

Abstract
Phenotypic variability in the presence of an identical molecular defect is a recurrent feature in heritable disorders and it was also reported in osteogenesis imperfecta (OI). OI is a prototype for skeletal dysplasias mainly caused by mutations in the two genes coding for type I collagen. No definitive cure is available for this disorder, but the understanding of molecular basis in OI phenotypic modulation will have a pivotal role in identifying possible targets to develop novel drug therapy.
We used a functional proteomic approach to address the study of phenotypic variability using the skin of the OI murine model Brtl. Brtl mice reproduce the molecular defect, dominant transmission and phenotypic variability of human OI patients. In the presence of a Gly349Cys substitution in α1(I)-collagen Brtl mice can have a lethal or a moderately severe outcome. Differential expression of chaperones, proteasomal subunits, metabolic enzymes, and proteins related to cellular fate demonstrated that a different ability to adapt to cellular stress distinguished mutant from wild-type mice and mutant lethal from surviving mutant animals. Interestingly, class discovery analysis identified clusters of differentially expressed proteins associated with a specific outcome, and functional analysis contributed to a deeper investigation into biochemical and cellular pathways affected by the disease. This article is part of a Special Issue entitled: Translational Proteomics.
Keywords: Functional proteomics, Pathway analysis, Hierarchical clustering, Osteogenesis imperfecta, Chaperones, Proteasomal
Graphical Abstract
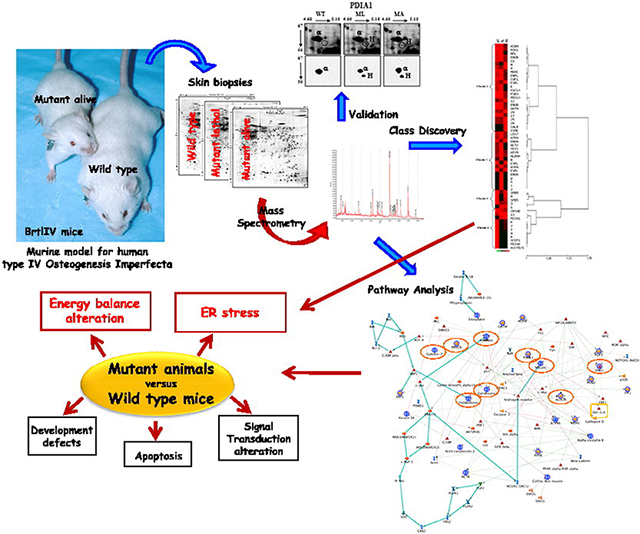
1. Introduction
The classical form of the “brittle bone disease” osteogenesis imperfecta (OI), classified by Sillence in four different types, from very mild to lethal [1], is a dominant negative disorder caused by mutations in the two genes, COL1A1 and COL1A2, coding for the α chains of type I collagen, the major structural protein of the extracellular matrix of bone, skin and tendon [2,3]. The typical clinical features of this pathology are bone fragility, skeletal deformities and growth deficiency, within a broad phenotypic range. Other symptoms of OI include short stature, blue sclerae, joint laxity, dentinogenesis imperfecta, and hearing loss [2].
At a biochemical level, classical OI is characterized by either quantitative or qualitative defects in the synthesis of type I collagen. The very mild form of the disease (OI type I) is generally caused by the presence of a null COL1A1 allele, due mainly to mutations which induce premature chain termination. In OI type I cases only half of the normal amount of type I procollagen is synthesized, although its structure is normal.
The more severe phenotypes (OI types II, III, and IV) are due to structural defects in the protein. The great majority (85%) of causative mutations are single nucleotide changes responsible for a glycine substitution with bulky, polar, or charged amino acids, in the collagen triple helical domain characterized by the triplet (Gly-X-Y)338. Exon skipping, insertions, and large deletions were also reported [4]. Although over 1500 independent mutations had been identified [5,6], a clear relationship between genotype and phenotype is still puzzling the research field [4] and even less clear are the molecular bases of the OI phenotypic variability reported in patients with identical molecular defect [4,7,8]. The OI clinical variability is still largely unknown mainly due to the limited availability of human samples and to the fact that the murine models of the disease are often inbred, thus lacking the heterogeneity of the genetic background characteristic of human patients.
Among the several available murine models for classical dominant OI [9-11], the Brittle (Brtl) mouse is the only knock-in murine model for the moderate severe form of osteogenesis imperfecta (human OI Type IV) [12]. Brtl reproduces the typical molecular defect, a glycine substitution in half of the α1(I) chains of type I collagen, the autosomic type of transmission and the clinical outcome reported for classical OI type IV patients. In the presence of the same Gly349Cys substitution, Brtl mice show both the lethal and the moderately severe phenotype [12]. This phenotypic variability reproduces the variability described in some OI patients. Furthermore Brtl mice have the advantage to be outbred, thus reproducing the heterogeneity of the human genetic background.
We recently used microarray and proteomic approaches to investigate OI phenotypic variability using calvarial bone samples from 1 day old Brtl mice with lethal or moderately severe outcome and WT littermate [13]. We found that both extracellular and intracellular factors were involved in the modulation of the clinical outcome in Brtl mice. In particular in mice with different phenotypes we identified a differential cellular response to the ER stress, known to be caused by mutant collagen retention [14]. Brtl mice with lethal phenotype showed a bone specific up-regulation of the pro-apoptotic protein Gadd153/Chop, whereas mutant mice with moderate severe outcome revealed an increased level of the heat shock protein αB-crystallin. Although the ability to handle cellular malfunction seemed to be of primary relevance in the modulation of OI phenotype, a difference in the expression of some extracellular matrix proteins was also showed in mutant animals. In lethal Brtl mice we identified an increased expression of the cartilaginous proteins Prelp, Bmp6, and Bmp7 and a lower expression of the bone matrix proteins matrilin 4, microfibril-associated glycoprotein 2, and thrombospondin 3, which suggested that both a delay in skeletal development and an alteration in extracellular matrix composition influenced OI outcomes [13].
OI is a bone disease, but skin outcome was reported both in patients and in murine models [15,16]. Furthermore, the great majority of human OI studies are based on fibroblasts culture data and skin biopsies are easier to obtain from OI patients [17]. Thus detailed information on OI dermal protein patterns, still missing at the present, will be a useful tool to translate and compare mice data to human samples.
In the present study we used a proteomic approach to define the expression pattern of skin proteins in wild type and Brtl mice.
Whole skin samples, including epithelial and non-epithelial components, from wild-type, mutant lethal and mutant alive mice were separated by high-resolution 2-D electrophoresis. The resulting electropherograms of skin proteins were subsequently analyzed by image analysis dedicated software and the biological reliability of the obtained data was assured by accurate statistics.
Significant qualitative as well as quantitative differences were observed between the protein expression profiles of skin samples from the three murine groups and identified by mass spectrometry. All the identified protein differences were functionally processed using specific software for class discovery and pathway analysis. This functional data processing allowed a deeper insight into the biochemical mechanisms as well as into cellular pathways affected by the disease.
2. Materials and methods
2.1. Animals
Brtl mice and wild-type littermates were kept in the animal facility of the Dept. of Experimental Medicine of the University of Pavia, Pavia, Italy [12]. The animals were maintained under standard experimental animal care protocol following the Italian Laws. One day old mice were used for the present study.
2.2. 2-D gel electrophoresis
Skin biopsies were obtained from 1 day old mutant lethal (ML) and moderate severe type IV (MA) Brtl mice and wild-type (WT) littermates (at least 7 different animals were analysed for each mouse type). Skin samples were thoroughly washed in PBS, minced and immediately frozen in liquid nitrogen, and kept at −80 °C until processed.
Protein extracts were obtained by pestle-pounding minced samples in the presence of a conventional 2-D lysis buffer: 8 M Urea, 4% (w/v) CHAPS, 1% (w/v) dithioerythritol (DTE), and samples were stored at −80 °C. Total protein quantitation was determined according to standard procedure [18].
2-DE was performed using the Immobiline-polyacrylamide system as described [19,20]. IEF was carried out on non-linear wide-range immobilized pH gradients (pH 3–10; 18 cm long IPG strips; GE Healthcare, Uppsala, Sweden) and performed using the Ettan™ IPGphor™ system (GE Healthcare). Strips for analytical and for 2-D Western blot preparative runs were rehydrated with 60 μg and 120 μg of protein, respectively, in 350 μl of lysis buffer and 0.2% (v/v) carrier ampholyte for 1 h at 0 V and for 8 h at 30 V, at 16 °C. The strips were then focused according to the following electrical conditions at 16 °C: 200 V for 1 h, from 300 V to 3500 V in 30 min, 3500 V for 3 h, from 3500 V to 8000 V in 30 min, 8000 V until a total of 80,000 Vh was reached.
MS-preparative IPG-strips were instead rehydrated with 350 μl of lysis buffer and 2% (v/v) carrier ampholyte, for 12 h at room temperature. Sample load, 800 μg per strip, was successively performed by cup loading in the IPGphor Cup Loading Strip Holders (GE Healthcare), and, to obtain a successful focusing on basic pH values, it was applied at the cathodic end of the strip. IEF was then achieved according to the following voltage steps at 16 °C: 30 V for 30 min, 200 V for 2 h, 500 V for 2 h, from 500 V to 3500 V in 30 min, 3500 V for 5 h, from 3500 V to 5000 V in 30 min, 5000 V for 4 h, from 5000 V to 8000 V in 30 min, 8000 V until a total of 95,000 Vh was reached.
