

ABSTRACT
RNautophagy and DNautophagy (RDA) are unconventional autophagic pathways where nucleic acids are directly transported through the lysosomal membrane, then degraded inside lysosomes. We have previously shown that bitopic protein LAMP2C and putative RNA transporter SIDT2, both lysosomal membrane proteins, mediate the direct transport of nucleic acids into lysosomes and that LAMP2C interacts with the nucleic acids and functions as a receptor during RDA. Because SIDT2-mediated RDA occurs in isolated lysosomes that lack LAMP2C, in this study, we tested the hypothesis that SIDT2 itself could also interact with the nucleic acids. Our results show that SIDT2 directly binds RNA and DNA through an arginine-rich motif (ARM) located within its main cytosolic domain, and disruption of this motif dramatically impairs SIDT2-mediated RNautophagic activity. We also found that SIDT2 interacts with exon 1 of HTT (huntingtin) transcript through the ARM in a CAG-dependent manner. Moreover, overexpression of SIDT2 promoted degradation of HTT mRNA and reduced the levels of polyglutamine-expanded HTT aggregates, hallmarks of Huntington disease. In addition, a comparative analysis of LAMP2C and SIDT2 functions at the cellular level revealed that the two proteins exert a synergistic effect on RNautophagic activity and that the ARMs which mediate the interactions of SIDT2 and LAMP2C with RNA are essential for the synergy. Together, our results point out the importance of nucleic acid-binding capacity of SIDT2 for its function in translocating nucleic acids through the lipid bilayer and suggests a potential application of RNautophagy activation to reduce the expression levels of disease-causing toxic proteins.
Abbreviations: ACTB/β-actin: actin beta; ARM: arginine-rich motif; CBB: Coomassie Brilliant Blue; CD: cytosolic domain; COX4I1/COX4: cytochrome c oxidase subunit 4I1; E. coli: Escherichia coli; EGFP: enhanced green fluorescent protein; EtBr: ethidium bromide; FITC: fluorescein isothiocyanate; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GOLGA2/GM130: golgin A2; GST: glutathione S-transferase; HRP: horseradish peroxidase; HSPA5/GRP78: heat shock protein family A (Hsp70) member 5; HTT: huntingtin; HTTex1: exon 1 of the HTT gene; LAMP2: lysosomal associated membrane protein 2; LMNA: lamin A/C; PAGE: polyacrylamide gel electrophoresis; PBS: phosphate-buffered saline; PEI: polyethyleneimine; polyQ: polyglutamine; qPCR: quantitative PCR; RAB5A: RAB5A, member RAS oncogene family; RDA: RNautophagy and DNautophagy; SCARB2/LIMP2: scavenger receptor class B member 2; SDS: sodium dodecyl sulfate; SID-1: systemic RNA interference deficient-1; SIDT2: SID1 transmembrane family member 2; WT: wild type.
KEYWORDS: Arginine-rich motif, autophagy, DNA, DNautophagy, LAMP2C, lysosome, RNA, RNautophagy, SIDT2
Introduction
RNautophagy and DNautophagy (RDA) are unconventional types of autophagy where RNA and DNA are directly transported through the lysosomal membrane in the presence of ATP, then degraded inside lysosomes [1–3]. We have reported that LAMP2C (lysosomal associated membrane protein 2 isoform C) and SIDT2 (SID1 transmembrane family member 2), transmembrane proteins that mainly localize to the lysosomal membrane [4–8], mediate the transport of nucleic acids into lysosomes in the process of RDA and also interact with each other [1,2,8–10].
LAMP2C is one of the 3 alternative splice isoforms of LAMP2 [11,12] and comprises a highly-glycosylated lumenal region, a single transmembrane region, and a short cytoplasmic segment of 11 amino acids [7,13]. The cytoplasmic sequence of LAMP2C directly binds to RNA and DNA via its arginine-rich motif (ARM) [14], and LAMP2C functions as a receptor during RDA [1,2]. In a previous study, we investigated the nucleic acid sequences that interact with LAMP2C. We found that the cytosolic tail of LAMP2C directly binds to poly-G and poly-dG but does not to poly-A, poly-C, poly-U, poly-dA, poly-dC or poly-dT [15]. Using isolated lysosomes, we further showed that poly-G/dG is transported to lysosomes in the presence of ATP, whereas poly-A/dA, poly-C/dC, poly-U, and poly-dT are not [15]. The binding selectivity of LAMP2C to the oligonucleotides perfectly correlated with the substrate selectivity of RDA in vitro, suggesting that substrate nucleic acids first interact with mediating membrane proteins to be transported across the lysosomal membrane into the lumen.
SIDT2 is the lysosomal vertebrate ortholog of the Caenorhabditis elegans RNA channel SID-1 (systemic RNA interference deficient-1) [16] and, therefore, a putative nucleic acid transporter on the lysosomal membrane. We have previously shown that Sidt2 knockdown inhibits up to 50% of RNA degradation at the cellular level in mouse embryonic fibroblasts. Suppression of the lysosomal function reverses the inhibition, strongly suggesting that SIDT2-mediated RNautophagy is a major RNA degradation pathway [8]. We have also found that SIDT2 overexpression notably increases intracellular RNA degradation in mouse neuroblastoma Neuro2a cells, and SIDT2 localization to the lysosomal membrane is necessary for SIDT2 function in the process of RNautophagy [17]. In addition, we reported that activation of RNautophagy by SIDT2 overexpression is observed in isolated lysosomes derived from lamp2 knockout cells [8] and that RDA occurs in isolated lysosomes derived from the brains of lamp2 knockout mice [1,2], suggesting that other lysosomal membrane proteins besides LAMP2C also function as receptors during RDA. Based on the above research results, and taking into consideration that it is unclear whether nucleic acids interact with SIDT2 during RDA, we hypothesized that SIDT2 could also bind to nucleic acids. SIDT2 is predicted to comprise an N-terminal glycosylated luminal region, a main cytosolic domain (CD), nine transmembrane helices, and a short C-terminal cytoplasmic tail [6]. Here, we investigated whether the CD of SIDT2 interacts with nucleic acids.
Results
The main CD of SIDT2 is capable of binding nucleic acids
The main CD of SIDT2 is located between transmembrane domains 1 and 2 (Figure 1A) and consists of amino acid residues 316 to 447. To investigate whether CD binds to nucleic acids, glutathione S-transferase (GST)-tagged CD (GST-CD) was expressed in Escherichia coli (E. coli), then purified using GST-tag (Figure 1B). Then we performed an affinity-isolation assay using GST-CD and purified total RNA derived from mouse brain (total RNA) or purified plasmid DNA. We found that both RNA and DNA were affinity-isolated with GST-CD, but not with GST alone (Figure 1C,D), indicating that SIDT2 CD directly binds to nucleic acids.
Figure 1.
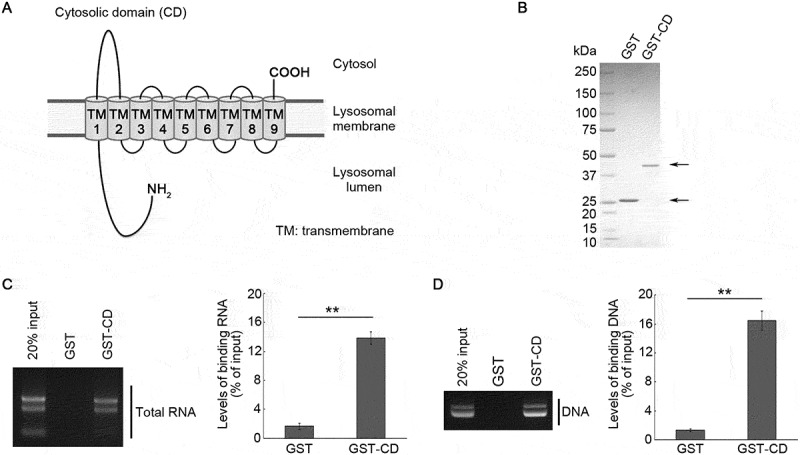
Interaction of SIDT2 CD with RNA and DNA. (A) Schematic diagram of the SIDT2 structure. (B) GST and GST-CD purified from Rosetta 2 competent cells were separated by SDS-PAGE. Following separation, we stained the resolved proteins with CBB. (C, D) Affinity-isolation assays were performed using GST or GST-CD and 1 μg of total RNA extracted from mice brains (C) or 1 μg of plasmid DNA (D). After elution, levels of affinity-isolated RNA or DNA were analyzed by agarose gel electrophoresis, followed by EtBr staining. The intensities of nucleic acid signals were quantified. **, P < 0.01 (n = 3)
To assess whether SIDT2 is involved in the in vitro RDA-substrate selectivity mentioned in the introduction, we constructed fluorescein isothiocyanate (FITC)-labeled poly-G/dG, poly-A/dA, poly-C/dC, poly-U and poly-dT (Figure 2A,B). We tested the interaction between CD and these oligonucleotides. CD bound to poly-G and poly-dG but did not bind to poly-A, poly-C, poly-U, poly-dA, poly-dC, and poly-dT (Figure 2C,D).
