

ABSTRACT
There is an increasing unmet need for successful immunotherapeutic interventions. Lymphocyte extravasation via tumor tissue endothelial cells (TECs) is required for lymphocyte infiltration into tumor sites. This study aimed to investigate the clinical significance of dysfunctional TECs in pancreatic ductal adenocarcinoma (PDAC) and identify chemical compounds that boost tumor-infiltrating lymphocyte (TIL) numbers. We performed immunohistochemical detection and clinicopathological analysis of VCAM-1 on TECs, which is essential for lymphocyte trafficking. We characterized the gene expression profiles of TECs from fresh PDAC tissues. We isolated compounds that upregulated VCAM-1 and E-selectin expression in TECs and examined their biological activities. Compared to endothelial cells from chronic pancreatitis tissue, TECs showed significantly lower VCAM-1 and E-selectin expression and significant weaknesses in lymphocyte adhesion and transmigration, resulting in decreased T cell infiltration around vessels. Patients with a relatively high percentage of VCAM-1+ vessels among all vessels in PDAC tissue had an improved prognosis. A bioinformatics survey demonstrated that TNFR1 signaling was involved in abnormal VCAM-1 and E-selectin expression in TECs. We screened compounds affecting TNFR1 signaling, and the IAP inhibitor, Embelin, induced these molecules on TECs and enhanced T cell adhesion to TECs and transmigration. Furthermore, in vivo, Embelin enhanced tumor-infiltrating T cell numbers, leading to an antitumor immune response. Embelin accelerates TIL infiltration and the antitumor immune response by recovering VCAM-1 expression in TECs. Our strategy may be a therapeutic approach for accelerating the immunotherapeutic response in immune-quiescent tumors, leading to clinical trials’ success.
KEYWORDS: pancreas cancer, tumor endothelial cells, lymphocyte infiltration, compound screening, immunotherapy
Introduction
Multiple mechanisms contribute to the heterogeneity of the immune response in the tumor microenvironment, leading to resistance to treatments.1–3 The immunological statuses of the tumor microenvironment can be largely classified into two categories: the immune-active ‘hot’ tumor status and immune-quiescent ‘cold’ tumor status. By this classification, pancreatic ductal adenocarcinoma (PDAC) has been reported to be a typical immune-quiescent tumor, and the efficacy of immunotherapies has been limited .4–6
To characterize the treatment-resistant immune microenvironment of PDAC, we performed two large-scale cohort studies in which elevated levels of tumor-infiltrating lymphocytes (TILs) and the formation of intratumoral lymphoid organs were significantly associated with prolonged survival .7,8 Based on these results and considering that the cancer vasculature acts as an immune gatekeeper in neoplastic tissue, we next focused on the endothelial cells in the tumor tissue (TECs). Through the endothelium, lymphocytes access and infiltrate into tumor tissues to generate a successful cellular immune response. Nevertheless, TECs possess a distinct resistance to effector lymphocyte infiltration. It has been reported that TECs express molecules that accelerate and select for regulatory T cell extravasation and infiltration into tumor tissues .9,10 Also, TECs express apoptosis-inducing molecules to kill T lymphocytes .11 Additionally, TECs aberrantly express lymphocyte adhesion/homing-associated molecules, leading to abrogated immune recognition of tumor cells. Here, it was hypothesized that the pharmacological reprogramming of TECs might induce therapeutic T cell infiltration and produce a dynamic change in the tumor immune microenvironment.
For the first time in this study, we showed an integrated compound repositioning approach to enhance lymphocyte infiltration and identify possible agents to produce a beneficial immune response.
Materials and methods
Patients and samples
This study was approved by the Institutional Review Board of the National Cancer Center, Japan. Informed consent was obtained from all the participants involved in the study, and all clinical investigations were conducted in line with the principles of the Declaration of Helsinki. Clinical and pathological data and the specimens used for immunohistochemical analysis were obtained through a detailed retrospective review of the medical records of 104 patients with PDAC: 104 patients who had undergone surgical resection between 2006 and 2008 at the National Cancer Center Hospital, Tokyo, and had available fresh-frozen tissues from surgically resected specimens were included in the study. None of the patients had received any therapy before surgery. All patients included in this study underwent macroscopic curative resection, and all cases involved conventional ductal carcinomas. The clinicopathological characteristics of the patients are summarized in Table 1. The median follow-up period after surgery for the patients as a whole and that for the living patients were 21.1 (3.6–109.2) and 39.85 (5.4–109.2) months, respectively. Each patient received follow-up care in the outpatient clinic every one to three months during the first postoperative year and every six to twelve months thereafter. Unless there was confirmation of disease relapse, the patients underwent physical examination, laboratory tests, chest radiography, abdominal computed tomography, and/or ultrasonography. The levels of the tumor markers carcinoembryonic antigen and carbohydrate antigen 19–9 were also measured until relapse. Recurrence was suspected when a new local or distant metastatic lesion was found on serial images and an increase in tumor marker levels was recognized. When disease progression was confirmed by repeated imaging studies, the date of the first suspicious radiological finding was used as the date of initial disease recurrence. At the census date (September 2017), we checked whether the patients were dead or alive; 27 patients (26.0%) were alive, 72 (69.2%) had died of pancreatic cancer, and 5 (4.8%) had died of other causes. All M1 (based on TNM classification12) patients had nodal metastasis around the abdominal aorta without any other form of metastasis.
Table 1.
Relationship between clinicopathological characteristics and the ratio of vessels composed of VCAM-1 expressing endothelial cells in total vessels (%VCAM1+V) in pancreatic ductal adenocarcinoma tissues (n = 104)
| |
|
|
%VCAM1+V |
|
|
|---|---|---|---|---|---|
| Characteristics | Total | High | Low | P | |
| Age, years | |||||
| <65 | 54 | 31 | 23 | ||
| ≥65 | 50 | 21 | 29 | 0.169 | |
| Sex | |||||
| Male | 64 | 29 | 35 | ||
| Female | 40 | 23 | 17 | 0.314 | |
| Pathologic tumor status | |||||
| T1, T2 | 61 | 34 | 27 | ||
| T3, T4 | 43 | 18 | 25 | 0.232 | |
| Pathologic node status | |||||
| N0 | 24 | 14 | 10 | ||
| N1, N2 | 80 | 38 | 42 | 0.486 | |
| Pathologic metastasis status | |||||
| M0 | 89 | 45 | 44 | ||
| M1 | 15 | 7 | 8 | 1.00 | |
| Tumor histological grade | |||||
| G1 | 14 | 10 | 4 | ||
| G2, G3 | 90 | 42 | 48 | 0.149 | |
| Tumor margin status | |||||
| Negative | 50 | 28 | 22 | ||
| Positive | 54 | 24 | 30 | 0.327 | |
| Nerve plexus invasion* | |||||
| Absence | 21 | 17 | 4 | ||
| Presence | 83 | 35 | 48 | 0.003 | |
| Lymphatic invasion* | |||||
| 0, 1 | 27 | 14 | 13 | ||
| 2, 3 | 77 | 38 | 39 | 1.00 | |
| Venous invasion* | |||||
| 0, 1 | 24 | 15 | 9 | ||
| 2, 3 | 80 | 37 | 43 | 0.244 | |
| Intrapancreatic neural invasion* | |||||
| 0, 1 | 31 | 21 | 10 | ||
| 2, 3 | 73 | 31 | 42 | 0.031 | |
| Adjuvant chemotherapy | |||||
| Presence | 20 | 9 | 11 | ||
| Absence | 84 | 43 | 41 | 0.804 | |
| Total | 52 | 52 | |||
*: Classified according to the classification of pancreatic carcinoma of Japan Pancreas Society.