After focusing, analytical and preparative IPG strips were equilibrated for 12 min in 6 M urea, 30% (v/v) glycerol, 2% (w/v) SDS, 0.05 M Tris–HCl, pH 6.8, 2% (w/v) DTE; and for further 5 min in the same urea/SDS/Tris buffer solution but substituting the 2% (w/v) DTE with 2.5% (w/v) iodoacetamide. The second dimension was carried out, at 10 °C, on 9–16% polyacrylamide linear gradient gels at 40 mA/gel constant current, until the dye front reached the bottom of the gel. Analytical gels were stained with ammoniacal silver nitrate, as previously described [21,22], while MS-preparative gels were stained according to a silver staining protocol compatible with MS [23] or using the SYPRO Ruby (Bio-Rad Headquarters, Hercules, CA) procedure, which was performed following the manufacturer’s protocol.
Silver stained gels were digitalized using a Molecular Dynamics 300S laser densitometer (4000×5000 pixels, 12 bits/ pixel; Sunnyvale, CA). Images of preparative-gels stained with SYPRO Ruby were instead digitalized with a Typhoon 9400 laser densitometer (GE Healthcare). Computer-aided 2-D image analysis was carried out using ImageMaster 2-D Platinum 7.0 software (GE Healthcare). For each tested condition image analysis was performed on 5 different maps from 5 different mice, with every produced spot map representing the skin protein expression profile of a single mouse.
Experimental pI and Mr (Da) values were determined by comigration with human serum as internal standard [24,25].
2.3. Statistical analysis
According to a fold change, between the three sample sets, of at least ±2 in relative volume (%V) ratio (Vol: integration of optical density over the spot area; %V: Vol of a single spot divided by the total volume of spots computed over the whole image and expressed in percentage form), and to the statistical analysis, with 1-way ANOVA at 95% level of significance (p<0.05), proteins with significant differences in expression were highlighted in the three different tested conditions. Pairwise comparison of experimental groups was also performed for the abundance significantly changing spots by the Tukey’s posthoc multiple comparison test using the Exel Template interSTAT-a 2.0.
In addition to the univariate, an unsupervised multivariate analysis, based on differential protein expression profiles, was carried out using the statistics and analytics software package STATISTICA 7.0 (StatSoft, Tulsa, Oklahoma). The overall correlations occurring between different spot maps were visualized by a principal components analysis (PCA). Spot maps were actually plotted in the two-dimensional space resulting in the PCA1 and PCA2 principal components that orthogonally divide the analyzed samples according to the two principal sources of variation in the data set. Matched spots considered for the multivariate analysis were selected according to volume ratio and 1-way ANOVA p values, as described above.
Finally, an unsupervised pattern analysis, Hierarchical Clustering (HC), was obtained using the EPCLUST tool for clustering and analysis of gene-expression data within Expression Profiler. Expression Profiler is a set of tool for microarray analysis developed at the European Bioinformatics Institute (EBI), available on line at the following web site: http://www.bioinf.ebc.ee/EP/EP, and maintained by The Bioinformatics, Algorithmics, and Data Mining group BITT. Hierarchical Clustering, similarly to PCA, performs sample clustering based on expression similarities occurring between proteins of interest. Filter criteria applied to matched spots, for the selection of proteins of interest to submit to the cluster analysis, were the same described above. HC expression matrix was computed using Absolute value of linear correlation based distance (Pearson) and average linkage (weighted group average, WPGMA).
2.4. Protein identification by mass spectrometry
Protein identification was carried out by peptide mass fingerprinting on an Ettan MALDI-ToF Pro mass spectrometer (GE Healthcare) [26,27] and by nano-LC-ESI-MS/MS peptide sequencing using a Micro-HPLC Pump Phoenix 40 (Thermo) and a LCQ DECA IT mass spectrometer (Thermo) [28].
Silver stained spots were manually excised, destained [29] and acetonitrile dehydrated. SYPRO Ruby stained spots were instead automatically excised, by an ETTAN spot-picker (GE Healthcare), destained in 2.5 mM ammonium bicarbonate and 50% acetonitrile, and finally dehydrated in acetonitrile. After spot rehydration in trypsin solution (Sigma Aldrich, St. Louis, MO), in-gel protein digestion was performed by an overnight incubation at 37 °C. From each spot, 0.75 μl of recovered digested peptides was prepared for MALDI-ToF MS by spotting them onto the MALDI target, allowed to dry, and then mixed to 0.75 μl of matrix solution (saturated solution of alphacyano-4-hydroxycinnamic acid in 50% (v/v) acetonitrile and 0.5% (v/v) trifluoroacetic acid). After matrix application to the dried sample and its own drying, tryptic peptide masses were acquired on an Ettan MALDI-ToF Pro mass spectrometer (GE Healthcare) [26,27]. Recorded spectra were calibrated using, as internal standard, the 842.509 and 2211.105 m/z peptides arising from trypsin autoproteolysis. The resulting mass lists were filtered for contaminant removal: mass matrix-related ions, trypsin auto-lysis and keratin peaks. Then, mass fingerprinting searching was carried out in UniProtKB database using Mascot (Matrix Science Ltd., London, UK, http://www.matrixscience.com) on-line-available software. Protein identification was achieved on the basis of corresponding experimental and theoretical peptide-fingerprinting patterns with a Δmass less than 100 ppm and the number of accepted missed cleavage sites set to one. The database search was taxonomically limited to Mus musculus and alkylation of cysteine by carbamidomethylation was assumed as fixed modification, while oxidation of methionine was considered as a possible one. The criteria used to accept identifications included the extent of sequence coverage, number of matched peptides, and probabilistic score, as detailed in Tables 1 and 2. Spots that were not unambiguously identified by MALDI-ToF MS were subsequently subjected to peptide sequencing on a nanoscale LC-ESI-MS/MS system using a Micro-HPLC Pump Phoenix 40 (Thermo) and a LCQ DECA IT mass spectrometer (Thermo). After tryptic peptide extraction in 35 μl of 50% (v/v) acetonitrile and 0.1% (v/v) TFA solution, samples were concentrated by Speed-Vac (SC110A Savant Speed-Vac, Thermo) and the nanoscale LC-ESI-MS/MS peptide sequencing was performed as previously described [28]. Briefly, 5 μl of sample solution was manually injected in a six-port valve and the peptides were trapped in a reversed-phase C18 trapping column (20 mm×100 μm ID×360 μm OD, Nanoseparations, Nieuwkoop, NL) using solvent A (0.1% formic acid in HPLC grade water) under a flow rate of 5 μl/min for 10 min. A linear gradient to 60% solvent B (0.1% formic acid in acetonitrile) in 30 min was used for analytical separation of peptides and a column flow-rate of 100–125 nl/min was obtained on a successive reversed-phase C18 analytical column (30cm×50 μm ID×360 μm OD, Nanoseparations) by a precolumn splitter restrictor. The ESI emitter, a gold-coated fused silica needle (5cm×25 μm ID×360 μm OD, Nanoseparations), was heated to 195 °C and an electric field strengths of up to 2 kV was applied for stable spray operation. The LC pump, the mass spectrometer as well as the automatic mass spectra acquisitions were controlled using the Excalibur software (Thermo). The mass spectrometer operated in data-dependent mode: a total of 3×106 ions were targeted per MS scan and 1×105 precursor ions were targeted for isolation for MS/MS spectral acquisition. The MS/MS database searching was carried out in the UniProtKB database using Mascot MS/MS ion search software. The taxonomy was limited to Mus musculus, peptide precursor charge was set to +2 or +3, mass tolerance of ±1.2 Da for precursor peptide and ±0.6 Da for fragment peptides were allowed and the number of accepted missed cleavage sites was set to one. Alkylation of cysteine by carbamidomethylation was assumed as fixed modification, while oxidation was considered as possible modification. We consider significant peptides with individual ion scores p <0.05.
