

ABSTRACT
Lipotoxicity, induced by saturated fatty acid (SFA)-mediated cell death, plays an important role in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). The KEAP1 (kelch like ECH associated protein 1)-NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2) pathway is a pivotal defense mechanism against lipotoxicity. We previously reported that SQSTM1/p62 has a cytoprotective role against lipotoxicity through activation of the noncanonical KEAP1- NFE2L2 pathway in hepatocytes. However, the underlying mechanisms and physiological relevance of this pathway have not been clearly defined. Here, we demonstrate that NFE2L2-mediated induction of SQSTM1 activates the noncanonical KEAP1-NFE2L2 pathway under lipotoxic conditions. Furthermore, we identified that SQSTM1 induces ULK1 (unc-51 like autophagy activating kinase 1) phosphorylation by facilitating the interaction between AMPK (AMP-activated protein kinase) and ULK1, leading to macroautophagy/autophagy induction, followed by KEAP1 degradation and NFE2L2 activation. Accordingly, the activity of this SQSTM1-mediated noncanonical KEAP1-NFE2L2 pathway conferred hepatoprotection against lipotoxicity in the livers of conventional sqstm1- and liver-specific sqstm1-knockout mice. Moreover, this pathway activity was evident in the livers of patients with nonalcoholic fatty liver. This axis could thus represent a novel target for NAFLD treatment.
Abbreviations: ACACA: acetyl-CoA carboxylase alpha; ACTB: actin beta; BafA1: bafilomycin A1; CM-H2DCFDA:5-(and-6)-chloromethyl-2ʹ,7ʹ-dichlorodihydrofluorescein diacetate; CQ: chloroquine; CUL3: cullin 3; DMSO: dimethyl sulfoxide; FASN: fatty acid synthase; GSTA1: glutathione S-transferase A1; HA: hemagglutinin; Hepa1c1c7: mouse hepatoma cells; HMOX1/HO-1: heme oxygenase 1; KEAP1: kelch like ECH associated protein 1; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3; MEF: mouse embryonic fibroblast; MTORC1: mechanistic target of rapamycin kinase complex 1; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NAC: N-acetyl-L-cysteine; NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis; NFE2L2/NRF2: nuclear factor, erythroid 2 like 2; NQO1: NAD(P)H quinone dehydrogenase 1; PA: palmitic acid; PARP: poly (ADP-ribose) polymerase 1; PRKAA1/2: protein kinase AMP-activated catalytic subunits alpha1/2; RBX1: ring-box 1; ROS: reactive oxygen species; SESN2: sestrin 2; SFA: saturated fatty acid; siRNA: small interfering RNA; SQSTM1/p62: sequestosome 1; SREBF1: sterol regulatory element binding transcription factor 1; TBK1: TANK binding kinase 1; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling; ULK1: unc-51 like autophagy activating kinase.
KEYWORDS: KEAP1-NFE2L2 pathway, lipotoxicity, NAFLD, SQSTM1, ULK1
Introduction
Nonalcoholic fatty liver disease (NAFLD) comprises a spectrum of diseases ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) with inflammation and fibrosis, which can develop into cirrhosis and hepatic cellular carcinoma [1]. The overload of saturated fatty acid (SFA) provokes oxidative stress-mediated cell death, referred to as lipotoxicity. This process is crucial for the pathogenesis of NASH [1,2]. Accordingly, studies of the mechanisms associated with the removal of oxidative stress could result in targeted therapeutic strategies for NASH [3,4].
The major cellular antioxidant mechanism involves the KEAP1 (kelch like ECH associated protein 1)-NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2) pathway. It comprises a pivotal system of cytoprotective responses to oxidative stresses, in which NFE2L2 is a master transcription factor that controls antioxidant enzymes [5]. Additionally, NFE2L2 is negatively regulated by the cysteine-rich protein KEAP1 via proteasomal degradation mediated by the CUL3 (cullin 3)-E3 ubiquitin ligase RBX1 (ring-box 1) complex under non-stress conditions [6,7]. However, the modification of cysteine residues in KEAP1, caused by oxidation, leads to NFE2L2 dissociation from KEAP1 in response to oxidative stress. Consequently, NFE2L2 translocates to the nucleus and activates the expression of cytoprotective genes [5,8]. This canonical NFE2L2 activation mechanism has been the target for the development of potential therapeutic strategies against oxidative stress [9].
However, this process potentially has nonspecific activity, which can result in the oxidation of different cysteine residues in other proteins. To address these concerns, approaches to activate noncanonical NFE2L2 with greater potency and higher clinical efficacy are being developed to treat NAFLD [9,10]. NFE2L2 activation by SQSTM1/p62 comprises the most well-known noncanonical KEAP1-NFE2L2 pathway [9]. This SQSTM1-mediated noncanonical KEAP1-NFE2L2 pathway increases that increase SQSTM1, and its phosphorylation (S351) can disrupt KEAP1-NFE2L2 interactions, thereby activating the noncanonical KEAP1-NFE2L2 pathway without KEAP1 oxidation [11,12]. In addition, SQSTM1-mediated NFE2L2 activation through autophagic KEAP1 degradation is part of this pathway [13].
Recently, we reported that SQSTM1 has anti-lipotoxic functions through autophagic KEAP1 degradation and NFE2L2 activation in hepatocytes [14]. However, the activation and molecular mechanisms of the aforementioned SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway and its physiological relevance with respect to lipotoxicity remain largely unknown. In this study, we demonstrate that the AMPK (AMP-activated protein kinase)-ULK1 (unc-51 like autophagy activating kinase 1) axis has an important role in which activation of the SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway protects against lipotoxicity. Additionally, we identify the clinical significance of this pathway using liver samples from human patients with NAFLD.
Results
SQSTM1 activates autophagy in response to lipotoxicity
To examine whether the SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway activates in response to lipotoxicity, we treated Hepa1c1c7 cells with saturated fatty acid (SFA), palmitic acid (PA). We observed that PA increased the levels of SQSTM1 and autophagic KEAP1 degradation, followed by NFE2L2 activation (Fig. S1A-D). Sqstm1 is known as an NFE2L2 target gene [15]. We tested whether NFE2L2 activation caused by reactive oxygen species (ROS) accumulation mediated the induction of SQSTM1. We observed that PA-mediated induction of SQSTM1 and NFE2L2 target genes were attenuated by co-treatment with N-acetyl-L-cysteine (NAC), ROS scavenger (Fig. S1E-G). Furthermore, to clarify that the induction of SQSTM1 is mainly dependent on ROS-induced NFE2L2 activation, we treated Nfe2l2 wild-type (WT) and nfe2l2-deficient (nfe2l2 KO) mouse embryonic fibroblast (MEF) cells with PA. The results showed that PA increased the induction of SQSTM1 and NFE2L2-target genes in Nfe2l2 WT MEFs, whereas PA partly blocked the expression of these target genes in nfe2l2 KO MEFs (Fig. S1H-K). Together, the results suggest that the induction of SQSTM1 is dependent on ROS-mediated NFE2L2 activation in response to lipotoxicity.
Consistent with our previous reports [14], we observed that KEAP1 degradation and NFE2L2 activation in response to lipotoxicity required SQSTM1 (Figure 1A–C). Collectively, these results indicate that the activation of the noncanonical KEAP1-NFE2L2 pathway in response to lipotoxicity requires the induction of SQSTM1. Indeed, to test whether autophagic KEAP1 degradation required an increase in SQSTM1, we transfected HEK-293 cells with HA-SQSTM1 and FLAG-KEAP1 and treated them with the proteasome inhibitor (MG132) or the autophagy inhibitor chloroquine (CQ). Immunoblot analysis revealed that the SQSTM1-induced KEAP1 degradation was significantly blocked in CQ-treated cells (Fig. S2A-B), and autophagy-defective cells (Fig. S2C-E). To further determine whether SQSTM1-mediated autophagy activation induces KEAP1 degradation in response to lipotoxicity, we co-treated Sqstm1 WT and sqstm1 KO MEFs with PA and bafilomycin A1 (BafA1). Our results showed that autophagic KEAP1 degradation was blocked in BafA1-treated sqstm1 KO MEFs. Furthermore, we found a further increase in the levels of LC3B-II in BafA1-treated Sqstm1 WT MEFs compared with those in sqstm1 KO MEFs (Figure 1D–E and S3). Based on these results, we suggest that two possible mechanisms can explain this SQSTM1-dependent autophagic KEAP1 degradation. SQSTM1 is well-established as an autophagy receptor protein, and bind KEAP1 via a domain designated the KEAP1 interaction region [11,15,16]. Consistently, the KEAP1 interaction region can undergo degradation in association with SQSTM1. A recent study reported that SQSTM1-mediated autophagy activation could improve Alzheimer disease-like pathology [17]. Consistent with these reports, SQSTM1 activates autophagy, followed by autophagic KEAP1 degradation and NFE2L2 activation. Together, the results suggest that this aforementioned SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway is active in response to lipotoxicity.
Figure 1.
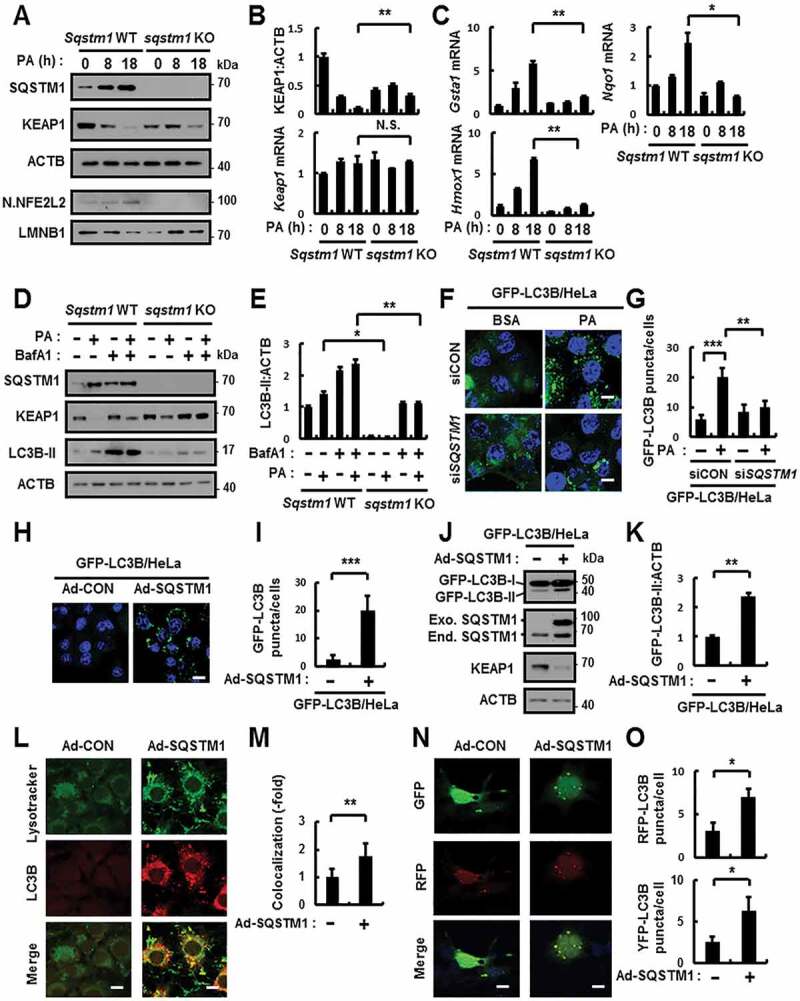
SQSTM1 activates autophagy in response to lipotoxicity. (A) Sqstm1 WT or sqstm1 KO MEFs were incubated with PA (500 μM) for indicated times and subjected to immunoblot analysis with antibodies against SQSTM1, KEAP1, ACTB (loading control), nuclear (N) NFE2L2, and LMNB1 (nuclear marker). (B) The densitometric analysis of KEAP1 immunoblots. (B–C) Total mRNA isolation from cells were treated as described in (A) and subjected to qRT-PCR analysis for relative mRNA expression of Keap1 (B), Gsta1, Hmox1, and Nqo1 (C). (D) Sqstm1 WT or sqstm1 KO MEFs were incubated with PA (500 μM) and BafA1 (10 nM) and subjected to immunoblot analysis with antibodies against SQSTM1, KEAP1, LC3B, and ACTB (loading control). (E) Densitometric analysis of LC3B-II immunoblots. (F) GFP-LC3B fluorescence analysis of puncta by confocal microscopy using GFP-LC3B HeLa cells transfected with control siRNA (siCON) or SQSTM1 siRNA and treated with PA for 18 h. The representative single optical sections and merge images are shown. Scale bar: 10 μm. (G) Quantitative analysis of GFP-LC3B puncta. (H) GFP-LC3B fluorescence analysis of puncta by confocal microscopy using GFP-LC3B HeLa cells infected with Ad-SQSTM1 for 18 h. The representative single optical sections and merge images are shown. Scale bar: 20 μm. (I) Quantitative analysis of GFP-LC3B puncta. (J) Immunoblot analysis with antibodies against GFP, SQSTM1, KEAP1 and ACTB. (K) Densitometric analysis of GFP-LC3B-II immunoblots. (L) The colocalization of Lysotracker and LC3B by confocal microscopy using Ad-SQSTM1-infected cells. The representative single optical sections and merge images are shown. Scale bar: 10 μm. (M) Quantitative analysis of colocalization. (N) Hepa1c1c7 cells were transfected with mRFP-GFP-LC3B plasmids and infected with Ad-SQSTM1. The representative single optical sections and merge images are shown. Scale bar: 10 μm. (O) Quantitative analysis of RFP-LC3B and YFP-LC3B. Data are presented as the mean ± SD from 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, and N.S., not significant
To further examine whether autophagy activation in response to lipotoxicity requires SQSTM1, we transfected HeLa cells stably expressing GFP-LC3B (GFP-LC3B HeLa) with SQSTM1 siRNA and treated them with PA. We observed that the number of GFP-LC3B puncta decreased in SQSTM1 knockdown cells in response to lipotoxicity (Figure 1F,G). We infected GFP-LC3B HeLa cells with FLAG-tagged SQSTM1 using adenovirus vector (Ad-SQSTM1) to determine whether SQSTM1 induced autophagy activation. We showed that GFP-LC3B puncta and GFP-LC3B-II levels increased in Ad-SQSTM1-infected cells, resulting in degradation of KEAP1 (Figure 1H–K). We further observed that the co-localization of lysotracker and LC3B partly increased in Ad-SQSTM1-infected cells (Figure 1L,M). Indeed, to determine whether SQSTM1 induces autophagy flux, we transfected cells with GFP-RFP-LC3B-expressing vectors and infected them with Ad-SQSTM1, which served to increase autophagic flux (Figure 1N,O). Together, the results suggest that SQSTM1 is possible to activate autophagy in response to lipotoxicity.