Figure 2.
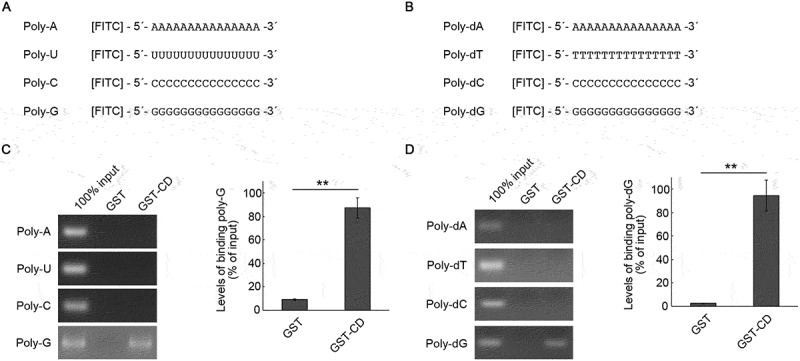
Interaction of SIDT2 CD with poly-G and poly-dG oligonucleotides. (A, B) Schematic diagrams of FITC-conjugated RNA (A) and DNA (B) used for affinity-isolation assays. (C) Affinity-isolation assays were performed using 1 pmol of poly-A, poly-U, poly-C, and poly-G and GST or GST-CD. (D) Affinity-isolation assays were performed using 1 pmol of poly-dA, poly-dT, poly-dC, and poly-dG and GST or GST-CD. **, P < 0.01 (n = 3)
The second half region of SIDT2 CD composed of amino acids 378 to 447 binds to RNA/DNA
We next sought to determine the specific region of CD that interacts with nucleic acids. CD was divided into two regions (Figure 3A). The first half region, comprising amino acids 316 to 377, and the second half region, comprising amino acids 378 to 447, were referred to as CD-1 and CD-2, respectively (Figure 3A). GST-tagged CD-1 (GST-CD-1) and CD-2 (GST-CD-2) were constructed and purified using E. coli (Figure 3B), then an affinity-isolation assay was performed using these recombinant proteins and total RNA. As a result, GST-CD-1 did not bind to RNA, while GST-CD-2 did (Figure 3C). Furthermore, as a result of a similar experiment using plasmid DNA, GST-CD-1 did not bind to DNA, but GST-CD-2 did (Figure 3D). These results indicate that the RNA/DNA binding domain is present in the second half region of SIDT2 CD.
Figure 3.
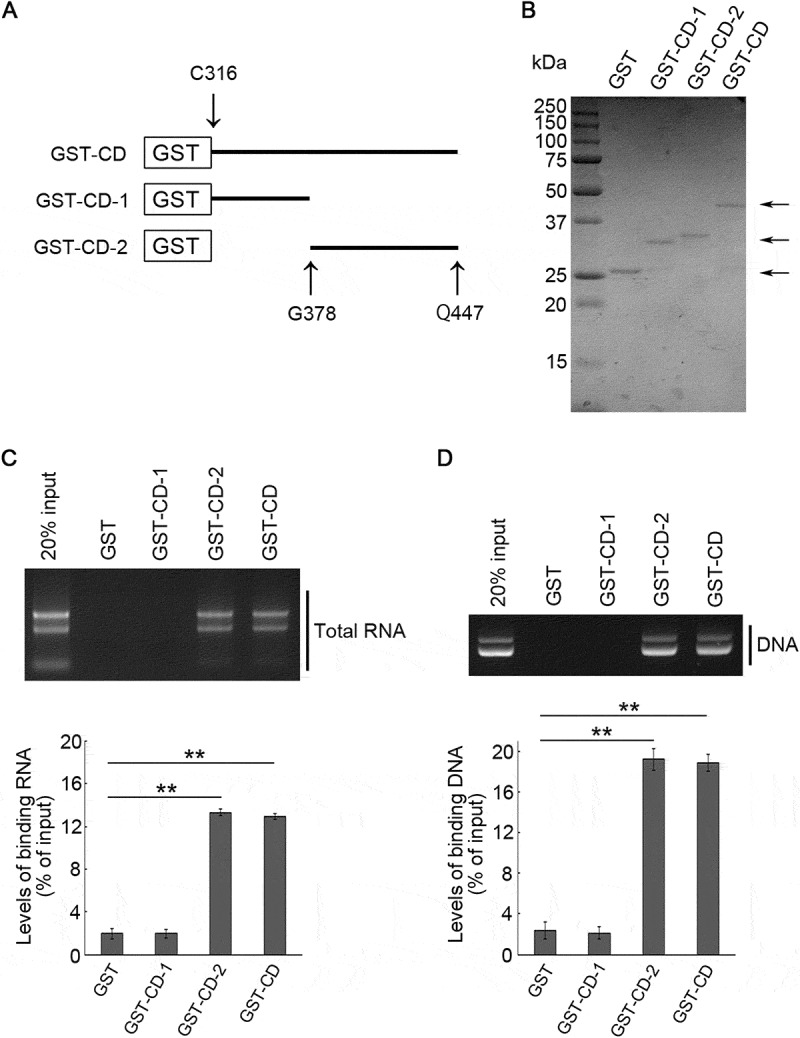
Interaction of the second half region of SIDT2 CD with RNA and DNA. (A) Schematic diagram of the recombinant GST-CD deletion constructs. (B) GST-fused proteins purified from Rosetta 2 competent cells were stained with CBB. (C, D) Affinity-isolation assays were performed using GST or GST-fused proteins and 1 μg of purified total RNA (C) or 1 μg of plasmid DNA (D). After elution, levels of affinity-isolated RNA and DNA were analyzed by agarose gel electrophoresis, followed by EtBr staining. Intensities of nucleic acids signals were quantified. **, P < 0.01 (n = 3)
Arginine residues at positions 436, 440 and 443 are necessary for binding of SIDT2 CD and RNA/DNA
Subsequently, amino acid residues required for binding of SIDT2 CD and nucleic acids were analyzed. Because SIDT2 possesses a nucleic acid binding selectivity similar to LAMP2C (Figure 2) [15], we predicted SIDT2 to possess a nucleic acid recognition motif that is similar to LAMP2C. We have reported that the C-terminal cytoplasmic region of LAMP2C binds to nucleic acids via an ARM commonly known as an RNA binding motif [14]. In ARMs, arginine residues are densely present in the nucleic acid binding domain, and basic side chains of arginine residues bind to nucleic acids [18,19]. In addition, lysine residues, which are also basic amino acid residues, assist in binding to nucleic acids [18]. We searched for arginine and lysine residues present in SIDT2 CD-2 and found that arginine and lysine residues were densely present on the C-terminal side of CD-2 (Figure 4A). Therefore, we predicted that the arginine residues at positions 436, 440, and 443 in this region, conserved among species (Figure 4B), are important for binding with nucleic acids.
Figure 4.
