125 μL Opti-MEM and transfected using FuGENE HD (Promega) according to the manufacturer’s protocol. Unless indicated otherwise, 0.02 μg CARD8 (or CARD8 chimera) was used. After 16-20 h, the cells were treated as described. For microscopy experiments, cells were plated directly into Nunc Lab-Tek II Chamber slide w/Cover sterile glass slides (Thermo Fisher Scientific, 154534) at 2.5 × 104 cells/well and treated with 25 μL transfection master mix dropwise.
LDH cytotoxicity, immunoblotting, and FLAG immunoprecipitation
HEK293T cells were transiently transfected and inhibitor treated as indicated. OCI-AML2 and MV-4-11 cells were plated in 6-well culture plates at 1.0 × 106 cells/well and treated with VbP as indicated. Supernatants were analyzed for LDH levels using the Pierce LDH Cytotoxicity Assay Kit (Life Technologies). LDH levels were quantified relative to a lysis control where cells were lysed in 80 μL of a 9% Triton X-100 solution. For FLAG immunoprecipitation, cell pellets were lysed in Tris-Buffered Saline (TBS) with 0.5% NP-40 on ice for 20 min and centrifuged at 20000 x g for 10 min. The soluble fraction was retained, and protein concentrations were normalized to 1 mg/mL using the DCA Protein Assay kit (Bio-Rad). Lysates were incubated with 40 μL of anti-FLAG-M2 agarose resin (Sigma) overnight at 4°C. After washing 3 × 1 mL with PBS, bound proteins were eluted by incubating resin with 100 μL of PBS containing 150 ng/ μL 3 × -FLAG peptide for 1 h at 4°C. An equal volume of 2 × sample loading dye was added to the eluate and incubated at 95°C for 10 min. For immunoblotting, cells were washed 2 × in PBS (pH = 7.4), resuspended in PBS, and lysed by sonication. Protein concentrations were determined and normalized using the DCA Protein Assay kit (Bio-Rad). The samples were separated by SDS-PAGE, immunoblotted, and visualized using the Odyssey Imaging System (Li-Cor).
Tandem mass tag labeling for mass spectrometry
HEK293T cells were seeded at 3 × 106 cells in 10 cm tissue culture dishes. The following day, cells were treated with either DMSO or VbP (10 μM). After 48 h, cells were harvested and lysed in 1 mL TBS containing 0.5% NP-40 per 10 cm dish on ice for 30 min. Lysates were then clarified at 20000 x g for 10 min. The soluble fraction was retained, and protein concentrations were normalized using the DCA Protein Assay kit (Bio-Rad). The samples were then reduced with 10 mM tris(2-carboxyethyl)phosphine (TCEP) for 1 h at 56°C while shaking and alkylated with 20 mM iodoacetamide for 30 min at room temperature, protected from light. Proteins were then precipitated by adding six volumes of pre-chilled acetone and incubated at −20°C overnight. Samples were then centrifuged at 8000 x g for 10 min at 4°C and carefully inverted to decant acetone. Acetone-precipitated pellets were then resuspended with 100 μL of 50 mM triethylammonium bicarbonate (TEAB) buffer per sample and total protein concentration was measured by DCA Protein Assay kit. Proteins were then digested overnight at 37°C with 2.5 μg trypsin per 100 μg of sample.
TMTsixplex Isobaric Label Reagents (ThermoFisher Scientific), 0.8 mg per label, were equilibrated to room temperature, dissolved in 41 μL of dry acetonitrile and mixed by vortex briefly before use. 41 μL of each TMT label reagent was carefully added to each sample (126, 127, and 128 = DMSO; 129, 130, and 131 = VbP) and incubated at room temperature for 1 h. 8 μL of 5% hydroxylamine was then added to each sample and incubated for 15 min to quench the labeling reaction. Samples were then combined in equal quantities, purified using the High pH Reversed-Phase Peptide Fractionation Kit (Pierce) and dried with a Genevac EZ-2 evaporator to give peptide pellets for tandem LC-MS/MS/MS analysis.
Tandem LC-MS/MS/MS
Mass spectrometry data was collected on an Orbitrap Fusion Lumos mass spectrometer coupled to an Easy-nLC 1200 (Thermo Fisher Scientific). Peptides were separated over a 220 min gradient of 0 to 50% acetonitrile in water with 0.1% formic acid at a flow rate of 300 nL/min on a 50 cm long PepMap RSLC C18 column (2 mm, 100 Å, 75 μm, x 50 cm). The full MS spectra were acquired in the Orbitrap at a resolution of 120,000. The 10 most intense MS1 ions were selected for MS2 analysis. The isolation width was set at 0.7 m/z and isolated precursors were fragmented by CID (35% CE). Following acquisition of each MS2 spectrum, a synchronous precursor selection (SPS) MS3 scan was collected on the top 10 most intense ions in the MS2 spectrum. The isolation width was set at 1.2 m/z and isolated precursors were fragmented using HCD. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez-Riverol et al., 2019) with the dataset identifier PXD015978.