Table 1 –
MS identified qualitative differences.
| Spot letter |
Protein description |
Metacore name a |
UniProt ID/name b |
HCA cluster c |
Subcellular location |
Theoretical pI/Mr (kDa) d |
Experimental pI/Mr (kDa) e |
Mascot search results |
Mean %V±SD×10−4 f |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | No. of matched peptides |
Sequence coverage (%) |
Wild type | Mutant lethal |
Mutant alive |
||||||||
| Cytoskeleton and cellular organization | |||||||||||||
| A | Keratin, type I cytoskeletal 10 | Keratin10 | P02535/K1C10 | 1 | Cytoplasm | 5.04/57.77 | 5.30/75.66 | 247 | 22/35 | 42 | 422±199 | 824±231 | - |
| C | α-Centractin | - | P61164/ACTZ | 2 | Cytoplasm | 6.19/42.61 | 6.66/49.28 | 107 | 6/6 | 26 | 68±9 | 203±40 | - |
| e | β-Actin γ-Actin |
ACTB Actin cytoplasmic 2 |
P60710/AXTB P63260/AXTG |
4 | Cytoplasm | 5.29/41.74 5.31/41.79 |
5.51/40.95 | 87 | 5/5 | 21 | 81±20 | - | - |
| l | Vimentin | Vimentin | P20152/VIME | 1 | Cytoplasm | 5.06/53.69 | 4.73/54.78 | 215 | 18/23 | 38 | - | - | 584±320 |
| L | Envoplakin | Envoplakin | Q9D952/EVPL | 2 | Cytoplasm | 6.16/232.20 | 6.08/161.26 | 91 | 16/27 | 9 | 165±36 | - | 152±20 |
| Protein fate (synthesis, folding, modification and destination) | |||||||||||||
| k | Calreticulin | Calreticulin | P14211/CALR | 1 | Endoplasmic reticulum | 4.33/47.99 | 5.35/63.34 | 150 | 11/18 | 39 | - | - | 1565±366 |
| H | Protein disulfide-isomerase A1 | P4HB | P09103/PDIA1 | 4 | Endoplasmic reticulum/cell surface | 4.77/57.06 | 4.84/57.70 | 107 | 8/12 | 17 | - | 387±54 | 536±43 |
| d | Protein disulfide-isomerase A6 | ERP5 | Q63081/PDIA6 | 4 | Endoplasmic reticulum | 5.00/48.17 | 6.53/44.99 | 134 | 8/9 | 22 | 410±151 | - | - |
| Metabolism | |||||||||||||
| c | Succinyl-CoA:3-ketoacid-coenzyme A transferase 1 | - | Q9D0K2/SCOT1 | 4 | Mitochondrion | 8.73/55.99 | 8.70/52.84 | MISSYVGENAEFER DGSVAIASKPR GMGGAMDLVSSSK | 142±18 | - | - | ||
| D | N-acetylneuraminate lyase | - | Q9DCJ9/NPL | 1 | Cytoplasm | 7.75/35.13 | 8.76/34.55 | 146 | 8/8 | 34 | 1247±176 | 1718±356 | - |
| f | α-enolase | ENO1 | P17182/ENOA | 2 | Cytoplasm | 6.37/47.14 | 6.47/53.10 | 112 | 8/12 | 29 | - | 87±27 | - |
| F | Triosephosphate isomerase | TPI1 | P17751/TPIS | 2 | Cytoplasm | 7.09/26.58 | 7.46/24.74 | 141 | 10/15 | 44 | 104±37 | 317±164 | - |
| Cellular fate | |||||||||||||
| O | Maspin | Maspin | P70124/SPB5 | 3 | Extracellular space | 5.55/42.11 | 5.43/41.83 | 175 | 11/13 | 38 | 175±20 | - | 95±44 |
Protein name in MetaCore pathway. Three proteins were excluded from the path according to the set parameters (−).
UniProt ID and entry name.
Cluster number in which proteins have been grouped according to their expression pattern as highlighted by the Hierarchical Clustering Analysis (HCA) (Fig. 3).
Predicted pI and Mr according to protein sequence as computed by the Compute pI/Mw tool (http://web.expasy.org/compute_pi/).
Experimentally determined pI and Mr using human serum as internal standard.
Each value represents the mean±SD of individually computed %V in spot maps from wild-type, mutant lethal and mutant alive animals.
Table 2 –
MS identified quantitative differences.
| Spot no |
Protein description |
MetaCore name a |
UniProt ID/ name b |
HCA Cluster c | Subcellular location |
Theoretical pI/Mr (kDa) d |
Experimental pI/Mr (kDa) e |
Mascot search results |
Mean %V±SD×10−4 f |
1-way ANOVA p value |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | No. of matched peptides |
Sequence coverage (%) |
Wild type | Mutant lethal |
Mutant alive |
|||||||||
| Cytoskeleton and cellular organization | ||||||||||||||
| 1 | Envoplakin | Envoplakin | Q9D952/EVPL | 1 | Cell junction, cytoplasm, cornified envelope | 6.16/232.32 | 6.20/161.70 | 145 | 18/22 | 10 | 144±42* | ↓57±8*§ | 183±7§ g | 1.27E–05 |
| 2 | Envoplakin | Envoplakin | Q9D952/EVPL | 2 | Cell junction, cytoplasm, cornified envelope | 6.16/232.32 | 6.48/161.70 | 107 | 17/26 | 11 | 174±94* | ↓29±10*§ | 126±29§ | 0.005 |
| 24 | Cofilin-1 | Cofilin, non muscle | P18760/COF1 | 1 | Nucleus matrix, cytoplasm | 8.22/18.56 | 9.18/15.11 | 107 | 6/9 | 46 | 1865±269¥ | 2850±504§ | ↓701±158¥§ | 1.59E–06 |
| Protein fate (synthesis, folding, modification and destination) | ||||||||||||||
| 19 | 26S protease regulatory subunit S10B | PSMC6 | P62334/PRS10 | 1 | Cytoplasm, nucleus | 7.09/44.17 | 8.07/47.02 | 159 | 12/14 | 33 | 530±66¥ | 473±89§ | ↓168±36¥§ | 3.88E–06 |
| 22 | Proteasome subunit β type 3 | PSMB3 | Q9R1P1/PSB3 | 1 | Cytoplasm, nucleus | 6.15/22.96 | 6.25/24.20 | 110 | 7/7 | 38 | 560±33¥ | 508±135§ | ↓195±69¥§ | 6.04E–05 |
| 23 | α-crystallin B chain | Alpha crystallin B | P23927/CRYAB | 4 | Cytoplasm, nucleus | 6.76/20.07 | 7.87/17.39 | 116 | 8/13 | 50 | 656±174¥ | 520±224§ | ↓242±2¥§ | 0.005 |
| 27 | Endoplasmin | Endoplasmin | P08113/ENPL | 1 | Endoplasmic reticulum | 4.74/92.48 | 4.72/59.11 N-fragment | TDDEVVQR FAFQAEVNR ELISNASDALDK SGTSEFLNK EEASDYLELDTIK | 1107±26¥ | 1812±60§ | ↓170±27¥§ | 6.48E–16 | ||
| Metabolism | ||||||||||||||
| 5 | α-Enolase | ENO1 | P17182/ENOA | 2 | Cytoplasm, cell membrane | 6.36/47.01 | 6.06/50.34 | 165 | 14/20 | 44 | 1215±226* | –324±11*§ | 1550±9§ | 1.32E–08 |
| 6 | Pyruvate kinase isozymes M1/M2 | PKM2 | P52480/KPYM | 2 | Cytoplasm, nucleus | 7.17/57.85 | 6.03/45.78 N-fragment | 100 | 8/12 | 23 | 164±47* | ↑550±86*§ | 108±50§ | 2.11E–07 |
| 7 | Fructose-bisphosphate aldolase A | ALDOA | P05064/ALDOA | 2 | Cytoplasm | 8.30/39.35 | 8.39/34.14 | GILAADESTGSIAK LQSIGTENTEENR RLQSIGTENTEENR QLLLTADDR FSNEEIAMATVTALR | 206±43* | ↑539±46*§ | 181±29§ | 9.37E–09 | ||
| 8 | ATP synthase α chain | ATP5A | Q03265/ATPA | 2 | Mitochondrion, cell membrane | 9.22/59.75 | 7.71/22.36 C-fragment | 114 | 8/8 | 20 | 423±7* | ↑933±93*§ | 269±55§ | 2.72E–09 |
| 9 | ATP synthase α chain | ATP5A | Q03265/ATPA | 2 | Mitochondrion, cell membrane | 9.22/59.75 | 6.82/22.06 C-fragment | 105 | 8/9 | 20 | 173±17* | f491±76*§ | 155±28§ | 1.31E–07 |
| 14 | β-Enolase | - | P21550/ENOB | 1 | Cytoplasm | 6.81/46.89 | 7.36/51.22 | 126 | 11/20 | 37 | 1199±235¥ | 1577±395§ | ↓480±169¥§ | 0.0002 |
| 15 | β-Enolase | - | P21550/ENOB | 1 | Cytoplasm | 6.81/46.89 | 6.95/51.22 | 115 | 9/13 | 31 | 520±200¥ | 583±365§ | ↓111±5¥§ | 0.041 |
| 16 | Glycine amidinotransferase | GATM | Q9D964/GATM | 1 | Mitochondrion | 8.00/48.30 | 7.14/50.24 | 112 | 9/14 | 20 | 250±65¥ | 269±13§ | ↓56±4¥§ | 2.04E–06 |
| 18 | Phosphoglycerate kinase 1 | PGK1 | P09411/PGK1 | 1 | Cytoplasm | 7.52/44.40 | 8.33/48.45 | 110 | 9/13 | 35 | 487±117¥ | 502±70§ | ↓164±57¥§ | 5.21E–05 |
| 20 | Creatine kinase, M chain | KCRM | P07310/KCRM | 1 | Cytoplasm | 6.58/43.04 | 7.32/46.12 | 165 | 11/16 | 32 | 455±63¥ | 544±71§ | ↓150±41¥§ | 5.69E–07 |
| 21 | Malate dehydrogenase | MDH1 | P14152/MDHC | 1 | Cytoplasm | 6.16/36.34 | 6.18/34.62 | 116 | 9/15 | 35 | 650±26¥ | 1037±152¥ | f140±11¥§ | 8.42E–09 |
| Cellular fate | ||||||||||||||
| 10 | Galectin-7 | Galectin-7 | O54974/LEG7 | 2 | Cytoplasm, nucleus, secreated | 6.70/15.17 | 7.00/12.62 | TSLPQGVR LDTSEVVFNTK | 451±190* | ↑916±25*§ | 274±87§ | 7.14E–06 | ||
| 26 | Maspin | Maspin | P70124/SPB5 | 3 | Extracellular space | 5.55/42.11 | 5.45/46.50 | 179 | 12/16 | 45 | 3286±144* | ↓702±22*§ | 2524±498§ | 3.64E–08 |
Protein name in MetaCore pathway. One protein was excluded from the path according to the set parameters (−).