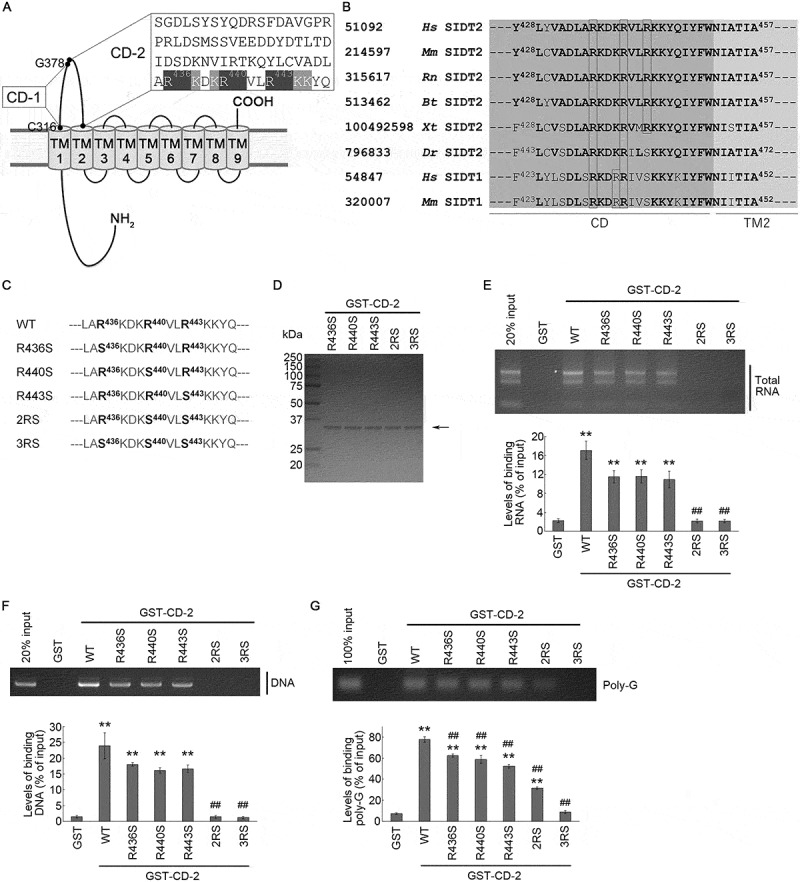
Requirement of arginine residues for interaction of the CD-2 of SIDT2 with RNA and DNA. (A) Amino acid sequence of SIDT2 CD-2. (B) Amino acid sequences in the proximity of transmembrane domain 2 of SIDT2 and SIDT1 from various species. On the outer left are the NCBI gene identification numbers, then the species names, followed by the common names of the proteins. Hs, Homo sapiens; Mm, Mus musculus; Rn, Rattus norvegicus; Bt, Bos taurus; Xt, Xenopus tropicalis; Dr, Danio rerio. Amino acids conserved among species are shown in bold letters. Arginine residues are boxed in black. TM2 stands for transmembrane domain 2. (C) Partial amino acid sequence of WT SIDT2 and mutants used in this study. (D) Purified GST-fused proteins expressed in Rosetta 2 competent cells were stained with CBB. (E, F) Affinity-isolation assays were performed using GST or GST-fused proteins and 1 μg of purified total RNA (E) or (F) 1 μg of plasmid DNA. After elution, levels of affinity-isolated RNA/DNA were analyzed by agarose gel electrophoresis, followed by EtBr staining. Intensities of nucleic acids signals were quantified. (G) Affinity-isolation assays were performed using 1 pmol of poly-G and GST-fused proteins. Intensities of nucleic acids signals were quantified. **, P < 0.01 vs GST; ##, P < 0.01 vs WT (n = 3)
To test this hypothesis, we constructed R436S, R440S, and R443S GST-CD-2, and we substituted each arginine residue with a serine residue, a 2RS GST-CD-2, which contains both R440S and R443S mutations, and a 3RS GST-CD-2, which contains R436S, R440S and R443S mutations (Figure 4C). Then, we prepared these GST-tagged peptides using E. coli (Figure 4D), and the nucleic acid binding activity was analyzed. As a result of the affinity-isolation assay using total RNA, binding levels of RNA with R436S, R440S, or R443S mutants were reduced by approximately 40% compared with wild type (WT) CD-2 (Figure 4E). Furthermore, we found that the 2RS or 3RS mutants eliminated the RNA-binding ability (Figure 4E). In the affinity-isolation assay using plasmid DNA, R436S, R440S, or R443S mutations also decreased DNA binding activity, and completely inhibited by 2RS or 3RS mutations (Figure 4F). An affinity-isolation assay using poly-G revealed that the binding activity of SIDT2 CD-2 was partially inhibited by R436S, R440S, R443S, or 2RS mutations, and completely by 3RS mutations (Figure 4G). These results indicate that the binding of SIDT2 CD and RNA or DNA requires arginine residues at positions 436, 440, and 443.
Mutations of arginine residues at positions 436, 440, and 443 of SIDT2 inhibit RNautophagic activity exerted by SIDT2 in vitro
We next determined whether the RNA binding ability of SIDT2 CD is required for SIDT2 function during RNautophagy by using lysosomes isolated from homogenates of Neuro2a cells (Fig. S1). We overexpressed WT, 2RS, and 3RS SIDT2 in Neuro2a cells, then lysosomes were isolated and finally performed an RNA uptake assay. Isolated lysosomes were mixed with purified total RNA and incubated in the presence of ATP. Then, we pelleted lysosomes by centrifugation, and RNA levels in the solution outside the lysosomes were analyzed. Consistent with our previous studies, overexpression of WT SIDT2 increased the RNA uptake activity of lysosomes compared with the control lysosomes (Figure 5A). 2RS mutations partially inhibited RNautophagic activity exerted by overexpression of SIDT2 and were almost completely inhibited by 3RS mutations (Figure 5A). Western blot analysis showed that in the isolated lysosomes, SIDT2 variants were overexpressed and that the levels of lysosomal marker protein SCARB2/LIMP2 did not differ among samples (Figure 5B). Thus, arginine residues at positions 436, 440, and 443 in the CD of SIDT2 are necessary for SIDT2-mediated RNautophagy in vitro.
Figure 5.