Bold letters: significant difference.
Primary culture of TEC
Tumor tissue samples were obtained from PDAC patients who underwent surgical resection at the National Cancer Center Hospital, Tokyo. Fresh tissue samples were obtained from tumor or nontumor areas, as determined by a certified surgical pathologist, and used for primary culture of TECs. The tissue samples were minced into approximately 10-mm3 pieces with scissors and digested with 1% type I collagenase (Wako, Osaka, Japan) for 15–20 min. Then, the tissue samples were passed through a metal mesh. The filtered cells were centrifuged, and the supernatant was discarded. The cell pellet was dissociated in phosphate-buffered saline (PBS), extensively pipetted, and filtered through a nylon cell strainer with a 100-µM pore size (BD Biosciences, San Jose, CA). CD45+ cells including macrophages were removed by adding CD45 Dynabeads (Invitrogen/Life Technologies, Carlsbad, CA) to the dissociated cells. Based on the endothelial marker statuses of TECs in the PDAC tissue samples (Supplementary Figure S1), CD31 showed mostly diffuse expression in the TECs, so we selected it for TEC isolation. Next, CD31 Dynabeads (Invitrogen/Life technologies) were added to the precleaned cell suspension and incubated for 20 min at room temperature with rotation. The target cells that interacted with the beads were magnetically isolated and extensively washed at least 5 times with PBS to minimize cellular contamination due to nonspecific binding. The washed beads and adhered cells were dissociated on fibronectin-coated dishes with endothelial culture medium (ECM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin-glutamine (Invitrogen), endothelial cell growth supplement (ECGS) purchased from Sciencell (Carlsbad, CA) and 100 ng/ml b-FGF (Roche/Sigma, Basel, Switzerland). The isolated cells were further expanded in the same culture medium at 37°C in a humidified incubator with 5% CO2, and morphologically distinct spindle-shaped fibroblast-like cell colonies were manually removed with a tiny cell scraper under a microscope. After reaching confluence, the monolayers were trypsinized and passaged 1:2. The purity of the TECs was assessed by evaluating the expression of CD31, ERG1, and von Willebrand factor (vWF) by immunofluorescence, and the purity was more than 95%. Cell populations from passage 2 were used for the study. As a control, endothelial cells extracted from normal pancreatic tissue were purchased from Sciencell and cultured on fibronectin-coated dishes with ECM supplemented with 10% FBS, 1% penicillin-streptomycin-glutamine, and ECGS according to the manufacturer’s instructions.
Microarray analysis
Microarray analyses were carried out as described previously .13 Briefly, total RNA was extracted from TECs using an RNeasy-Mini kit (QIAGEN, Valencia, CA) and quantified using a NanoDrop 8000 instrument (Thermo Fisher Scientific, Waltham, MA). The RNA integrity number (RIN) was evaluated by a 2100 Bioanalyzer (Agilent, Santa Clara, CA). Cy3-labeled complementary RNA was prepared from total RNA and hybridized to the SurePrint-G3-HumanGE8x60K-Microarray ver.2.0 (Agilent). The following analyses were performed at the Chemical Evaluation Research Institute (CERI, Tokyo, Japan). Briefly, signals were normalized by Gene Spring GX 14.5 (Agilent) with global normalization. To compare 2 groups of samples, i.e., endothelial cells derived from PDAC tissue samples and those derived from chronic pancreatitis (CP) tissue samples, Welch’s t-test was used for statistical significance. The statistically significant genes were classified into an upregulated gene expression group or a downregulated gene expression group. Then, the data were separately imported to Ingenuity Pathway Analysis (IPA) ver. 43605602 (QIAGEN), and canonical pathway analysis was performed by using the Core Analysis program.
Drug screening
A customized drug library targeting TNFR1 signaling was created, and the agents are summarized in Supplementary Table S1. Compounds were resolved in DMSO, and added to culture medium (final concentration, 0.05% DMSO). To eliminate cytotoxic compounds, normal pancreatic endothelial cells were seeded in a 96-well plate at 5 × 103 cells/well in ECM supplemented with 10% FBS and incubated at 37°C in a humidified atmosphere of 5% CO2. The following day, the cells were treated with 10 µM compound targeting TNFR1 signaling, 10 ng/ml TNF-α (PeproTech, Rocky Hill, NJ) as positive control, or 0.05% DMSO as negative control for 48 hours. Cell survival was evaluated using the CCK-8 reagent (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). CCK-8 was added for 1 hour, and the absorbance at 450 nm was measured using a microplate reader. To perform qPCR-based drug screening, total RNA was extracted from endothelial cells in a 96-well cell culture plate by using Cells-to-CT 1-step TaqMan (Invitrogen/Thermo Scientific). Following the manufacturer’s protocols, qPCR was performed with QuantStudio3 (Thermo Scientific) and the following TaqMan gene expression assays: probe# Hs02786624_g1 for GAPDH, probe# Hs00174057_m1 for SELE, and probe# Hs01003372_m1 for VCAM1. CT values were normalized to the CT value of GAPDH, and the ∆∆CT method was used to compare the expression levels of the genes. To test drug effect, relative expression of genes is calculated based on the comparison with negative control as adding DMSO instead of compounds.
Effects of Embelin concentration on the expression of adhesion molecules
After cultivation of TECs in a 48-well plate supplemented with 0, 0.01, 0.1, 1.0, or 10.0 µM of Embelin (final concentration) for 48 hours, gene expression was measured quantitatively using qPCR methods as mentioned above.
Tumor transplantation experiments
Female C57/B6 mice, 4 to 6 weeks old, were purchased from Charles River Laboratories Japan (Kanagawa, Japan) and maintained in our facilities under standard conditions. KMPC9-A6 cells were established from a pancreatic cancer tumor formed in Ptf1aCre/+;LSL-K-RasG12D/+ mice (Ptf1aCre/+ mice were obtained from Mutant Mouse Regional Resource Centers, Bar Harbor, ME, and LSL-K-RasG12D/+ mice were obtained from the National Cancer Institute, Mouse Models of Human Cancers Consortium, Rockville, MD).14 KMPC9-A6 cells cultivated in RPMI-1640 supplemented with 5% FBS and 1% penicillin-streptomycin-glutamine (Invitrogen, Carlsbad, CA) at 37°C in a humidified incubator with 5% CO2 were trypsinized, washed twice in PBS, and injected subcutaneously into the right hind flank of C57/B6 mice at a dose of 2 × 106 cells/mouse in 100 µl of sterile PBS. Tumor size was measured with a digital caliper 2–3 times per week, and tumor volume was calculated according to the following formula: volume = length x width2/2. For antibody depletion, 0.2 mg of anti-CD8 antibody (53–6.72) or isotype immunoglobulin (BioCell, Irvine, CA) was injected intraperitoneally in 100 µl of PBS every 3 days during the course of experiments. Four microliters of Embelin [50 mg/ml in dimethyl sulfoxide (DMSO)] or DMSO diluted in 100 µl of PBS was injected intraperitoneally every 3 days during the course of experiments. Mice were sacrificed 30 days after transplantation, and the tumors were excised. Then, morphology, expression of VCAM-1 and ICAM-1 by endothelial cells, and tumor-infiltrating CD3+ T cells, CD8+ T cells, Foxp3+ regulatory T cells (Tregs), and Arg1+ M2 macrophages were analyzed by immunohistochemistry. The experiments were repeated three times. The animal experiments were conducted according to the guidelines of the Ethics Committee on Animal Experimentation and the Animal Welfare Regulations of the National Cancer Center.