UniProt ID and entry name.
Cluster number in which proteins have been grouped according to their expression pattern as highlighted by the Hierarchical Clustering Analysis (HCA) (Fig. 3).
Predicted pI and Mr according to protein sequence as computed by the Compute pI/Mw tool (http://web.expasy.org/compute_pi/).
Measured pI and Mr values were experimentally determined using human serum as internal standard.
Each value represents the mean±SD of individually computed %V in spot maps from wild-type, mutant lethal and mutant alive mice.
Multiple pair-wise comparison was performed using a Tukey post hoc test (p≤0.05).
Only proteins showing both statistical reliability and at least two fold change in expression are listed as significant differences: wild-type means that significantly differ from mutant lethal means are visualized by
wild-type means significantly differing from mutant alive ones are visualized by; and
significant differences occurring between mutant lethal and mutant alive mice are shown by.
Finally, arrows indicate the significant up (↑) or down (↓) variation of protein expression in mutants compared to WT animals.
2.5. Protein network analyses
Proteins changing in expression that were identified by MS were further analyzed by pathway analysis using the MetaCore network building tool (GeneGo Inc., St. Joneph, MI). MetaCore includes a manually annotated database of protein interactions and metabolic reactions obtained by scientific literature. Gene names of the all identified proteins and their corresponding mean fold change values were imported into MetaCore and processed using the shortest path algorithm. Hypothetical networks were built among the experimental proteins and the MetaCore protein database. The relevant pathway maps were then prioritized according to their statistical significance (p<0.001) and networks were graphically visualized as nodes (proteins) and edges (the relationship between proteins).
2.6. Western blot
For 1-D Western blot, skin and calvarial bone protein extracts from 1 day old ML and MA Brtl mice, and WT littermates (at least n=3 different animals for each mouse type) were obtained by pestle-pounding minced samples in the presence of the Laemmli buffer: 100 mM Tris-HCl pH 6.8, 2% (w/v) SDS, 20% (v/v) glycerol, 4% (v/v) β-mercaptoethanol, and heated at 95 °C for 5 min [30]. For each sample, 25 μg of proteins was separated on 12% polyacrylamide gel and then transferred onto nitrocellulose (Hybond ECL, GE Healthcare) according to Towbin [31].
Also 2-D Western blots were performed by standard procedure. After the second dimension, Western blot preparative gels were electroblotted onto nitrocellulose by overnight transfer via a total current of 2 Å, at 4 °C.
Before immunodetection, gel sample loading, in 1-D and 2-D Western blots, was preliminary proved to be equivalent by Ponceau Red staining (0.2% w/v Ponceau S in 3% w/v trichloroacetic acid) of nitrocellulose.
The immunodetection was achieved using the following antibodies: antβ-Tubulin, anti-Endoplasmin, anti-Maspin, anti-Galectin-7, anti-PRS10, anti-PSB3, anti-Oct3/4, and anti-Vimentin antibodies from Santa Cruz Biotechnology, Inc. (San Jose, CA); anti-PDI from Stressgen Bioreagents (Victoria, BC Canada); and anti-Mouse, anti-Rat, anti-Goat, and anti-Rabbit secondary antibodies from Sigma Aldrich.
Hybridization with primary antibodies was performed overnight at room temperature using proper dilutions as suggested by the manufacturer’s. Incubation with specific HRP-conjugated secondary antibodies was then performed for 2 h at room temperature, and immunostained bands and spots were visualized by chemiluminescence in Hyperfilm ECL sheets (GE Healthcare) using ECL detection reagents (GE Healthcare). Exposed films were digitized by a Molecular Dynamics 300S laser densitometer (4000×5000 pixels, 12 bits/ pixel; Sunnyvale, CA, USA). The obtained 1-D Western blot images were then analysed with the ImageQuant 3.0 software (Molecular Dynamics), while the 2-D ones were processed using the Image Master Platinum version 7.0 software (GE Healthcare). beta-tubulin immunoblotting ensured equal loading of samples in each lane.
3. Results
3.1. Image analysis and mass spectrometry
Skin proteins of mutant Brtl mice with lethal (ML) or moderately severe (MA) outcome and WT littermates were resolved by 2-DE and representative silver stained electropherograms are reported in Fig. 1A, B, C.
Fig. 1 –
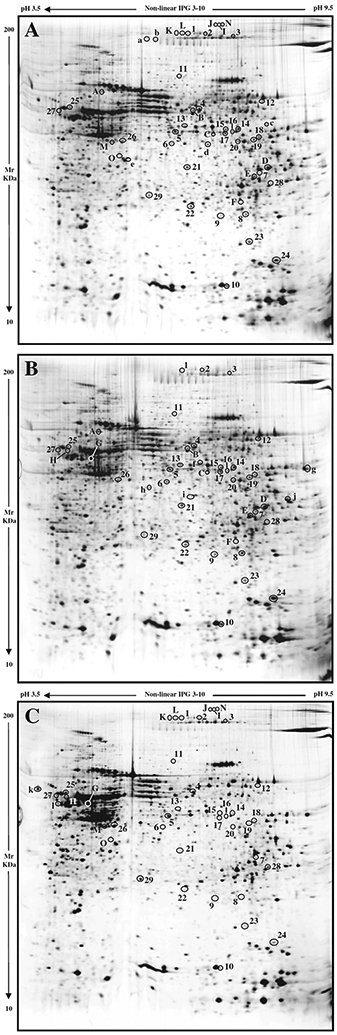
Silver stained electropherograms of WT (A), ML (B), and MA (C) skin samples. Numbers visualize quantitative differences found among the three mouse types, while letters show the qualitative ones.
At least five different gels were analysed for each studied condition and three synthetic images were elaborated by computer matching, with each synthetic image representing all spots constantly present in all spot maps from the same sample type.
Obtained 2-DE spot maps showed a high level of reproducibility intra and inter set of biological replicates, as proved by spot number, about 3000 protein spots were constantly detected in each gel, and by spot intensity values. According to statistics and to relative volume (%V) ratio threshold, as detailed in the Materials and methods, 56 proteins were found as qualitative and/or quantitative significant differences occurring among the three biological conditions.
Thirty-three of such differentially expressed proteins were unambiguously identified by MALDI-ToF MS and nano-LC-ESI-MS/MS (Tables 1-2).
The identified proteins are involved in cytoskeletal and cellular organization, metabolic activities, protein folding and protein degradation, and in cellular fate.
3.2. Qualitative expression profile comparison of lethal and non lethal Brtl mice and WT littermates
The performed proteomic analysis pointed out, as allowed by the analytical applied tool limits, the presence of 27 qualitative differences between the skin maps of WT, ML and MA mice, 13 of which were identified (Table 1). In Fig. 1, spots detected in only one type of sample are indicated by small letters (from “a” to “l”), while protein spots found in two different sample types are showed with capitals (from “A” to “O”).
Interestingly, the expression of the cytoskeletal proteins keratin (spot A), α-centractin (spot C), and β/γ-actin (spot e) were reduced in MA mice, whereas the spot l, identified as the intermediate filament vimentin, was only detected in MA (Table 1). The chaperones calreticulin (spot k) and protein disulfide isomerase A1 (spot H) revealed a higher expression in both ML and MA animals, although it showed a higher level in MA mice. An isoform of maspin (spot O), a member of the serpin family with anti-angiogenic and anti-protease function, was absent in ML mice.
Down-regulation of metabolic proteins was detected in samples from MA mice. An isoform of N-acetylneuraminate lyase (spot D) and an isoform of triosophosphate isomerase (spot F) were undetectable in MA. On the other hand, these two proteins resulted up-regulated in ML mice, where also an isoform of α-enolase, not detected in WT and in MA, was found up-regulated.
3.3. Quantitative expression profile comparison of lethal and non lethal Brtl mice and WT littermates
Quantitative analysis of the three synthetic spot maps revealed 29 significant variations occurring between the three mouse types (Fig. 1). Among them, 20 protein spots were identified by MS (Table 2).
Of particular interest among the proteins less expressed in MA mice, we detected the cytoskeletal protein cofilin-1 (spot 24), the ER chaperone endoplasmin (spot 27), and two members of the proteosomal complex involved in protein degradation: the 26S proteasomal regulatory subunit S10B (spot 19) and the proteasomal subunit β type 3 (spot 22). Based on sequence coverage and experimental Mr value the endoplasmin spot 27 seemed the result of a degradation process, as supported further by western blot (Fig. 4).
