

ABSTRACT
Cancer immunotherapy based on Immune checkpoint blockade (ICB) is a promising strategy to treat patients with advanced highly aggressive therapy-resistant tumors. Unfortunately, the clinical reality is that only a small number of patients benefit from the remarkable clinical remissions achieved by ICB. Experimental and clinical evidence claimed that durable clinical benefit observed using ICB depends on the immune status of tumors, notably the presence of cytotoxic effector immune cells. In our paper, we revealed that genetically targeting the autophagy-related protein PIK3C3/VPS34 in melanoma and colorectal tumor cells, or treating tumor-bearing mice with selective inhibitors of the PIK3C3/VPS34 kinase activity, reprograms cold immune desert tumors into hot, inflamed immune infiltrated tumors. Such reprograming results from the establishment of a proinflammatory signature characterized by the release of CCL5 and CXCL10 in the tumor microenvironment, and the subsequent recruitment of natural killer (NK) and CD8+ T cells into the tumor bed. Furthermore, we reported that combining pharmacological inhibitors of PIK3C3/VPS34 improves the therapeutic benefit of anti-PD-1/PD-L1 immunotherapy. Our results provided the proof-of-concept to set-up innovative clinical trials for cold ICB-unresponsive tumors by combining PIK3C3/VPS34 inhibitors with anti-PDCD1/PD-1 and anti-CD274/PD-L1.
KEYWORDS: Autophagy, cancer immunotherapy, cold/hot tumors, colorectal cancer, immune checkpoints, immune infiltration, melanoma, NK and T CD8 cells, proinflammatory cytokines, VPS34
The use of immune checkpoint blockades (ICBs), notably anti-CTLA4 (cytotoxic T-lymphocyte associated protein 4), anti-PDCD1/PD-1 (programmed cell death 1) and anti-CD274/PD-L1 (CD274 molecule), has emerged as a revolutionary treatment for cancer patients in whom conventional therapies have failed. The clinical benefit of ICB is only observed in a minority of patients, while many of them do not benefit or relapse after a short-term benefit. The immune landscape of tumors, which refers to the type, density, functionality and location of different subsets of immune cells in the tumor microenvironment, is a major factor predicting the response to ICB. Indeed, tumors not infiltrated by cytotoxic immune cells are clinically classified as non-eligible or not responding to ICB. Instead, tumors infiltrated with preexisting T cells are more likely to respond to ICBs. This has led to the concept of “cold” immune desert tumors and “hot” inflamed immune infiltrated tumors. In line with this concept, it is tempting to speculate that strategies that are able to switch immune desert or cold tumors to hot or immune infiltrated ones would significantly improve the efficacy of ICBs. To extend further the clinical benefits of ICBs, immuno-oncology research pushes toward combining ICBs with other immune-modulating drugs. Whereas the immuno-oncology field delivered a wealth of knowledge and plethora of drugs for combinations, testing all possible potential combinations remains very challenging. In addition, not all combinations tested in clinical trials will necessarily meet the clinical expectations and patient needs. Therefore, designing innovative ICB combinations should carefully consider the following issues: what to combine? how to combine? and when to combine? We strongly think that ICBs should be combined with drugs able to i) drive cytotoxic immune cells (NK and CD8+ T cells) into the tumor microenvironment; ii) limit the infiltration and the function of immune suppressive cells, such as regulatory T cells (Treg), pro-tumoral macrophages and/or myeloid-derived suppressor cells (MSDCs); and iii) promote the release of proinflammatory chemokines, including CCL2, CCL5, CXCL9, CXCL10, CXCL11 and CXCL13, into the tumor microenvironment.
Several clinical studies attempted to combine anti-cancer therapies with nonselective macroautophagy/autophagy inhibitors such as chloroquine (CQ) or hydroxychloroquine (HCQ) with mitigated success. The failure in providing a clear statement about the therapeutic benefit of combining autophagy inhibitors with other therapies in cancer patients could be related to the lack of selective drugs inhibiting autophagy. More selective and druggable autophagy target proteins have been proposed which include the early autophagy protein BECN1/beclin1 and its interacting protein PIK3C3/VPS34 (a component of the class III phosphatidylinositol 3-kinase complex). We therefore evaluated the impact of genetic targeting of PIK3C3/VPS34 in tumor cells or pharmacological treatment of tumor-bearing mice with drugs inhibiting the PIK3C3/VPS34 kinase activity, on tumor growth and the immune landscape of B16-F10 melanoma and CT26 colorectal cancer [1].
Genetic targeting of PIK3C3/VPS34 in tumor cells or pharmacologically inhibiting its activity in tumors using two PIK3C3/VPS34 inhibitors (PIK3C3/VPS34i) SB02024 (Sprint Bioscience) or SAR405 (Sanofi), significantly decrease tumor growth and tumor weight, and improved mice survival in both B16-F10 melanoma and CT26 colorectal cancer. Similar results were obtained in Renca renal cell carcinoma and genetically engineered melanoma mouse (GEMM) tumors transplanted in immunocompetent mice. Consistent with the fact that targeting PIK3C3/VPS34 (either genetically or pharmacologically) fails to show any effect on tumor growth in immunocompromised NOD SCID Gamma (NSG) mice, our results indicate that PIK3C3/VPS34-dependent inhibition of tumor growth involves the host immune system. Comprehensive phenotyping of lymphoid and myeloid subsets reveals an increased infiltration of major immune effector cells (NK, CD8+ and CD4+ T cells, DC and M1 macrophages) in PIK3C3/VPS34i-treated B16-F10 and CT26 tumors compared to control tumors. We discovered that the major immune cell subsets mediating the inhibition of tumor growth in PIK3C3/VPS34i-treated mice are NK and CD8+ T cells, as such inhibition is no longer observed in NK- and CD8-depleted tumor-bearing mice. By assessing the expression of several proinflammatory chemokines/cytokines involved in the recruitment of CD8+ T cells into the tumor microenvironment, we showed that PIK3C3/VPS34 inhibition induces the release, most likely by tumor cells, of proinflammatory CCL5 and CXCL10 in the tumor microenvironment. Mechanistically, we demonstrated that PIK3C3/VPS34 inhibitors activate signaling pathways mediated by the STAT1-IRF7 axis in tumor cells (Figure 1).