SQSTM1-mediated autophagy activation is dependent on ULK1
To examine how SQSTM1 activates autophagy in response to lipotoxicity, we treated Sqstm1 WT and sqstm1 KO MEFs with PA. Interestingly, we found marked inhibition of the phosphorylation of ULK1 at S317 in sqstm1 KO in response to lipotoxicity (Figure 2A–D). It is well-established that various phosphorylation of ULK1 can regulate autophagy, including phosphorylation sites located at S317, S555 (autophagy activation), and S757 (autophagy inhibition) [18–20]. Correlating with this, we observed partial inhibition of the PA-induced phosphorylation of ULK1 at S555 in sqstm1 KO MEFs. However, the phosphorylation of ULK1 at S757, which inhibits autophagy, was independent of SQSTM1 in response to lipotoxicity (Fig. S4). Collectively, the results suggest that SQSTM1-induced autophagy activation is dependent on AMPK-mediated ULK1 phosphorylation at specific autophagy activation sites (S317 and S555). A recent study reported the requirement of autophagy for the AMPK-ULK1 axis, which can induce autophagy through ULK1 phosphorylation at S317 by AMPK in response to nutrient starvation [18,19]. Furthermore, we recently reported that ulk1 KO MEFs mainly block autophagic KEAP1 degradation in response to lipotoxicity. We further observed that PA-induced ULK1 degradation is dependent on autophagy [21]. To investigate whether autophagic ULK1 degradation requires overexpression of SQSTM1, we transfected Atg5 WT and atg5 KO MEFs with HA-SQSTM1 and FLAG-ULK1. Immunoblot analysis showed a significant block in the SQSTM1-induced ULK1 degradation in autophagy-defective cells (Fig. S5). These results demonstrate that PA-induced SQSTM1 leads to autophagic ULK1 degradation. Based on these results, we transfected GFP-LC3B HeLa cells with ULK1 siRNA and overproduced Ad-SQSTM1 to investigate whether SQSTM1-induced autophagic KEAP1 degradation required ULK1. We showed that SQSTM1-mediated autophagy activation was mainly blocked, thereby led to inhibit autophagic KEAP1 degradation in ULK1-knockdown cells (Figure 2E–I). To further confirm that SQSTM1-mediated autophagy activation is dependent on ULK1, we infected Ulk1 WT and ulk1 KO MEFs with Ad-SQSTM1. We found that autolysosome formation caused by SQSTM1 was diminished in ulk1 KO MEFs (Figure 2J,K). To further determine whether SQSTM1 induces phosphorylation of ULK1, we infected GFP-LC3B HeLa cells with Ad-SQSTM1 and treated them with a ULK1 activity inhibitor (SBI-0206965). We observed that SQSTM1 induced phosphorylation of ULK1 at S317, resulting in degradation of KEAP1. However, SQSTM1-mediated phosphorylation of ULK1 significantly decreased, resulting in blocked degradation of KEAP1 in ULK1 inhibitor-treated cells (Fig. S6).
Figure 2.
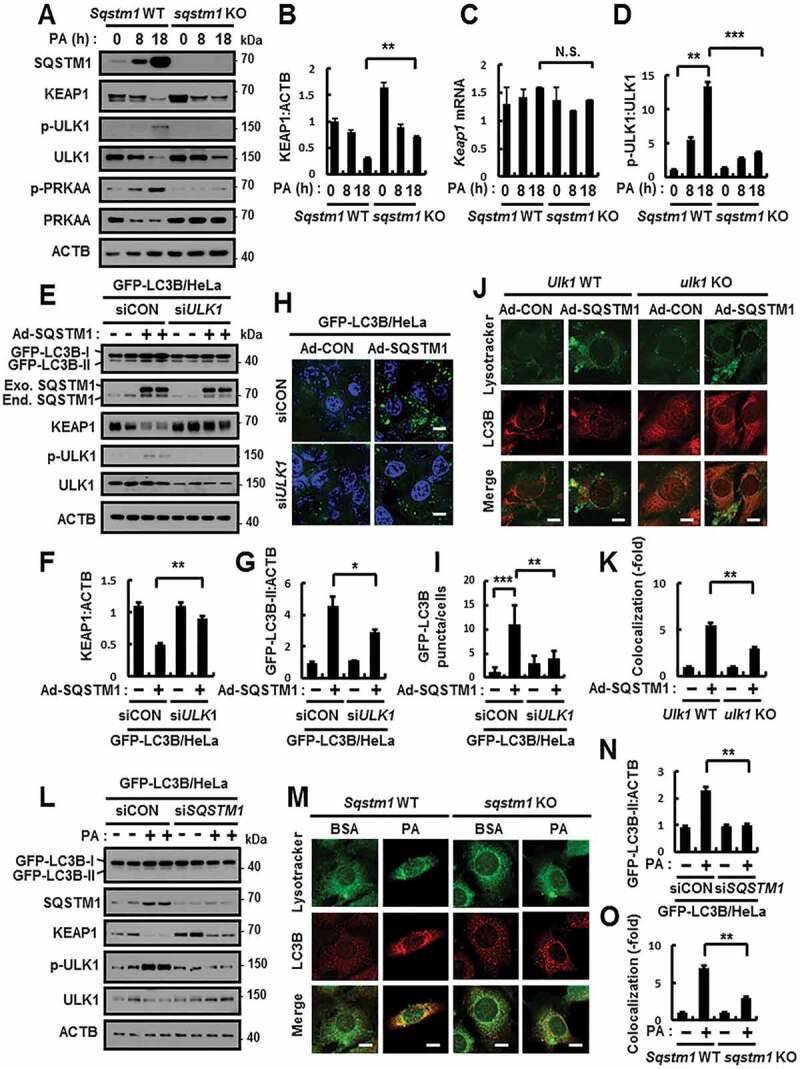
SQSTM1-mediated autophagy activation is dependent on ULK1. (A) Sqstm1 WT or sqstm1 KO MEFs were incubated with PA (500 μM) for indicated times and subjected to immunoblot analysis with antibodies against SQSTM1, KEAP1, p-ULK1(S317), ULK1, p-PRKAA(T172), PRKAA, and ACTB (loading control). (B, D) Densitometric analysis of KEAP1 (B) and p-ULK1:ULK1 (D) immunoblots. (C) Total mRNA isolation from cells were treated as described in (A) and subjected to qRT-PCR analysis for relative mRNA expression of Keap1. (E) GFP-LC3B HeLa cells transfected with control siRNA (siCON) or ULK1 siRNA and infected with Ad-SQSTM1 for 18 h and subjected to immunoblot analysis with antibodies against GFP, SQSTM1, KEAP1, p-ULK1, ULK1, and ACTB. (F-G) Densitometric analysis of KEAP1 (F) and GFP-LC3B-II (G) immunoblots. (H–I) GFP-LC3B fluorescence analysis of puncta by confocal microscopy as described in (E), and (I) quantitative analysis of GFP-LC3B puncta. The representative single optical sections and merge images are shown. Scale bar: 10 μm. (J) Ulk1 WT or ulk1 KO MEFs were infected with Ad-SQSTM1 for 18 h, and confocal microscopy analysis of colocalization of Lysotracker and LC3B. (K) Quantitative analysis of colocalization. The representative single optical sections and merge images are shown. Scale bar: 5 μm. (L) GFP-LC3B HeLa cells were transfected with SQSTM1 siRNA and treated with PA (500 μM) for 18 h. Immunoblot analysis with antibodies against GFP, SQSTM1, KEAP1, p-ULK1(S317), ULK1, and ACTB (loading control). (N) Densitometric analysis of GFP-LC3B-II immunoblots. (M) Sqstm1 WT or sqstm1 KO MEFs were treated with PA (500 μM) for 18 h, and confocal microscopy analysis of colocalization of Lysotracker and LC3B. The representative single optical sections and merge images are shown. Scale bar: 5 μm. (O) Quantitative analysis of colocalization. Data are presented as the mean ± SD from 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, and N.S., not significant
We further examined whether SQSTM1 induces autophagy activation in response to lipotoxicity. Our results showed significant blockage in GFP-LC3B-II levels and ULK1 phosphorylation in SQSTM1 knockdown cells (Figure 2L,N, and S7). Moreover, our results showed a partial decrease in the autolysosome formation in sqstm1 KO MEFs in response to lipotoxicity (Figure 2M,O). Together, these findings suggest that SQSTM1 facilitates autophagy activation, thereby leading to ULK1-mediated autophagic KEAP1 degradation in response to lipotoxicity.
SQSTM1-mediated phosphorylation of ULK1 protects cells from lipotoxicity
To further examine whether the phosphorylation of ULK1 at S317 protects cells against lipotoxicity, we transfected ulk1 KO MEFs with expressing vector of the phosphorylation-defective mutant of ULK1 (ULK1S317A) and WT ULK1 in response to lipotoxicity. We observed partial inhibition in the ULK1-mediated autophagic KEAP1 degradation and NFE2L2 activation in ULK1S317A-transfected cells under lipotoxic conditions (Figure 3A–E), and partial diminishment in PA-mediated autolysosome formation in ULK1S317A-transfected cells (Figure 3F,G). Accordingly, we found that ROS-mediated cell death was further increased in ULK1 S317A-transfected cells compared with WT ULK1 in response to lipotoxicity (Figure 3H–L). In addition, to verify whether SQSTM1-mediated phosphorylation of ULK1 positively regulates autophagy activation, we transfected ulk1 KO MEFs with HA-tagged ULK1 S317A and WT ULK1 and infected them with Ad-SQSTM1. We observed that the SQSTM1-mediated autophagy activation, as measured by the levels of LC3B-II and autolysosome formation, was increased in WT ULK1-transfected cells compared with ULK1S317A-transfected cells (Figure 4A–D), thereby it led to autophagic KEAP1 degradation and NFE2L2 activation (Figure 4A–B, E–H). Together, these results demonstrate that SQSTM1-mediated phosphorylation of ULK1 at S317 has an essential cytoprotective role against lipotoxicity.
Figure 3.
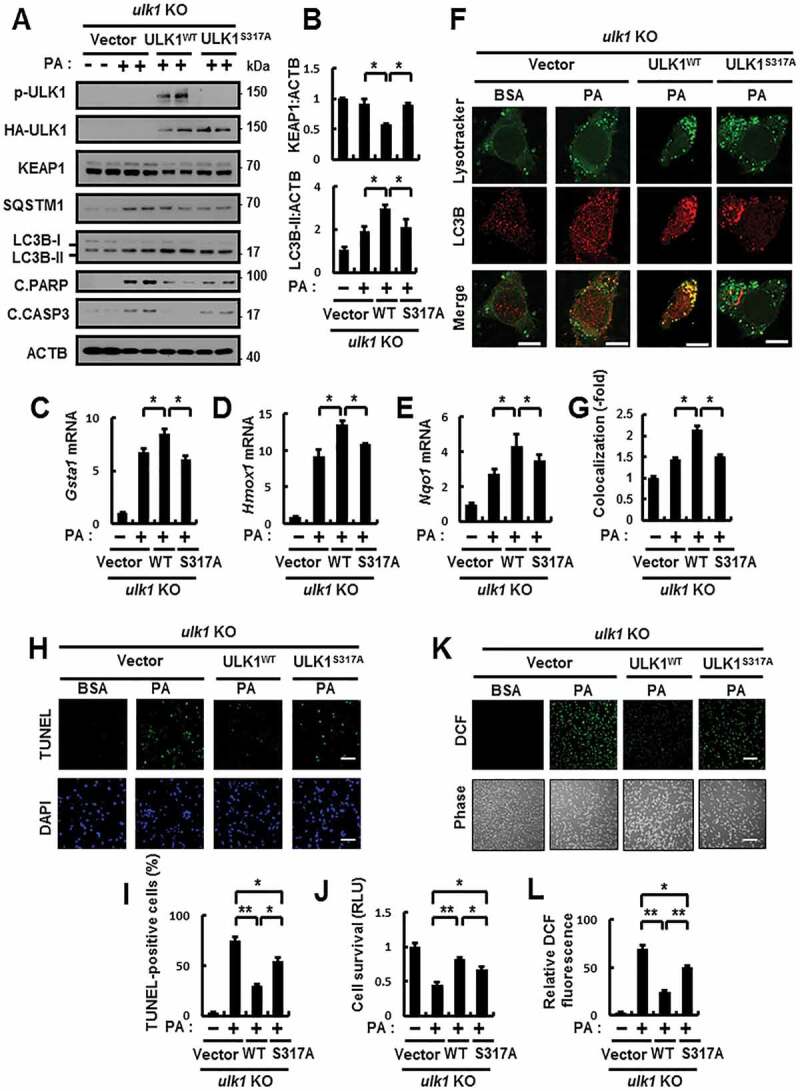
The phosphorylation of ULK1 is required for autophagy activation in response to lipotoxicity. (A) ulk1 KO MEFs co-transfected with vectors encoding HA-WT ULK1 or the HA-ULK1S317A mutant were incubated with PA (500 μM) for 18 h and subjected to immunoblot analysis with antibodies against p-ULK1(S317), HA-ULK1, KEAP1, SQSTM1, LC3B, C.PARP, C.CASP3, and ACTB (loading control). (B) Densitometric analysis of KEAP1 and LC3B-II immunoblots. (C–E) Total mRNA isolation from cells treated as described in (A) and subjected to qRT-PCR analysis for relative mRNA expression of Gsta1 (C), Hmox1 (D), Nqo1 (E). (F) Confocal microscopy analysis of colocalization of Lysotracker and LC3B the cells treated as described in (A). The representative single optical sections and merge images are shown. Scale bar: 10 μm. (G) Quantitative analysis of colocalization. (H) TUNEL analysis of cells treated as in (A). Scale bar: 100 μm. (I) Quantification of TUNEL-positive cells. (J) Cell viability was estimated using a Cell titer-Glo assay kit. Live cell numbers were expressed as absorbance at luminescence. (K) Reactive oxygen species (ROS) were determined using CM-H2DCFH-DA. The representative images are shown. Scale bar: 100 μm. (L) Quantification of relative DCF fluorescence. Data are presented as the mean ± SD from 3 independent experiments. *p < 0.05, **p < 0.01
Figure 4.