Fig. 4 –
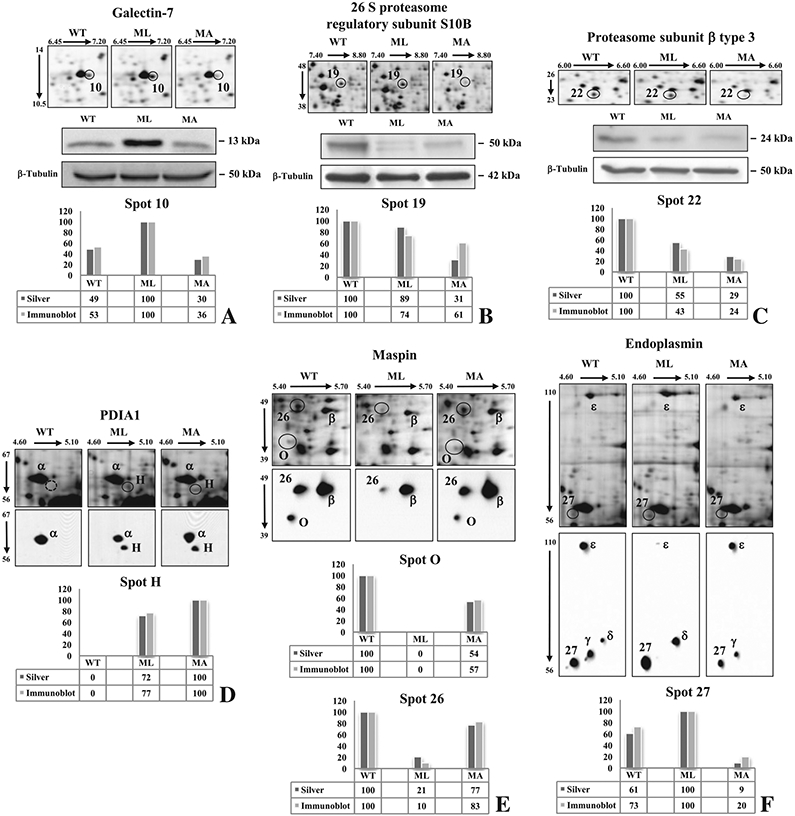
Validation of six differentially expressed proteins by immunostaining. Galectin-7 (A), PRS10 (B), and PSB3 (C) were confirmed by 1-D Western blots, while PDIA1 (D), maspin (E), and endoplasmin (F) were validated by 2-D Western blots using proper antibodies. Normalized relative-integrated-density values of detected bands (A, B, C) and normalized spot optical-density values (D, E, F) are visualized by histograms. Corresponding silver stained spots are highlighted by circles. Greek letters in 2-D blots indicate immunostained spots that were not detected in silver stained gels or that do not correspond to significant differences.
To our surprise, samples from MA mice showed a down regulation of the chaperon αB-crystallin (spot 23) that in our previous work, on bone samples from the same mice was on the contrary increased [13].
Some metabolic enzymes were found decreased in MA mice with respect both WT and ML animals such as, β-enolase (spots 14 and 15), glycine amidinotransferase (spot 16), phosphoglycerate kinase I (spot 18), creatine kinase M chain (spot 20), and malate dehydrogenase (spot 21).
Besides the qualitative difference founded for maspin (spot O), another maspin isoform (spot 26) was detected showing a consistent down-regulation in ML mice. On the contrary, among the identified proteins involved in cellular fate processes, a significant increase was reported for Galectin-7 in ML animals.
3.4. Multivariate analysis
To examine the relationships existing between WT and mutant mice in relation to the significant differences detected in their protein expression profiles, and to validate the biological significance of our 2-DE analyses, the acquired data were unsupervised processed using two different multivariate analysis methods: the Principal Component Analysis (PCA) and the Hierarchical Clustering (HC). PCA revealed distinct expression patterns for the three tested conditions (Fig. 2) and demonstrated a consistent reproducibility among the biological replicates. In fact spot maps properly segregate in three distinct experimental groups (encircled by different colors). The first principal component (PC1) resulted to account for the 52.55% of the variance in the data, while the second principal component (PC2) distinguished an additional 27.47% of variation. The WT cluster turned out roughly interposed between the ML and MA clusters. Interestingly, PC1 proved a highest level of differences to occur between WT and ML mice.
Fig. 2 –

Unsupervised multivariate analysis: principal components analysis. PCA clearly clustered the 15 spot maps into three groups. Each group corresponds exactly to one of the analyzed sample set: wild type (blue), mutant lethal (red), and mutant alive (green).
The Hierarchical Clustering analysis highlighted expression profiles of qualitative and quantitative differences found between WT and mutant mice. The normalized mean relative intensity (%V) of each difference was plotted against the three mouse types and clustered into four main groups, whose individual number of corresponding spots is indicated as cluster size (Fig. 3, Tables 1-2: “HCA cluster” column). The parameters for cluster analysis were accurately selected in order to emphasize the expression differences occurring between one type and the other two types of mice. Actually, each cluster contains protein spots that are up or down regulated in one mouse condition (cluster 1: MA; cluster 2: ML; clusters 3 and 4: WT) in comparison to the other two, consequently there are different and complementary expression profiles within the same cluster. Cluster 1 grouped 24 significant differences whose expression pattern was similar in WT and ML mice and distinct in MA. Interestingly the cytoskeletal proteins (keratin, cofilin 1 and vimentin), the chaperones (calreticulin and endoplasmin), the proteasomal components (PRS10 and PBS3) and various metabolic enzymes belonged to this cluster. Cluster 2 consisted of 16 protein spots showing similar expression in WT and MA animals and different in ML and it manly grouped metabolic enzymes, but it also included the cellular fate protein galectin-7. Cluster 3 and Cluster 4 consisted of 16 spots with a protein trend approximately similar in mutant mice. In particular they included the chaperones PD1A1, PD1A6 and αB-crystallin, and the cellular fate protein maspin. The class discovery analysis helped to identify, among the 2-DE detected differentially expressed proteins, those whose change in expression resulted in alive or lethal outcome, as well as a set of proteins that characterized the mutant status. Interestingly, the up- or down-regulation of Cluster 1 proteins, with respect to the WT expression levels, seems to act as positive modulator of the phenotype, whereas, the up- or down-regulation of Cluster 2 proteins, with respect to the WT, appears related to the lethal effect of the mutation.
Fig. 3 –

Unsupervised multivariate analysis: Hierarchical Clustering. Normalized %V mean values of qualitative and quantitative differences, as computed in WT, ML, and MA spot maps, are visualized by expression matrix, where each reported %V value is proportional to the intensity of the color. Corresponding spot identity (protein name, as listed in Tables 1-2, and number or letter for not identified spots) is annotated on the right of the diagram, near the dendrogram of the average linkage clustering based on Pearson distance. The proteins were grouped into four major clusters, which are numbered from top to bottom. Expression profiles of proteins from each cluster are shown on the right of the dendrogram.
3.5. Western blot validation
To confirm some of the differences highlighted by proteomic investigation, Western blots were performed on independent protein extracts from murine skin samples.
For Galectin-7 as well as for the two identified proteasomal components, PRS10 and PSB3, we confirmed the 2-DE data by 1-D Western blots via normalized relative integrated density values of detected bands (Fig. 4A, B, C histograms).
Expression variations of PDIA1, both maspin isoforms, and endoplasmin (GRP94) were instead validated by 2-D Western blots using normalized spot optical density value (Fig. 4D, E, F histograms). For these proteins, the presence of different immunostained isoforms with relatively close Mr values allowed a better understanding and clarification of the reported 2-DE differences. Of particular interest, the up-regulation of a low molecular weight form of the chaperon endoplasmin (spot 27) was confirmed in ML mice where it was also found an evident down-regulation of another endoplasmin isoform (spot ε), which, based on computed Mr value of the full-length protein sequence, probably corresponds to the entire protein. The spot ε was not detected as significant difference by image analysis on silver stained gels because its protein signal was definitely masked by the hypoxia up-regulated protein-1 presence, as we proved by MS analysis (data not shown), and that do not change in expression among the samples. In ML mice endoplasmin seemed to be subjected to a high degradation process that could, at least in part, result in the signal increase of spots 27 and δ and decrease of spot ε. On the contrary, the spot ε had a similar expression level in MA and WT. Anyway, the overall endoplasmin pattern of expression resulted altered in isoform/degradation products presence and/or in their signal intensity in mutant mice.
3.6. Pathway analysis
The functional correlation existing between the identified expression-profile-differences was assessed using the MetaCore program. The performed pathway analysis allowed the functional integration of the found differences with known protein-factors acting in characterized cellular pathways. This analysis suggested the biochemical contest in which the proteins of interest act, and how their aberrant expression may alter cellular and/or tissue biology in the disease status. All the expression changing proteins were processed by the shortest path algorithm and consequently only those proteins known to be closely related were included in the resulting path. The most significant relevant map is shown in Fig. 5A. It is especially noteworthy that the 85% of the identified differences closely clustered together in the produced network suggesting a high probative value of the produced data and, particularly, of the biological meaning of their aberrant expression. Moreover, vimentin, maspin, endoplasmin, calreticulin, α-enolase, and the two proteasomal components, PRS10 and PSB3, that have reasonable implications in modulating the mutant phenotype (see Discussion), are central “functional hubs” in the map.