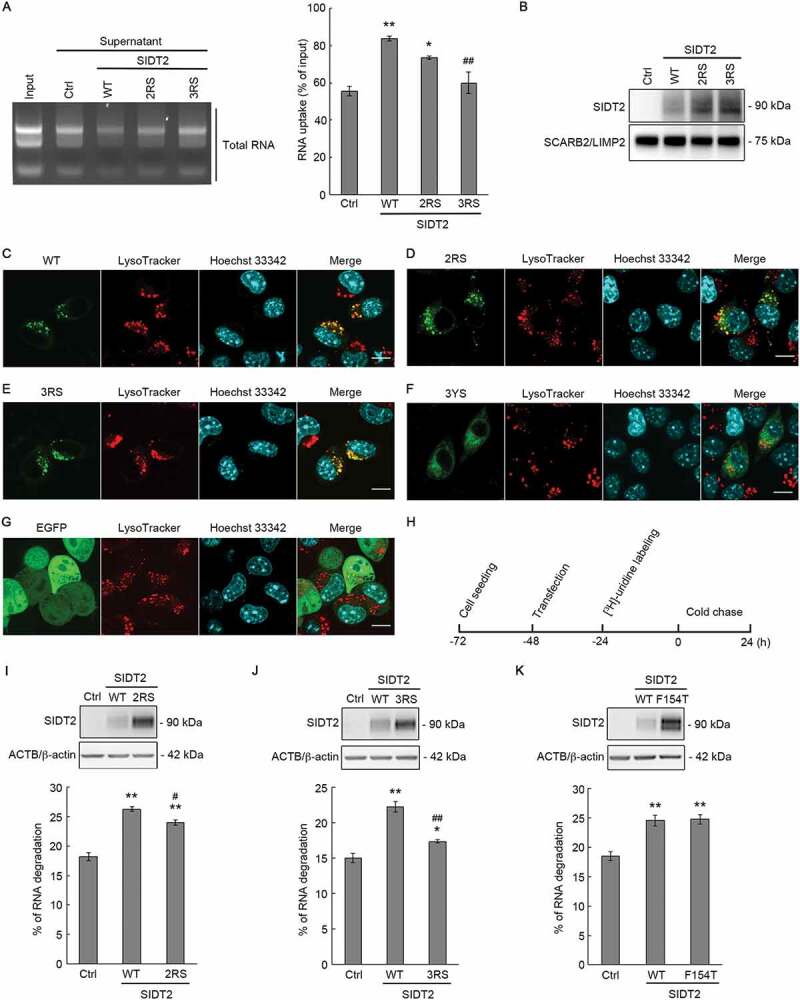
Requirement of arginine residues of SIDT2 CD for RNautophagy in vitro and at the cellular level. (A) Lysosomes were isolated from Neuro2a cells overexpressing each SIDT2 variant or control cells overexpressing EGFP, mixed with purified mouse total RNA, and incubated at 37°C for 3 min in the presence of ATP. After incubation, centrifugation was performed to separate supernatant and pellet, and RNA was detected by agarose gel electrophoresis and EtBr staining. Intensities of nucleic acid signals were quantified. **, P < 0.01 vs Ctrl; *, P < 0.05 vs Ctrl; ##, P < 0.01 vs WT (n = 3). (B) Protein levels of isolated lysosomes were analyzed by western blotting using anti-SIDT2 and anti-SCARB2/LIMP2 antibodies. (C-G) EGFP of EGFP-tagged SIDT2 variants were expressed in Neuro2a cells, and the subcellular localization was analyzed by confocal microscopy. Lysosomes were stained with LysoTracker and the nucleus with Hoechst 33,342. Scale bars: 10 μm. (H) Schematic diagram of the pulse-chase experiment performed to measure the degradation levels of endogenous RNA. (I-K, upper panels) Neuro2a cells seeded in 24-well culture plates were co-transfected with 5 ng of each vector together with carrier DNA (pCI-neo vector) up to 0.1 μg DNA/well using 0.4 μl of 1 μg/μl PEI/well and after 28 h, cells were harvested with lysis buffer and western blotting was performed using anti-SIDT2 and anti-ACTB/β-actin antibodies. (I-K, lower panels) Degradation of endogenous RNA was assessed in control and WT, 2RS, 3RS or F154T SIDT2-overexpressing Neuro2a cells by pulse-chase analysis. Cells were labeled with [3H]-uridine 24 h post-transfection. Radioactivity was expressed as a percentage of degraded RNA. n = 7 (I). n = 8 (J). n = 4 (K). **, P < 0.01 vs Ctrl, *, P < 0.05 vs Ctrl, #, P < 0.05 vs WT ##, P < 0.01 vs WT
Mutations of arginine residues at positions 436, 440, and 443 of SIDT2 inhibit SIDT2-mediated RNA degradation at the cellular level
We further investigated whether the RNA-binding capacity of SIDT2 through CD is necessary for SIDT2-mediated RNautophagy at the cellular level. First, we confirmed whether the mutations that change arginine to serine do not affect the lysosomal localization of SIDT2. We have previously reported that SIDT2 fused with EGFP at the C-terminus localizes correctly to lysosomes [8,17]. Therefore, we fused EGFP to the C-terminus of SIDT2 mutants, and the intracellular localization of these proteins was analyzed in Neuro2a cells by confocal microscopy. As a result, WT, as well as 2RS and 3RS SIDT2 colocalized with the LysoTracker-positive lysosomes (Figure 5C–E). We also used the 3YS SIDT2, a SIDT2 mutant where the three YxxΦ lysosomal targeting signals in the CD are disrupted [17]. We confirmed that EGFP-fused 3YS SIDT2 did not localize to the lysosomes in our experimental conditions (Figure 5F). We also visualized the EGFP expression pattern in Neuro2a cells and observed EGFP itself does not colocalize with lysosomes (Figure 5G).
Next, we investigated the effect of arginine to serine mutations on SIDT2-mediated RNA degradation at the cellular level using a pulse-chase assay (Figure 5H). First, we confirmed the overexpression of WT or 2RS SIDT2 in Neuro2a cells under our experimental conditions (Figure 5I, upper panel). Then, endogenous RNA of control cells and cells overexpressing WT or 2RS SIDT2 were labeled with [3H]-uridine, and the radioactivity of the labeled RNA at 0 and 24 h post-labeling was measured. Consistent with our previous study [17], the RNA degradation level was significantly increased in cells overexpressing WT SIDT2 compared with control cells. 2RS mutations decreased WT SIDT2-mediated increase in intracellular RNA degradation by approximately 30% (Figure 5I, lower panel). We performed the same assay using 3RS SIDT2 and found that the RNA degradation level in cells overexpressing 3RS SIDT2 was remarkably lower compared with that of cells overexpressing WT SIDT2 (Figure 5J). 3RS mutations inhibited the SIDT2 function by approximately 70% (Figure 5J). The side chain of arginine is positively charged while the side chain of serine is polar but not charged. To confirm that the change in the electrical charge that occurs because of arginine-to-serine substitutions does not affect the SIDT2 function, we also investigated the effect of arginine-to-lysine mutations (3RK) on the SIDT2 function by pulse-chase analysis (Fig. S2A). 3RK mutations almost completely inhibited SIDT2 function (Fig. S2B), just like 3RS mutations. This result suggests that the positive charges of the binding sites are insufficient for SIDT2 to function during RNautophagy and that arginine residues at positions 436, 440, and 443 are essential functional residues. As a negative control, we substituted valine at position 441 with leucine and leucine at position 442 with isoleucine (VLLI), then we investigated the effect of VLLI SIDT2 overexpression on RNautophagic activity (Fig. S2A). Our results showed that VLLI SIDT2 promoted intracellular RNA degradation as readily as WT SIDT2 (Fig. S2C). Together, these results indicate that arginine residues at positions 436, 440, and 443 are necessary for SIDT2-mediated RNautophagy at the cellular level.
Mutation of a phenylalanine residue at position 154 of SIDT2 did not inhibit RNA degradation mediated by SIDT2
The N-terminal region of SIDT2, located in the lysosomal lumen, can bind to RNA and a mutation that changes phenylalanine residue to threonine at position 154 (F154T) inhibits the binding capacity [20]. To further understand the nucleic acid transport mechanism of SIDT2, we also investigated whether the F154T mutation affects SIDT2-mediated RNA degradation at the cellular level. As a result, the F154T mutation did not affect the RNA degradation exerted by overexpression of SIDT2 (Figure 5K), suggesting that SIDT2 function during RNautophagy does not require the RNA binding ability of the lysosomal lumenal domain of SIDT2.
SIDT2 CD interacts with huntington disease (HD)-associated CAG RNA repeats
Under our experimental conditions, SIDT2 CD interacts exclusively with poly-G/dG (Figure 2), and almost all trinucleotides the repeats of which cause neurodegenerative disorders comprise guanine in their structures [21]. CAG repeats are the most common repeats known to cause triplet expansion disorders. To explore a possible biological significance of our findings, we assessed whether SIDT2 CD interacts with CAG repeats. We focused on the HTT (huntingtin) gene, in which the expansion of CAG repeat in exon 1 is the cause for HD. Expansion of CAG repeats cause neurodegeneration mainly through the expanded polyglutamine (polyQ) containing mutant proteins that exhibit cytotoxicity [22]. Here, we utilized a model that contains exon 1 of the HTT gene (HTTex1) generally used in HD research [23]. WT and mutant HTTex1 RNA containing 22 (HTTex1-CAG-22) and 110 (HTTex1-CAG-110) CAG repeats, respectively (Fig. S3A), were prepared and examined the interaction with SIDT2 CD-2 (Fig. S3B) by affinity-isolation assay. Approximately 3.5% of HTTex1-CAG-22 and 12.7% of HTTex1-CAG-110 (% of input) interacted with WT CD-2, while we observed no significant interaction between 3RS CD-2 and HTTex1-CAG-22 or HTTex1-CAG-110 (Figure 6A,B). These results indicate that SIDT2 interacts with HTTex1 RNA in a CAG repeat-dependent manner.
Figure 6.