Prior to the transplantation assays, we determined the concentration of Embelin that can kill the murine PDAC cell line KMPC9-A6 by MTT assay. MTT assay was performed using MTT cell count kit (Nacalai Tesque, Inc. Kyoto, Japan).
Accession number
The gene microarray data have been deposited and National Center for Biotechnology Information’s Gene Expression Omnibus data accession number is GSE151945.
Statistical analysis
Comparisons of qualitative variables were performed using Fisher’s exact test. Pairwise comparisons of subgroups were performed using the Mann-Whitney U test. Postoperative overall survival (OS) and disease-free survival (DFS) rates were calculated by the Kaplan-Meier method. Univariate analysis was performed for prognostic factors using the log-rank test. The factors found to be significant by univariate analysis were subjected to multivariate analysis using the Cox proportional hazards model (the backward elimination method). For cell experiments, significant differences were determined by a two-sided t-test. Differences with P < .05 were considered statistically significant. Statistical analyses were performed with StatView-J 5.0 software (Abacus Concepts, Berkeley, CA).
The following subsections are included in “Supplementary Methods”: pathological examination, immunofluorescence, tube formation assay, immunohistochemistry, TUNEL assay, lymphocyte adhesion assay, lymphocyte transmigration assay, and measurement of Embelin concentration in blood of an animal tumor transplantation model.
Results
Endothelial cells in PDAC tissue are potentially dysfunctional for lymphocyte trafficking
The extravasation of lymphocytes through the endothelium is a multistep mechanism .15–17 Multiple molecular choices (i.e., available adhesion molecules and chemokines) at each step provide a large degree of combinational diversity, whereby different lymphocyte subsets can be recruited to appropriate sites. For the recruitment of effector T cells into cancer tissues, some adhesion molecules expressed on endothelial cells, including E-selectin, ICAM-1, and VCAM-1, are involved .18 To determine whether endothelial cells are potentially dysfunctional for lymphocyte trafficking in PDAC tissue, we performed double immunohistochemistry to detect the expression of VCAM-1 in ERG1+ TECs (Figure 1a) using 104 samples of PDAC tissue and examined the infiltration of CD8+ T cells and their clinicopathological influences. The percentage of vessels composed of VCAM-1-expressing endothelial cells among all vessels (%VCAM1+V) in the PDAC tissue samples was lower than that in chronic pancreatitis (CP) tissue samples (Figure 1b). Compared to those surrounding vessels lacking VCAM-1+ endothelial cells, tumor-infiltrating CD8+ T cells surrounding the vessels composed of VCAM-1+ endothelial cells were abundant in PDAC tissues (Figure 1c and Supplementary Figure S2); this was also observed in CP tissue. These results suggest that TECs are potentially dysfunctional for lymphocyte trafficking in PDAC tissue. Furthermore, Kaplan-Meier survival analysis revealed that PDAC patients with a higher %VCAM1+V exhibited longer OS than those with a lower %VCAM1+V (P = .029) (Figure 1d). A similar tendency was found for DFS, although no statistically significant result was obtained (Figure 1e). When the variables significant by univariate Cox analysis were subjected to multivariate Cox analysis, increased lymphatic invasion and a reduced %VCAM1+V were closely associated with reduced OS (hazard ratio 1.871, 95% confidential interval 1.162–3.012, P = .010) (Table 2). A decreased %VCAM1+V was significantly correlated with increased intrapancreatic neural invasion and the presence of nerve plexus invasion among the various clinicopathological factors examined (Table 1).
Figure 1.
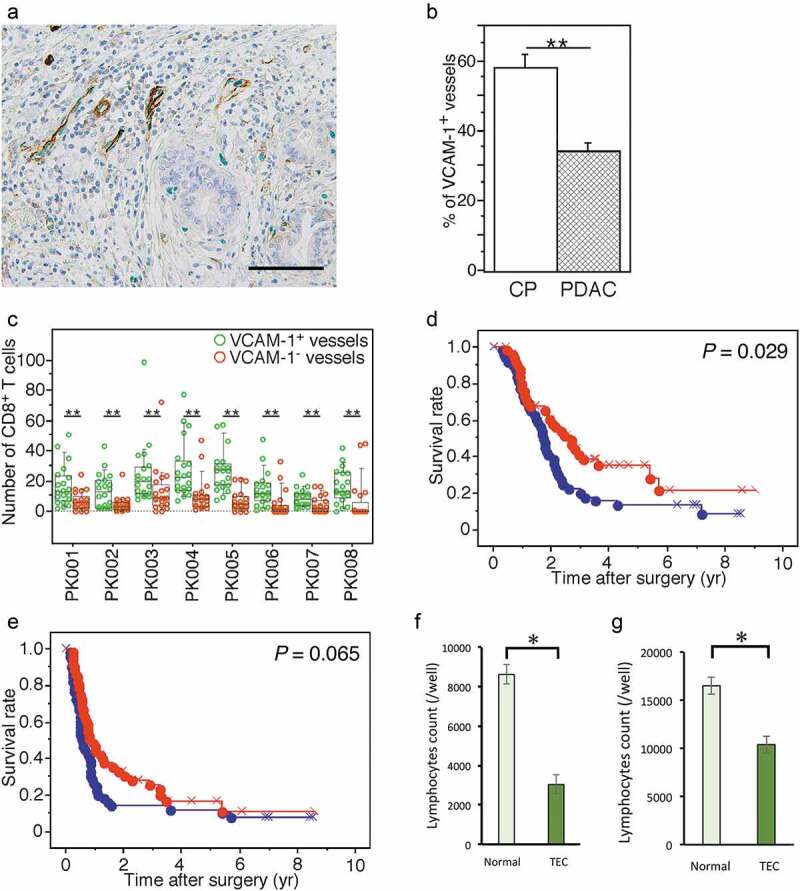
Endothelial cells in PDAC tissue are potentially dysfunctional for lymphocyte trafficking. (a) Double immunohistochemical detection of VCAM-1 (brown) and ERG1 (green) in PDAC tissue. High-power view. The bar indicates 100 µm. (b) Comparison of the percentage of vessels composed of VCAM-1+ endothelial cells among all vessels (%VCAM1+V) detected by evaluating ERG1+ endothelial cells in PDAC (n = 104) and CP tissue samples (n = 10). (c) Comparison of the numbers of tumor-infiltrating CD8+ T cells surrounding blood vessels composed of VCAM-1+ or VCAM-1− endothelial cells in PDAC tissue samples. Box plots and the scatter blots are combined. Whisker heights extend to 1.5 times the height of the box or, if no case/row has a value in that range, to the minimum and maximum values. The horizontal lines within the box indicate the median values. (d, e) Kaplan-Meier survival curves showing the comparison of OS (d) or DFS (e) between the high (red, n = 52) and low (blue, n = 52) %VCAM1+V groups of PDAC patients (n = 104). P-values were obtained from log-rank tests. “x” and “+” represent censoring and failure, respectively. (f, g) Adhesion assay evaluating the interaction between lymphocytes and TECs (f). Transmigration assay for lymphocytes migrating through an endothelial cell layer (g). Data are represented as the mean ± SD. Differences were analyzed by using the Mann-Whitney U test. *, P < .05 and **, P < .01
Table 2.