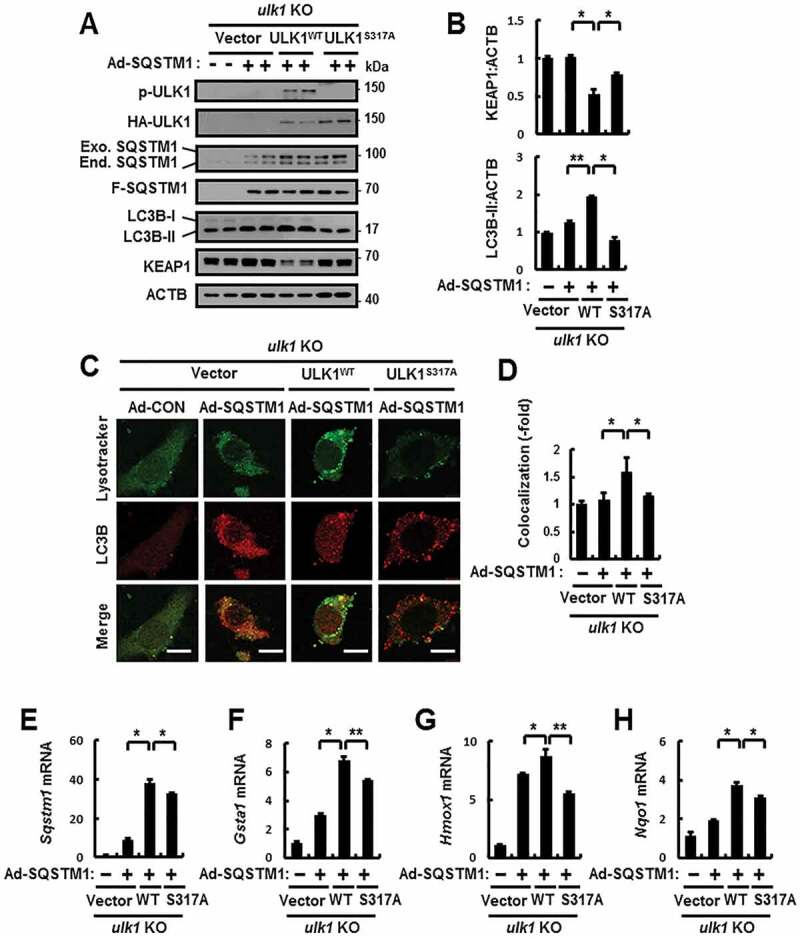
SQSTM1-mediated ULK1 phosphorylation induces autophagy activation. (A) ulk1 KO MEFs co-transfected with vectors encoding HA-WT ULK1 or the HA-ULK1S317A mutant were infected with Ad-SQSTM1 for 18 h and subjected to immunoblot analysis with antibodies against p-ULK1(S317), HA-ULK1, SQSTM1, FLAG-SQSTM1, LC3B, KEAP1, and ACTB (loading control). (B) Densitometric analysis of KEAP1 and LC3B-II immunoblots. (C) Confocal microscopy analysis of colocalization of Lysotracker and LC3B the cells treated as described in (A). The representative single optical sections and merge images are shown. Scale bar: 10 μm. (D) Quantitative analysis of colocalization. (E–H) Total mRNA isolation from cells treated as described in (a) and subjected to qRT-PCR analysis for relative mRNA expression of sqstm1 (E), Gsta1 (F), Hmox1 (G), and Nqo1 (H). Data are presented as the mean ± SD from 3 independent experiments. *p < 0.05, **p < 0.01
SQSTM1-mediated autophagy activation is dependent on the AMPK-ULK1 axis
AMPK positively regulates ULK1-mediated autophagy [18]. Consistent with these notions, to verify whether ULK1-mediated autophagic KEAP1 degradation in response to lipotoxicity requires AMPK, we treated Prkaa1/2 WT and prkaa1/2 KO MEFs with PA. Our results showed that the AMPK-ULK1 axis was blocked in prkaa1/2 KO MEFs, resulting in inhibition of KEAP1 degradation (Figure 5A–C). Furthermore, we observed that this mechanism also was inhibited by compound C (C.C), pan inhibitor of AMPK (Fig. S8). We observed significant blockage in the phosphorylation of AMPK in sqstm1 KO MEFs in response to lipotoxicity (Figure 2A). Based on these notions, to further examine whether AMPK is involved in SQSTM1-mediated ULK1 phosphorylation, resulting in induced autophagic KEAP1 degradation, we transfected GFP-LC3B HeLa cells with PRKAA1 siRNA and infected them with Ad-SQSTM1. We found a partial decrease in GFP-LC3B-II expression and GFP-LC3B puncta formation, followed by inhibition of ULK1-mediated autophagic KEAP1 degradation in AMPK knockdown cells (Figure 5D–G, I). Furthermore, we observed mainly reduced Ad-SQSTM1-induced autolysosomes formation in prkaa1/2 KO MEFs (Figure 5H,J). Together, these findings suggest that SQSTM1 can induce autophagy by activation of the AMPK-ULK1 axis in response to lipotoxicity.
Figure 5.
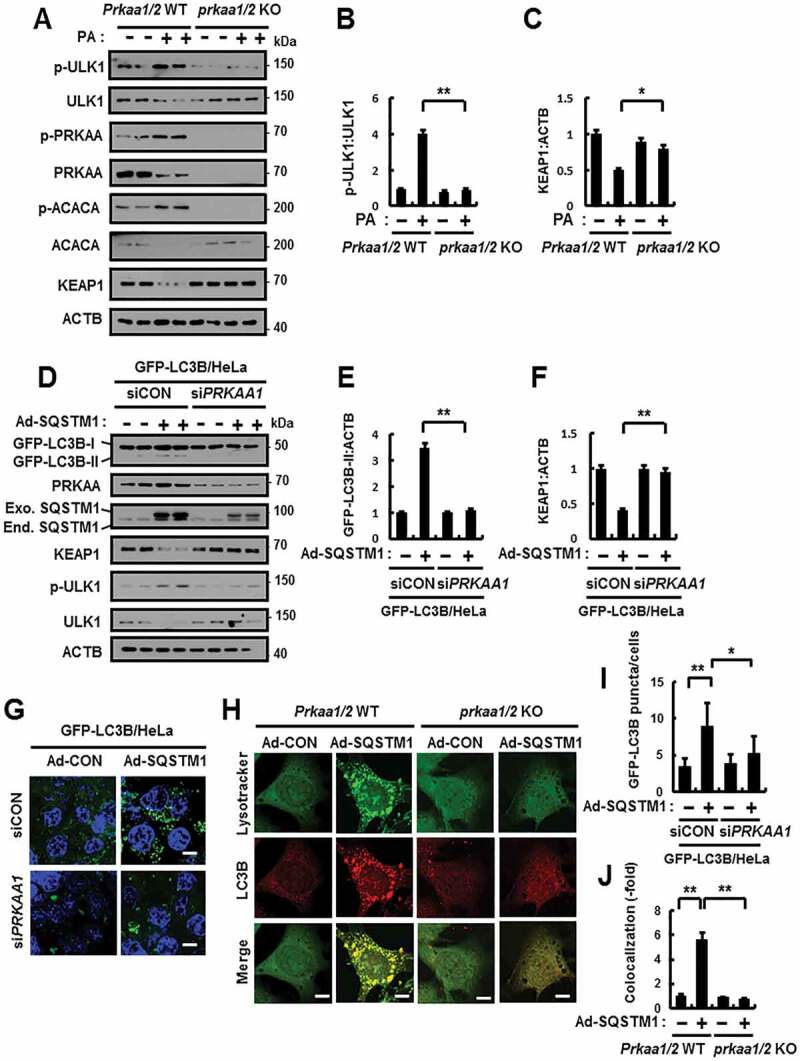
SQSTM1-mediated autophagy activation is dependent on ULK1-AMPK axis. (A) Prkaa1/2 WT or prkaa1/2 KO MEFs were incubated with PA (500 μM) for 18 h and subjected to immunoblot analysis with antibodies against p-ULK1(S317), ULK1, p-PRKAA(T172), PRKAA, p-ACACA(S79), ACACA KEAP1 and ACTB. (B–C) Densitometric analysis of p-ULK1:ULK1 (B) and KEAP1 (C) immunoblots. (D) GFP-LC3B HeLa cells transfected with control siRNA (siCON) or PRKAA1 siRNA (siPRKAA1) and infected with Ad-SQSTM1 for 18 h and subjected to immunoblot analysis with antibodies against GFP, PRKAA, SQSTM1, KEAP1, p-ULK1(S317), ULK1, and ACTB. (E–F) Densitometric analysis of GFP-LC3B-II (E) and KEAP1 (F) immunoblots. (G) Confocal microscopy analysis of GFP-LC3B puncta and representative single optical sections and merge images are shown. Scale bar: 10 μm. (H) Prkaa1/2 WT or prkaa1/2 KO MEFs were infected with Ad-SQSTM1 for 18 h, and confocal microscopy analysis of Lysotracker and LC3B, and representative single optical sections and merge images are shown. Scale bar: 5 μm. (I) Quantitative analysis of GFP-LC3B puncta. (J) Quantitative analysis of colocalization. Data are presented as the mean ± SD from 3 independent experiments. *p < 0.05, **p < 0.01
SQSTM1 mediates the ULK1 phosphorylation by regulation of the AMPK-ULK1 axis
To understand whether SQSTM1 mediates phosphorylation of ULK1, we transfected cells with HA-SQSTM1, MYC-PRKAA1, and FLAG-ULK1 expression vectors. FLAG-ULK1 was immunoprecipitated, and we examined its phosphorylation with phospho-specific antibodies. The ectopic expression of SQSTM1 dramatically increased the phosphorylation of ULK1 (Figure 6A,B). Furthermore, we showed a strong block in the phosphorylation of ULK1 in sqstm1 KO MEFs (Figure 6C,D). Previously, we reported SQSTM1 interacts with AMPK via its PB1 domain [22]. Also, our previous reports suggested that SQSTM1 binds ULK1 without a specific domain [21]. To explore whether SQSTM1 could enhance the interacts with AMPK-ULK1 complex, we transfected HEK-293 cells with MYC-PRKAA1 or FLAG-ULK1 expression vectors together with HA-SQSTM1 and then subjected cell lysates to co-immunoprecipitation analysis. Results showed significant enhancement in the interactions by HA-SQSTM1 (Figure 6E,F). To further evaluate the association between SQSTM1 and the AMPK-ULK1 complex, we examined these interactions in Sqstm1 WT or sqstm1 KO MEFs. Results demonstrated that the interaction was significantly diminished in sqstm1 KO MEFs (Figure 6G–J). We further observed that the endogenous SQSTM1-AMPK-ULK1 complex strongly interacted in response to lipotoxicity (Figure 6K–M). These results indicate that SQSTM1 has an essential role in facilitating enhanced interactions between AMPK and ULK1.
Figure 6.
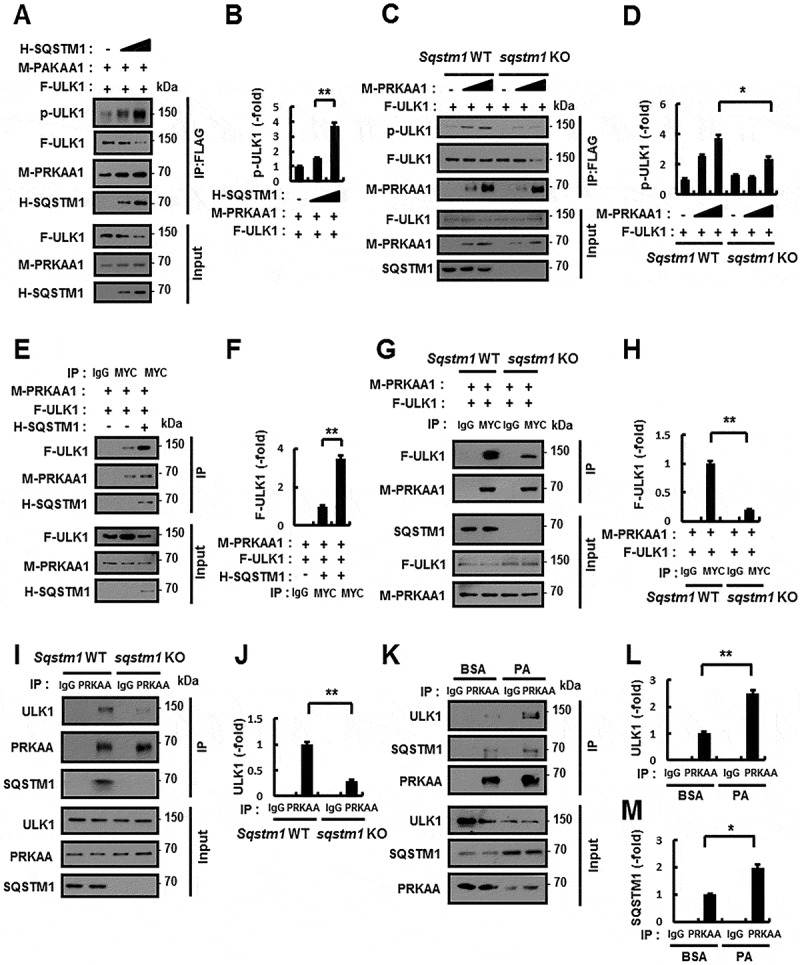
SQSTM1-mediated phosphorylation of ULK1 via enhanced interactions between PRKAA1/2 and ULK1. (A) Lysates from HEK-293 cells transfected with vectors encoding MYC-PRKAA1 (M-PRKAA1) and FLAG-ULK1 (F-ULK1) together with those expressing HA-SQSTM1 (H-SQSTM1) were subjected to immunoprecipitation with antibodies against FLAG, and the resulting precipitates (IPs) as well as whole cell lysates (WCLs) were subjected to immunoblot analysis with antibodies against p-ULK1, FLAG, MYC, and HA. (B) Densitometric analysis was obtained. (C) Lysates from Sqstm1 WT or sqstm1 KO MEFs were transfected with vectors encoding FLAG-ULK1 together with those expressing MYC-PRKAA1, and subjected to immunoprecipitation with antibodies to FLAG, and the resulting IPs and WCLs were subjected to immunoblot analysis with antibodies against p-ULK1(S317), FLAG, and MYC. (D) Densitometric analysis was obtained. (E) Lysates from HEK-293 cells transfected with vectors encoding MYC-PRKAA1, FLAG-ULK1, and HA-SQSTM1 were subjected to immunoprecipitation with antibodies against MYC, and the resulting IPs and WCLs were subjected to immunoblot analysis with antibodies against FLAG, MYC, and HA. (F) Densitometric analysis was obtained. (G) Sqstm1 WT or sqstm1 KO MEFs transfected with vectors encoding MYC-PRKAA1 and FLAG-ULK1 were subjected to immunoprecipitation with antibodies against MYC, and the resulting IPs and WCLs were subjected to immunoblot analysis with antibodies against FLAG, MYC, and SQSTM1. (H) Densitometric analysis was obtained. (I) Lysates from Sqstm1 WT or sqstm1 KO MEFs were subjected to immunoprecipitation with antibodies against PRKAA, and the resulting IPs and WCLs were subjected to immunoblot analysis with antibodies against ULK1, PRKAA, and SQSTM1. (J) Densitometric analysis was obtained. (K) Lysates from Hepa1c1c7 cells were treated with PA (500 μM) and subjected to immunoprecipitation with antibodies against PRKAA, and the resulting IPs and WCLs were subjected to immunoblot analysis with antibodies against ULK1, SQSTM1, and PRKAA. (L-M) Densitometric analysis of ULK1 (L) and SQSTM1 (M) immunoblots. Data are presented as the mean ± SD from 3 independent experiments. *p < 0.05, **p < 0.01
We further examined whether SQSTM1 affected activation of AMPK, as well as enhancement of the interaction between AMPK and ULK1. We assessed phosphorylation of ACACA (acetyl-CoA carboxylase alpha), which is a marker of AMPK activity, was significantly blocked in sqstm1 KO MEFs in response to lipotoxicity (Figure 2A and S9). We further elucidated how SQSTM1 activates AMPK. Several studies have reported that AMPK is phosphorylated by SESN2 (sestrin 2), which is a stress-inducible protein that directly phosphorylates AMPK [23,24]. Furthermore, SESN2 can directly bind to SQSTM1, and thereby it led to autophagic KEAP1 degradation and NFE2L2 activation [13]. Based on these notions, to identify whether SQSTM1 may activate the SESN2-AMPK axis, we transfected Sqstm1 WT and sqstm1 KO MEFs with MYC-PRKAA1 and FLAG-SESN2. We observed that the ablation of SQSTM1 diminished the phosphorylation of AMPK (Fig. S10A and B). We further observed that the interaction of AMPK and SESN2 decreased in sqstm1 KO MEFs in response to lipotoxicity (Fig. S10C and D). To further identify whether SQSTM1 may activate AMPK through direct interaction with SESN2, we co-expressed either MYC-PRKAA1, FLAG-SESN2 or HA-SQSTM1, and analyzed immunoprecipitates. We observed that SQSTM1 could enhance the interaction between AMPK and SESN2, thereby leading to the induction of AMPK phosphorylation (Fig. S10E and F). To investigate whether SQSTM1-induced AMPK phosphorylation is dependent on SESN2, we transfected Sesn2 WT and sesn2 KO MEFs with FLAG-SQSTM1 and MYC-PRKAA1 expression vectors and then subjected cell lysates to co-immunoprecipitation analysis. We observed that SQSTM1-mediated the phosphorylation of AMPK was significantly diminished in sesn2 KO MEFs (Fig. S11A and B). To further verify that SQSTM1-mediated AMPK-ULK1 activation is dependent on SESN2 in response to lipotoxicity, we treated Sesn2 WT and sesn2 KO MEFs with PA. We observed a partial inhibition in the activation of AMPK in sesn2 KO MEFs and Sesn2 knockdown cells in response to lipotoxicity. Moreover, we observed that autophagic KEAP1 degradation by the SESN2-AMPK-ULK1 axis induced NFE2L2 activation (Fig. S11C-H). Together, these findings suggest that SQSTM1 can activate SESN2-mediated phosphorylation of AMPK as well as enhance the interaction between AMPK and ULK1 in response to lipotoxicity.
