

Abstract
In the current issue of Molecular cell, Liu et al. (2020) show that the secretion of cancer-linked forms of mutant calreticulin allow cancer cells to escape protective immune responses induced by chemotherapeutic and immunotherapeutic drugs, thereby promoting tumor growth.
Calreticulin is an endoplasmic reticulum (ER) localized lectin chaperone that assists in the folding of glycoprotein substrates. Calreticulin also helps maintain cellular calcium homeostasis by sequestering calcium ions via its C-terminal acidic domain. Calreticulin’s role in cellular physiology, however, is not just limited to the prevention of protein aggregation, maintenance of cellular homeostasis and ensuring quality control. It is a multifunctional protein that regulates cell-matrix adhesion, cell migration and phagocytosis of apoptotic cells (reviewed in Gold et al., 2010). Display of calreticulin as an ‘eat me’ signal on the surface of dying cells imprints these cells for removal by phagocytosis (Gardai et al., 2005). The C-terminal acidic domain of calreticulin is important for ER retention of calreticulin (along with its KDEL motif; (Sönnichsen et al., 1994)) and for calcium-dependent interactions with phospholipids on the surface of apoptotic cells (Wijeyesakere et al., 2016). Calreticulin exposure and subsequent calreticulin-dependent phagocytosis of dying tumor cells by dendritic cells are thought to precede the induction of strong CD8+ anti-tumor T cell responses, a type of immunogenic cell death (ICD) which is activated by various chemotherapeutic drugs (reviewed in Galluzzi et al., 2015). Recent studies showed that somatic frameshift mutations in exon 9 of the calreticulin gene are drivers of oncogenic transformation in a subset of patients with myeloproliferative neoplasms (MPN) (Klampfl et al., 2013; Nangalia et al., 2013). The most frequent mutations, type 1 (a 52 base-pair deletion, called Del52), and type 2 (5 base-pair insertion, called Ins5) cause frameshifts, resulting in the replacement of C-terminal acidic residues of calreticulin with basic residues, along with loss of the KDEL sequence. Not surprisingly, the mutant calreticulin proteins expressed ectopically in cell lines are secreted and mutant calreticulin proteins are also detectable in vivo in the plasma of Del52 knock-in mice and in MPN patients (reviewed in How et al., 2019). In this issue, Liu et al., (2020) have established an immunosuppressive role for the secreted form of mutant calreticulin, that equips cancer cells with the ability to avert phagocytic uptake by immune cells.
The authors used a technique called retention using selective hooks (RUSH) to confirm the differential localization of wild type and mutant calreticulin. This technique provides a useful tool to control the secretory trafficking of cargo proteins in cells. A reporter protein fused to streptavidin binding protein (SBP) is confined in a selected compartment via interaction with streptavidin carrying retention hooks for localization within that compartment (the ER being the compartment of interest in this study). Biotin-mediated competitive inhibition of SBP-streptavidin binding allows the release of SBP-reporter protein chimera that can be tracked and quantified (Boncompain et al., 2012). Wild type or MPN mutant calreticulin proteins were expressed in fusion with the SBP-GFP and retained within the ER via binding to the streptavidin-KDEL hook. Addition of biotin triggered the loss of the GFP signal for mutant calreticulin proteins from the cells and this phenotype was reversed by pretreatment with Brefeldin A (BFA), confirming trafficking of the mutant proteins via the secretory pathway. In contrast, wild type calreticulin was localized to ER even in the presence of biotin, due to its intact ER retention signal. The pathophysiological secretion of mutant calreticulin in vivo was verified by the detection of mutant calreticulin in plasma and on the surface of monocytes and lymphocytes of MPN patients.
Liu et al. (2020) further demonstrate the immunosuppressive role of secreted calreticulin. Oxaliplatin (OXA) and other platinum-based chemotherapeutic drugs can induce ICD by triggering surface calreticulin exposure (reviewed in Galluzzi et al., 2015). Here, the authors show that OXA treatment of MCA205 cells enhances their phagocytosis by CD11c+ dendritic cells, which is inhibited by treatment with recombinant wild type calreticulin protein. Inhibition of phagocytosis by recombinant calreticulin was replicated in vivo when OXA-treated MCA205 cells were injected into the spleen of C57Bl/6 mice, although the same effect was not tested with recombinant Del52 instead of wild type protein. Interestingly, however, knock-in mice expressing the Del52 mutant in hematopoietic cells also show reduced phagocytosis of OXA-treated cancer cells in the absence of any exogenous calreticulin. These findings indicate that extracellular forms of recombinant wild type calreticulin or MPN mutant calreticulin inhibit the phagocytic uptake of drug treated cancer cells, presumably by competing with receptor-binding sites on the phagocytes.