Univariate and multivariate analyses of prognostic factors associated with overall survival in patients with pancreatic ductal adenocarcinoma (n = 104)
| |
Univariate analysis |
Multivariate analysis |
||
|---|---|---|---|---|
| Variables | HR (95% CI) | P value | HR (95% CI) | P value |
| Age (< 65 years/≥65 years) | 0.743 (0.465–1.187) | 0.214 | ||
| Gender (female/male) | 0.968 (0.598–1.568) | 0.896 | ||
| Pathologic tumor status (T1+ T2/T3+ T4) | 1.633 (1.027–2.596) | 0.038 | ||
| Pathologic node status (N0/N1) | 2.533 (1.345–4.771) | 0.004 | ||
| Pathologic metastasis status (M0/M1) | 1.784 (0.957–3.326) | 0.069 | ||
| Histological grade (G1/G2+ G3) | 2.460 (1.062–5.698) | 0.036 | ||
| Tumor margin status (negative/positive) | 1.549 (0.969–2.474) | 0.067 | ||
| Nerve plexus invasion (absence/presence)* | 2.281 (1.132–4.598) | 0.021 | ||
| Lymphatic invasion (0, 1/2, 3)* | 3.502 (1.855–6.614) | 0.0001 | 3.765 (1.983–7.148) | <0.0001 |
| Venous invasion (0, 1/2, 3)* | 2.292 (1.213–4.330) | 0.011 | ||
| Intrapancreatic neural invasion (0, 1/2, 3)* | 2.201 (1.241–3.903) | 0.007 | ||
| Adjuvant chemotherapy (presence/absence) | 0.908 (0.487–1.694) | 0.762 | ||
| %VCAM1+V (high/low) | 1.680 (1.050–2.688) | 0.031 | 1.871 (1.162–3.012) | 0.010 |
*Classified according to the classification of pancreatic carcinoma of Japan Pancreas Society.
HR: hazard ratio, CI: confidential interval, Bold letters: significant difference.
The unusual phenotype of TECs from PDAC tissue samples
To investigate the characteristics of TECs, we isolated CD31+CD45− cells from fresh PDAC tissue samples. Morphologically, these cells showed a cobblestone-like appearance under confluent cell conditions with a large cytoplasm and small nucleus, and TECs composed >95% of all nucleated cells (Supplementary Figure S3A). Endothelial cell markers, CD31 and ERG1, were intensely expressed in these cells (Supplementary Figure S3B), consistent with the pathological findings (Supplementary Figure S1). Next, to examine the cellular phenotype of the CD31 and ERG1 double-positive cells, a tube formation assay was performed, and angiogenic potential was analyzed in TECs compared with endothelial cells isolated from a normal pancreas. TECs had the angiogenic ability (Supplementary Figure S3C). Automatic image analysis showed that compared to normal pancreatic endothelial cells, TECs tended to form short vessels, a narrow capillary network area, and increased numbers/lengths of isolated branches, indicating that the TECs had some different angiogenic characteristics than the endothelial cells from normal pancreas tissue (Supplementary Figures S3D and S3E).
To evaluate the function of TECs in lymphocyte trafficking, we examined the adherence of lymphocytes to TECs and the transmigration of lymphocytes through a TEC monolayer. An adhesion assay showed that lymphocytes adhered significantly less to TECs than normal pancreatic endothelial cells (Figure 1f). A transmigration assay also showed that TECs exhibited weaker supporting activities in lymphocyte transmigration than normal pancreatic endothelial cells (Figure 1g). These results implied that the TECs had dysfunctional characteristics for lymphocyte trafficking.
Delineating a signaling shift in the TNFR1 pathway in TECs
To find a molecular target for compound identification to enhance lymphocyte trafficking, we needed to clarify what molecules and pathways were distinct between TECs and endothelial cells from CP tissue. The gene expression profiles of endothelial cells from PDAC and CP tissue samples were analyzed using a microarray technique and compared. Scatter plot analysis graphed the gene expression of the two different cell types. In total, 2,748 genes exhibited significantly different expression between the two cell types (Figure 2a). Next, we employed bioinformatics software, termed Ingenuity Pathway Analysis (IPA), and visualized the pathways of interest. The analysis identified and ranked the distinct signaling pathways between TECs and endothelial cells from CP tissue, and 47 pathways were identified (P < .05): 31 pathways were downregulated, and 16 pathways were upregulated (Figure 2b, Supplementary Table S2, and Supplementary Figure S4A).
Figure 2.
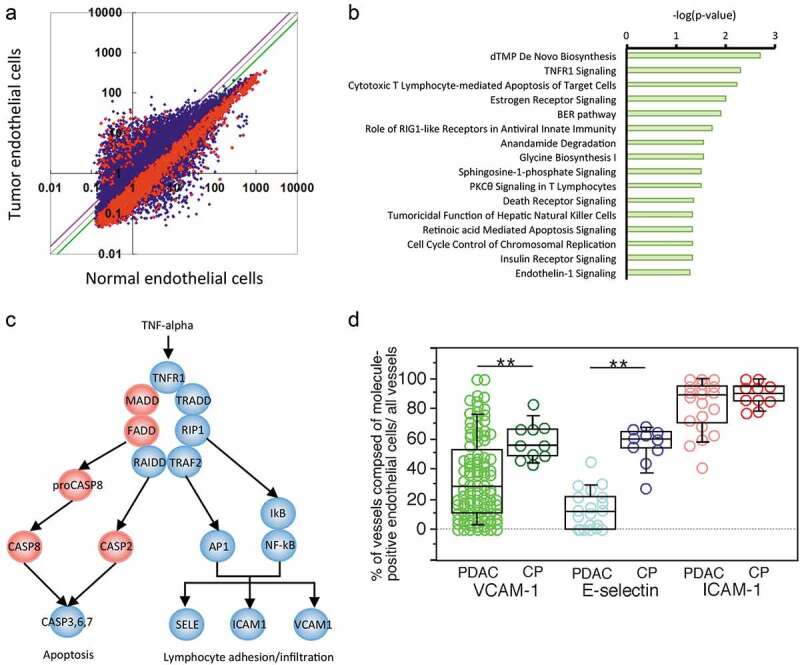
Gene expression and pathway profiles of TECs. (a) Scatter plot analysis of microarray datasets depicting the different expression profiles of TECs and endothelial cells isolated from CP tissue. The X- and Y-axes indicate averaged signal values. In total, 24,456 probes were examined, and the red dots indicate genes with expression that was significantly different between the two groups, whereas the blue dots indicate genes with no significant difference in expression. The purple and green lines indicate borders of 1.5- and 0.67-fold changes, respectively; 326 genes exhibited upregulated expression, and 2,422 genes exhibited downregulated expression in the TECs compared to the CP endothelial cells. (b) Ingenuity pathway analysis (IPA) of the altered pathways between TECs and CP endothelial cells. The pathways are ranked from the lowest P-value (top) to the highest (bottom). The green bars indicate the – log(P-value) of each pathway, so the strength of each statistical association is represented by the length of the corresponding bars. (c) Visualization of altered TNFR1 signaling in TECs. IPA canonical pathway analysis reconstructed TNFR1 signaling. The proposed intracellular signaling is illustrated. The genes with significantly upregulated expression in TECs are colored red, and those with significantly downregulated expression in TECs are colored blue. (d) The expression status of TECs in PDAC tissue samples compared to that in CP tissues determined by immunohistochemistry. Box plots show the value distribution of the percentage of vessels composed of VCAM-1+, E-selectin+, and ICAM-1+ endothelial cells among all vessels detected by evaluating ERG1+ or vVF+ endothelial cells in PDAC [VCAM-1 (n = 104), E-selectin (n = 20), and ICAM-1 (n = 20)] and CP tissue (n = 10). The scatter blots are combined. Whisker heights extend to 1.5 times the height of the box or, if no case/row has a value in that range, to the minimum and maximum values. The horizontal lines within the box indicate the median values. Differences were analyzed by using the Mann-WhitneyU test. *, P < .05 and **, P < .01
We found downregulation of several genes in the TNFR1 signaling pathway encoding major molecules involved in lymphocyte trafficking19 (Figure 2c), and we focused on TNFR1 signaling, which ranked second among the 31 downregulated pathways (Figure 2b). Since TNFR1 is involved in a variety of cellular responses based on cellular origins ,20 it was important to integrate the general information in the IPA database and the current understanding of TNFR signaling in endothelial cells19 to delineate an underlying interaction (Figure 2c).
