

ABSTRACT
Recent years have seen a surge of methylome-wide association studies (MWAS). We observed that many of these studies suffer from test statistic inflation that is most likely caused by commonly used quality control (QC) pipelines not going far enough to remove technical artefacts. To support this claim, we reanalysed GEO datasets with an improved QC pipeline that reduced test-statistic inflation parameter lambda from the original mean/median of 20.16/15.17 to 3.07/1.14. Furthermore, the mean/median number of methylome-wide significant findings was reduced by 65,688/57,805 loci after more thorough QC. To avoid such false positives we argue for more extensive QC and that reporting the test-statistic inflation parameter lambda become standard for all MWAS allowing readers to better assess the risk of false discoveries.
KEYWORDS: DNA methylation, epigenetics, reproducibility
Brief report
In high-dimensional investigations where many biological markers are tested for association with an outcome, it is critical that the p-values used to declare statistical significance are accurate. Of particular concern are situations where the p-values of markers without true effects are systematically smaller than expected. Due to the large number of markers tested, this may result in a considerable number of false discoveries. In the literature, this problem is known as test-statistic inflation.
Recent years have seen a surge of methylome-wide association studies (MWAS). Surprisingly, test-statistic inflation in MWAS often appears to be inadequately addressed. To support this observation, we carried out a systematic review of array-based MWAS data deposited into the NIH Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo, accessed November 2018). a summary of our selection process is given in Supplementary Figure 1. We included datasets that resulted in a peer-reviewed publication and had a minimum of 300 samples to ensure that possible test-statistic inflation was not simply the result of deviations from the assumed test-statistic distribution caused by small sample sizes. Datasets were excluded for i) twin or cancer studies, ii) not providing the variables needed to reproduce the published results, or iii) being from tissues for which cell type proportions could not be estimated from existing DNA methylation reference panels.
A common way to assess test-statistic inflation is to calculate lambda (λ), which is the ratio of the median observed test-statistic distribution to the expected median test-statistic distribution under the assumed null. Table 1 shows the studies that were included in our review and the source of lambda for each. Frequently, λ was not reported in the published studies. In these instances, we instead calculated λ from the downloaded MWAS p-values. If p-values were not provided, we downloaded the processed data from GEO and performed MWAS with the same set of covariates as reported in the initial publications. Overall, of the 16 studies in Table 1 (Initial Lambda), three had a λ < 1.5, seven had λ between 5–10, and three had λ ranging from 15–64.
Table 1.
GEO Datasets included in systematic review
| GEO Accession | PMID | n | Platform | Tissue | Outcome | Initial Lambda | Recalculated Lambda | Permutation Median Lambda (95% CI) | Bonferroni Δ | Source of Initial Lambda |
|---|---|---|---|---|---|---|---|---|---|---|
| GSE56046 | 25404168 | 1202 | 450 k | Blood | age | 3.12 | - | - | MWAS of processed data | |
| GSE87571 | 23826282 | 732 | 450 k | Blood | age | 17.68 | 7.69 | 0.97 (0.86–1.38) | 92,591 | Calculated from summary statistics |
| GSE72774 | 26655927 | 508 | 450 k | Blood (all samples) | age | 5.48 | - | - | MWAS of processed data | |
| Blood (controls only) | age | 2.16 | - | - | ||||||
| GSE72778 | 27479945 | 475 | 450 k | Brain (meta-analysis) | age | 7.30 | - | - | Reported in manuscript | |
| Brain (frontal lobe) | age | 3.50 | - | - | ||||||
| Brain (parietal lobe) | age | 3.00 | - | - | ||||||
| Brain (occipital lobe) | age | 2.20 | - | - | ||||||
| GSE55763 | 25853392 | 2711 | 450 k | Blood | Case-control | 1.00 | - | - | Reported in manuscript | |
| GSE84727 | 27572077 | 847 | 450 k | Blood | Case-control | 1.53 | - | - | MWAS of processed data | |
| GSE42861 | 23334450 | 689 | 450 k | Blood | Case-control | 15.17 | 1.14 | 1.00 (0.96–1.08) | 57,805 | MWAS of processed data |
| GSE74193 | 26619358 | 675 | 450 k | Brain | Case-control | 64.65 | 4.37 | 0.98 (0.78–1.35) | 178,241 | Calculated from summary statistics |
| GSE80417 | 27572077 | 675 | 450 k | Blood | Case-control | 1.32 | - | - | MWAS of processed data | |
| GSE111629 | 28851441 | 572 | 450 k | Blood | Case-control | 1.77 | 1.01 | 1.00 (0.93–1.11) | 7 | MWAS of processed data |
| GSE87648 | 27886173 | 384 | 450 k | Blood | Case-control | 1.54 | 1.13 | 1.01 (0.82–1.35) | −202 | MWAS of processed data |
| GSE100264 | 29269866 | 386 | 450 k | Blood | Ordinal (drug use) | 1.08 | - | - | Reported in manuscript |
Two factors potentially explain why the lambdas are much higher compared to what is expected under the null hypothesis (λ = 1). First, because λ is based on the median test-statistic, the presence of a small number of effects, relative to the total number of tests, will not result in values that differ from one. However, when the outcome of interest is associated with many effects, λ may be greater than one and suggest test-statistic inflation [1]. This latter explanation is plausible in the context of MWAS as the methylome is dynamic and can be altered by many different biological (e.g, ageing, cellular heterogeneity) and environmental factors (e.g., lifestyle, diet, medication) that may differ between cases and controls.