Figure 1.
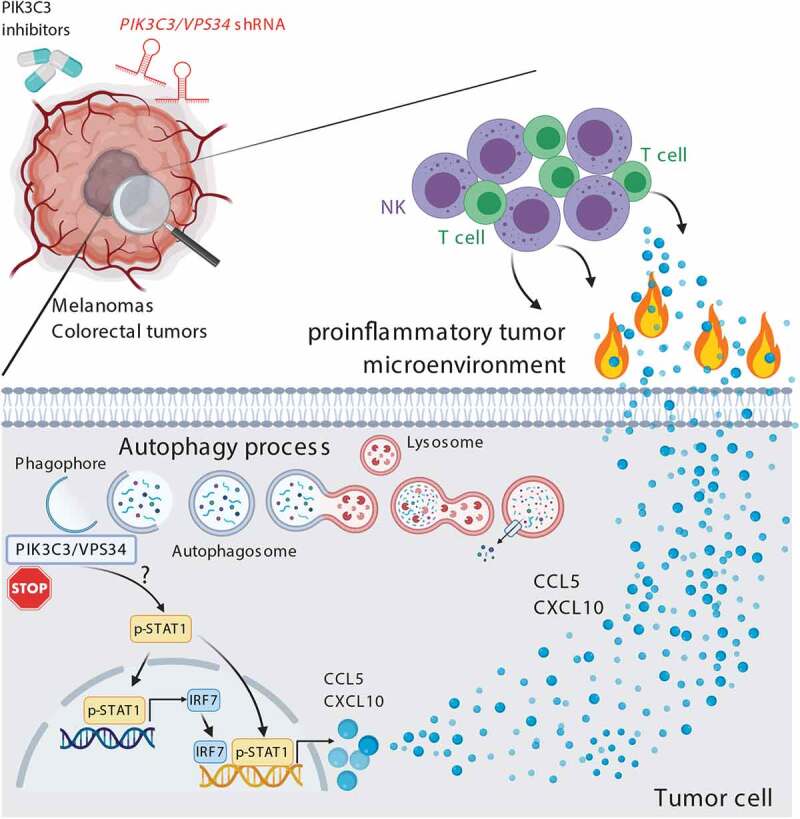
Genetic inhibition of the autophagy-related protein PIK3C3/VPS34 by shRNA in melanoma and colorectal tumor cells or pharmacological treatment of tumor-bearing mice with selective PIK3C3/VPS34 inhibitors (SB02024 or SAR405) induces the phosphorylation of STAT1 by a mechanism that is not yet defined. Phosphorylated STAT1 directly, or indirectly via the activation of IRF7 (interferon regulatory factor 7), induces the expression of the pro-inflammatory chemokines CCL5 and CXCL10. These chemokines induce an inflammatory tumor microenvironment and result in driving more NK and CD8+ T cells into the tumor bed. PIK3C3/VPS34i-treated tumors become hot and therefore susceptible to anti-PDCD1- and anti-CD274-based immunotherapy. Combining PIK3C3/VPS34i with anti-PDCD1 and anti-CD274 improves the therapeutic benefit of immunotherapy, and significantly decreases the tumor growth
Our data described above strongly argue that PIK3C3/VPS34i-treated B16-F10 and CT26 tumors display all characteristics to be susceptible to ICBs. Indeed, we showed that combining PIK3C3/VPS34i improves the therapeutic benefit of anti-PDCD1 and anti-CD274 immunotherapy in both melanoma and colorectal cancer. Collectively, our results demonstrate that cancer immunotherapy would benefit from the ability of PIK3C3/VPS34 inhibition to decrease tumor growth and modulate the tumor immune landscape most likely by switching the cold immune desert tumor microenvironment into a hot highly infiltrated immune supportive tumor microenvironment. An important issue that arises from our study is to determine whether the effects described here are specific to PIK3C3/VPS34 or could also be observed by targeting genes encoding other autophagy-related proteins. It would be interesting to investigate whether targeting several genes, involved in multiple steps of autophagy, would elicit similar effects, thus providing additional pharmacological targets to be combined with ICBs.
Acknowledgments
This work was supported by grants from Luxembourg National Research Fund C18/BM/12670304/COMBATIC; FNRS Televie (grants 7.4606.18); Fondation Cancer Luxembourg (FC/2018/06); Kriibskrank Kanner Foundation (2019); Janssen Cilag Pharma; Roche pharma and Action LIONS Vaincre le Cancer Luxembourg.
Disclosure statement
SP and ADM are employees and shareholders at Sprint Bioscience. All other authors declare that they have no competing interests.
Reference
- [1].Noman MZ, Parpal S, Moer KV, et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci Adv. 2020;6(18):eaax7881. [DOI] [PMC free article] [PubMed] [Google Scholar]