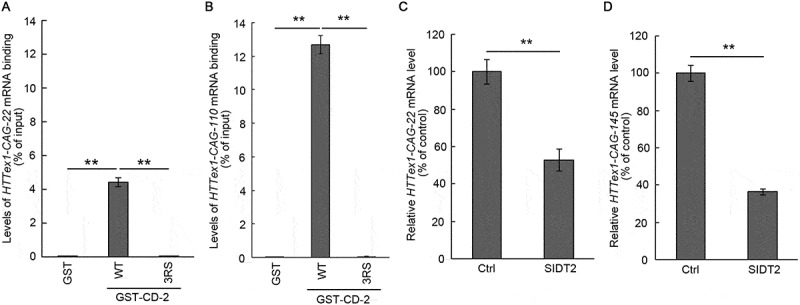
SIDT2 CD interacts with Huntington disease (HD)-associated CAG repeat RNA. (A, B) Affinity-isolation assays were performed using WT or 3RS GST-CD-2 and 1 μg of HTT RNA containing 22 (A) or 110 (B) CAG repeats. Relative levels of affinity-isolated and input RNA were analyzed by qPCR. Interaction levels were expressed as a percentage of input RNA. **, P < 0.01 (n = 3). (C, D) Effects of SIDT2 overexpression on HTTex1-CAG-22 mRNA (C) and HTTex1-CAG-145 mRNA (D) were assessed in Neuro2a cells. Quantification was performed by qPCR. RNA levels were expressed as percentage of control (cells transfected with empty vector). **, P < 0.01 (n = 3)
SIDT2 overexpression accelerated the degradation of HTTex1 mRNA and reduced the protein levels of HTTex1 with a 145 glutamine repeat tract
Because SIDT2 CD interacts with HTTex1 mRNA, we hypothesized that RNautophagy could degrade HTTex1 mRNA and reduce its expression levels. Therefore, SIDT2 and HTTex1 constructs (CAG-22 and CAG-145) were co-transfected in Neuro2a cells, and we examined the effect of SIDT2 overexpression on HTTex1 mRNA levels. We extracted RNA from the cells and analyzed expression levels of HTTex1 mRNA by quantitative PCR (qPCR). Overexpression of SIDT2 reduced the levels of HTTex1-CAG-22 and HTTex1-CAG-145 mRNA to ~53% and ~36%, respectively (% of control) (Figure 6C,D).
Next, we investigated the effect of SIDT2 overexpression on the degradation of HTTex1-CAG-145 mRNA by using a Tet-Off system. Doxycycline treatment suppressed transcription of HTTex1 in Neuro2a cells. Then, cells were harvested and analyzed for HTTex1-CAG-145 mRNA levels by qPCR. We found that overexpression of SIDT2 efficiently promoted the degradation of HTTex1-CAG-145 mRNA (Fig. S4).
We also examined the effects of SIDT2 overexpression on HTTex1 protein containing a tract of 145 glutamines (HTTex1-Q145). Mutant proteins containing expanded polyQ, including mutant HTT, are generally accepted to exert cytotoxicity via toxic gain-of-function [22]. Mutant HTT forms insoluble aggregates in cells, and aggregation correlates with toxicity in cell models as well as pathology in animal models of HD [24,25]. Soluble expanded polyQ, including monomers, have also been reported to be toxic [26]. Therefore, we analyzed levels of both sodium dodecyl sulfate (SDS)-soluble HTTex1-Q145 as well as of the insoluble aggregates. First, we confirmed that HTTex1-Q145 forms insoluble aggregates in Neuro2a cells by filter-trap assay (Figure 7A). Then, we overexpressed Neuro2a cells with HTTex1-Q145 and SIDT2, and the levels of soluble and insoluble aggregates of HTTex1-Q145 were analyzed by western blotting and filter-trap assay, respectively. We found that overexpression of SIDT2 remarkably decreased the levels of soluble HTTex1-Q145 (to 18% of control) (Figure 7B). Overexpression of SIDT2 also reduced the level of HTTex1-Q145 aggregates (Figure 7C). We performed similar experiments to analyze the effect of SIDT2 overexpression on the protein levels of SDS-soluble nonpathogenic HTTex1-Q22. SIDT2 overexpression also decreased the levels of HTTex1-Q22 (to 53% of control), but the level of decrease was less dramatic than in the case of HTTex1-Q145 (Figure 7D).
Figure 7.
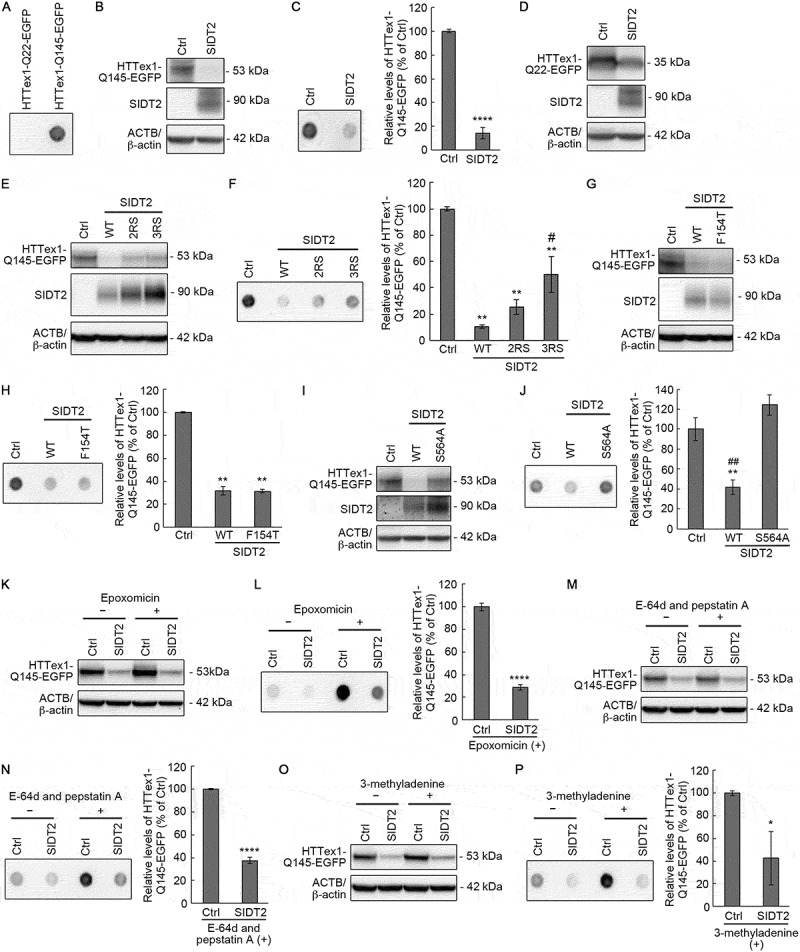
SIDT2 reduces mutant HTT levels through RNautophagy. (A) Neuro2a cells expressing HTTex1-Q22-EGFP or HTTex1-Q145-EGFP (48 h post-transfection) were subjected to filter-trap assay. (B, C) HTTex1-Q145-EGFP-expressing control or SIDT2-overexpressing Neuro2a cells (48 h post-transfection) were harvested with lysis buffer and cell lysates were subjected to western blotting using anti-GFP, anti-SIDT2 and anti-ACTB/β-actin antibodies (B) and to filter-trap assay (C). ****, P < 0.0001 (n = 3). (D) HTTex1-Q22-EGFP-expressing control or SIDT2-overexpressing Neuro2a cells (48 h post-transfection) were harvested with lysis buffer and cell lysates was subjected to western blotting using anti-GFP, anti-SIDT2 and anti-ACTB/β-actin antibodies. (E, F) HTTex1-Q145-EGFP-expressing control or WT, 2RS or 3RS SIDT2-overexpressing Neuro2a cells (48 h post-transfection) were harvested with lysis buffer and cell lysates were subjected to western blotting using anti-GFP, anti-SIDT2 and anti-β-actin antibodies (E) and to filter-trap assay (F). **, P < 0.01 vs Ctrl; #, P < 0.05 vs WT (n = 3). (G, H) HTTex1-Q145-EGFP-expressing control or WT or SIDT2F154T-overexpressing Neuro2a cells (48 h post-transfection) were harvested with lysis buffer and cell lysates were subjected to western blotting using anti-GFP, anti-SIDT2 and anti-ACTB/β-actin antibodies (G) and to filter-trap assay (H). **, P < 0.01 (n = 3). (I, J) HTTex1-Q145-EGFP-expressing control or WT or SIDT2S564A-overexpressing Neuro2a cells (48 h post-transfection) were harvested with lysis buffer and cell lysates were subjected to western blotting using anti-GFP, anti-SIDT2 and anti-ACTB/β-actin antibodies (I) and to filter-trap assay (J). **, P < 0.01 vs Ctrl; ##, P < 0.01 vs S564A (n = 3). (K, L) HTTex1-Q145-EGFP-expressing control or SIDT2-overexpressing Neuro2a cells (24 h post-transfection) were treated with 50 nM of epoxomicin for 24 h. Then, cells were harvested with lysis buffer and cell lysates were subjected to western blotting using anti-GFP, anti-SIDT2 and anti-ACTB/β-actin antibodies (K) and to filter-trap assay (L). ****, P < 0.0001 (n = 3). (M, N) HTTex1-Q145-EGFP-expressing control or SIDT2-overexpressing Neuro2a cells (24 h post-transfection) were treated with 50 μg/mL of E-64d and 50 μg/mL of pepstatin A for 24 h. Then, cells were harvested with lysis buffer and cell lysates were subjected to western blotting using anti-GFP, anti-SIDT2 and anti-ACTB/β-actin antibodies (M) and to filter-trap assay (N). ****, P < 0.0001 (n = 3). (O, P) HTTex1-Q145-EGFP-expressing control or SIDT2-overexpressing Neuro2a cells (24 h post-transfection) were treated with 5 mM of 3-methyladenine for 24 h. Then, cells were harvested with lysis buffer and cell lysates were subjected to western blotting using anti-GFP, anti-SIDT2 and anti-ACTB/β-actin antibodies (O) and to filter-trap assay (P). *, P < 0.05 (n = 3)