SQSTM1-mediated noncanonical activation of the KEAP1-NFE2L2 pathway is required for cytoprotection against lipotoxicity
To further verify the cytoprotective roles of SQSTM1 in response to lipotoxicity, we transfected sqstm1 KO MEFs with MYC-SQSTM1 and treated them with PA. Results showed that overexpression of SQSTM1 could prevent the cells against lipotoxicity through activation of the SQSTM1-mediated noncanonical KEAP1-NFE2L2 pathway, which was associated with ULK1-dependent autophagic KEAP1 degradation (Figure 7A–C). As a result, this pathway could protect cells against lipotoxicity (Figure 7D–F) through the elimination of ROS (Figure 7G,H). Collectively, our results show that SQSTM1-mediated noncanonical activation of the KEAP1-NFE2L2 pathway has an anti-lipotoxic function in hepatocytes.
Figure 7.
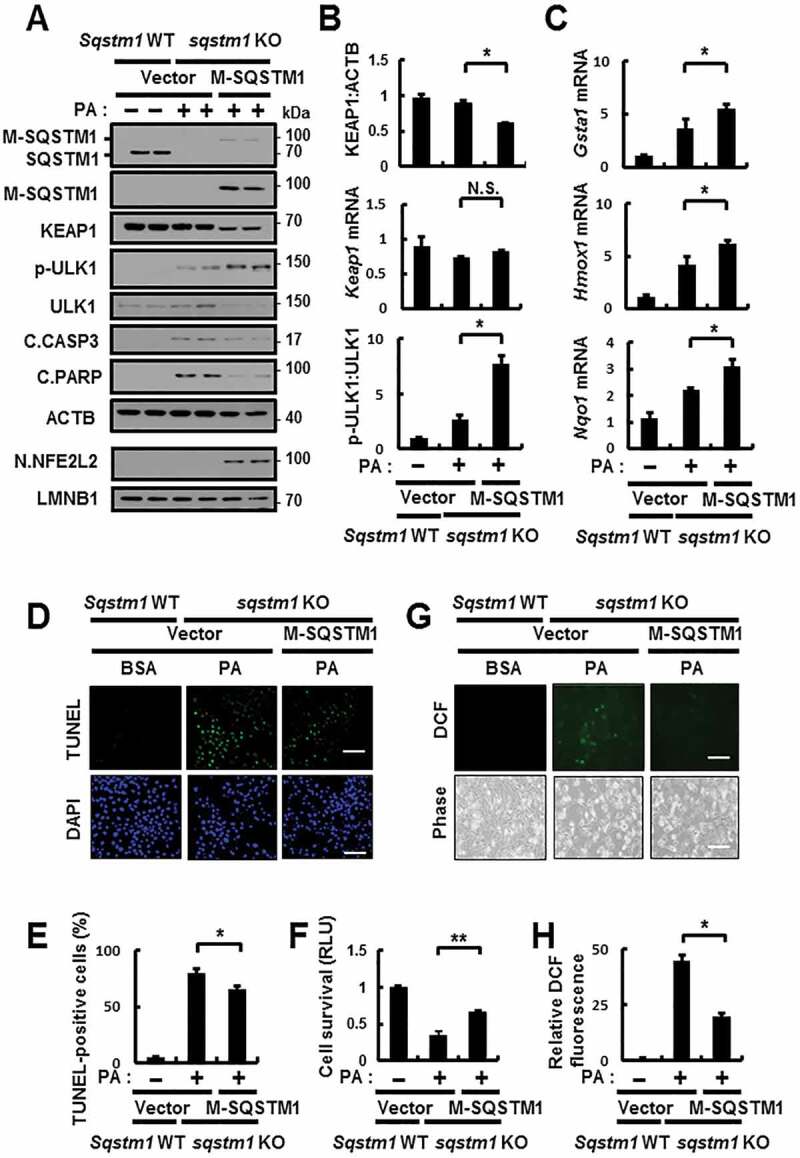
SQSTM1-mediated noncanonical activation of the KEAP1-NFE2L2 pathway is required for cytoprotection against lipotoxicity. (A) sqstm1 KO MEFs were transfected with MYC-SQSTM1 and treated with PA (500 μM) for 18 h, and subjected to immunoblot analysis of SQSTM1, MYC, KEAP1, p-ULK1(S317), ULK1, C.CASP3, C.PARP, ACTB (loading control), nuclear NFE2L2, and LMNB1 (nuclear marker). (B) Densitometric analysis of KEAP1 and p-ULK1:ULK1 immunoblots. (B–C) Total mRNA isolation from cells were treated as described in (A) and subjected to qRT-PCR analysis for relative mRNA expression of Keap1 (B), Gsta1, Hmox1, Nqo1 (C). (D) TUNEL analysis of cells treated as in (A). Scale bar: 100 μm. (E) Quantification of TUNEL-positive cells. (F) Cell viability was estimated using a Cell titer-Glo assay kit. Live cell numbers were expressed as absorbance at luminescence. (G) Reactive oxygen species (ROS) were determined using CM-H2DCFH-DA. The representative images are shown. Scale bar: 100 μm. (H) Quantification of relative DCF fluorescence. Data are presented as the mean ± SD from 3 independent experiments. *p < 0.05, **p < 0.01, and N.S., not significant
SQSTM1 activates autophagy in mouse livers in response to acute physiological lipotoxicity
Regarding its physiological relevance concerning lipotoxicity, we first examined the role of SQSTM1 in a physiological setting. We fed mice a high-fat diet (HFD) as a chronic model and fasted them overnight, which was followed by refeeding with a high-carbohydrate, fat-free diet (R) as an acute model [13,25]. We found activation of the SQSTM1-dependent noncanonical of the KEAP1-NFE2L2 pathway under acute physiological lipotoxicity. Thus, our results showed that induction of SQSTM1 leads to activation of the noncanonical KEAP1-NFE2L2 pathway, and phosphorylated ULK1 in response to physiological lipotoxicity (Fig. S12A-D). However, our results showed that this mechanism was not affected in HFD-fed mice (Fig. S12E-H).
Moreover, physiological lipotoxic conditions increased triglyceride synthesis, which was accompanied by an increase in lipogenesis [13]. Consistently, we observed that refeeding elicited an acute physiological lipotoxicity in the liver as indicated by marked increases in the expression of lipogenic genes including Srebf1 (sterol regulatory element binding transcription factor 1), Fasn (fatty acid synthase), and Acaca (acetyl-CoA carboxylase alpha) (Fig. S13A-C). Therefore, we profiled fatty acids from mouse livers by performing gas chromatography-mass spectrometry or liquid chromatography-mass spectrometry (Fig. S13D). After refeeding, mouse livers showed a marked increase in PA (Fig. S13E). Consistent with these results, we observed increases in ULK1-mediated autophagic KEAP1 degradation and NFE2L2 activation and exaggerated liver injury in an acute physiological lipotoxicity model (Figure 8A–J). Consistent with in vitro findings, we also found that the induction of SQSTM1 was dependent on NFE2L2 in response to acute physiological lipotoxicity (Fig. S14). Based on these results, we used this model to explore the mechanism underlying the SQSTM1-mediated noncanonical KEAP1-NFE2L2 pathway. In addition, to investigate whether SQSTM1 could induce autophagy through this mechanism in a physiological setting, we used GFP-LC3B transgenic mice with refeeding. We observed increases in GFP-LC3B-II levels and GFP-LC3B puncta in the livers of re-fed GFP-LC3B transgenic mice accompanied by SQSTM1-mediated ULK1 phosphorylation and KEAP1 degradation (Figure 9A–D). To further study whether SQSTM1 induced autophagy activation in physiological conditions, we injected using GFP-LC3B transgenic mice with Ad-SQSTM1 via the tail vein to transduce their livers. We found that SQSTM1-induced autophagy activation led to phosphorylation of ULK1-mediated autophagic KEAP1 degradation (Figure 9E–H). Together, our results suggested that SQSTM1-induced autophagy activation could contribute to ULK1-mediated autophagic KEAP1 degradation in response to physiological lipotoxicity, resulting in noncanonical NFE2L2 activation.
Figure 8.
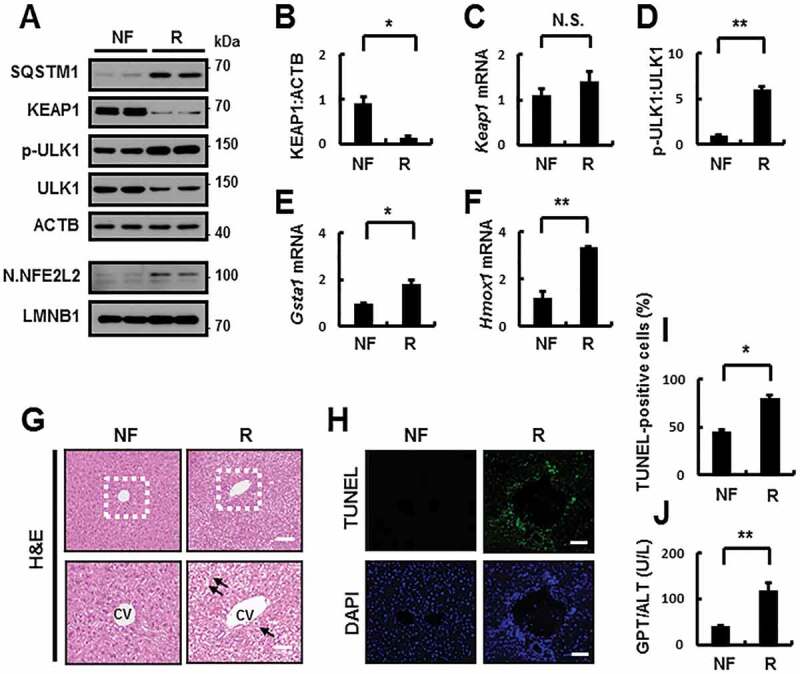
Activation of the SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway in mouse liver under lipotoxic stress. C57BL/6J mice were maintained in a non-fasted state (NF) or fasted overnight and then re-fed a high-carbohydrate, fat-free diet (R). These animals were randomly assigned to 2 groups (8–9 mice in each group). (A) Immunoblot analysis of SQSTM1, KEAP1, p-ULK1(S317), ULK1, ACTB (loading control), nuclear NFE2L2, and LMNB1 (nuclear marker). (B, D) Densitometric analysis of KEAP1 (B) and p-ULK1:ULK1 (D) immunoblots. (C, E-F) Total mRNA isolation from tissue and subjected to qRT-PCR analysis for relative mRNA expression of Keap1 (C), Gsta1 (E), Hmox1 (F). (G) Liver sections of mice were stained with hematoxylin and eosin (H&E). CV, central vein. Scale bar: 200 μm. (H) Images from TUNEL analysis of liver sections. Scale bar: 200 μm. (I-J) Quantification of TUNEL-positive cells (I) and serum GPT/ALT (glutamic pyruvic transaminase, soluble) levels (J) in mice. Data are presented relative to corresponding values in non-fasted mice and are means ± standard errors for 8–9 mice per group. *p < 0.05, **p < 0.01, and N.S., not significant
Figure 9.
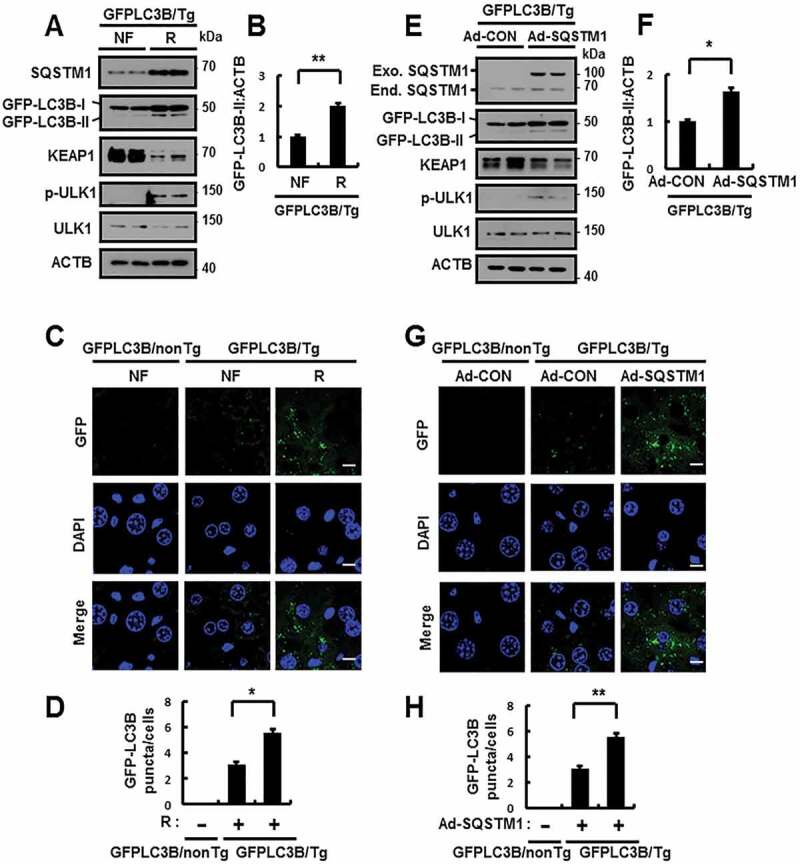
SQSTM1 activates autophagy in mouse liver under lipotoxic stress. GFP-LC3B transgenic mice were maintained in a non-fasted state (NF) or fasted overnight and then re-fed a high-carbohydrate, fat-free diet (R). These animals were randomly assigned to 2 groups (3–4 mice in each group). (A) Immunoblot analysis of SQSTM1, GFP, KEAP1, p-ULK1(S317), ULK1, and ACTB (loading control). (B) Densitometric analysis of GFP-LC3B-II immunoblots. (C) GFP-LC3B fluorescence analysis of puncta by confocal microscopy. Scale bar: 10 μm. (D) Quantitative analysis of GFP-LC3B puncta. GFP-LC3B transgenic mice were injected with empty vector (Ad-CON)- or SQSTM1 (Ad-SQSTM1)-expressing adenovirus. (E) Immunoblot analysis of SQSTM1, GFP, KEAP1, p-ULK1(S317), ULK1, and ACTB (loading control). (F) Densitometric analysis of GFP-LC3B-II immunoblots. (G) GFP-LC3B fluorescence analysis of puncta by confocal microscopy. Scale bar: 10 μm. (H) Quantitative analysis of GFP-LC3B puncta. Data are presented relative to corresponding values in non-fasted mice and are means ± standard errors for 3–4 mice per group. *p < 0.05 and **p < 0.01
SQSTM1 has a hepatoprotective role against acute lipotoxicity in the mouse liver
To further examine whether SQSTM1 has a hepatoprotective role via noncanonical KEAP1-NFE2L2 activation under physiological lipotoxic conditions, we subjected Sqstm1 WT or sqstm1 KO mice to fasting, followed by refeeding. We observed a decrease in the SQSTM1-mediated phosphorylation of ULK1 in sqstm1 KO MEFs, and we consistently found a marked inhibition in ULK1 phosphorylation in sqstm1 KO mice, thereby inhibiting activation of the SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway under physiological lipotoxic conditions (Figure 10A–G). Furthermore, liver injury was exaggerated in sqstm1 KO mice under lipotoxic conditions, as determined by hematoxylin and eosin (H&E) staining (Figure 10H), serum GPT/ALT levels (Figure 10I), and TUNEL assays (Figure 10J,K). Consequently, this mechanism is critical for the protection of the liver against acute lipotoxicity.