Proteomic analysis
MS raw files were converted into MGF and processed using Proteome Discoverer version 2.2.0.388 (Thermo Scientific) by searching against the Uniprot human database supplemented with common contaminant protein sequences and quantifying according to SPS MS3 reporter ions. Identification was based on CID spectra using SequestHT. Search criteria included: 20 ppm mass tolerance for MS spectra, 0.5 Da fragment mass tolerance for MS/MS spectra, a maximum of two allowed missed cleavages, static carbamidomethylation (+57.021 Da) of cysteine and TMTsixplex (+229.163 Da) of lysine and of peptide N terminus, dynamic modifications of methionine oxidation (+15.995 Da), N-terminal protein acetylation (+42.011 Da), asparagine or glutamine deamidation (+0.984 Da), and serine, tyrosine or tryptophan phosphorylation (+79.966 Da) and a false discovery rate of < 0.01.
RNA-Seq and analysis
7 × 106 HEK293T cells were plated in 10 cm tissue culture plates. The following day, cells were treated with Val-boroPro (10 μM) or DMSO for 48 h. Cellular RNA was harvested using the RNeasy Mini Kit (QIAGEN). After RiboGreen quantification and quality control by Agilent BioAnalyzer, 500 ng of total RNA underwent polyA selection and TruSeq library preparation according to instructions provided by Illumina (TruSeq Stranded mRNA LT Kit, Cat. No. RS-122-2102) with 8 cycles of PCR. Samples were barcoded and run on a HiSeq 4000 in a 50 bp/50 bp paired end run, using the HiSeq 3000/4000 SBS Kit (Illumina). An average of 41 × 106 paired reads was generated per sample. At the most, the ribosomal reads represented 3.5% of the total reads generated and the percent of mRNA bases averaged 70%. FASTQ output files were aligned to the human genome using rnaStar aligner software, then 2-pass mapping method was performed as described in Engstrom et al., 2013. Expression count matrix R/Bioconductor package DESeq (http://www.huber.embl.de/users/anders/DESeq/). The RNA-Seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE153744 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153744).
Limited proteolysis
HEK293T cells were transiently transfected with 2 μg of indicated constructs. After 16-20 h, cells were harvested. Cells were washed 2 × in PBS (pH = 7.4), resuspended in PBS, and lysed by sonication. Protein concentrations were determined using the DCA Protein Assay kit (Bio-Rad) and normalized to 1 mg/mL. 48 μL of lysate was mixed with 12 μL of trypsin or proteinase K at the final indicated concentrations for 30 min at room temperature. The reaction was stopped by adding 60 μL of 2 × SDS-PAGE sample loading buffer and incubating samples for 10 min at 95°C. The samples were separated by SDS-PAGE, immunoblotted, and visualized using the Odyssey Imaging System (Li-Cor).
Fluorescence microscopy
Imaging was performed on a Zeiss Axio Observer.Z1 inverted widefield microscope using 40x/0.95NA air objective. Cells were plated on LabTek 8-well chambered cover glass (ThermoFisher Scientific, 155409) with #1.5 coverslip. For each chamber, 10 positions were imaged on brightfield, red, and green fluorescence channels at a single time point from a given experiment. Data was exported as raw .czi files and analyzed using custom macro written in ImageJ/FIJI. Total cell area was estimated from RFP-positive signal.
Generation of stable cell lines
For generating HEK293T ectopically expressing caspase-1 and gasdermin D, the expression plasmid was packaged into lentivirus in HEK293T cells using the Fugene HD transfection reagent (Promega) and 2 μg of the vector, 2 μg psPAX2, and 1 μg pMD2.G. After 2 days, HEK293T caspase-1-expressing cells were selected with hygromycin (200 μg/mL) and HEK293T gasdermin D-expressing cells were selected with puromycin (1 μg/mL).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism 7.0 software. Statistical significance was determined using two-sided Students t tests. Statistical significance was determined using two-sided Students t tests.