Fig. 5 –

MetaCore Pathway and validation of Oct3/4 and vimentin. (A) MetaCore pathway analysis was used to map identified proteins on existing mammalian pathways and networks, in accordance with known protein–protein interactions and other features established in the literature. Network proteins are visualized by proper symbols, which specify the functional nature of the protein (network caption). The edges define the relationships existing between individual proteins, and the arrowheads represent the direction of the interaction. The following line colors designate the nature of the interaction: red=negative effect, green=positive effect, gray=unspecified effect. Links highlighted in thick cyan line refer to known canonical pathways which are significantly related to the path built by the program.
Skin protein lysates from wild type and mutant mice were immunoblotted with anti-Oct3/4 (B) and anti-Vimentin (C). beta-Tubulin immunoblotting ensured equal loading of samples in each lane. * and § symbols indicate the significativity of expression changes occurring between WT and ML, and ML and MA mice, respectively.
Interestingly, several of the program added factors are proteins whose alteration in expression has been previously described in development defects, such as Oct3/4, Notch, NFIC and ERR-1 [32-35]. Furthermore, allowing the program to show canonical pathways associated to our proteomic data, proteins involved in signal transduction pathways (MAPK members) important for cell proliferation [36] as well as proteins involved in apoptosis (Bcl2, Bak, Bax) [37] entered in the built path.
Analyzing the pathway, transcription factors attracted our attention since these proteins are usually present at very low levels in cells and they are classically not detectable in 2-D gels using conventional staining protocols. One of the most interesting was Oct3/4, a core component of the transcriptional network controlling pluripotency [34,38], whose over-expression had been associated to hypoxia as well [39]. A mono-dimensional Western blot was thus performed to evaluate the expression of Oct3/4 in lethal or alive Brtl mice and WT littermates. Worthy of note, as shown in Fig. 5B and consistent with mean relative integrated density±SD, an evident up-regulation of Oct3/4 was observed in skin samples from ML animals, in comparison to WT and MA mice.
In addition, according to its characterization as a central hub in the elaborated path and consistent with the MS identification of spot l, we also decided to verify by Western blot the expression of vimentin. Even if the expression pattern of this protein is usually complex [40], due to its numerous post-translational modifications, we decided to attempt a 1-D Western blot to firstly test changes of vimentin expression profile in the three mouse types. Interestingly, according to normalized relative integrated density values, a general vimentin down-regulation was proved to occur in mutant mice and specifically in ML at least for isoform 3 (Fig. 5C).
3.7. Skin versus bone
Since OI is a bone disease we decided to test some of the differences found in skin samples also in bone tissues. Calvarial bone samples were obtained from 1 day old mutant mice with lethal and non lethal outcome and WT littermates, and expression variations of PRS10, PSB3, maspin, Oct3/4 and vimentin were tested (Fig. 6). At least three different mice were independently tested for each condition and expression variations were evaluated between mean relative integrated density±SD values (Fig. 6 histograms).
Fig. 6 –
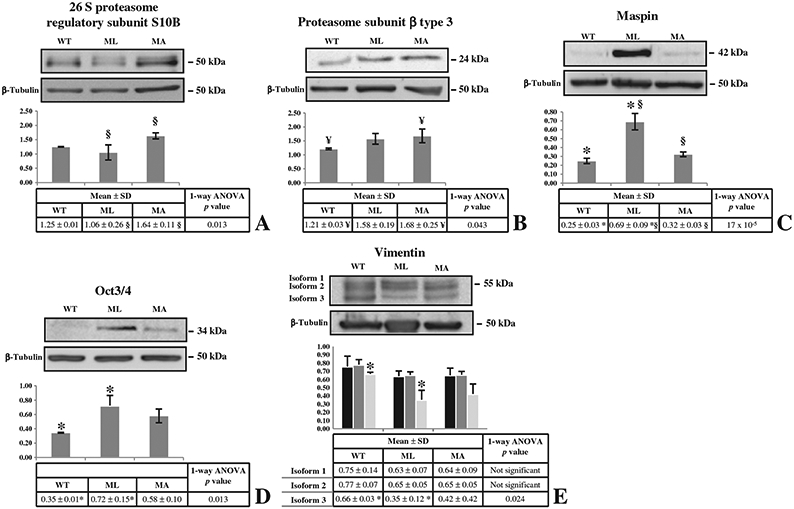
Western blots on calvarial bone samples. Bone protein extracts from 1 day old mutant mice with lethal and non lethal outcome and WT littermates were analyzed by Western blot to evaluate expression variations of PRS10 (A), PSB3 (B), maspin (C), Oct3/4 (D), and vimentin (E), as proved to occur in skin samples or suggested by the pathway analysis. Histograms visualize normalized mean relative-integrated-density±SD values. *, ¥, and § symbols indicate the significativity of expression changes occurring between WT and ML, WT and MA, and ML and MA mice, respectively.
An opposite pattern with respect to the one found in skin samples was detected for the two proteasomal components and for maspin. In MA mice the proteasomal subunits were up-regulated: PRS10 expression was increased with respect to ML and PBS3 with respect to WT animals. Maspin resulted up-regulated in ML mice with respect to both MA and WT mice.
Expression of Oct3/4 in bone samples reproduced the up-regulation detected in skin of ML mice with respect to the WT (Fig. 6D). Vimentin in calvarial samples showed an expression pattern similar to the skin specimens, even if its down-regulation was less evident (Fig. 6E).
4. Discussion
The development of different clinical outcomes associated to an identical molecular defect, known as “phenotypic variability”, is a common feature of various heritable diseases and its molecular basis is mainly still unclear [41]. Among patients affected by the classical form of osteogenesis imperfecta (OI) dozen of independent occurrences of the same substitution at a particular glycine residue leads to divergent clinical outcomes. Understanding the molecular basis of phenotypic variability is relevant not only to better investigate OI pathophysiology, but also to identify potential novel target for developing a new therapy for this disorder.
The outbred OI murine model Brittle IV (Brtl) represents an invaluable tool to address the study of phenotypic variability in OI, since it reproduces the heterogeneity of the genetic background typical of human patients.
In the present study we investigated the OI phenotypic variability using protein extracts from the skin of mutant Brtl and WT mice in order to be able to extend the proteomic data from the OI murine model to human patients. Skin biopsies are indeed more accessible than bone.
The 2-DE spot maps obtained from skin proteins extracted from Brtl mice carrying lethal and moderately severe outcome and WT littermates were compared. According to stringent statistical analysis, numerous differentially expressed proteins were detected and several of them were identified by MS. High reproducibility of biological replica and distinct expression patterns of the three tested conditions were proved by unsupervised multivariate analyses: PCA and HC. Of particular interest, as clearly shown by HC, peculiar differences in expression profiles of specific proteins were detected to occur between mutants with alive or lethal outcome: clusters 1 and 2.
In the MetaCore produced network the presence of vimentin, endoplasmin, calreticulin, and, less in extent, of proteosomal subunits as central hubs highly stresses the occurrence of aberrant protein folding and degradation processes in mutant mice. Moreover, as visualized by HC, their different, and often opposite, expression pattern in mutants may reasonably justify, at least in part, the different outcome of the mutation. Also anomalous energy supply and metabolic processes are proved to specifically characterize the mutant condition. Actually, not only α-enolase and pyruvate kinase isozyme M1/M2 are essential hubs, but almost all the metabolic identified proteins map in the path. Moreover, in line with the previously described extracellular-matrix defects in OI, maspin is a principal functional hub in the path. All these findings in addition to the functional integration of identified differentially expressed proteins with specific protein factors, known to characterize precise cellular pathways, suggest that the main biological functions affected by the mutant status are related to ER stress and energy balance, but also to development, signal transduction, and apoptosis. The different capability of single mutant to overcome aberrant functionalities in such networks may result in lethal or alive outcome.
Actually, in skin, as was for calvarial bone samples, a different cellular response to ER stress was evident in mice with different outcome, and indeed we already described the presence of ER stress in Brtl skin fibroblasts by electron microscopy [14]. In samples from surviving mice we identified a specific expression pattern of the chaperones calreticulin, protein disulfide isomerase-associated 1 (PDIA1), and endoplasmin (GRP94) together with a reduction of the two proteasomal components: 26S protease regulatory subunit S10B (PRS10) and Proteasome subunit β type 3 (PBS3).
The function of chaperones for type I collagen folding does not follow the classical roles of globular proteins [42]. Type I collagen folding in the ER begins with the recognition of the C-propeptides of two α1 and one α2 chains followed by the formation of specific disulfide bonds between cysteine residues [43,44]. General ER chaperones, such as calnexin, calreticulin, PDI, GRP94 and Bip, bind to the globular C-propeptide to discourage its misfolding, but other more specific chaperones, specifically bound to the folded triple helix, are necessary to make collagen folding thermodynamically favourable [43]. It is known that glycine substitution in the triple helical domain of type I collagen are not responsible for a classical unfolded protein response activation, rather they cause the formation of misfolded collagen vesicles addressed to autophagy or lysosome degradation [45,46].