To determine the protein status of downstream molecules in the TNFR1 signaling pathway in TECs from PDAC tissue, we detected the expression levels of E-selectin, VCAM-1, and ICAM-1 (Supplementary Figure S1E). The ratios of designated molecule-expressing vessels to all vessels were then calculated. Box plots showed that E-selectin and VCAM-1 expression was reduced in most TECs compared to that in CP (Figure 2d), implying insufficient activation of TNFR1 signaling in the TECs. This finding also implied that the downregulation of E-selectin or VCAM-1 expression might be responsible for the reduced adherence and transmigration characteristics of TECs and that E-selectin and VCAM-1 could be therapeutic targets to enhance lymphocyte adhesion. Meanwhile, ICAM-1 was expressed in most TECs, although its staining intensity was often weaker than that in CP (Supplementary Figure S5). It is possible that ICAM-1 gene expression levels might be reduced in TECs.
The gene expression profiles also showed that FADD, MADD, proCASP8, CASP8, and CASP2, which are apoptosis-associated molecules, exhibited upregulated expression in TECs (Figure 2c and Supplementary Figure S6), although few apoptotic endothelial cells were found in PDAC tissues (data not shown). This finding might imply a signaling shift in the TNFR1 pathway in the TECs but does not accelerate endothelial cell apoptosis.
Compound screening for candidates targeting TNFR1 signaling to enhance immune cell infiltration
Based on the results shown above, we planned a drug screen to identify a compound that boosts lymphocyte infiltration (Supplementary Figure S4B). As a first step, we recruited various compounds, inhibitors, and activators targeting TNFR1 signaling and created a customized drug library (Supplementary Table S1). Next, to exclude harmful compounds, a cytotoxicity test was performed, and the cutoff value was defined as >80% cell survival; consequently, 36 nontoxic agents were selected (Supplementary Figure S4C). To further identify E-selectin- or VCAM1-inducing compounds, qPCR-based screening was performed. Since different endothelial signaling pathways may be activated under confluent cellular conditions versus sparse conditions due to cell-to-cell interactions ,21 we performed drug screening under these two experimental conditions to evaluate endothelial cell responses to the compounds. We also used mixed endothelial cells derived from three PDAC samples selected randomly to represent the average characteristics of TECs, since TECs are potentially heterogeneous. As expected, the results showed that different pharmacological reactions were observed. Overall, four drugs in group C, which are inhibitors of apoptosis protein (IAP) inhibitors, tended to upregulate VCAM-1 expression under both confluent and sparse conditions (Figure 3a). Therefore, the 4 IAP inhibitors – AZD5582, BV-6, Embelin, and LCL161, were evaluated in lymphocyte adhesion and transmigration assays. To mimic the clinical conditions of the immune microenvironment of PDAC, we further prepared TILs from patient surgical samples and used these cells in the cellular assays. Representative results revealed that the addition of AZD5582, BV-6, Embelin, or LCL161 largely increased the adhesion between the TILs and TECs and that Embelin significantly upregulated this interaction (Figure 3b). Also, Embelin significantly enhanced the transmigration of TILs (Figure 3c). Thus, our integrated compound screening study might have identified a possible agent to enhance lymphocyte infiltration to produce a beneficial immune response. Upon analyzing the relationship between the induction of adhesion molecules and the concentration of Embelin in TECs, Embelin concentration of more than 0.1 µM could effectively induce the expression of VCAM1 (Figure 3d) and SELE (Figure 3e) genes, with stronger expression at higher concentrations.
Figure 3.