Figure 10.
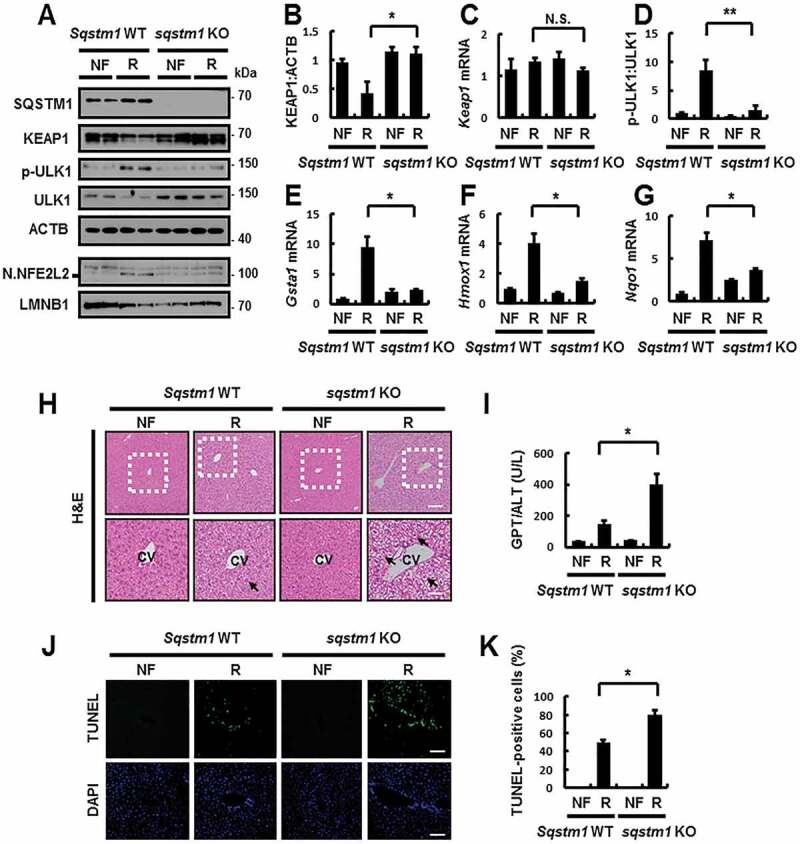
SQSTM1 has a hepatoprotective role against acute lipotoxicity in the mouse liver. Sqstm1 WT or sqstm1 KO mice were maintained in a non-fasted state (NF) or fasted overnight and then re-fed a high-carbohydrate, fat-free diet (R). These animals were randomly assigned to 4 groups (5–6 mice in each group). (A) Immunoblot analysis of SQSTM1, KEAP1, p-ULK1(S317), ULK1, ACTB (loading control), nuclear NFE2L2, and LMNB1 (nuclear marker). (B, D) Densitometric analysis of KEAP1 (B) and p-ULK1:ULK1 (D) immunoblots. (C, E-G) Total mRNA isolation from tissue and subjected to qRT-PCR analysis for relative mRNA expression of Keap1 (C), Gsta1 (E), Hmox1 (F) and Nqo1 (G). (H) Liver sections of mice were stained with H&E. CV, central vein. Scale bar: 200 μm. (I) Serum GPT/ALT levels in mice. (J) Images from TUNEL analysis of liver sections. Scale bar: 200 μm. (K) Quantification of TUNEL-positive cells in liver sections. Data are presented relative to corresponding values in non-fasted mice and are means ± standard errors for 5–6 mice per group. *p < 0.05, **p < 0.01, and N.S., not significant
To test whether SQSTM1 overexpression could rescue the damaging effects of SQSTM1 ablation, we performed tail vein injection using sqstm1 KO mice to transduce their livers with SQSTM1-expressing adenovirus (Ad-SQSTM1) and then subjected the mice to refeeding. Results showed that the overexpression of SQSTM1 induced ULK1-mediated autophagic KEAP1 degradation, thereby leading to NFE2L2 activation (Figure 11A–G). We also observed that the overexpression of SQSTM1 alleviated liver damage in sqstm1 KO mice, as determined by H&E staining (Figure 11H), serum GPT/ALT levels (Figure 11I), and TUNEL assays (Figure 11J,K) in response to physiological lipotoxicity. Collectively, our results show that hepatoprotection against physiological lipotoxic stress requires SQSTM1, and functions in the activation of the noncanonical NFE2L2 activation by facilitating enhanced ULK1-mediated autophagic KEAP1 degradation.
Figure 11.
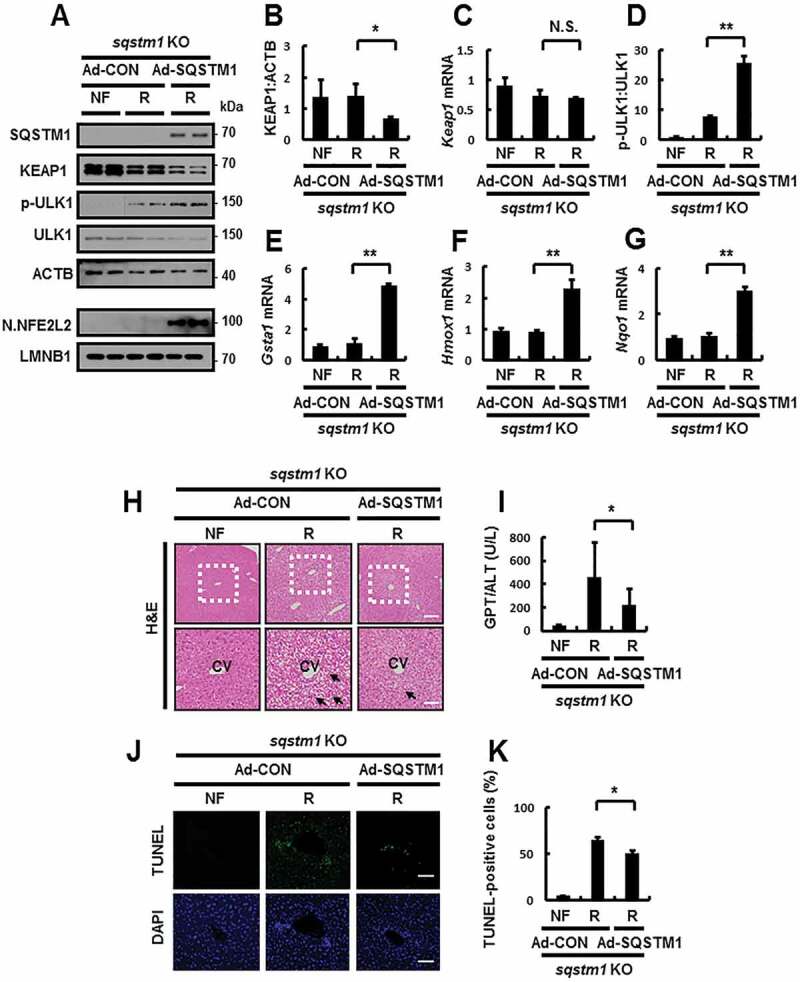
Reinforcement of SQSTM1 ameliorates acute lipotoxicity-induced liver injury in sqstm1 KO mice. sqstm1 KO mice were injected with control vector (Ad-CON)- or SQSTM1 (Ad-SQSTM1)-expressing adenovirus and maintained in a non-fasted state (NF) or fasted overnight and then re-fed a high-carbohydrate, fat-free diet (R). These animals were randomly assigned to 3 groups (5–6 mice in each group). (A) Immunoblot analysis of SQSTM1, KEAP1, p-ULK1(S317), ULK1, ACTB (loading control), nuclear NFE2L2, and LMNB1 (nuclear marker). (B, D) Densitometric analysis of KEAP1 (B) and p-ULK1:ULK1 (D). (C, E-G) qRT-PCR analysis for relative mRNA expression of Keap1 (C), Gsta1 (E), Hmox1 (F), and Nqo1 (G). (H) Liver sections from mice were stained with H&E. CV, central vein. Scale bar: 200 μm. (I) Serum GPT/ALT levels in mice. (J) Images from TUNEL analysis of liver sections. Scale bar: 200 μm. (K) Quantitative analysis of TUNEL-positive cells in liver sections. Data are presented relative to the corresponding value for non-fasted mice and are means ± standard errors for 5–6 mice per group. *p < 0.05, **p < 0.01, and N.S., not significant
Hepatic SQSTM1 protects against acute lipotoxicity in the mouse liver
To further investigate whether hepatic SQSTM1 protects the mouse liver in response to physiological lipotoxic stress, we generated liver-specific sqstm1 KO mice, hereafter referred to as sqstm1 Alb mice by crossing Sqstm1-floxed mice (Sqstm1f/f) with Alb-Cre transgenic mice. When we conducted immunoblot analysis and qRT-PCR using mRNA from various organs of sqstm1 Alb mice, we observed Sqstm1 expression, but not in the liver (Fig. S15). Accordingly, we subjected Sqstm1f/f or sqstm1 Alb mice to fasting, followed by refeeding. We found a marked inhibition in ULK1 phosphorylation in sqstm1 Alb mice, thereby inhibiting activation of the SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway under physiological lipotoxic conditions (Figure 12A–G). As a result, liver injury was exaggerated in sqstm1 Alb mice under lipotoxic conditions, as determined by H&E staining (Figure 12H), serum GPT/ALT levels (Figure 12I), and TUNEL assays (Figure 12J,K). Together, our results showed that hepatic SQSTM1 promotes ULK1-mediated autophagic KEAP1 degradation under physiological lipotoxic conditions, and thereby protecting the liver from lipotoxicity-induced oxidative damage. These findings are consistent with our suggested mechanism.
Figure 12.
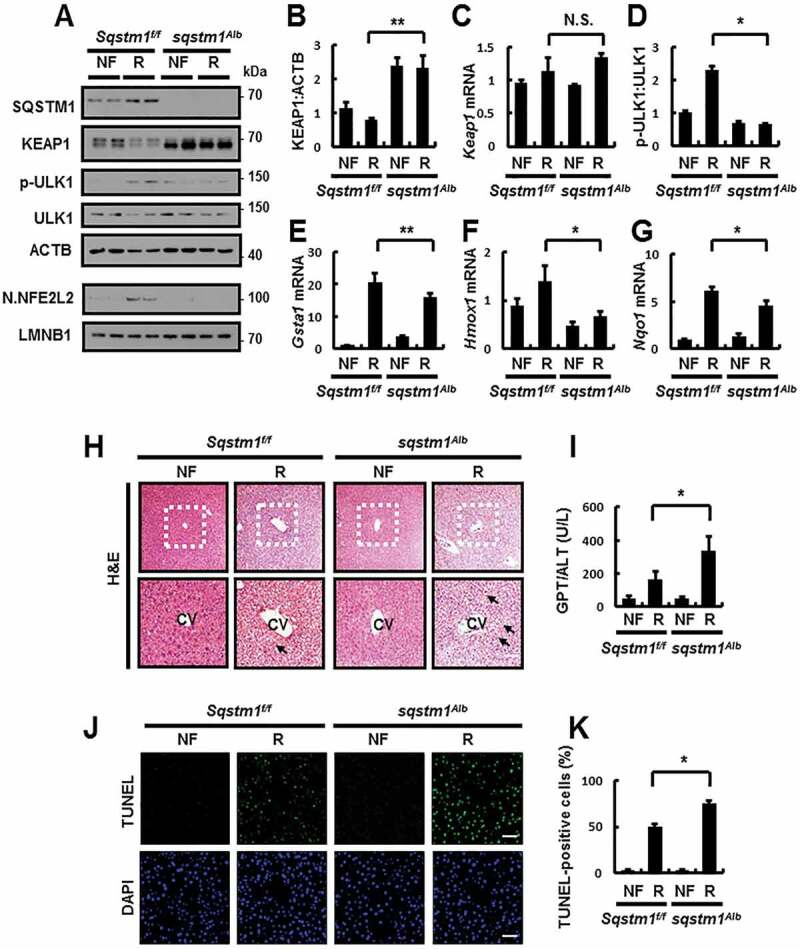
Hepatic SQSTM1 protects against acute lipotoxicity in the mouse liver. Sqstm1 f/f or sqstm1Alb mice were maintained in a non-fasted state (NF) or fasted overnight and then re-fed a high-carbohydrate, fat-free diet (R). These animals were randomly assigned to 4 groups (5–6 mice in each group). (A) Immunoblot analysis of SQSTM1, KEAP1, p-ULK1(S317), ULK1, ACTB (loading control), nuclear NFE2L2, and LMNB1 (nuclear marker). (B, D) Densitometric analysis of KEAP1 (B) and p-ULK1:ULK1 (D) immunoblots. (C, E–G) qRT-PCR analysis for relative mRNA expression of Keap1 (C), Gsta1 (E), Hmox1 (F), and Nqo1 (G). (H) Liver sections of mice were stained with H&E. CV, central vein. Scale bar: 200 μm. (I) Serum GPT/ALT levels in mice. (J) Images from TUNEL analysis of liver sections. Scale bar: 200 μm. (K) Quantitative analysis of TUNEL-positive cells in liver sections. Data are presented relative to corresponding values in non-fasted mice and are means ± standard errors for 5–6 mice per group. *p < 0.05, **p < 0.01, and N.S., not significant
To determine if hepatic SQSTM1 could rescue the damaging effects in sqstm1 Alb mice, we performed tail vein injection to transduce their livers with Ad-SQSTM1, before subjecting the mice to refeeding. Results showed that the overexpression of SQSTM1 induced ULK1 phosphorylation-mediated autophagic KEAP1 degradation, and thereby activating NFE2L2 (Figure 13A–G). We also observed that the overexpression of SQSTM1 alleviated liver damage in sqstm1 Alb mice, as determined by H&E staining (Figure 13H), serum GPT/ALT levels (Figure 13I), and TUNEL assays (Figure 13J,K) in response to physiological lipotoxicity. Collectively, our results showed that hepatic SQSTM1 is essential for protection against lipotoxicity through activation of the ULK1-KEAP1-NFE2L2 axis.