CD-2- but not N-terminal-mediated nucleic acids-binding ability of SIDT2 were required to decrease mutant HTTex1 protein expression
Next, we investigated the relationship between the decrease of soluble HTTex1-Q145 and aggregated forms by SIDT2 overexpression and SIDT2 ability to bind nucleic acids. First, we examined the effects of arginine-to-serine substitutions in SIDT2 CD-2. The same experiments, as described in the previous section, were performed using 2RS and 3RS SIDT2. Our results showed that the replacement of arginine residues suppressed the decrease of soluble and insoluble HTTex1-Q145 mediated by WT SIDT2 (Figure 7E,F). Next, we investigated the effect of an F154T mutation on the SIDT2 function, and our results showed that SIDT2F154T decreased HTTex1-Q145 levels as readily as WT SIDT2 (Figure 7G,H). These results indicate that CD-2- but not N-terminal-mediated nucleic acids-binding ability of SIDT2 is required to decrease HTTex1-Q145 levels and strongly suggest that SIDT2 reduces HTTex1-Q145 levels due to degradation of HTTex1-CAG-145 mRNA.
Nucleic acid transport activity of SIDT2 was required to decrease mutant HTTex1 protein expression
Previous studies have reported that S564A mutation completely inhibits nucleic acid transport activity of SIDT2 [9,27]. Therefore, we investigated the effect of S564A mutation on SIDT2 ability to suppress HTTex1-Q145 levels. SIDT2S564A failed to decrease HTTex1-Q145 levels in both soluble and aggregated forms (Figure 7I,J). The effect was striking in the case of HTTex1-Q145 aggregates (Figure 7J). Therefore, the reduction of HTTex1-Q145 by overexpression of SIDT2 was dependent on the nucleic acid transport activity of SIDT2.
Together, these results strongly suggest that activation of RNautophagy by SIDT2 overexpression enhances the degradation of HTTex1-CAG-145 mRNA, thereby reducing the levels of HTTex1-Q145 protein.
Decreasing of HTTex1-Q145 levels by SIDT2 overexpression was independent of the proteasomal or lysosomal proteolysis
Pathogenic HTT proteins are substrates for both the ubiquitin-proteasome system and lysosomal proteolysis [22]. To support our theory that the SIDT2-mediated decrease in HTTex1-Q145 protein level is due to degradation of HTTex1-CAG-145 mRNA by RNautophagy, we also investigated the effect of SIDT2 overexpression on HTTex1-Q145 expression level when we inhibited proteasomal or lysosomal proteolysis. To this end, we used a selective inhibitor of the proteasome, epoxomicin, selective inhibitors of lysosomal proteolysis, E-64d, and pepstatin A, and a generally used inhibitor of macroautophagy, 3-methyladenine. As a result, HTTex1-Q145 levels were also decreased by SIDT2 overexpression in epoxomicin-treated cells (Figure 7K,L). In contrast, epoxomicin treatment increased HTTex1-Q145 levels (Figure 7K,L). Overexpression of SIDT2 also decreased HTTex1-Q145 levels in E-64d and pepstatin A-, or 3-methyladenine-treated cells, whereas treatment with E-64d and pepstatin A, or 3-methyladenine increased HTTex1-Q145 levels (Figure7M–P). Together, these data indicate that the SIDT2-mediated decrease in HTTex1-Q145 protein expression levels is independent of the proteasomal and lysosomal proteolysis.
SIDT2 and LAMP2C act synergistically during RNautophagy
Until now, we identified SIDT2 and LAMP2C as mediators in the direct transport of RNA across the lysosomal membrane in the process of RNautophagy. We sought to elucidate if SIDT2 and LAMP2C function independently or coordinately during RNautophagy. SIDT2 and LAMP2C were overexpressed individually or jointly in Neuro2a cells and confirmed overexpression by western blotting (Figure 8A, left panel). Next, we measured RNA degradation at the cellular level by pulse-chase analysis. The level of RNA degradation in LAMP2C-overexpressing cells was slightly higher (~1.10-fold increase) compared with that of control cells, although the increase was not statistically significant (Figure 8A, right panel). The level of RNA degradation in SIDT2-overexpressing cells was significantly higher than that of control cells (~1.32-fold increase) and LAMP2C-overexpressing cells and (~1.20-fold increase) (Figure 8A, right panel). In addition, co-overexpression of SIDT2 and LAMP2C induced a synergistic effect on enhancing the RNA degradation level, which was significantly higher compared with the levels of both control cells and SIDT2-overexpressing cells (~1.52- and ~1.15-fold increases, respectively) (Figure 8A, right panel), indicating that SIDT2 and LAMP2C can function synergistically during RNautophagy.
Figure 8.