Second, MWAS may be susceptible to technical artefacts not adequately removed by commonly used quality control procedures. To explore this possibility, we reanalysed the five GEO datasets that provided raw IDATs for control probe extraction. In addition to the reported covariates used for association testing in the original publication, we used the control probes to add the following lab technical covariates to the analysis: 1) Bisulphite conversion percentages estimated from control probes; 2) Median signal intensities for methylated and unmethylated channels; 3) Slide and well effects that refer to the individual BeadChip arrays (slide) and the positional effects on each array (well) which hold 12 samples each (eight samples for the EPIC array); 4) principal components (PCs) of control probe values as measures of technical variation among individual samples; and 5) PCs of the methylation beta values which capture any remaining unmeasured confounders. The effect of these technical covariates on variation in each of the reanalysed datasets is included in Supplementary File 1. From these reanalysed datasets, we find that items two through five above are most often significantly associated with variation in the data. a user-friendly vignette showing how we implemented these additional QC steps through the RaMWAS [2] software can be found at https://bioconductor.org/packages/devel/bioc/vignettes/ramwas/inst/doc/RW5a_matrix.html.
Using these additional QC steps reduced lambda (Recalculated Lambda in Table 1) from a median/mean λ = 20.16/15.17 to a median/mean λ = 3.07/1.14. We also compared the number of methylome-wide significant findings before and after reanalysis with more thorough QC. Using a Bonferroni correction for multiple testing (type I error of alpha = 0.05), we observe a mean/median decrease of the number of significant findings of 65,688/57,805 loci. As these sites no longer reach significance after better QC, this suggest that technical artefact contributed substantially to test-statistic inflation in these previously published datasets leading to many false-positive results being reported.
Even after improved QC, multiple λs remained larger than 1 (Table 1). We explored three possible reasons. First, we performed 2,000 MWAS with the outcome being randomly permuted. The average λ was close to one (Supplementary Figure 2) suggesting that the increased lambda was not caused by inaccurate assumptions about the test statistic distribution. Second, we rely on the variables reported in the five GEO data sets, our list of QC variables may have been be incomplete. To explore this, we performed MWAS on two outcomes (case-control status and age) in a new study of our own that focuses on postpartum depression [3]. For the case-control MWAS (Figure 1A), λ decreased from 29.1 to 1.01 when following our proposed QC. This provides some evidence that key QC variables may be missing for the analysis of the GEO datasets, resulting in the residual λs larger than 1 in Table 1. For analyses with age as an outcome (Figure 1B), λ remained relatively higher (λ = 1.84), though not to the same degree as other published studies (Table 1). As the effect of age on the methylome is pervasive, this provided also some evidence that in MWAS part of the ‘inflation’ may be due to true signal.
Figure 1.
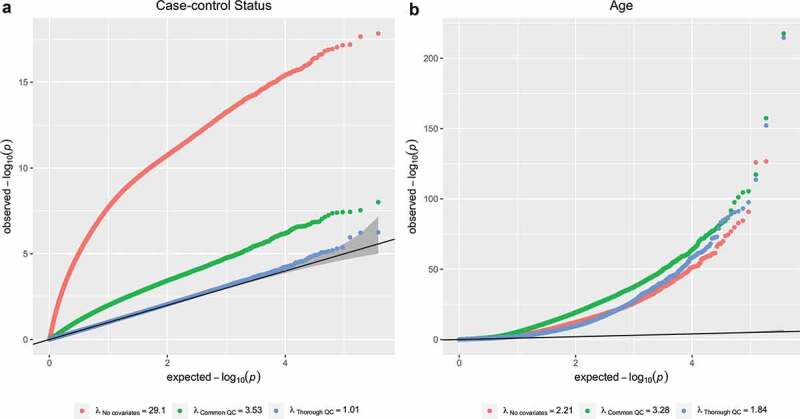
Effects of covariate inclusion on test-statistic inflation. quantile-quantile (QQ) plots for the outcomes postpartum depression case status (a) and age (b). Each colour denotes a different level of quality control used in association testing: no covariates (red), commonly used pipeline (green), and our proposed iterative quality control process (blue)
In conclusion, we observed that for published MWASs the calculated lambdas were often substantially greater than one and that technical artefacts account for a considerable proportion of this inflation. Because a large number of markers are tested, this suggests that many false discoveries may reside within the current MWAS literature. For example, for the five reanalysed GEO datasets, the number of methylome-wide significant findings reduced by a mean of 65,688 loci after better QC. To avoid such false positives, we propose the use of additional QC steps that we implemented in a user-friendly analysis pipeline/vignette. Further, we recommend that reporting the test-statistic inflation parameter λ should become standard for all published MWAS allowing readers to better assess the risk of false discoveries.
Supplementary Material
Funding Statement
This work was supported by the National Institute of Mental Health [MH095992].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Yang J, Weedon MN, Purcell S, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19:807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shabalin AA, Hattab MW, Clark SL, et al. RaMWAS: fast methylome-wide association study pipeline for enrichment platforms. Bioinformatics. 2018;34:2283–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Guintivano J, Sullivan PF, Stuebe AM, et al. Adverse life events, psychiatric history, and biological predictors of postpartum depression in an ethnically diverse sample of postpartum women. Psychol Med. 2018;48:1190–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.