Figure 13.
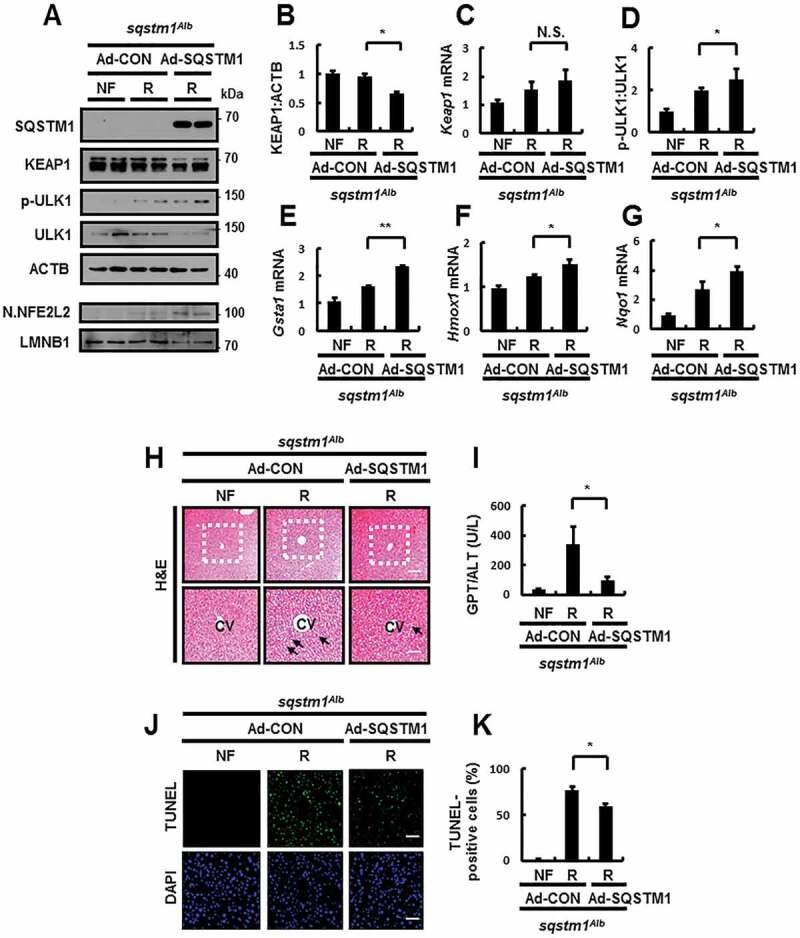
Reinforcement of SQSTM1 ameliorates acute lipotoxicity in liver-specific sqstm1 KO mouse livers. sqstm1Alb mice were injected with control vector (Ad-CON)- or SQSTM1 (Ad-SQSTM1)-expressing adenovirus and maintained in a non-fasted state (NF) or fasted overnight and then re-fed a high-carbohydrate, fat-free diet (R). These animals were randomly assigned to 3 groups (5–6 mice in each group). (A) Immunoblot analysis of SQSTM1, KEAP1, p-ULK1(S317), ULK1, ACTB (loading control), nuclear NFE2L2, and LMNB1 (nuclear marker). (B, D) Densitometric analysis of KEAP1 (B) and p-ULK1:ULK1 (D). (C, E-G) qRT-PCR analysis for relative mRNA expression of Keap1 (C), Gsta1 (E), Hmox1 (F), and Nqo1 (G). (H) Liver sections from mice were stained with H&E. CV, central vein. Scale bar: 200 μm. (I) Serum GPT/ALT levels in mice. (J) Images from TUNEL analysis of liver sections. Scale bar: 200 μm. (K) Quantitative analysis of TUNEL-positive cells in liver sections. Data are presented relative to the corresponding value for non-fasted mice and are means ± standard errors for 5–6 mice per group. *p < 0.05, **p < 0.01, and N.S., not significant
Activation of the SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway in human patients with nonalcoholic fatty liver disease
To identify the clinical significance of our proposed mechanism, we performed a series of experiments using the livers of NAFLD patients. We first examined whether the SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway is activated in patients with NAFLD. Further, immunoblot analysis revealed increases in the levels of SQSTM1 and LC3B-II in NAFLD specimens compared to that in normal specimens. Additionally, enhanced autophagic KEAP1 degradation was induced through the phosphorylation of ULK1, thereby resulting in NFE2L2 activation in patients with NAFLD, as compared to that in samples from normal individuals. Thus, the levels of nuclear NFE2L2 and its target genes were increased in the livers of NAFLD patients (Figure 14A–E).
Figure 14.
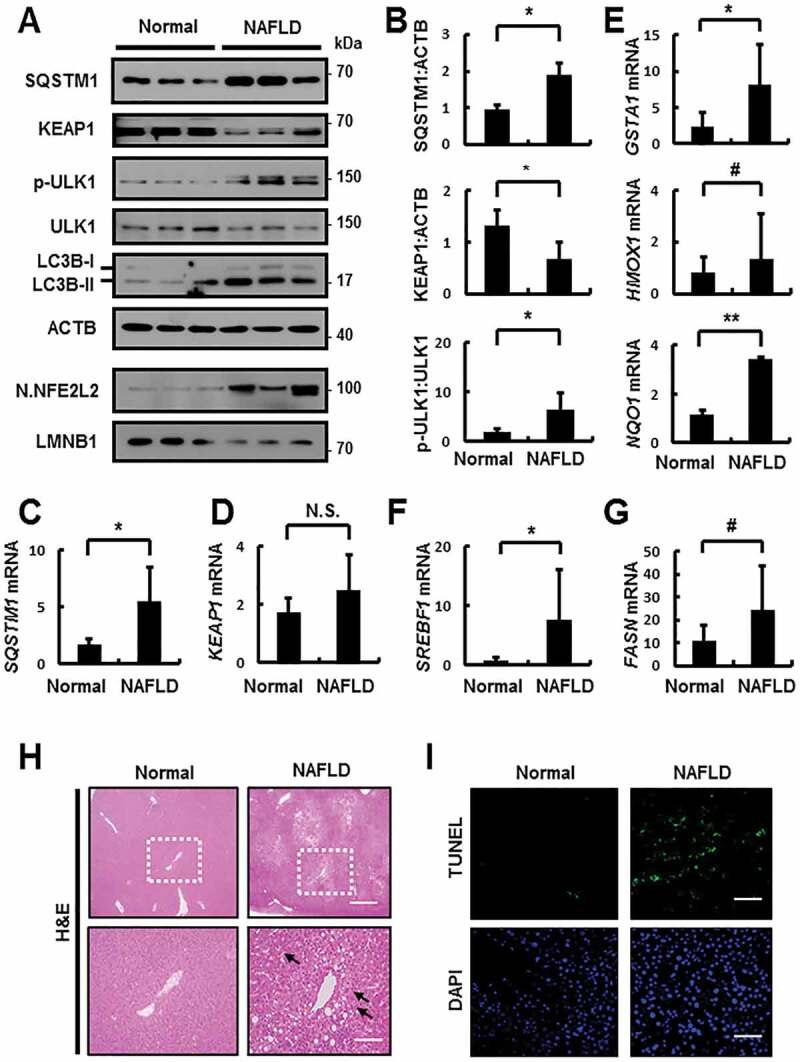
Activation of the SQSTM1-dependent noncanonical KEAP1-NFE2L2 pathway through ULK1 phosphorylation in patients with nonalcoholic fatty liver disease (NAFLD). (A) Immunoblotting for SQSTM1, KEAP1, p-ULK1(S317), ULK1, LC3B, ACTB (loading control), nuclear NFE2L2, and LMNB1 (nuclear marker) from human liver samples. (B) Densitometric analysis of SQSTM1, KEAP1, and p-ULK1:ULK1 immunoblots. (C–G) qRT-PCR analysis for relative mRNA expression of SQSTM1 (C), KEAP1 (D), GSTA1, HMOX1, NQO1 (E), SREBF1 (F), and FASN (G). (H) Representative images of H&E staining. Scale bar: 200 μm. (I) TUNEL analysis of liver sections from patients with NAFLD. Scale bar: 200 μm. Data are means ± standard errors for 3 per group. *p < 0.05, **p < 0.01, #p = 0.1, and N.S., not significant
Indeed, we investigated whether oxidative liver injury was increased in patients with NAFLD. Our results showed that oxidative stress-induced liver injury accompanied by an increase in lipogenesis-target genes was upregulated in patients with NAFLD, as compared to that in normal patients (Figure 14F,G), based on H&E staining (Figure 14H) and TUNEL analysis (Figure 14I). Thus, our findings provide important insights into the clinical significance of the SQSTM1-mediated ULK1-KEAP1-NFE2L2 pathway in the livers of patients with NAFLD.
Discussion
The complexity of and various factors involved in the pathogenesis of NAFLD make the development of effective targeted strategies for treatment extremely difficult. Recently, several studies have reported that suppression of autophagy function contributes to the pathogenesis of NAFLD [26,27]. Furthermore, the lipid accumulation-mediated damaged mitochondria lead to oxidative stress-induced cell death in the livers of NAFLD patients [28]. Consistent with these reports, our data showed that the LC3B-II:LC3B-I ratio increased in the livers of NAFLD patients compared with that in normal patients (Figure 14A). However, autophagic flux can be detected by the conversion of LC3B-I to LC3B-II following treatment with lysosomal inhibitors, but these experiments were not possible in the livers of NAFLD patients [29–31]. However, the possible contribution of autophagy to the development of NASH has not yet fully determined. Our results show that the activation of phosphorylation of the ULK1-mediated KEAP1-NFE2L2 pathway accompanied the suppression of autophagy in NAFLD patients. Thus, we suggest that this is a compensation mechanism for defense against the progression of NASH. As such, characterizations like these are key factors that must be considered when developing therapeutic target strategies [3].
SQSTM1/p62 acts as an autophagy receptor protein and activator of the noncanonical KEAP1 (kelch like ECH associated protein 1)-NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2) pathway [11,12,16]. There are multiple reported functions of SQSTM1 with respect to hepatocellular carcinomas (HCCs). Therefore, this protein contributes to the progression of this disease [12,32]. Moreover, a recent study reported that the expression of SQSTM1 in hepatic stellate cells (HSCs) inhibits liver fibrosis and liver cancer through binding to the vitamin D receptor [33]. Furthermore, SQSTM1 is aggregated as forms of Mallory-Denk bodies and intracellular hyaline bodies in alcoholic hepatitis tissue [34]. However, the role of SQSTM1 in NAFLD progression remains unclear.
Our previous results demonstrate that SQSTM1 has anti-lipotoxic functions in hepatocytes through activation of the noncanonical KEAP1-NFE2L2 pathway [14]. KEAP1 is well-known as a repressor of NFE2L2 activation through direct binding to NFE2L2, which can lead to NFE2L2 degradation under normal conditions. According to the hinge-latch mechanism, KEAP1 has binding domains for NFE2L2, including latch (low affinity, DLG motif) and hinge (high affinity, ETGE motif). However, the oxidation of KEAP1 with modification of cysteine induces canonical NFE2L2 activation in response to oxidative stress or electrophilic stress through competitively disrupting the KEAP1-NFE2L2 interaction [5,6,35,36]. This canonical mechanism might induce SQSTM1 as a NFE2L2 target gene [15]. Sequentially, the induction of SQSTM1 by canonical NFE2L2 activation can activate the noncanonical KEAP1- NFE2L2 pathway through binding to specific DLG motifs of KEAP1, resulting in interruption of the interaction between NFE2L2 and KEAP1 without cysteine modification of KEAP1, which can increase the stabilization of NFE2L2 [11]. This SQSTM1-mediated noncanonical KEAP1-NFE2L2 pathway is an effective defense mechanism against lipotoxicity [14]. However, the molecular mechanism underlying the SQSTM1-dependent activation of this pathway and its physiological relevance in response to lipotoxicity were not clearly defined.
Several studies have shown that a positive feedback loop upregulates SQSTM1 induction in an NFE2L2-dependent manner in response to oxidative stress [11,15]. Consistently, we found that noncanonical NFE2L2 activation, facilitated by the induction of SQSTM1, is dependent on reactive oxygen species (ROS)-mediated NFE2L2 activation in response to lipotoxicity. We suggest that the induction of SQSTM1 can enhance the noncanonical KEAP1-NFE2L2 pathway by disrupting the interaction between KEAP1 and NFE2L2.
Furthermore, SQSTM1 has multiple phosphorylation sites that may be targeted by various kinases, and its phosphorylation has diverse functions. The removal of ubiquitinated proteins through autophagic clearance requires the activation of TBK1 (TANK binding kinase 1)-mediated SQSTM1 phosphorylation (serine 403) [37,38] in response to lipotoxicity [39]. The phosphorylation of SQSTM1 at S351 regulates the noncanonical KEAP1-NFE2L2 pathway positively [12,22]. Based on these findings, we suggest that SQSTM1 could be a key regulator for this pathway, as well as for autophagy.
We previously reported SQSTM1 as an autophagy receptor protein required for autophagic KEAP1 degradation [14]. Here, we found that induction of SQSTM1 facilitates autophagy activation through the phosphorylation of ULK1 (unc-51 like autophagy activating kinase 1), which is followed by KEAP1 degradation in response to lipotoxic stress. Consistent with our observation, a recent study reported that increased SQSTM1 expression could activate autophagy [17]. Moreover, the formation of autophagosomes via self-oligomerization required SQSTM1 mediated by its PB1 domain [40]. Furthermore, our results showed that SQSTM1 increases the formation of autophagosomes (AP) and autolysosomes (AL) through activation of the AMPK-ULK1 axis by facilitating the enhancement of interaction between AMPK and ULK1 in response to lipotoxicity. We further found that SQSTM1 could enhance AMPK and ULK1 as well as activation of AMPK in response to lipotoxicity. This mechanism is possible to SESN2-mediated AMPK activation. Consequently, these findings suggest that SQSTM1 could activate the noncanonical KEAP1-NFE2L2 pathway through the SESN2-AMPK-ULK1 axis in response to lipotoxicity.
However, SQSTM1 could negatively regulate autophagy through activation of MTORC1 (mechanistic target of rapamycin kinase complex 1) under nutrient starvation conditions [41,42]. Conversely, autophagy activation caused by SFA is independent of MTORC1 [43]. Accordingly, we suggest that the induction of SQSTM1 activates MTORC1-independent autophagy in response to lipotoxicity. Collectively, SQSTM1 inhibits the activation of autophagy under nutrient starvation conditions.
Conversely, it could facilitate the activation of autophagy under lipotoxic stress. These phenomena reveal that multiple mechanisms regulate SQSTM1-mediated autophagy under various conditions. Based on these findings, we suggest the implication of SQSTM1-mediated autophagy activation in pathophysiological lipotoxicity. Therefore, SQSTM1 is an effective target for the treatment of NAFLD, which may be accompanied by the suppression of autophagy. In this study, we found that SQSTM1 acts as an autophagy receptor protein and can activate the initiation of this process. These findings suggest that the SQSTM1 has dual hepatoprotective roles against NAFLD progression.