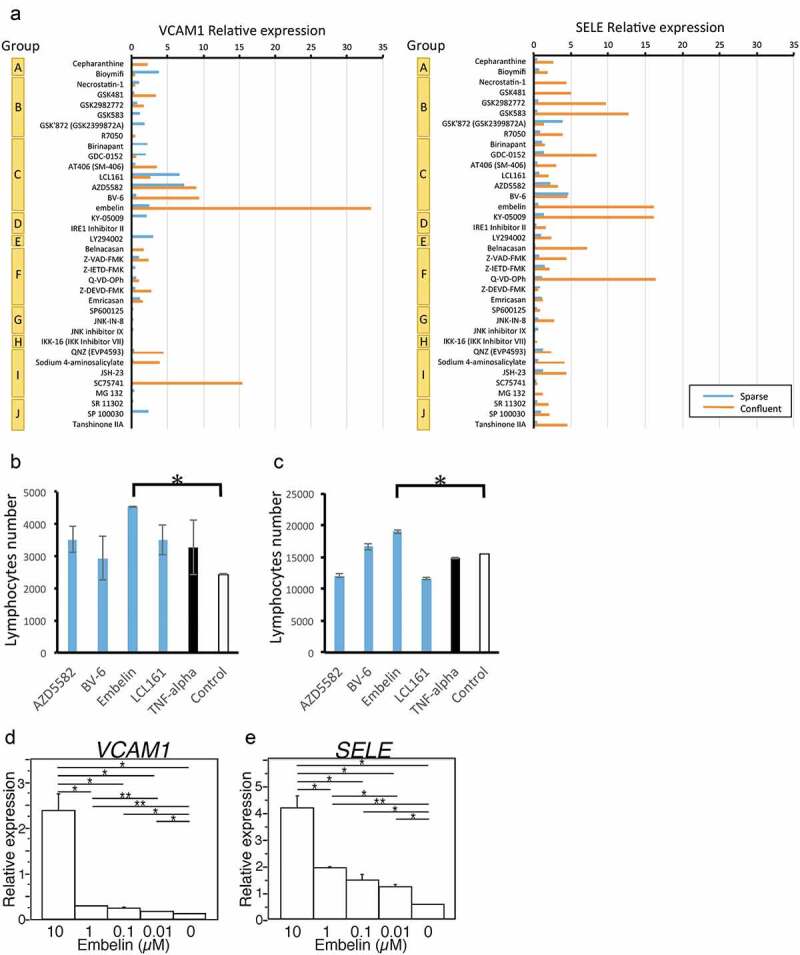
Compound screening for candidates targeting TNFR1 signaling in TECs. (a) qPCR-based compound screening. Relative expression is calculated based on the comparison with negative control as treatment with DMSO instead of compounds. (b, c) Experimental adhesion transmigration assay with an endothelial cell layer. TILs isolated from surgical samples were fluorescence-labeled and added to confluent cell layers of TECs. The numbers of adhered or transmigrated lymphocytes were measured. Distribution of the numbers of lymphocytes adhered to (b)/transmigrated through (c) the endothelial cell layer. Data are shown as the mean ± SD. *, P < .05. (d, e) Embelin treatment increases gene expression of VCAM1 (d) and SELE (e) in TECs. 48 hours after various concentrations of Embelin treatment for TECs, gene expressions are measured by the RT-qPCR method. Data is normalized by GAPDH gene expression. Data are shown as the mean ± SD. Student’s t-test analyzed differences. *, P < .05 and **, P < .01
Embelin facilitates T cell recruitment in murine PDAC tissue to induce an antitumor immune response
Next, we examined whether Embelin treatment facilitates tumor-infiltrating T cells and induces an antitumor immune response in vivo. We injected the murine PDAC cell line KMPC9-A6, which was established from a PDAC tumor formed in an aged Ptf1aCre/+;LSL-K-RasG12D/+ mouse, into the right hind flank of syngeneic C57BL/6 mice. Since IAPs inhibit apoptosis and their expression is elevated in many cancer cells, IAP inhibitors, including Embelin, induce cell death and have antitumor activity, as reported earlier .22–24 To avoid the direct tumor-cell-killing effect of Embelin, we determined the Embelin concentration that could kill KMPC9-A6 cells in vitro, which was more than 3.13 µM (Supplementary Figure S4D). We set Embelin injection amounts and timing (Materials and Methods, and Supplementary Figure S7) because Embelin has tumor-killing activity, relatively rapid blood clearance, and effective induction of VCAM-1 and E-selectin at low concentrations (Figure 3d and e). Indeed, three hours after the intraperitoneal injection of Embelin, the Embelin blood concentration dropped below 3 µM and was kept within the effective concentration for more than 48 hours in this model (Supplementary Figure S4E). Tumor growth was significantly delayed by Embelin treatment compared with no treatment (Figure 4a and b). Immunohistochemical and immunofluorescence analyses revealed that the %VCAM1+V and %ICAM1+V increased significantly in the Embelin-treated tumors compared with those in the untreated tumors (Figure 4c and d and Supplementary Figures S8A-S8D).
Figure 4.
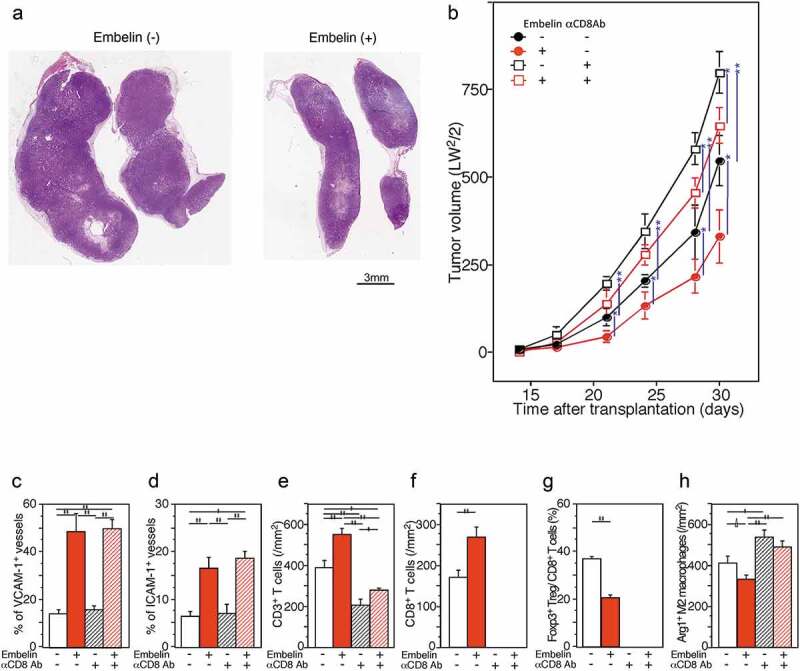
Embelin treatment slowed tumor growth through increasing T cell infiltration by restoring VCAM-1 expression in TECs. (a, b) Embelin treatment slowed tumor growth. The loupe feature of the representative tumors excised on day 30 after transplantation (hematoxylin and eosin stain) (a). C57BL/6 mice were injected with KMPC9-A6 cells, and Embelin (red line) or DMSO (black line) diluted in PBS was injected (b). For CD8+ T cell depletion, an anti-CD8 antibody (square) or isotype immunoglobulin (circle) was injected. Each data point represents the mean tumor size ± SE of eight mice. Differences were analyzed by using the Mann-Whitney U test. *, P < .05 and **, P < .01. (c-h) Immunohistochemical evaluation of the expression of VCAM-1, ICAM-1, CD3, CD8, Foxp3, and Arg1 in tumors excised from mice sacrificed on day 30 after transplantation. Comparison of the %VCAM1+V (c) and %ICAM1+V (d) detected as ERG1+ endothelial cells among the tumors from mice with different treatment conditions. Densities of tumor-infiltrating CD3+ T cells (e), CD8+ T cells (f), Arg1+ M2 macrophages (h), and ratio of tumor-infiltrating Foxp3+ Tregs/CD8+ T cells (g) in tumor tissues were compared. Student’s t-test analyzed differences. ∆, P < .10, *, P < .05 and **, P < .01
Additionally, significant CD3+ and CD8+ T cell infiltration was observed in the Embelin-treated tumors (Figure 4e and f, and Supplementary Figures S8E-S8H). In contrast, tumor-infiltrating immune-suppressive cells were reduced in the Embelin-treated tumors (Figure 4g and h): the ratio of tumor-infiltrating Foxp3+ Treg/CD8+ T cells was significantly lower and Arg1+ M2 macrophage infiltration tended to be lower in the Embelin-treated tumors compared to untreated tumors. Next, we investigated the effect of depleting CD8+ T cells from animals on tumor growth. CD8+ T cell depletion effectively blocked the tumor growth inhibition mediated by treatment with Embelin (Figure 4a). These findings suggested that Embelin efficiently induced T cell migration in vivo and promoted an antitumor responsive microenvironment, and that tumor growth was inhibited through an adaptive immune reaction.