To further examine the functional outcomes of calreticulin-mediated phagocytosis inhibition, the authors vaccinated mice with cancer cells killed by treatment with another chemotherapeutic drug mitoxantrone (MTX; previously shown to induce ICD (reviewed in Galluzzi et al., 2015)) with or without intravenously administered recombinant calreticulin. Remarkably, the administration of recombinant purified calreticulin impaired the efficacy of vaccine-mediated tumor protection in C57Bl/6 mice. Upon re-challenge with live cancer cells, both the naïve mice and those with wild type calreticulin-expressing bone marrow transplants exhibited a reduction in the tumor incidence owing to the protective effects of vaccination, whereas mice bearing Del52 bone marrow transplants developed tumors at higher frequencies.
The authors also used the RUSH technique in vivo by injecting mice with live MCA205 cells expressing SBP-tagged wild type or mutant calreticulin proteins, followed by chemotherapy with MTX with or without biotin administration. As expected, biotin treatment reduced the intensity of cellular SBP-mutant calreticulin staining but not SBP-wild type calreticulin staining. Correspondingly, biotin administration along with mitoxantrone did not diminish the tumor protective effect of mitoxantrone in the SBP-wild type calreticulin context, but biotin treatment abrogated tumor protection conferred by MTX in the SBP-mutant calreticulin contexts. MTX treatment resulted in enhanced ratios of CD8+ T cells to T-regulatory cells (an immunosuppressive T cell subset) in wild type calreticulin-expressing tumors, and this ratio was reduced in tumors bearing mutant calreticulin proteins, correlating with their reduced in vivo phagocytic uptake by CD11c+ dendritic cells. Although, not fully elucidated from a mechanistic viewpoint, Liu et al. (2020) suggest a link between impaired phagocytosis, reduction in effector T cell numbers and tumor progression triggered by the secretion of mutant calreticulin proteins. Additionally, the presence of secreted forms of calreticulin suppressed the protective immunity furnished by immune checkpoint blockade therapy.
Based on the current models, mutant calreticulin proteins expressed in MPNs induce oncogenic transformation through aberrant and preferential interactions with the thrombopoietin (TPO) receptor MPL (reviewed in How et al., 2019). In the study by Liu et al (2020), MPN mutant calreticulin secretion is shown to provide an additional advantage to tumor growth by inhibiting tumor cell phagocytic uptake and reducing the activation of adaptive anti-tumor immune responses (Figures 1A and 1B). Since the C-terminus of calreticulin mediates calcium-dependent binding to apoptotic cells (Wijeyesakere et al., 2016), the level or the orientation of cell-surface calreticulin on dying tumor cells is also predicted to be different for the wild type compared to mutant calreticulin (Figure 1B), which requires further assessments. Additionally, cellular calreticulin insufficiency and dysfunction in tumor cells expressing mutant calreticulin can directly impact antigen presentation by tumor cells (Arshad and Cresswell, 2018). Interestingly, Liu et al. (2020), by analysis of cancer genomic databases, found calreticulin mutations in different types of solid tumors, with a high frequency of C-terminally located mutations, some of which were predicted to enhance their secretion. In mice, tumors expressing such mutants were also found to be refractory to the protective effects of MTX and OXA. Thus, mutations inducing calreticulin secretion are prevalent among other cancers besides MPNs, and can help cancer cells evade immune recognition.
Figure 1: Mutant calreticulin secreted by cancer cells inhibits their phagocytosis by dendritic cells.
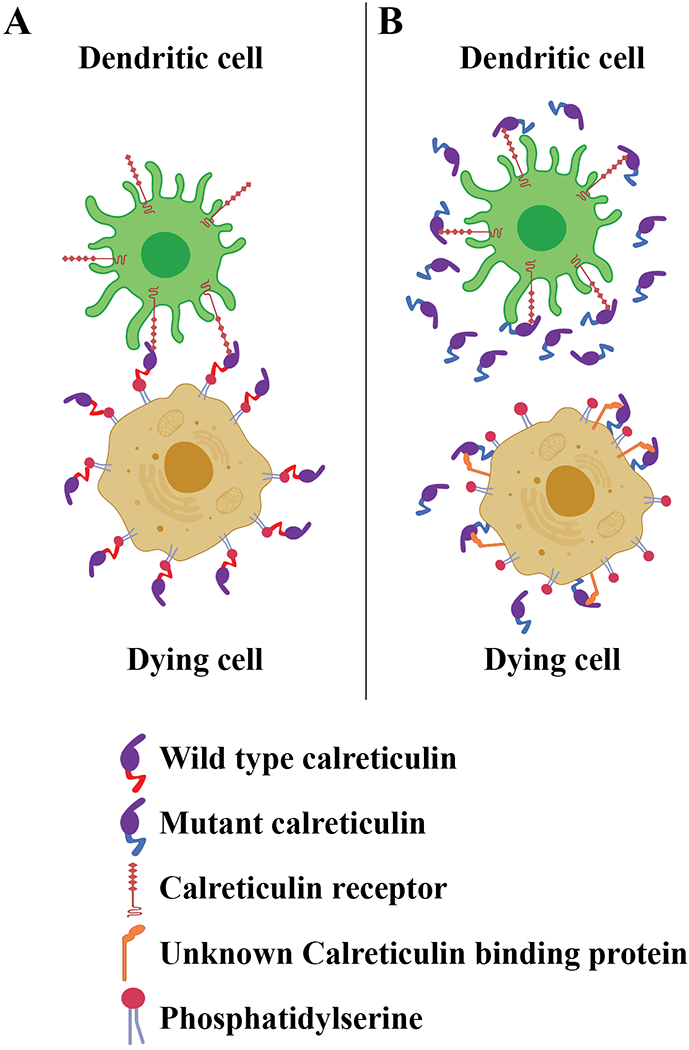
A) Dying cancer cells express cell-surface calreticulin under immunogenic cell death conditions, such as those induced by certain types of drug treatments (Galluzzi et al., 2015). The acidic C-terminal domain of wild type calreticulin is known to engage surface phosphatidylserine of apoptotic cells in a calcium-dependent manner, which is one mechanism of calreticulin anchoring to the cell surface (Wijeyesakere et al., 2016). Cell-surface calreticulin binds phagocyte receptors such as the lipoprotein-related receptor I (LRP-1) (Gardai et al 2005), to enhance phagocytic uptake and induction of protective anti-tumor T cell responses (Galluzzi et al., 2015).