The anti-lipotoxic functions of SQSTM1 in a physiological setting are unknown. Our findings show that the upregulation of SQSTM1 is accompanied by KEAP1 degradation in physiological lipotoxic conditions, resulting in NFE2L2 activation. In addition, we found that hepatic SQSTM1 activates autophagy through the activation of ULK1 in the livers of GFP-LC3B transgenic mice. Furthermore, we demonstrate that SQSTM1 has a hepatoprotective role against physiological lipotoxic stress through activation of the ULK1-KEAP1-NFE2L2 pathway in mouse livers. Indeed, we observed that the SQSTM1-dependent noncanonical KEAP1- NFE2L2 pathway is activated and can rescue Sqstm1 deletions, which protect mouse livers from lipotoxic stress in both conventional sqstm1- and liver-specific sqstm1-knockout mice.
In summary, SQSTM1-mediated ULK1 phosphorylation activates autophagy and noncanonical KEAP1-NFE2L2 pathway by facilitating the enhanced formation of an AMPK-ULK1-SQSTM1 complex (Fig. S16). Consistent with our observations, increased phosphorylation of ULK1 is associated with KEAP1 degradation and NFE2L2 activation in the livers of NAFLD patients. Together, our data show that SQSTM1 promotes activation of the noncanonical KEAP1-NFE2L2 pathway and is, therefore, a promising therapeutic target for the treatment of NAFLD.
Materials and methods
Cell culture
HEK-293 cells (Korean Cell Line Bank, 21573), mouse embryonic fibroblasts (MEFs), and green fluorescent protein (GFP)-conjugated LC3B (GFP-LC3B)-expressing HeLa (GFP-LC3B HeLa) cells were maintained in an atmosphere of 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, HS3243.01) supplemented with 10% fetal bovine serum (Hyclone, SV30087.02), 1% penicillin-streptomycin (Biowest, L0022). Hepa1c1c7 cells (Korean Cell Line Bank, 22026) were maintained in an atmosphere of 5% CO2 at 37°C in Minimum Essential Media (Welgene, LM007-01) supplemented with 10% fetal bovine serum, 1% penicillin, and streptomycin. Sqstm1 MEFs were kindly provided by Dr. J. Shin (Samsung Biomedical Research Institute, Sungkyunkwan University School of Medicine, Suwon, Republic of Korea) and Dr. Komatsu (Department of Biochemistry, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan; Department of Physiology, Juntendo University Graduate School of Medicine, Tokyo, Japan). Atg5 MEFs were provided by Dr. D.S.Min (Department of Molecular Biology, College of Natural Science, Pusan National University, Pusan, Republic of Korea) and Dr. Mizhushima (Department of Biochemistry and Molecular Biology, Graduate School and Faculty of Medicine, The University of Tokyo, Tokyo, Japan). Prkaa1/2 MEFs were provided by Dr. J.Ha (Department of Biochemistry and Molecular Biology, Graduate School, Kyung Hee University, Seoul, Republic of Korea). Sesn2 MEFs were provided by Dr. S.G.Rhee (Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Republic of Korea). Ulk1 MEFs were provided by Dr. J.Kim (Department of Oral Biochemistry and Molecular Biology, School of Dentistry, Kyung Hee University, Seoul, Republic of Korea). GFP-LC3B HeLa cells were provided by Dr. J.Min (Department of Biochemistry and Biomedical Sciences, Seoul National University, College of Medicine, Seoul, Republic of Korea).
Small interfering RNA (siRNA) transfection
GFP-LC3B HeLa cells were transfected with siRNAs targeting SQSTM1 (GGACCCAUCUGUCUUCAAATT), ULK1 (AAGGACCGCAUGGACUUUGAU), and PRKAA1 (Santa Cruz Biotechnology, sc-45312) or control siRNA. Hepa1c1c7 cells were transfected with Sesn2 (GCAUCAGAUACGAUGACUATT) or control siRNA. The cells were transfected with siRNAs using Lipofectamine RNA/iMAX following the manufacturer’s instructions (Invitrogen, P/N 56532).
Immunoprecipitation (IP) and immunoblot analysis
For immunoprecipitation, the cells were lysed in lysis buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma Aldrich, 10837091001), 10% glycerol (Duchefa Biochemie, G1345.1000), 0.1% Triton X-100 (Sigma Aldrich, T8787), 1 mM ethylenediaminetetraacetic acid (EDTA; Sigma Aldrich, 03690), 0.5% IGEPAL® CA-630 (Sigma Aldrich, 56741), 10 mM β-glycerophosphate (Sigma Aldrich, G9422), 1 mM Na3V04 (Sigma Aldrich, 450243), 5 mM NaF (Sigma Aldrich, 201154), and 1 mg/ml of aprotinin (Sigma Aldrich, 10236624001) and leupeptin (Sigma Aldrich, L8511). The cell lysates were centrifuged, and the resulting supernatants were subjected to IP with antibodies against FLAG, HA, or MYC using protein G-Sepharose beads (GE Healthcare, 17-0618-01). For immunoblot analysis, cell lysates or immunoprecipitates were subjected to sodium dodecyl sulfate polyacrylamide (SDS-PAGE) gel electrophoresis; the separated proteins were transferred to polyvinylidene difluoride membranes (PVDF; Merk Millipore, L-IPVH 00010), which were then incubated with primary antibodies at 4°C for overnight, followed by incubation with horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h. The proteins were visualized with enhanced chemiluminescence lightning solution (Thermo Fisher Scientific, 34580).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) analysis
To analyze apoptosis in mouse livers and cells, a Click-iT Plus TUNEL assay kit (Promega Coporation, G3250) was used according to the manufacturer’s instructions. The fluorescence signals were detected with a confocal microscope (Carl Zeiss, LSM700). The frequency of apoptotic cells in liver sections was quantified by determining the percentage of TUNEL-positive cells in five random microscopic fields per specimen.
Cell cytotoxicity assay and treatment of palmitic acid (PA)
Cells were seeded at a density of 2 × 103 cells/well in a final volume of 100 μL in 96-well plates. After 24 h, the cells were treated with bovine serum albumin (BSA; MPBiomedicals, 160,069) or palmitic acid (PA; Sigma Aldrich, P0500) with for 18 h. The cells were treated with PA, as previously described [21,44]. Cell viability was estimated using the CellTiter-Glo Luminescent cell viability assay kit (Promega Corporation, G7570) according to the manufacturer’s protocol. PA was dissolved in isopropyl alcohol at a stock concentration of 160 mM. This solution was added to DMEM containing 1% BSA to ensure a physiological ratio between bound and unbound FFA in the medium [14,44].
Measurement of reactive oxygen species (ROS)
Intracellular ROS generation was measured by 5,6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA; Molecular Probes, C6827). The cells (3 × 105) were plated in 35 mm dishes. After 18 h, the cells were treated with BSA or PA, rinsed once with 2 ml Hanks balanced salt solution (HBSS) (Biowest, L0607), and incubated for 5 min with CM-H2DCFDA. The cells were then washed again with HBSS, and images were obtained using a fluorescence microscope (Carl Zeiss, Axiovert 200 Basic standard). The relative dichlorofluorescein (DCF) fluorescence was calculated by averaging the levels of fluorescence from 80 to100 cells after subtracting the background fluorescence.
Immunofluorescence analysis
GFP-LC3B HeLa cells were cultured in 12 well plates. Cells were washed with dulbecco’s phosphate-buffered saline (DPBS; Biowest, X0515-500), fixed with 4% paraformaldehyde (Biosesang, P2031) for 10 min, and washed 3 times with PBS. To analyze autolysosomes, Hepa1c1c7 cells and MEFs were stained with LysoTracker™ Green DND-26 (Thermo Fisher Scientific, L7526) used according to the manufacturer’s instructions. The fluorescence signals were detected with a confocal microscope (Carl Zeiss, LSM 700).
Subcellular fractionation
Cells or liver tissues were lysed in cytoplasmic extraction reagents containing 10 mM HEPES (bioWORLD, 40820042-1), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT) (Sigma Aldrich, 10197777001), 0.05% 0.5% IGEPAL® CA-630 (Sigma Aldrich, 56741), PMSF (Sigma Aldrich, 10837091001), and 1 mg/mL aprotinin and leupeptin. The lysates were centrifuged (900 × g for 10 min), and the resulting pellet was resuspended on ice in nuclear extraction reagents containing 5 mM HEPES, pH 8.0, 0.2 mM EDTA, 1.5 mM MgCl2, 0.5 mM DTT, 26% glycerol. The lysates were then centrifuged (24,000 × g for 20 min) and the resulting supernatants were subjected to immunoblot analysis.
Animals and in vivo transfection
sqstm1 knockout (KO) mice in a C57BL/6 background and littermates of Sqstm1 WT and sqstm1 KO mice that were used for the experiments were kindly provided by Dr. J. Shin (Samsung Biomedical Research Institute, Sungkyunkwan University School of Medicine, Suwon, Republic of Korea) and Dr. J.W. Ryu (Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Republic of Korea) [45]. Male C57BL/6J mice at 7 weeks of age were purchased from Japan SLC, Inc. (SLC-M-0133). Alb-Cre transgenic mice of a C57BL/6 background (provided by Dr. Y.H. Lee; Division of Endocrinology and Metabolism, Department of Internal Medicine, Yonsei University College of Medicine, Seoul, Republic of Korea) were bred with Sqstm1 floxed (Sqstm1f/f) mice of a C57BL/6 background provided by Dr. Yanagawa (Department of Oral and Maxillofacial Suregy, Faculty of Medicine, Tsukuba University, Tsukuba-shi, Japan) and M.S.Lee (Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Republic of Korea) [46,47]. GFP-LC3B transgenic mice that were used for the experiments were provided by Dr. Y.H. Lee and J.W. Ryu [48,49]. nfe2l2 KO mice were obtained from RIKEN BioResoruce Center (RBRC00984) permitted by Dr. M. Yamamoto (Department of Medical Biochemistry, Tohoku University Graduate School of Medicine, Sendai, Japan) and provided by Dr. S. Rhee (Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Republic of Korea) and W. Jeong (Department of Life Science and Research Center for Cellular Homeostasis, Ewha Woman’s University, Seoul, Republic of Korea). Mice were fed a normal chow diet (LabDiet, 5053) without fasting and a high-carbohydrate diet (HCD; Dyets, 102235) after 24 h of fasting, and infected with the Ad-CON and Ad-SQSTM1 vector. Ad-SQSTM1 was a generous gift from M. Komatsu (Department of Biochemistry, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan; Department of Physiology, Juntendo University Graduate School of Medicine, Tokyo, Japan). Recombinant adenovirus (2 × 109 plaque-forming units) was delivered by tail vein injection to mice. Other mice were fed a chow diet or high-fat diet (HFD; Research Diets, D12492) for 12 weeks. These animals were randomly assigned to 1 to 4 groups (3–9 mice in each group). All mice had free access to water and food in rooms maintained at 23 ± 2°C with a 12-h light/12-h dark cycle and 50–70% humidity. After 12 h of refeeding, mice were sacrificed. GPT/ALT (glutamic pyruvic transaminase, soluble) levels were quantified by colorimetric determination using an activity assay kit (FUJIFILM, 3250). All animal experiments were approved by the Animal Care and Use Committee of the Yonsei University College of Medicine.
Histological analysis
Liver tissues of mice were fixed in 10% neutral-buffered formalin solution, embedded in paraffin, and sectioned (5 μm thickness). Liver sections were subjected to hematoxylin and eosin (H&E) staining and TUNEL analysis, as indicated.
Human NAFLD samples
Liver specimens were collected from 6 samples (3 histologically normal and 3 simple NAFLD) from patients who underwent hepatomy due to metastatic lesions in the liver. Only remnant non-tumor areas were isolated and used for further analysis. Inclusion criteria for patients with NAFLD were based on pathologic diagnosis [22,48]. The protocol for this study was approved by the Institutional Review Board at Severance Hospital (IRB No 4-2014-0674), and all patients provided informed consent for participation in the study.
Antibodies and reagents
The following antibodies were used: anti-KEAP1 (Proteintech, 10503-2-AP); anti-ACTB (Cell Signaling Technology, sc-47778); FLAG (Sigma Aldrich, F1804); anti-p-ULK1 (S317; Cell Signaling Technology, 12753S); anti-p-ULK1 (S555; Cell Signaling Technology, 5869S), anti-p-ULK1 (S757; Cell Signaling Technology, 14202S), anti-ULK1 (Sigma Aldrich, A7481), anti-SQSTM1 (Abnova, H00008878-M01), anti-LC3B (Novus, NB-100-2220), anti-SESN2 (Proteintech, 10795-1-AP), anti-MYC (Merk Millipore, 05–419), anti-cleaved CASP3 (Cell Signaling Technology, 9661S), anti-cleaved PARP (Cell Signaling Technology, 9544S), anti-NFE2L2 (Santa Cruz Biotechnology, sc-13032), anti-LMNB1 (Proteintech, 12987-1-AP), anti-HA (Bethyl Laboratory, A190-108A), anti-p-PRKAA (T172; Cell Signaling Technology, 2531S), anti-PRKAA (Cell Signaling Technology, 2532S), anti-p-ACACA(S79; Cell Signaling Technology, 3661S), anti-ACACA (Cell Signaling Technology, 3662S), and anti-GFP (Santa Cruz Biotechnology, sc-9996). Palmitic acid (PA; Sigma Aldrich, P0500), dimethyl sulfoxide (DMSO; Sigma Aldrich, 67-68-5), bafilomycin A1 (BafA1; Sigma Aldrich, B1793), chloroquine (CQ; Sigma Aldrich, C6628), MG132 (Sigma Aldrich, C8699), and N-acetyl-L-cysteine (NAC; Sigma Aldrich, A7250), compound C (Sigma Aldrich, 171260) were purchased from Sigma Aldrich. Flag-tagged SQSTM1 adenovirus vectors (3 × 108 plaque-forming units) was a generous gift from M. Komatsu, and used to infect cells and mice for 24 h. SBI-0206965 (Selleck Chemicals, S7885) was purchased from Selleck Chemicals.