Discussion
To date, various compound screening/repositioning methods have been reported to target malignant features of cancer, e.g., proliferation, motility, invasion, and metastasis. This study is the first compound identification study showing boosted lymphocyte infiltration. So far, only limited blocking antibodies targeting TECs for acceleration and selection of T cell extravasation and infiltration have been reported .9,10 We identified a novel compound that induced T cell infiltration, leading to tumor shrinkage.
Lymphocytes cross the vascular endothelium by multiple steps consisting of tethering, rolling, arrest, crawling, and diapedesis .15–17,25 In lymphocyte extravasation, underlying cooperative molecular participants expressed on the endothelium, including selectins, ICAMs, VCAM-1, VE-cadherin, CD31 (PECAM1), CD99, and JAMA, are involved .15–17 Endothelial cells are activated with the transcriptional induction of numerous leukocyte-trafficking molecules, termed type II activation, mostly driven by cytokines such as TNF-α and IL-1β. Among the adhesion molecules, E-selectin, ICAM-1, and VCAM-1 are induced in type II activation, which are downstream effector molecules of the TNFR1 pathway. In endothelial cells, TNFR1 signaling produces two distinct cellular events: cell apoptosis and lymphocyte extravasation support .19,20,25 In contrast to the endothelial cells derived from CP, whose phenotype was similar to that of the endothelial cells with type II activation in this study, the phenotype of TECs was different and rather dysfunctional. In the TNFR1 pathway, TNF-α is an upstream ligand; however, TECs insufficiently transmit downstream effectors for lymphocyte infiltration. There may be several possible causes: 1) a deficient TNF-α supply in the tumor microenvironment of PDAC, 2) TNFR downregulation on TECs, or 3) abrogated downstream signaling in TECs. Higher TNF-α levels have been reported in PDAC patients than in healthy individuals .26 For TNFR downregulation on TECs, there were no significant differences in the TNFR expression levels between the endothelial cells from the noncancerous pancreas and TECs in our experiment (data not shown). Therefore, we focused on abrogated downstream signaling in TECs.
In general, IAPs are a family of proteins consisting of cIAP1, cIAP2, and XIAP, which inhibit apoptosis by blocking caspase activation or activity in the TNFR1 signaling pathway, through limiting non-canonical NF-κB signaling, promoting canonical NF-κB and mitogen-activated protein kinase (MAPK) signaling, and inhibiting both caspase-dependent and -independent cell death .27 IAPs can also regulate innate immune responses and the Toll-like receptor signaling pathways ,27 although the regulation is not simple as the regulatory roles are different among members of IAPs and cell types. Also, XIAP has an essential role in nucleotide-binding oligomerization domain (NOD) signaling, connected from cellular recognition of bacterial peptidoglycan derivatives through NOD1/2, leading to the production of inflammatory mediators. XIAP-deficient patients and animals are affected by a range of immunological defects, including hemophagocytic lymphohistiocytosis and inflammatory bowel disease like Crohn’s disease .28,29
In endothelial cells, IAPs are crucial regulators of vascular integrity, survival, angiogenesis, and homeostasis .30 Specifically, cIAP1 and cIAP2 regulate the activation of downstream signaling, that is, NF-κB signaling .31 XIAP also participates in the NF-κB activation cascade ,32 which may lead to enhanced migration and adhesion of physiologically normal endothelial cells .33 Thus, IAPs and their subfamily molecules are associated with endothelial function, although the details of their roles in the molecular pathway remain to be elucidated. In this study, we observed increased adhesion and transmigration behavior between lymphocytes and TECs via VCAM-1 upregulation induced by the addition of an XIAP inhibitor. These changes may be caused by unique intracellular signaling cascades or crosstalk among TECs, as shown in this study. At least in part, inhibiting XIAP may contribute to the vascular normalization by TECs for inducing lymphocyte extravasation. Further mechanistic studies are needed to understand the endothelial functional alterations in TECs that become more profound during treatment.
Recently, studies targeting IAPs for cancer treatment have been actively performed, in which IAP inhibitors have been used for the induction of tumor cell death .34–36 Targeting all three IAPs is highly inflammatory and dead tumor cells can help trigger the immune system to attack cancer cells. Such events allow the enhance the antitumor effect .34–36 Indeed, IAP inhibitors can increase the production of death ligands, IFNγ, and IL-2 by immune cells, sensitizing cancer cells to the death ligands produced, enhancing cytotoxic lymphocyte killing of tumor cells, and reducing immune suppressive T cell functions .37–41 Thus, vascular normalization activity, a new additional activity of IAP inhibitors found in this study, will provide an excellent tool for developing a combination therapy.
Recovering the dysfunctional TECs by Embelin occurred at a lower concentration compared to that of the tumor-cell-killing effect. Embelin has been shown to have an antitumor effect on glioma, leukemia, and pancreatic cancers in in vitro assays using cell lines ,22–24 where more than 20–30 µM of Embelin was needed in contrast to the 0.1 µM concentration required for inducing the lymphocyte-trafficking-related adhesion molecules in TECs, as shown in this study. Although Embelin was used at a relatively low dose to avoid its direct tumor-killing effect in the animal model in this study, if a high dose of Embelin is administered, higher antitumor activity is expected through direct tumor killing, vascular normalization, and some additional immunological effects, as described above.
Other than PDAC, some colorectal cancer, non-small cell lung cancer, and squamous cell carcinoma of the head and neck are also immune-quiescent ‘cold’ tumors .4–6 Our methodology may be similarly applicable to these immunologically quiescent tumors and could identify a compound to reprogram TECs for a successful antitumor response. Although many immunotherapeutic modalities, such as cancer vaccines, activated autologous lymphocyte therapies, checkpoint inhibitors, and genetically engineered T cells, have been developed in the past decade to halt PDAC progression, some previous attempts have not resulted in beneficial clinical outcomes for unknown reasons .4–6 Here, we suggest that these failures were due in part to not considering lymphocyte-TEC interactions and propose a novel modality of combination therapy. Combination therapy targeting TECs could be essential for improving immunotherapy efficacy and should be considered in future clinical trials. Otherwise, immunotherapies that do not consider TECs could fail in clinical trials.
In conclusion, our strategy may be added to the therapeutic arsenal for treating the immune-quiescent tumor microenvironment to produce a beneficial immune response.
Supplementary Material
Acknowledgments
The authors thank Ms. Sachiko Miura, Toshiko Sakaguchi, and Chizu Kina for excellent techniques. We are grateful to the National Cancer Center Biobank for the tissue samples used in this study.