B) Cancer cells express somatic mutants of calreticulin with basic C-terminal domains and lacking a KDEL sequence (Klampfl et al., 2013; Nangalia et al., 2013), and such mutants are known to be secreted (reviewed in How et al., 2019). Secreted forms of calreticulin are shown by Liu et al. to inhibit the phagocytosis of dying cancer cells by dendritic cells, presumably by saturating the calreticulin binding sites on the surface of dendritic cells. This results in reduced T cell-mediated anti-tumor immunity (Liu et al., 2020). The mode of calreticulin binding to the surface of dying cancer cells is also predicted to be altered by mutations of the C-terminal acidic domain of calreticulin to basic sequences, which need further assessment.
Acknowledgements:
This work is supported by NIH grants AI123957 and AI044115 (to MR) and by the University of Michigan Fast Forward Protein Folding Diseases Initiative.
Bibliography
- Arshad N, Cresswell P, 2018. J. Biol. Chem 293, 9555–9569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompain G, Divoux S, Gareil N, De Forges H, Lescure A, Latreche L, Mercanti V, Jollivet F, Raposo G, Perez F, 2012. Nat. Methods 9, 493–498. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G, 2015. Cancer Cell 28, 690–714. [DOI] [PubMed] [Google Scholar]
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM, 2005. Cell 123, 321–334. [DOI] [PubMed] [Google Scholar]
- Gold LI, Eggleton P, Sweetwyne MT, Van Duyn LB, Greives MR, Naylor SM, Michalak M, Murphy-Ullrich JE, 2010. FASEB J. 24, 665–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- How J, Hobbs G, Mullally A, 2019. Blood 134, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Carola Casetti I, Sant E, Ferretti V, Elena C, Schischlik F, Cleary C, Six Melanie, Schalling M, Schönegger A, Bock C, Malcovati L, Pascutto C, Superti-Furga G, Cazzola M, Kralovics R, Six M, Ricovero Cura Carattere Scientifico Policlinico San Matteo, di E., 2013. N Engl J Med 369, 2379–90. [DOI] [PubMed] [Google Scholar]
- Liu P, Zhao L, Loos F, Marty C, Xie W, Martins I, Lachkar S, Qu B, Waeckel-Énée E, Plo I, Vainchenker W, Perez F, Rodriguez D, López-Otin C, van Endert P, Zitvogel L, Kepp O, Kroemer G, 2020. Mol. Cell 77, 1–13. [DOI] [PubMed] [Google Scholar]
- Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, Aziz A, Godfrey AL, Hinton J, Martincorena I, Van Loo P, Jones AV, Guglielmelli P, Tarpey P, Harding HP, Fitzpatrick JD, Goudie CT, Ortmann CA, Loughran SJ, Raine K, Jones DR, Butler AP, Teague JW, O’Meara S, McLaren S, Bianchi M, Silber Y, Dimitropoulou D, Bloxham D, Mudie L, Maddison M, Robinson B, Keohane C, Maclean C, Hill K, Orchard K, Tauro S, Du MQ, Greaves M, Bowen D, Huntly BJP, Harrison CN, Cross NCP, Ron D, Vannucchi AM, Papaemmanuil E, Campbell PJ, Green AR, 2013. N. Engl. J. Med 369, 2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sönnichsen B, Füllekrug J, Nguyen Van P, Diekmann W, Robinson DG, Mieskes G, 1994. J. Cell Sci 107, 2705–2717. [DOI] [PubMed] [Google Scholar]
- Wijeyesakere SJ, Bedi SK, Huynh D, Raghavan M, 2016. J. Immunol 196, 3896–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]