Proteomic statistical analysis was performed with pFIND. GraphPad Prism 7 software was used for SEM calculations. Clustal Omega was used to generate CARD8 species sequence alignment. For in vitro cell assays, n refers to number of replicate wells analyzed for each treatment group. Error bars for in vitro assay figures represent SEM from mean.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GSDMD Rabbit polyclonal Ab | Novus Biologicals | Cat# NBP2-33422; RRID:AB_2687913 |
| GSDMD Rabbit monoclonal Ab | Abcam | Cat# ab209845; RRID:AB_2783550 |
| FLAG® M2 monoclonal Ab | Sigma | Cat# F3165; RRID:AB_259529 |
| CARD8 C terminus Rabbit polyclonal Ab | Abcam | Cat# ab24186; RRID:AB_2275096 |
| CARD8 N-terminal Rabbit polyclonal Ab | Abcam | Ab194585 |
| GAPDH Rabbit monoclonal Ab | Cell Signaling Tech | Cat# 2118; RRID:AB_561053 |
| MTMR1 | Abcam | Ab240569 |
| D2HGDH | Abcam | Ab233516 |
| ATF-3 | Cell Signaling Tech | Cat# 33593; RRID:AB_2799039 |
| IGBP1 | Cell Signaling Tech | Cat# 5699; RRID:AB_10897328 |
| SMAD9 | Abcam | Cat# ab124094; RRID:AB_10971189 |
| ARF1 | Abcam | Cat# ab58578; RRID:AB_879566 |
| ACTR5 | Proteintech | Cat# 21505-1-AP; RRID:AB_10733470 |
| AKAP8 | Abcam | Cat# ab72196; RRID:AB_1267650 |
| GSTP1 | Cell Signaling Tech | Cat# 3369; RRID:AB_2279558 |
| SET | Abcam | Cat# ab1183; RRID:AB_298611 |
| NLRP1B | Vance Laboratory | 2A12 |
| IRDye 800CW anti-rabbit | LICOR | Cat# 925-32211; RRID:AB_2651127 |
| IRDye 800CW anti-mouse | LICOR | Cat# 925-32210; RRID:AB_2687825 |
| IRDye 680CW anti-rabbit | LICOR | Cat# 925-68073; RRID:AB_2716687 |
| IRDye 680CW anti-mouse | LICOR | Cat# 925-68072; RRID:AB_2814912 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Val-boroPro (VbP) | Okondo et al., 2017 | N/A |
| Bortezomib (Bort) | MilliporeSigma | 504314 |
| MLN4924 | Cayman | 15217 |
| bestatin methyl ester (Me-Bs) | Sigma | 200485 |
| Sequencing grade modified trypsin | Promega | V5113 |
| Proteinase K (PROK) | Invitrogen | 25530049 |
| FuGENE HD | Promega | E2311 |
| TMTsixplex Isobaric Label Reagents | ThermoFisher Scientific | 90061 |
| Critical Commercial Assays | ||
| MycoAlert Mycoplasma Detection Kit | Lonza | LT07-318 |
| DCA Protein Assay kit | Bio-Rad | 5000111 |
| Pierce LDH Cytotoxicity Assay Kit | Life Technologies | PI88953 |
| Deposited Data | ||
| TMT proteomics raw data | This paper | ProteomeXchange PXD015978 |
| RNA-seq raw data | This paper | GEO GSE153744 |
| Experimental Models: Cell Lines | ||
| Human HEK293T | ATCC | CRL-3216 |
| Human HEK293T CASP1/GSDMD | Johnson et al., 2018 | N/A |
| Human THP-1 | ATCC | TIB-202 |
| Human THP-1 CARD8 KO | Johnson et al., 2018 | N/A |
| Human THP-1 CARD8 KO + GFP | This paper | N/A |
| Human THP-1 CARD8 KO + CARD8 | This paper | N/A |
| Human THP-1 CARD8 KO + CARD8 ZUC | This paper | N/A |
| Human THP-1 CARD8 KO + GFP-CARD8 ZUC | This paper | N/A |
| Human THP-1 CARD8 KO + V5-GFP-CARD8 ZUC | This paper | N/A |
| Human THP-1 CARD8 KO + MTMR1(M1-Q94)-CARD8 ZUC | This paper | N/A |
| Human THP-1 CARD8 KO + D2HGDH(M1-R51)-CARD8 ZUC | This paper | N/A |
| Human OCI-AML2 | DSMZ | ACC 99 |
| Human MV-4-11 | DSMZ | N/A |
| Mouse RAW 264.7 | ATCC | TIB-71 |
| Oligonucleotides | ||
| Primer for CARD8 FL: ATGGAAAAAAAGGAGTGTCCAG | This paper | N/A |
| Primer for CARD8 ZUC: atggggcctgaaggaaatgtggatg | This paper | N/A |