Funding Statement
This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science, #18K07008 (NH) and #18K16377 (KN), and the Japan Agency for Medical Research and Development, #17938501 (NH).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Gajewski TF, Schreiber H, Fu YX.. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakajima K, Nangia-Makker P, Hogan V, Raz A. Cancer self-defense: an immune stealth. Cancer Res. 2017;77:5441–5444. doi: 10.1158/0008-5472.CAN-17-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer. 2019;19:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balachandran VP, Beatty GL, Dougan SK. Broadening the impact of immunotherapy to pancreatic cancer: challenges and opportunities. Gastroenterology. 2019;156:2056–2072. doi: 10.1053/j.gastro.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016;381:244–251. doi: 10.1016/j.canlet.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu JW, Wang L, Cheng YG, Zhang GY, Hu SY, Zhou B, Zhan H-X. Immunotherapy for pancreatic cancer: A long and hopeful journey. Cancer Lett. 2018;425:143–151. doi: 10.1016/j.canlet.2018.03.040. [DOI] [PubMed] [Google Scholar]
- 7.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108(4):914–923. doi: 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer. 1782-90;2015:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O’Brien-Jenkins A, Gimotty PA, Coukos G, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]
- 10.Shetty S, Weston CJ, Oo YH, Westerlund N, Stamataki Z, Youster J, Hubscher SG, Salmi M, Jalkanen S, Lalor PF, et al. Common lymphatic endothelial and vascular endothelial receptor-1 mediates the transmigration of regulatory T cells across human hepatic sinusoidal endothelium. J Immunol. 2011;186(7):4147–4155. doi: 10.4049/jimmunol.1002961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20:607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brierley JD, Gospodarowicz MK, Wittekind C. TNM classification of malignant tumours. Hoboken (NJ): Wiley-Blackwell; 2017. [Google Scholar]
- 13.Hiraoka N, Yamazaki-Itoh R, Ino Y, Mizuguchi Y, Yamada T, Hirohashi S, Kanai Y. CXCL17 and ICAM2 are associated with a potential anti-tumor immune response in early intraepithelial stages of human pancreatic carcinogenesis. Gastroenterology. 2011;140:310–321. doi: 10.1053/j.gastro.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/S1535-6108(03)00309-X. [DOI] [PubMed] [Google Scholar]
- 15.Friedl P, Weigelin B. Interstitial leukocyte migration and immune function. Nat Immunol. 2008;9:960–969. [DOI] [PubMed] [Google Scholar]
- 16.Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 17.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 18.Hiraoka N. Tumor-infiltrating lymphocytes and hepatocellular carcinoma: molecular biology. Int J Clin Oncol. 2010;15:544–551. [DOI] [PubMed] [Google Scholar]
- 19.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. [DOI] [PubMed] [Google Scholar]
- 20.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. [DOI] [PubMed] [Google Scholar]
- 21.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 22.Siegelin MD, Gaiser T, Siegelin Y. The XIAP inhibitor Embelin enhances TRAIL-mediated apoptosis in malignant glioma cells by down-regulation of the short isoform of FLIP. Neurochem Int. 2009;55:423–430. doi: 10.1016/j.neuint.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 23.Hu R, Zhu K, Li Y, Yao K, Zhang R, Wang H, Yang W, Liu Z. Embelin induces apoptosis through down-regulation of XIAP in human leukemia cells. Med Oncol. 2011;28:1584–1588. doi: 10.1007/s12032-010-9601-5. [DOI] [PubMed] [Google Scholar]
- 24.Peng M, Huang B, Zhang Q, Fu S, Wang D, Cheng X, Wu X, Xue Z, Zhang L, Zhang D, et al. Embelin inhibits pancreatic cancer progression by directly inducing cancer cell apoptosis and indirectly restricting IL-6 associated inflammatory and immune suppressive cells. Cancer Lett. 2014;354:407–416. doi: 10.1016/j.canlet.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 25.Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41:694–707. doi: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 26.Blogowski W, Deskur A, Budkowska M, Salata D, Madej-Michniewicz A, Dabkowski K, Dołęgowska B, Starzyńska T. Selected cytokines in patients with pancreatic cancer: a preliminary report. PLoS One. 2014;9:e97613. doi: 10.1371/journal.pone.0097613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lalaoui NV. D. L. Recent advances in understanding inhibitor of apoptosis proteins. F1000 Faculty Rev. 2019;7:1889. [Google Scholar]
- 28.Bauler LD, Duckett CS, O’Riordan MX. XIAP regulates cytosol-specific innate immunity to Listeria infection. PLoS Pathog. 2008;4:e1000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latour S, Aguilar C. XIAP deficiency syndrome in humans. Semin Cell Dev Biol. 2015;39:115–123. doi: 10.1016/j.semcdb.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 30.Santoro MM, Samuel T, Mitchell T, Reed JC, Stainier DY. Birc2 (cIap1) regulates endothelial cell integrity and blood vessel homeostasis. Nat Genet. 2007;39:1397–1402. doi: 10.1038/ng.2007.8. [DOI] [PubMed] [Google Scholar]
- 31.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofer-Warbinek R, Schmid JA, Stehlik C, Binder BR, Lipp J, de Martin R. Activation of NF-kappa B by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J Biol Chem. 2000;275:22064–22068. [DOI] [PubMed] [Google Scholar]
- 33.Kim J, Ahn S, Ko YG, Boo YC, Chi SG, Ni CW, Go YM, Jo H, Park H.. X-linked inhibitor of apoptosis protein controls alpha5-integrin-mediated cell adhesion and migration. Am J Physiol Heart Circ Physiol. 2010;299:H300–9. doi: 10.1152/ajpheart.00180.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beug ST, Tang VA, LaCasse EC, Cheung HH, Beauregard CE, Brun J, Nuyens JP, Earl N, St-Jean M, Holbrook J, et al. Smac mimetics and innate immune stimuli synergize to promote tumor death. Nat Biotechnol. 2014;32(2):182–190. doi: 10.1038/nbt.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brumatti G, Ma C, Lalaoui N, Nguyen NY, Navarro M, Tanzer MC, Richmond J, Ghisi M, Salmon JM, Silke N, et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci Transl Med. 2016;8:339ra69. [DOI] [PubMed] [Google Scholar]
- 36.Lalaoui N, Hanggi K, Brumatti G, Chau D, Nguyen NY, Vasilikos L, Spilgies L, Heckmann D, Ma C, Ghisi M, et al. Targeting p38 or MK2 enhances the anti-leukemic activity of smac-mimetics. Cancer Cell. 2016;29(2):145–158. doi: 10.1016/j.ccell.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 37.Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, Akhmetzyanova I, Kosiorek HE, Calcinotto A, Riggs DL, et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat Med. 2016;22(12):1411–1420. doi: 10.1038/nm.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clancy-Thompson E, Ali L, Bruck PT, Exley MA, Blumberg RS, Dranoff G, Dougan SK. IAP antagonists enhance cytokine production from mouse and human iNKT cells. Cancer Immunol Res. 2018;6:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dougan M, Dougan S, Slisz J, Firestone B, Vanneman M, Draganov D, Goyal G, Li W, Neuberg D, Blumberg R, et al. IAP inhibitors enhance co-stimulation to promote tumor immunity. J Exp Med. 2010;207(10):2195–2206. doi: 10.1084/jem.20101123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim DS, Dastidar H, Zhang C, Zemp FJ, Lau K, Ernst M, Rakic A, Sikdar S, Rajwani J, Naumenko V, et al. Smac mimetics and oncolytic viruses synergize in driving anticancer T-cell responses through complementary mechanisms. Nat Commun. 2017;8:344. doi: 10.1038/s41467-017-00324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao R, Allen CT, Tran L, Patel P, Park SJ, Chen Z, Van Waes C, Schmitt NC. Antagonist of cIAP1/2 and XIAP enhances anti-tumor immunity when combined with radiation and PD-1 blockade in a syngeneic model of head and neck cancer. Oncoimmunology. 2018;7:e1471440. doi: 10.1080/2162402X.2018.1471440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.