Quantitative RT-PCR analysis
Total RNA was prepared from cultured cells using TRIzol® reagent (MRC, TR 118) and was treated with RNase (Sigma Aldrich, R6148). Some RNA (1 μg) was then subjected to reverse transcription with random-hexamer primers and a TAKARA cDNA synthesis kit (TaKaRa, RR036A). The resulting cDNA was subjected to quantitative PCR analysis with SYBR® Green (ABI, 4367659) and mouse-specific primer pairs (forward and reverse). The sequences of the primers for mouse cDNAs were as follows:
Keap1, 5′-GGCAGGACCAGTTGAACAGT-3′ and 5′-GGGTCACCTCACTCCAGGTA-3′; Hmox1, 5′-GAGCAGAACCAGCCTGAACTA-3′ and 5′-GGTACAAGGAAGCCATCACCA-3; Gsta1, 5′- TGCCCAATCATTTCAGTCAG-3′ and 5′-CCAGAGCCATTCTCAACTA-3′; Nqo1, 5′-TTCTCTGGCCGATTCAGAG-3′ and 5′- GGCTGCTTGGAGCAAAATAG-3′; Sqstm1, 5ʹ- CCGCCGCTTCAGCTTCTGCT-3′ and 5ʹ- GTTCCCGCCGGCACTCCTTC-3′; Atg5, 5′- ACTTGCTTTACTCTCTATCA-3′ and 5′-CATCTTCTTGTCTCATAACC-3′; Nfe2l2, 5′- TCTCCTCGCTGGAAAAAGAA-3′ and 5′-AATGTGCTGGCTGTGCTTTA-3′; Srebf1, 5′- GGAGCCATGGATTGCACATT-3′ and 5′- GGCCCGGGAAGTCACTGT-3′; Fasn, 5′-GCTGCGGAAACTTCAGGAAAT-3′ and 5′-AGAGACGTGTCACTCCTGGACTT-3′; Acaca, 5′-TGGACAGACTGATCGCAGAGAAAG-3′ and 5′-TGGAGAGCCCCACACACA-3′. Rn18S, 5′-CGCTCCCAAGATCCAACTAC-3′ and 5ʹ-CTGAGAAACGGCTACCACATC-3ʹ; Rn18S ribosomal RNA was used as an internal control. The sequences of the primers for human cDNA were as follows:
KEAP1, 5′- TGGCCAAGCAAGAGGAGTTC-3′ and 5′- GGCTGATGAGGGT CACCAGTT-3′; HMOX1, 5′- GCCAGGTGCTCAAAAAGATT-3′ and 5′- CCTGCAACTCCTCAAAAGAGC-3; GSTA1, 5′- GATTTGTTTTCATTAGGATCTGA-3′ and 5′- CATGGAGAAGATTGGAAATCTGAAT-3′; NQO1, 5ʹ- GGGCAAGTCCATCCCAACTG-3ʹ and 5ʹ- GCAAGTCAGGGAAGCCTGGA-3ʹ; SQSTM1, 5′- CAGAGAAGCCCATGGACAG-3′ and 5′- AGCTGCCTTGTACCCACATC-3′; SREBF1, 5′-AAACTCAAGCAGGAGAACCT-3′ and 5′-GTCAGTGTGTCCTCCACCTC-3′; FASN, 5′-TCGTGGGCTACAGCATGGT-3′ and 5′-GCCCTCTGAAGTCGAAGAAG-3′; GAPDH, 5′- CGACCACTTTGTCAAGCTCA-3′ and 5ʹ-AGGGGAGATTCAGTGTGGTG-3ʹ; GAPDH ribosomal RNA was used as an internal control.
Fatty acid profiling
Liver tissue (10 mg) from each aliquot was added to 1 mL of 7% methanolic HCl (Sigma-Aldrich) with 5 µg of nonadecanoic acid (C19:0, Sigma-Aldrich) as an internal standard [50]. Tissue samples were put on dry-ice and then homogenized by ultrasonication (30% amplitude; 4 s pulse/1 s pause) for 30 min using a Vibra-cell ultrasonic liquid processor (VCX130, Sonics & Materials, Inc.) [51]. Lipid hydrolysis and acidic transmethylation of fatty acids in homogenates were carried out at 100°C for 2 h. After cooling to room temperature, methylated fatty acids were extracted with 1 mL of hexane (3 repeats). The collected organic layer was evaporated under nitrogen and reconstituted in 100 µL of hexane. The fatty acids were then quantified using GC-MS (GCMS-QP2010, Shimadzu) equipped with a DB-5 ms capillary column (30 m × 0.25 mm, 0.25 µm, Agilent). The injection volume was 1 µL at 1:2 split mode and helium gas at a constant rate of 1 mL/min was used as a carrier gas. Injection temperature was 270°C and the column temperature program for separation was as follows: an initial temperature at 70°C for 1 min, an increase to 150°C at 20°C/min, an increase to 180°C at 6°C/min, an increase to 220°C at 20°C/min, a hold for 1 min, an increase to 240°C at 4°C/min, and a hold for 17 min, for a total of 35 min. Electron impact with 70 eV and a 200°C source temperature was used for compound ionization. The mass detection range was 40 to 500 m/z, with a scan rate of 2,500/s. For identification, the retention index was measured using an alkane mixture (C7-C40, Sigma-Aldrich) and the NIST mass spectral library (NIST08) was compared to the spectrum of each peak. The peak area of fatty acids was integrated using a total ion chromatogram and normalized based on that of the internal standard.
Statistical analysis
Data were analyzed using a two-tailed Student’s t-tests for comparisons between 2 groups or one-way analysis of variance with Tukey’s honest significant difference post-hoc tests for multiple comparisons (SPSS 21.0K for Windows; SPSS, Chicago, IL, USA) to determine statistical significance. Differences with P values less than 0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank J. Shin, and M. Komatsu for providing the Sqstm1 MEFs and the plasmid encoding human SQSTM1. FLAG-, HA-tagged ULK1 cDNAs and Ulk1 MEFs were kindly provided by J Kim. We thank D. S. Min and Mizhushima for providing the Atg5 MEFs. Green fluorescent protein (GFP)-conjugated LC3B (GFP-LC3B)-expressing HeLa (GFP-LC3B HeLa) cells were kindly provided by J.Min. We thank J. Ha for providing the Prkaa1/2 MEFs. FLAG-tagged KEAP1 cDNA and Nfe2l2 MEFs were kindly provided by M. Komatsu. Sesn2 MEFs and FLAG-tagged SESN2 cDNAs were kindly provided by S.G.Rhee. We thank T. Yanagawa and M.S.Lee for providing the Sqstm1 flox mouse. We thank to M. Komatsu, S.G. Rhee, S.H. Sung, S.Y. Oh, M.J. Lee, H.W. Ji, and E.J. Oh for providing encouragement and comments.
Funding Statement
This work was supported by the National Research Foundation of Korea (NRF-2017R1A2B4007400 to S.H. Bae; NRF-2017R1D1A1B03032808 to J.S. Park) and a Faculty Research Grant from the Yonsei University College of Medicine (6-2019-0068 to S.H. Bae). The study was also supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) (HI17C0913 and HI16C0257 to S.H.Bae).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Browning JD, Horton JD.. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest.2004 Jul;114(2):147–52. PubMed PMID: 15254578; PubMed Central PMCID: PMCPMC449757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fuchs M, Sanyal AJ.. Lipotoxicity in NASH. J Hepatol. 2012. January;56(1):291–293. PubMed PMID: 21741924. [DOI] [PubMed] [Google Scholar]
- [3].Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. 2016. April;15(4):249–274. PubMed PMID: 26794269. [DOI] [PubMed] [Google Scholar]
- [4].Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial oxidative stress and antioxidants balance in fatty liver disease. Hepatol Commun. 2018. December;2(12):1425–1439. PubMed PMID: 30556032; PubMed Central PMCID: PMCPMC6287487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. PubMed PMID: 16968214. [DOI] [PubMed] [Google Scholar]
- [6].Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011. February;16(2):123–140. PubMed PMID: 21251164. [DOI] [PubMed] [Google Scholar]
- [7].Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009. April;34(4):176–188. PubMed PMID: 19321346. [DOI] [PubMed] [Google Scholar]
- [8].Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010. December 1;13(11):1665–1678. PubMed PMID: 20446768. [DOI] [PubMed] [Google Scholar]
- [9].Silva-Islas CA, Maldonado PD. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol Res. 2018. August;134:92–99. PubMed PMID: 29913224. [DOI] [PubMed] [Google Scholar]
- [10].Musso G, Cassader M, Rosina F, et al. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia. 2012. April;55(4):885–904. PubMed PMID: 22278337. [DOI] [PubMed] [Google Scholar]
- [11].Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010. March;12(3):213–223. PubMed PMID: 20173742. [DOI] [PubMed] [Google Scholar]
- [12].Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013. September 12;51(5):618–631. PubMed PMID: 24011591. [DOI] [PubMed] [Google Scholar]
- [13].Bae SH, Sung SH, Oh SY, et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013. January 8;17(1):73–84. PubMed PMID: 23274085. [DOI] [PubMed] [Google Scholar]
- [14].Park JS, Kang DH, Lee DH, et al. Concerted action of p62 and Nrf2 protects cells from palmitic acid-induced lipotoxicity. Biochem Biophys Res Commun. 2015. October 9;466(1):131–137. PubMed PMID: 26325428. [DOI] [PubMed] [Google Scholar]
- [15].Jain A, Lamark T, Sjottem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010. July 16;285(29):22576–22591. PubMed PMID: 20452972; PubMed Central PMCID: PMCPMC2903417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. Febs J. 2015. December;282(24):4672–4678. PubMed PMID: 26432171. [DOI] [PubMed] [Google Scholar]
- [17].Caccamo A, Ferreira E, Branca C, et al. p62 improves AD-like pathology by increasing autophagy. Mol Psychiatry. 2017. June;22(6):865–873. PubMed PMID: 27573878; PubMed Central PMCID: PMCPMC5479312. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [18].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011. February;13(2):132–141. PubMed PMID: 21258367; PubMed Central PMCID: PMCPMC3987946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Egan D, Kim J, Shaw RJ, et al. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011. June;7(6):643–644. PubMed PMID: 21460621; PubMed Central PMCID: PMCPMC3359466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Laker RC, Drake JC, Wilson RJ, et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017. September 15;8(1):548. PubMed PMID: 28916822; PubMed Central PMCID: PMCPMC5601463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Park JS, Lee DH, Lee YS, et al. Dual roles of ULK1 (unc-51 like autophagy activating kinase 1) in cytoprotection against lipotoxicity. Autophagy. 2020 Jan;16(1):86–105. PubMed PMID: 30907226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee DH, Han DH, Nam KT, et al. Ezetimibe, an NPC1L1 inhibitor, is a potent Nrf2 activator that protects mice from diet-induced nonalcoholic steatohepatitis. Free Radic Biol Med. 2016. October;99:520–532. PubMed PMID: 27634173. [DOI] [PubMed] [Google Scholar]
- [23].Lee JH, Budanov AV, Talukdar S, et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012. September 5;16(3):311–321. PubMed PMID: 22958918; PubMed Central PMCID: PMCPMC3687365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008. August 8;134(3):451–460. PubMed PMID: 18692468; PubMed Central PMCID: PMCPMC2758522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McCurdy CE, Bishop JM, Williams SM, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest. 2009. February;119(2):323–335. PubMed PMID: 19147984; PubMed Central PMCID: PMCPMC2631287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yan S, Huda N, Khambu B, et al. Relevance of autophagy to fatty liver diseases and potential therapeutic applications. Amino Acids. 2017. December;49(12):1965–1979. PubMed PMID: 28478585; PubMed Central PMCID: PMCPMC5759960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011. April;5(2):159–166. PubMed PMID: 21476911; PubMed Central PMCID: PMCPMC3104297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Masarone M, Rosato V, Dallio M, et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid Med Cell Longev. 2018;2018:9547613. PubMed PMID: 29991976; PubMed Central PMCID: PMCPMC6016172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gonzalez-Rodriguez A, Mayoral R, Agra N, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014. April 17;5:e1179. PubMed PMID: 24743734; PubMed Central PMCID: PMCPMC4001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xie F, Jia L, Lin M, et al. ASPP2 attenuates triglycerides to protect against hepatocyte injury by reducing autophagy in a cell and mouse model of non-alcoholic fatty liver disease. J Cell Mol Med. 2015. January;19(1):155–164. PubMed PMID: 25256142; PubMed Central PMCID: PMCPMC4288359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010. February 5;140(3):313–326. PubMed PMID: 20144757; PubMed Central PMCID: PMCPMC2852113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Saito T, Ichimura Y, Taguchi K, et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun. 2016. June 27;7:12030. PubMed PMID: 27345495; PubMed Central PMCID: PMCPMC4931237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Duran A, Hernandez ED, Reina-Campos M, et al. p62/SQSTM1 by binding to vitamin D receptor inhibits hepatic stellate cell activity, fibrosis, and liver cancer. Cancer Cell. 2016. Oct 10;30(4):595–609. PubMed PMID: 27728806; PubMed Central PMCID: PMCPMC5081228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Taniguchi K, Yamachika S, He F, et al. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016. August;590(15):2375–2397. PubMed PMID: 27404485; PubMed Central PMCID: PMCPMC4983218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tong KI, Kobayashi A, Katsuoka F, et al. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006. Oct–Nov;387(10–11):1311–1320. PubMed PMID: 17081101. [DOI] [PubMed] [Google Scholar]
- [36].Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999. January 1;13(1):76–86. PubMed PMID: 9887101; PubMed Central PMCID: PMCPMC316370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Matsumoto G, Wada K, Okuno M, et al. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011. October 21;44(2):279–289. PubMed PMID: 22017874. [DOI] [PubMed] [Google Scholar]
- [38].Pilli M, Arko-Mensah J, Ponpuak M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012. August 24;37(2):223–234. PubMed PMID: 22921120; PubMed Central PMCID: PMCPMC3428731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cho CS, Park HW, Ho A, et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated p62/sequestosome 1 phosphorylation. Hepatology. 2018 Oct; 68(4):1331–1346. PubMed PMID: 29251796; PubMed Central PMCID: PMCPMC6005718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011. January 10;192(1):17–27. PubMed PMID: 21220506; PubMed Central PMCID: PMCPMC3019556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011. October 7;44(1):134–146. PubMed PMID: 21981924; PubMed Central PMCID: PMCPMC3190169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res. 2012. December;66(6):457–462. PubMed PMID: 22841931. [DOI] [PubMed] [Google Scholar]
- [43].Tan SH, Shui G, Zhou J, et al. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J Biol Chem. 2012. April 27;287(18):14364–14376. PubMed PMID: 22408252; PubMed Central PMCID: PMCPMC3340233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cazanave SC, Mott JL, Bronk SF, et al. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem. 2011. November 11;286(45):39336–39348. PubMed PMID: 21941003; PubMed Central PMCID: PMCPMC3234758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kwon J, Han E, Bui CB, et al. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 2012. February 1;13(2):150–156. PubMed PMID: 22222206; PubMed Central PMCID: PMCPMC3271336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kageyama S, Sou YS, Uemura T, et al. Proteasome dysfunction activates autophagy and the Keap1-Nrf2 pathway. J Biol Chem. 2014. September 5;289(36):24944–24955. PubMed PMID: 25049227; PubMed Central PMCID: PMCPMC4155663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Harada H, Warabi E, Matsuki T, et al. Deficiency of p62/Sequestosome 1 causes hyperphagia due to leptin resistance in the brain. J Neurosci. 2013. September 11;33(37):14767–14777. PubMed PMID: 24027277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kim SH, Kim G, Han DH, et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy. 2017. October 3;13(10):1767–1781. PubMed PMID: 28933629; PubMed Central PMCID: PMCPMC5640190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mizushima N, Kuma A. Autophagosomes in GFP-LC3 transgenic mice. Methods Mol Biol. 2008;445:119–124. PubMed PMID: 18425446. [DOI] [PubMed] [Google Scholar]
- [50].Ichihara K, Fukubayashi Y. Preparation of fatty acid methyl esters for gas-liquid chromatography. J Lipid Res. 2010. March;51(3):635–640. PubMed PMID: 19759389; PubMed Central PMCID: PMCPMC2817593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huang Q, Tan Y, Yin P, et al. Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res. 2013. August 15;73(16):4992–5002. PubMed PMID: 23824744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.