The identification in skin of MA Brtl mice of an up-regulation, in respect to WT, or ML, or ML and WT, of some isoforms of the general chaperones was indeed quite puzzling. It is possible that even in the presence of triple helical glycine substitution the folding of the single mutant chains is compromised, thus requiring a further help by the general chaperones in order to begin triple helix association.
We cannot anyway exclude that a general ER malfunction, caused by mutant collagen intracellular retention [14], could affect also the folding of other secreted proteins. Maybe, the changing in protein expression, degradation, and/or co-post-translational modification of the general chaperones acted primarily to help the cells to handle the correct folding of non-collagenous secreted proteins in an engorged environment [47].
Chaperones and the ATP-dependent ubiquitin-proteasome pathway are known to work together to prevent the accumulation of misfolded and potentially toxic proteins [48,49]. In the skin from MA Brtl mice the expression pattern “adjustment” of the molecular chaperones, in relation to ML and WT animals, seems to overcome the more energy demanding from the increased proteasome function, which is reasonably related to the ER stress associated to the mutant status. Actually, a significant reduction of a set of metabolic enzymes, mainly involved in energy production, support this interpretation.
Cytoskeletal proteins are well known to be involved in membrane trafficking, cell migration, and cell division [50]. The down-regulation of cofilin 1, keratin, α centractin as well as an actin isoform in MA mice indicated a reduced or altered intracellular trafficking and/or a cytoskeletal rearrangement, which can be an important factor in cell surviving ability.
Intermediate filaments (IFs), one of the major structural components of cytoskeleton in addition to actin microfilaments and microtubules, are known to be important contributors to cellular stress response, acting together with heat shock proteins [51]. Furthermore cytoplasmic IFs proteins tend to antagonize death-receptor induced apoptosis by attenuating the strength of the signals that feed into the formation of death-inducing complexes. Vimentin, a member of the IFs family [52,53], is expressed abundantly in fibroblasts and, as proved by the Western blot assay, its over-expression in MA Brtl mice, with respect to ML animals, again underlines the MA ability to better handle the ER stress.
In Brtl mice with lethal outcome, together with a general increase of the expression of metabolic enzymes and a down-regulation of some cytoskeletal proteins, a significant over-expression of galectin-7 and a down-regulation of maspin, two proteins related to cellular fate, were detected with respect to both WT and MA mice. Galectin-7 is a member of the galectins family, a group of soluble lectins sharing a unique carbohydrate recognition domain that confers specificity for β-galactoside derivatives and known to have diverse biological functions including regulation of cell adhesion, cell growth and apoptosis [54]. Interestingly over-expression of galectin-7 in cultured cells was associated with apoptosis activation as well as with up-regulation of several metabolic enzymes, extracellular matrix proteins, tumor antigens and secreted proteins involved in cell growth/development and differentiation [55]. The higher expression of galectin-7 in ML animals let us speculate that cells from these mice may be more sensible to apoptosis induced by the ER stress.
Maspin, initially identified as tumor suppressor protein in human breast epithelial cells, is a member of the serine proteases inhibitor (serpin) superfamily with tumor suppressive and anti-angiogenic functions [56]. It is mainly a cytosolic protein but it has been also found associated with secretory vesicles at the cell surface where it is known to interact with collagen I and III [57]. Thus its down regulation in ML can affect cell-extracellular matrix interaction.
Being OI a bone disease we decided to test the expression of some crucial proteins found differentially expressed in Brtl mice with different outcomes also in calvarial bone samples.
Interestingly, for the two proteasomal component PSB3 and PRS10 as well as for maspin differences were detected among the ML, MA and WT samples but with an opposite pattern in bone versus skin.
Of particular interest was the up-regulation of maspin detected in bone from ML mice. Since, in bone, maspin was reported to be actively expressed in osteoblasts located in site of active bone formation and remodelling, and to act as a TGFβ modulator [58], its over expression in ML bone cells can cause a disequilibrium between the active and latent form of TGFβ, thus playing a role in bone matrix formation.
Differences in skin versus bone collagen composition in OI had been reported in the past and still are of great interest especially to clarify why, being collagen the most abundant protein in both tissues, the disease is mainly a brittle bone disorder. The data are anyway limited due to the difficulty to obtain both skin and bone biopsies from the same patient. Mundlos et al. had the opportunity to investigate fibroblasts and osteoblasts as well as skin and bone, from an OI case carrying a 5 exons deletion in the α2(I) chain. Even if it was present in the cell fraction the mutant collagen was not secreted by fibroblasts and it was absent from the skin extracellular matrix. Interestingly the patient osteoblasts secreted the mutant protein that was indeed also identified in bone tissue [59]. This difference suggests that the molecular organization in bone could be more permissive for the incorporation of mutant collagen, still intracellularly both tissues and cell types had to handle the stress due to the presence of mutant protein. Similarly, in an in vitro study Sarafova et al. reported in four OI patients a higher level of mutant collagen glycosylation in osteoblasts compared to fibroblasts [60]. It seems likely that a tissue specificity exists in collagen metabolism and our data further support this conclusion. However our results suggest also the activation of common cellular responses. We demonstrated that the differential ability to respond to cellular malfunction is one of the factor modulating the OI phenotypic variability in skin as we previously reported for bone samples [14]. Tissue specific response seemed to be activated as confirmed by the differential expression of some proteins in skin versus bone, although common pathways can be modulated as well, as demonstrated by Oct3/4 data.
Our proteomic analysis supports the idea that the molecular basis of OI is not only based on abnormal bone extracellular matrix structure but also on a cellular machinery malfunction likely due to synthesis and intracellular retention and degradation of mutant collagen. The identification of different cellular adaptation to ER stress as a modulator factor for OI phenotypic variability pointed to this pathway as a potential target for novel OI therapeutic approaches.
Supplementary Material
Acknowledgments
This work was supported by PRIN 2008 (2008XA48SC), by Fondazione Cariplo 2007 and by Progetto Regione Lombardia (cod. SAL/45) “Dalla scienza dei materiali alla medicina molecolare” to Antonella Forlino, and by the FIRB project “Italian Human ProteomeNet” (BRN07BMCT_013-MIUR) to Luca Bini.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at doi:10.1016/j.jprot.2012.01.038.
This article is part of a Special Issue entitled: Translational Proteomics.
REFERENCES
- [1].Sillence DO, Rimoin DL. Classification of osteogenesis imperfect. Lancet 1978;1:1041–2. [DOI] [PubMed] [Google Scholar]
- [2].Byers PH, Cole WG. In: Royce PM, Steinmann B, editors. Connective Tissue and Heritable Disorders. New York: Wiley-Liss; 2002. p. 385–430. [Google Scholar]
- [3].Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 2011;7:540–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 2007;28:209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dalgleish R The human type I collagen mutation database. Nucleic Acids Res 1997;25:181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dalgleish R The Human Collagen Mutation Database 1998. Nucleic Acids Res 1998;26:253–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bodian DL, Chan TF, Poon A, Schwarze U, Yang K, Byers PH, et al. Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype-phenotype relationships. Hum Mol Genet 2009;18:463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Byers PH, Wallis GA, Willing MC. Osteogenesis imperfecta: translation of mutation to phenotype. J Med Genet 1991;28:433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kamoun-Goldrat AS, Le Merrer MF. Animal models of osteogenesis imperfecta and related syndromes. J Bone Miner Metab 2007;25:211–8. [DOI] [PubMed] [Google Scholar]
- [10].Lisse TS, Thiele F, Fuchs H, Hans W, Przemeck GK, Abe K, et al. ER stress-mediated apoptosis in a new mouse model of osteogenesis imperfecta. PLoS Genet 2008;4:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Daley E, Streeten EA, Sorkin JD, Kuznetsova N, Shapses SA, Carleton SM, et al. Variable bone fragility associated with an Amish COL1A2 variant and a knock-in mouse model. J Bone Miner Res 2009;13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Forlino A, Porter FD, Lee EJ, Westphal H, Marini JC. Use of the Cre/lox recombination system to develop a non-lethal knock-in murine model for osteogenesis imperfecta with an alpha1(I) G349C substitution. Variability in phenotype in BrtlIV mice. J Biol Chem 1999;274:37923–31. [DOI] [PubMed] [Google Scholar]