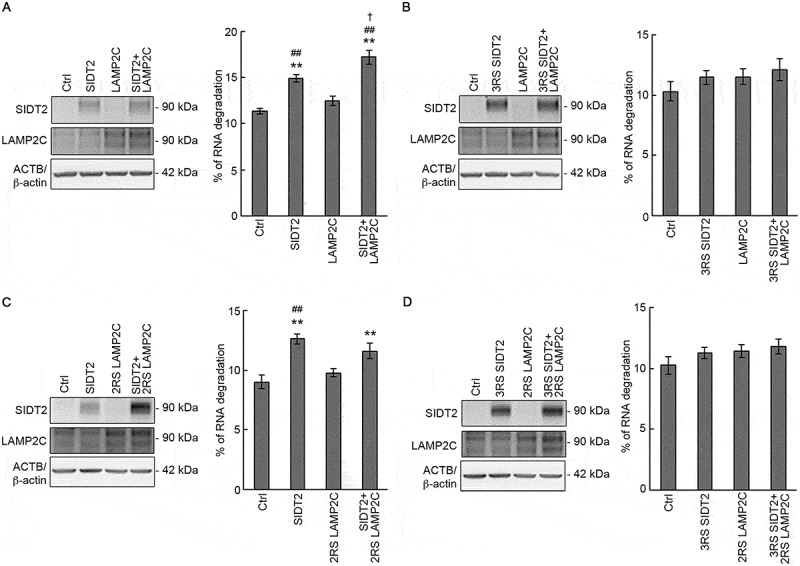
SIDT2 and LAMP2C function synergistically during RNautophagy. (A-D, upper panels) Neuro2a cells seeded in 24-well culture plates were co-transfected with 5 ng of each vector together with carrier DNA (pCI-neo vector) up to 0.1 μg DNA/well using 0.4 μl of 1 μg/μl PEI/well. After 28 h, cells were harvested with lysis buffer and western blotting was performed using anti-SIDT2, anti-LAMP2C and anti-ACTB/β-actin antibodies. (A, lower panel) Degradation of endogenous RNA was assessed in control and SIDT2, LAMP2C or SIDT2 and LAMP2C-overexpressing Neuro2a cells by pulse-chase analysis. Cells were labeled with [3H]-uridine 4 h post-transfection. Radioactivity was expressed as a percentage of degraded RNA. **, P < 0.01 vs Ctrl; ##, P < 0.01 vs LAMP2C, †, P < 0.05 vs SIDT2 (n = 12). (B, lower panel) Degradation of endogenous RNA was assessed in control and 3RS SIDT2, LAMP2C or 3RS SIDT2 and LAMP2C-overexpressing Neuro2a cells by pulse-chase analysis. Cells were labeled with [3H]-uridine 4 h post-transfection. Radioactivity was expressed as a percentage of degraded RNA. No significant difference was observed (n = 8). (C, lower panel) Degradation of endogenous RNA was assessed in control and SIDT2, 2RS LAMP2C or SIDT2 and 2RS LAMP2C-overexpressing Neuro2a cells by pulse-chase analysis. Cells were labeled with [3H]-uridine 4 h post-transfection. Radioactivity was expressed as a percentage of degraded RNA. **, P < 0.01 vs Ctrl; ##, P < 0.01 vs LAMP2C (n = 8). (D, lower panel) Degradation of endogenous RNA was assessed in control and 3RS SIDT2, 2RS LAMP2C or 3RS SIDT2 and 2RS LAMP2C-overexpressing Neuro2a cells by pulse-chase analysis. Cells were labeled with [3H]-uridine 4 h post-transfection. Radioactivity was expressed as a percentage of degraded RNA. No significant difference was observed (n = 8)
We next investigated whether the ARM within SIDT2 CD-2 is involved in the synergy. We co-overexpressed 3RS SIDT2 and WT LAMP2C in Neuro2a cells (Figure 8B, left panel), then performed pulse-chase analysis. We observed no synergistic effect between 3RS SIDT2 and WT LAMP2C. This result indicates that arginine residues at positions 436, 440, and 443 are necessary for the synergy between SIDT2 and LAMP2C.
We have previously reported that human LAMP2C interacts with nucleic acids, and the two consecutive arginine residues within its cytoplasmic tail (R401 and R402) mediate the interaction [14]. We constructed mutant murine LAMP2C in which we substituted arginine residues corresponding to R401 and R402 of human LAMP2C (R406 and R407) with serine (2RS LAMP2C). Then, we investigated the effect of WT SIDT2 and 2RS LAMP2C co-overexpression on RNautophagy activity. No synergistic effect was observed between WT SIDT2 and 2RS LAMP2C, indicating that the synergy between SIDT2 and LAMP2C needed R406 and R407 of LAMP2C (Figure 8C). 3RS SIDT2 and 2RS LAMP2C co-overexpression also had no synergistic effect on RNautophagy activity (Figure 8D). Together, these results indicate that SIDT2 and LAMP2C act synergistically during RNautophagy and that synergy is mediated by the capacities of both SIDT2 and LAMP2C to interact with nucleic acids.
Discussion
In general, transporters are transmembrane proteins that bind to and transport substrates [28]. For example, glucose transporters and ATP-binding cassette transporters bind to substrates and function in the selectivity of substrates to be transported [29,30]. However, it was unknown whether SIDT2 binds to substrate nucleic acids during RDA. In this study, we found that the main CD of SIDT2 binds to RNA and DNA and that arginine residues at positions 436, 440, and 443 in CD are necessary for this binding. Mutations in these arginines showed inhibition of RNautophagic activity induced by SIDT2 overexpression in vitro and at the cellular level, indicating that binding of SIDT2 CD to RNA is important during RNautophagy.
ARMs usually consist of under 20 amino acid residues, containing multiple arginine residues and other basic residues [18,31]. The ARM containing arginine residues at positions 436, 440, and 443 of SIDT2 also possess multiple lysine residues (Figure 4A), and therefore it is a typical ARM. The ARM of SIDT2 is located on the C-terminal side of CD and is adjacent to the transmembrane domain 2 (Figure 4A). We previously reported that LAMP2C possesses a typical ARM, which is located on the cytoplasmic side, adjacent to the transmembrane domain, and binds to RNA and DNA [14]. Our previous and present studies revealed that both the cytosolic sequence of LAMP2C and SIDT2 CD directly bind to poly-G and poly-dG, but does not to poly-A, poly-C, poly-U, poly-dA, poly-dC and poly-dT in vitro (Figure 2) [15]. Thus, SIDT2 and LAMP2C possess the same type of nucleic acid binding motif, and the binding selectivity for nucleic acids of the motifs is also similar. In addition, we have shown in the present study that SIDT2 and LAMP2C function in a synergistic way, and the ARMs of both SIDT2 and LAMP2C are essential for the synergy (Figure 8). Considering the findings of the present and previous studies, we proposed a current model of RDA: SIDT2 functions as both a transporter and a receptor for nucleic acids on the lysosomal membrane, and LAMP2C functions as a receptor through the ARM in its C-terminal tail, and enhances SIDT2 activity. We showed that the level of RNA degradation in SIDT2-overexpressing cells was significantly higher than in LAMP2C-overexpressing cells (Figure 8A), suggesting that SIDT2 plays a more important role than LAMP2C during RNautophagy. The current model is consistent with this result. We also showed that overexpression of LAMP2C alone did not significantly increase RNA degradation in Neuro2a cells but tended to increase degradation (Figure 8A). Because Neuro2a cells express a very low level of endogenous SIDT2 proteins compared with overexpressed SIDT2 [17], the function of LAMP2C in RDA may be dependent on the presence of SIDT2, and this notion is consistent with the current model. Mutation of the three arginine residues in SIDT2 completely abolished the nucleic acid binding capacity of SIDT2 (Figure 4) but did not completely inhibit overexpressed SIDT2-mediated RNautophagic activity in vitro (Figure 5A) and at the cellular level (Figure 5J). The possible explanation is that a receptor, such as LAMP2C, partially substitutes the nucleic acid binding ability of SIDT2. However, we cannot completely exclude the possibility that other cytoplasmic regions of SIDT2 bind to nucleic acids.
We previously reported that SIDT2 partly localizes to the plasma membrane and mediates the uptake of extracellularly added artificial nucleic acids, fully 2′-O-methylated RNA, by cells [27]. Although the physiological roles of this function remain unknown, the results of our previous studies suggest that SIDT2 can function as a bidirectional transporter for nucleic acids. This notion is consistent with the report showing that Caenorhabditis elegans SID-1 is a bidirectional RNA transporter [16]. In a previous study, we also showed that SIDT2-mediated uptake of extracellular nucleic acids is almost completely inhibited by the F154T mutation [27], which is located on the extracellular (lysosomal luminal) region of SIDT2 and inhibits the RNA binding activity of this region [20]. In the present study, the F154T mutation did not affect the SIDT2-mediated RNA degradation (Figure 5K). Collectively, the findings of the previous and present studies strongly suggest that SIDT2 function during RDA does not require F154-mediated interaction of SIDT2 with nucleic acids. We have also shown that the S564 of SIDT2 is necessary for the nucleic acid transport activity of SIDT2 both in the process of RDA and in the uptake of extracellular nucleic acids by cells; therefore, SIDT2S564A is inactive in the transport of nucleic acids in both directions [8,9,27]. Because the directionality of the SIDT2-mediated transport of nucleic acids can be distinguished by using SIDT2F154T, this mutant seems to be a useful tool for studying the SIDT2 function.