| Primer for CARD8 G131M-537: ATGGACATTTGCTCAGAAGAG | This paper | N/A |
| Primer for CARD8 K147M-537: atgGTCTGTTTTGAGATCGAAG | This paper | N/A |
| Primer for CARD8 F150M-537: ATGGAGATCGAAGAAGATTATAAAAATCG | This paper | N/A |
| Primer for CARD8 E153M-537: ATGGAAGATTATAAAAATCGTCAGTTTCTG | This paper | N/A |
| Primer for CARD8 Y156M-537: ATGAAAAATCGTCAGTTTCTGGGG | This paper | N/A |
| Primer for CARD8 K157M-537: atgAATCGTCAGTTTCTGGG | This paper | N/A |
| Primer for CARD8 UPA-CARD: atgcgcctctccatccccatcac | This paper | N/A |
| sgRNA target sequence for CARD8: TGACGATTGCGTTTGGTTCC | Johnson et al., 2018 | N/A |
| Recombinant DNA | ||
| pLEX_307 | Gift from David Root | Addgene Plasmid Cat #41392 |
| MTMR1 | Origene | RC212024 |
| D2HGDH | Genscript | OHu27566 |
| pEGFP-GFP11-Clathrin light chain | Gift from Bo Huang | Addgene Plasmid Cat #70217 |
| pcDNA3.1-GFP(1-10) | Gift from Bo Huang | Addgene Plasmid Cat #70219 |
| pLEX_307_CARD8 (and truncations) | This paper | N/A |
| pLEX_307_HA-GSSGnx-CARD8 ZUC | This paper | N/A |
| pLEX_307_GFP-CARD8 ZUC | This paper | N/A |
| pLEX_307_V5-GFP-CARD8 ZUC | This paper | N/A |
| pLEX_307_GFP(1-10)-CARD8 ZUC | This paper | N/A |
| pLEX_307_MTMR1(M1-Q94)-CARD8 ZUC | This paper | N/A |
| pLEX_307_MTMR1 | This paper | N/A |
| pLEX_307_MTMR1Δ1-94 | This paper | N/A |
| pLEX_307_D2HGDH | This paper | N/A |
| pLEX_307_D2HGDHΔ1-51 | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism Version 7 | GraphPad Software | https://www.graphpad.com/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Proteome Discoverer version 2.2.0.388 | Thermo Fisher Scientific | OPTON-30945 |
Highlights.
CARD8 has an N-terminal disordered region that is required for its activation
DPP8/9 inhibitors induce the degradation of disordered and misfolded proteins
CARD8 uses its disordered region to sense a perturbation induced by DPP8/9 inhibition
ACKNOWLEDGMENTS
We thank W. Bachovchin, W. Wu, and J. Lai for VbP; R. Vance for the α-NLRP1B antibody (2A12); D. Rufa for advising protein disorder prediction analysis; M. Miele, Z. Li, and R. Hendrickson for mass spectrometry assistance; and Mono Pirun for RNA-seq assistance. This work was supported by the Josie Robertson Foundation (to D.A.B.), a Stand Up to Cancer (SU2C) innovative research grant (grant SU2C-AACR-IRG11-17 to D.A.B.; SU2C is a program of the Entertainment Industry Foundation, and research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C). This work also was supported by the Pew Charitable Trusts (to D.A.B. is a Pew-Stewart Scholar in Cancer Research), the Pershing Square Sohn Cancer Research Alliance (to D.A.B.), the NIH (R01 AI137168 to D.A.B., T32 GM007739-Andersen to A.R.G., F30 CA243444 to A.R.G., and T32 GM115327-Tan to E.L.O.), an MSKCC core grant (P30 CA008748), an Alfred P. Sloan Foundation research fellowship (to D.A.B.), Gabrielle’s Angel Foundation (to D.A.B.); an American Cancer Society postdoctoral fellowship (PF-17-224-01-CCG to C.Y.T.), Mr. William H. and Mrs. Alice Goodwin, the Commonwealth Foundation for Cancer Research, the Center for Experimental Therapeutics of Memorial Sloan Kettering Cancer Center (to D.A.B.), and the Ludwig Center at Memorial Sloan Kettering Cancer Center (to D.A.B.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108264.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Li et al. (1999); Madeira et al. (2019); Romero et al. (1997).