- [13].Forlino A, Tani C, Rossi A, Lupi A, Campari E, Gualeni B, et al. Differential expression of both extracellular and intracellular proteins is involved in the lethal or nonlethal phenotypic variation of BrtlIV, a murine model for osteogenesis imperfecta. Proteomics 2007;7:1877–91. [DOI] [PubMed] [Google Scholar]
- [14].Forlino A, Kuznetsova NV, Marini JC, Leikin S. Selective retention and degradation of molecules with a single mutant alpha1(I) chain in the Brtl IV mouse model of OI. Matrix Biol 2007;26:604–14. [DOI] [PubMed] [Google Scholar]
- [15].Hansen B, Jemec GB. The mechanical properties of skin in osteogenesis imperfecta. Arch Dermatol 2002;138:909–11. [DOI] [PubMed] [Google Scholar]
- [16].Canuto HC, Fishbein KW, Huang A, Doty SB, Herbert RA, Peckham J, et al. Characterization of skin abnormalities in a mouse model of osteogenesis imperfecta using high resolution magnetic resonance imaging and Fourier transform infrared imaging spectroscopy. NMR Biomed 2012;25:169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cabral WA, Milgrom S, Letocha AD, Moriarty E, Marini JC. Biochemical screening of type I collagen in osteogenesis imperfecta: detection of glycine substitutions in the amino end of the alpha chains requires supplementation by molecular analysis. J Med Genet 2006;43:685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54. [DOI] [PubMed] [Google Scholar]
- [19].Gorg A, Postel W, Gunther S. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 1988;9:531–46. [DOI] [PubMed] [Google Scholar]
- [20].Bjellqvist B, Pasquali C, Ravier F, Sanchez JC, Hochstrasser D. A nonlinear wide-range immobilized pH gradient for two-dimensional electrophoresis and its definition in a relevant pH scale. Electrophoresis 1993;14:1357–65. [DOI] [PubMed] [Google Scholar]
- [21].Hochstrasser DF, Patchornik A, Merril CR. Development of polyacrylamide gels that improve the separation of proteins and their detection by silver staining. Anal Biochem 1988;173:412–23. [DOI] [PubMed] [Google Scholar]
- [22].Oakley BR, Kirsch DR, Morris NR. A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal Biochem 1980;105:361–3. [DOI] [PubMed] [Google Scholar]
- [23].Sinha P, Poland J, Schnolzer M, Rabilloud T. A new silver staining apparatus and procedure for matrix-assisted laser desorption/ionization-time of flight analysis of proteins after two-dimensional electrophoresis. Proteomics 2001;1:835–40. [DOI] [PubMed] [Google Scholar]
- [24].Bjellqvist B, Hughes GJ, Pasquali C, Paquet N, Ravier F, Sanchez JC, et al. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis 1993;14:1023–31. [DOI] [PubMed] [Google Scholar]
- [25].Bianchi L, Lorenzoni P, Bini L, Weber E, Tani C, Rossi A, et al. Protein expression profiles of Bos taurus blood and lymphatic vessel endothelial cells. Proteomics 2007;7:1600–14. [DOI] [PubMed] [Google Scholar]
- [26].Hellman U, Wernstedt C, Gonez J, Heldin CH. Improvement of an “In-Gel” digestion procedure for the micropreparation of internal protein fragments for amino acid sequencing. Anal Biochem 1995;224:451–5. [DOI] [PubMed] [Google Scholar]
- [27].Soskic V, Gorlach M, Poznanovic S, Boehmer FD, Godovac-Zimmermann J. Functional proteomics analysis of signal transduction pathways of the platelet-derived growth factor beta receptor. Biochemistry 1999;38:1757–64. [DOI] [PubMed] [Google Scholar]
- [28].Meiring HD, van der Heeft E, ten Hove GJ, de Jong APJM. Nanoscale LC-MS(n): technical design and applications to peptide and protein analysis. J Sep Sci 2002;25:557–68. [Google Scholar]
- [29].Gharahdaghi F, Weinberg CR, Meagher DA, Imai BS, Mische SM. Mass spectrometric identification of proteins from silver-stained polyacrylamide gel: a method for the removal of silver ions to enhance sensitivity. Electrophoresis 1999;20:601–5. [DOI] [PubMed] [Google Scholar]
- [30].Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–5. [DOI] [PubMed] [Google Scholar]
- [31].Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 1979;76:4350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zanotti S, Canalis E. Notch regulation of bone development and remodeling and related skeletal disorders. Calcif Tissue Int 2011;2011:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bonnelye E, Vanacker JM, Dittmar T, Begue A, Desbiens X, Denhardt DT, et al. The ERR-1 orphan receptor is a transcriptional activator expressed during bone development. Mol Endocrinol 1997;11:905–16. [DOI] [PubMed] [Google Scholar]
- [34].Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 2006;38:431–40. [DOI] [PubMed] [Google Scholar]
- [35].Park JC, Herr Y, Kim HJ, Gronostajski RM, Cho MI. Nfic gene disruption inhibits differentiation of odontoblasts responsible for root formation and results in formation of short and abnormal roots in mice. J Periodontol 2007;78:1795–802. [DOI] [PubMed] [Google Scholar]
- [36].Gehart H, Kumpf S, Ittner A, Ricci R. MAPK signalling in cellular metabolism: stress or wellness? EMBO Rep 2010;11:834–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Heath-Engel HM, Chang NC, Shore GC. The endoplasmic reticulum in apoptosis and autophagy: role of the BCL-2 protein family. Oncogene 2008;27:6419–33. [DOI] [PubMed] [Google Scholar]
- [38].Young JC, Major AT, Miyamoto Y, Loveland KL, Jans DA. Distinct effects of importin alpha2 and alpha4 on Oct3/4 localization and expression in mouse embryonic stem cells. FASEB J 2011;25:3958–65. [DOI] [PubMed] [Google Scholar]
- [39].Iida H, Suzuki M, Goitsuka R, Ueno H. Hypoxia induces CD133 expression in human lung cancer cells by up-regulation of OCT3/4 and SOX2. Int J Oncol 2011;40:71–9. [DOI] [PubMed] [Google Scholar]
- [40].Bianchi L, Bruzzese F, Leone A, Gagliardi A, Puglia M, Di Gennaro E, et al. Proteomic analysis identifies differentially expressed proteins after HDAC vorinostat and EGFR inhibitor gefitinib treatments in Hep-2 cancer cells. Proteomics 2011;11:3725–42. [DOI] [PubMed] [Google Scholar]
- [41].Girirajan S, Eichler EE. Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet 2011;19:R176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Makareeva E, Aviles NA, Leikin S. Chaperoning osteogenesis: new protein-folding disease paradigms. Trends Cell Biol 2011;21:168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Keide T, Nagata K, in: Springer-Verlag; (Ed.), Collagen 2005, pp. 85–114. [Google Scholar]
- [44].Engel J, Bachinger HP, in: Springer-Verlag; (Ed.), Collagen 2005, pp. 7–33. [Google Scholar]
- [45].Ishida Y, Yamamoto A, Kitamura A, Lamande SR, Yoshimori T, Bateman JF, et al. Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Biol Cell 2009;20:2744–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ishida Y, Nagata K. Autophagy eliminates a specific species of misfolded procollagen and plays a protective role in cell survival against ER stress. Autophagy 2009;5:1217–9. [DOI] [PubMed] [Google Scholar]
- [47].Vidal R, Caballero B, Couve A, Hetz C. Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington’s disease. Curr Mol Med 2011;11:1–12. [DOI] [PubMed] [Google Scholar]
- [48].Dahlmann B Role of proteasomes in disease. BMC Biochem 2007;8(Suppl. 1):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett 2007;581:3641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Firat-Karalar EN, Welch MD. New mechanisms and functions of actin nucleation. Curr Opin Cell Biol 2011;23:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Toivola DM, Strnad P, Habtezion A, Omary MB. Intermediate filaments take the heat as stress proteins. Trends Cell Biol 2011;20:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang N, Stamenovic D. Mechanics of vimentin intermediate filaments. J Muscle Res Cell Motil 2002;23:535–40. [DOI] [PubMed] [Google Scholar]
- [53].Kim S, Coulombe PA. Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes Dev 2007;21:1581–97. [DOI] [PubMed] [Google Scholar]
- [54].Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, et al. Galectins: a family of animal beta-galactoside-binding lectins. Cell 1994;76:597–8. [DOI] [PubMed] [Google Scholar]
- [55].Kuwabara I, Kuwabara Y, Yang RY, Schuler M, Green DR, Zuraw BL, et al. Galectin-7 (PIG1) exhibits pro-apoptotic function through JNK activation and mitochondrial cytochrome c release. J Biol Chem 2002;277:3487–97. [DOI] [PubMed] [Google Scholar]
- [56].Zhang M, Volpert O, Shi YH, Bouck N. Maspin is an angiogenesis inhibitor. Nat Med 2000;6:196–9. [DOI] [PubMed] [Google Scholar]
- [57].Blacque OE, Worrall DM. Evidence for a direct interaction between the tumor suppressor serpin, maspin, and types I and III collagen. J Biol Chem 2002;277:10783–8. [DOI] [PubMed] [Google Scholar]
- [58].Tokuyama R, Satomura K, Maeda E, Kudoh K, Yamasaki Y, Nagayama M. Maspin is involved in bone matrix maturation by enhancing the accumulation of latent TGF-beta. J Bone Miner Res 2007;22:1581–91. [DOI] [PubMed] [Google Scholar]
- [59].Mundlos S, Chan D, Weng YM, Sillence DO, Cole WG, Bateman JF. Multiexon deletions in the type I collagen COL1A2 gene in osteogenesis imperfecta type IB. Molecules containing the shortened alpha2(I) chains show differential incorporation into the bone and skin extracellular matrix. J Biol Chem 1996;271:21068–74. [DOI] [PubMed] [Google Scholar]
- [60].Sarafova AP, Choi H, Forlino A, Gajko A, Cabral WA, Tosi L, et al. Three novel type I collagen mutations in osteogenesis imperfecta type IV probands are associated with discrepancies between electrophoretic migration of osteoblast and fibroblast collagen. Hum Mutat 1998;11:395–403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.