REFERENCES
- Bagnall RD, Roberts RG, Mirza MM, Torigoe T, Prescott NJ, and Mathew CG (2008). Novel isoforms of the CARD8 (TUCAN) gene evade a nonsense mutation. Eur. J. Hum. Genet 16, 619–625. [DOI] [PubMed] [Google Scholar]
- Ball DP, Taabazuing CY, Griswold AR, Orth EL, Rao SD, Kotliar IB, Johnson DC, and Bachovchin DA (2019). Human caspase-1 autoproteolysis is required for ASC-dependent and -independent inflammasome activation. bioRxiv. [Google Scholar]
- Boyden ED, and Dietrich WF (2006). Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet 38, 240–244. [DOI] [PubMed] [Google Scholar]
- Breydo L, Wu JW, and Uversky VN (2012). A-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 1822, 261–285. [DOI] [PubMed] [Google Scholar]
- Broz P, and Dixit VM (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16, 407–420. [DOI] [PubMed] [Google Scholar]
- Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, Ball DP, Taabazuing CY, Orth EL, Vittimberga BA, and Bachovchin DA (2019). N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli KM, Gorfu G, Hassan MA, Printz M, Crown D, Leppla SH, Grigg ME, Saeij JP, and Moayeri M (2014). Inflammasome sensor NLRP1 controls rat macrophage susceptibility to Toxoplasma gondii. PLoS Pathog. 10, e1003927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, and Reed JC (2011). CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS ONE 6, e27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom PG, Steijger T, Sipos B, Grant GR, Kahles A, TheConsortium RGASP, Ratsch G, Hubbard TJ, Harrow J, Guigo R, and Bertone P (2013). Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 10, 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald SE, Chavarria-Smith J, and Boothroyd JC (2014). NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun 82, 460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Sekine S, Pessino V, Li H, Leonetti MD, and Huang B (2017). Improved split fluorescent proteins for endogenous protein labeling. Nat. Commun 8, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, Bertin J, and Gough PJ (2012). Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem 287, 25030–25037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew BC, Joag VR, and Mogridge J (2012). Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 8, e1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai K, Okondo MC, Rao SD, Chui AJ, Ball DP, Johnson DC, and Bachovchin DA (2019). DPP8/9 inhibitors are universal activators of functional NLRP1 alleles. Cell Death Dis. 10, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold AR, Ball DP, Bhattacharjee A, Chui AJ, Rao SD, Taabazuing CY, and Bachovchin DA (2019). DPP9’s Enzymatic Activity and Not Its Binding to CARD8 Inhibits Inflammasome Activation. ACS Chem. Biol 14, 2424–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson J, Yang Y, Paliwal K, and Zhou Y (2017). Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics 33, 685–692. [DOI] [PubMed] [Google Scholar]
- Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A, and Bachovchin DA (2018). DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med 24, 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, Ishikawa H, Leonetti MD, Marshall WF, Weissman JS, and Huang B (2016). Versatile protein tagging in cells with split fluorescent protein. Nat. Commun 7, 11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, and Dixit VM (2014). Mechanisms and functions of inflammasomes. Cell 157, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Lee JG, Takahama S, Zhang G, Tomarev SI, and Ye Y (2016). Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol 18, 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Romero P, Rani M, Dunker AK, and Obradovic Z (1999). Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform. Ser. Workshop Genome Inform 10, 30–40. [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, and Lopez R (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47 (W1), W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- Newman ZL, Printz MP, Liu S, Crown D, Breen L, Miller-Randolph S, Flodman P, Leppla SH, and Moayeri M (2010). Susceptibility to anthrax lethal toxin-induced rat death is controlled by a single chromosome 10 locus that includes rNlrp1. PLoS Pathog. 6, e1000906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, Poplawski SE, Wu W, Liu Y, Lai JH, et al. (2017). DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol 13, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P, Obradovic Z, and Dunker KA (1997). Sequence Data Analysis for Long Disordered Regions Prediction in the Calcineurin Family. Genome Inform. Ser. Workshop Genome Inform 8, 110–124. [PubMed] [Google Scholar]
- Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, and Vance RE (2019). Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364, eaau1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736. [DOI] [PubMed] [Google Scholar]
- Taabazuing CY, Griswold AR, and Bachovchin DA (2020). The NLRP1 and CARD8 inflammasomes. Immunol. Rev 297, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong FL, Robinson K, Teo DET, Tan KY, Lim C, Harapas CR, Yu CH, Xie WH, Sobota RM, Au VB, et al. (2018). Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem 293, 18864–18878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez-Riverol et al., 2019) with the dataset identifier PXD015978. The RNA-Seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE153744 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153744).