HD is a progressive brain degenerative disorder caused by abnormal CAG repeat expansion in exon 1 of the HTT gene. Expansion of CAG repeats cause neurodegeneration mainly through the expanded polyQ-containing mutant HTT proteins, which exhibit cytotoxicity [22]. In our experimental model, SIDT2 interacts with HTT exon 1 mRNA in a CAG-dependent manner, and SIDT2 overexpression promotes HTT exon 1 mRNA degradation, resulting in decreased levels of both soluble and insoluble HTT protein (Figures 6 and 7). Expansion of CAG repeats is the cause of at least nine neurodegenerative conditions generically named polyQ diseases [21], and the main pathologic mechanism of these diseases is not the loss-of-function of a protein due to gene mutation, but the toxic gain-of-function of a protein due to the extension of the polyQ region in its structure. Although additional research is needed, degradation of mRNA that encodes polyQ-associated pathogenic proteins by activation of RNautophagy may be an effective strategy to reduce the amount of pathogenic proteins in which the polyQ region is abnormally elongated.
In conclusion, the present study identifies the importance of nucleic acid-binding capacity of SIDT2 for its function in translocating nucleic acids into lysosomes and suggests a potential application of RNautophagy activation to reduce expression levels of polyQ diseases-causing toxic proteins.
Materials and methods
Cell culture
Murine neuroblastoma Neuro2a cells (provided by Dr. Y. Suzuki [32]) were grown in Dulbecco’s modified Eagle’s medium (Life Technologies, C11995500) supplemented with 10% fetal bovine serum (Hyclone, SH30910.03) at 37°C under humidified 5% CO2 atmosphere. Cell passage was performed every time the cells reached 80–100% confluency.
Antibodies
The primary antibodies used for immunoblotting were: rabbit polyclonal anti-SCARB2/LIMP2 (Abcam, ab176317), monoclonal mouse anti-ACTB/β-actin (Sigma-Aldrich, A1978), rabbit polyclonal anti-LAMP2C antibody prepared as previously described [1,2], rabbit polyclonal anti-SIDT2 prepared as previously described [17], monoclonal mouse anti-GOLGA2/GM130 antibody (BD Transduction Laboratories, 610823), rabbit polyclonal anti-LMNA/Lamin A/C (Santa Cruz Biotechnology, sc-20680), rabbit monoclonal anti-COX4I1/COX4 (Thermo Fisher Scientific, A21348), goat polyclonal anti-GAPDH (Santa Cruz Biotechnology, sc-20357), mouse monoclonal anti-KDEL (Enzo Life Science, SPA-827), mouse monoclonal anti-GFP (Santa Cruz Biotechnology, sc-9996), rabbit polyclonal anti-RAB5A (Santa Cruz Biotechnology, sc-309) and goat polyclonal anti-GST (GE Healthcare, 27-4577-01). The secondary antibodies used was horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Thermo Fisher Scientific, 31460), HRP-conjugated anti-mouse IgG (Thermo Fisher Scientific, 31430) and HRP-conjugated anti-goat IgG (Jackson ImmunoResearch, 305-036-003).
Generation of plasmid vectors
The previously prepared pCI-neo WT mouse Sidt2, pCI-neo WT mouse Lamp2c, pEGFP WT mouse Sidt2 and pEGFP 3YS mouse Sidt2 plasmids were used [1,8,9,17,27]. The DNA encoding SIDT2 CD, CD-1, and CD-2 were inserted into the EcoRI and XhoI sites of the pGEX-6P-1 vector (GE Healthcare, 28-9546-48). All constructs encoding SIDT2 and LAMP2C mutants were generated with the Quik-Change Mutagenesis Kit (Agilent Technologies, 200518) or PrimeSTAR Mutagenesis Basal Kit (Takara, R046A) according to the manufacturer’s instructions. The DNA corresponding to HTT exon 1 was prepared as previously described [33], and the expression plasmids pcDNA3-HTT containing exon 1 of WT (CAG-22) and mutant (CAG-110) HTT were constructed by ligating the corresponding DNA and pcDNA3 vector using BamHI sites. The regulatory expression plasmids pTRE-Tight-HTT-EGFP containing exon 1 of WT (CAG-22) and mutant (CAG-145) HTT with an EGFP tag at the C-terminus were constructed as follows: HTT exon 1 DNA was inserted into the pEGFP-N1 vector (Clontech, product number is unavailable) using Bam HI sites, then HTT-EGFP DNA was ligated into the pTRE-Tight vector (Clontech, 631059). All resulting constructs were confirmed by sequencing.
Purification of GST/GST-tagged proteins
GST and GST-tagged recombinant proteins were expressed in the E. coli strain Rosetta 2 (DE3) (Merck Millipore, 71400). Protein expression was induced by adding isopropyl-β-D-thiogalactopyranoside (FUJIFILM Wako, 094–05144) to a final concentration of 0.5 mM, followed by incubation for 17 h at 20°C. After induction, the cells were harvested and lysed by sonication in phosphate-buffered saline (PBS; Life Technologies, C14190500BT) containing 0.05% Triton X-100 (Sigma-Aldrich, T8787) and protease inhibitors (Roche Diagnostics, 11873580001). GST and GST-tagged proteins were purified using Glutathione Sepharose 4B (GE Healthcare, 17-0756-01). Purified proteins were added 5 M NaCl to a final concentration of 1 M, and then incubated for 10 min on ice. After incubation, the buffer was changed to PBS with Slide-A-Lyzer MINI Dialysis Devices, 20K MWCO, 0.5 mL (Thermo Fisher Scientific, 88402).
Total RNA extraction
Total RNA was isolated from the brains of 2 male C57BL/6J mice of 10-12-weeks old using TRI Reagent (Molecular Research Center, TR118), according to the manufacturer’s instructions. All animal experiments were approved by the animal experimentation committee of the National Center of Neurology and Psychiatry (K2017013R1).
Coomassie brilliant blue (CBB) staining
One microgram of purified proteins was separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), and then stained with CBB staining solution (0.04% CBB G250 [FUJIFILM Wako, 038-17932], 60% perchloric acid [FUJIFILM Wako, 162–00695] in water). The stained gel was washed with ultrapure water until background staining was fully removed.
Affinity-isolation assay
Glutathione Sepharose 4B beads were blocked overnight with 3% bovine serum albumin in PBS at 4°C. The blocked beads were incubated at 4°C for 1 h with 1 μg of RNA or DNA and 8 nmol of GST or GST-tagged proteins in incubation buffer (0.05% Triton-X 100 in PBS). In the affinity-isolation assay with oligonucleotides, 1 pmol of oligonucleotides was used instead of 1 μg of total RNA or DNA. After incubation, beads were washed 3 times with incubation buffer. Affinity-isolated RNA was extracted using TRI Reagent. For extraction of affinity-isolated DNA, beads were incubated for 60 min at 37°C in DNA extraction buffer (10 mM Tris–HCl pH 7.5, 1 mM ethylenediaminetetraacetic acid pH 8.0, 1% SDS, 0.1 mg/ml proteinase K [FUJIFILM Wako, 169-21041] in saline-sodium citrate buffer [15 mM sodium citrate, 150 mM NaCl]), followed by phenol–chloroform extraction. FITC-labeled ssRNA and ssDNA were subjected to agarose gel electrophoresis, then detected with a WSE-6100H LuminoGraph I Imaging System (Atto, Tokyo, Japan) and quantified by densitometry. HTT RNA levels were quantified by qPCR.
Western blot analysis
Lysosomes, Neuro2a cells or Neuro2a cell homogenates were lysed with a lysis buffer containing 3% SDS, 5% glycerol, 10 mM Tris-HCl, pH 7.8, 0.02% bromophenol blue, 2% 2-mercaptoethanol and protease inhibitors (lysis buffer). Equal amounts of proteins were separated by SDS-PAGE. The resolved proteins were then transferred to a polyvinylidene difluoride membrane (BIO-RAD, 1620177) and the membrane was blocked for 60 min with 1% skim milk (FUJIFILM Wako, 190–12865) in PBS-T, then incubated overnight at 4°C with primary antibodies diluted in PBS containing 3% bovine serum albumin (Iwai Chemicals, A001) or PBS-T containing 1% skim milk. After incubation with primary antibodies, each blot was probed with an HRP-conjugated secondary antibody. The blots were developed with ImmunoStar Zeta (FUJIFILM Wako, 295-72404) or ImmunoStar LD (FUJIFILM Wako, 290-69,904), then detected using FluorChem 8000 (Alpha Innotech, CA, USA).
Uptake of DNA and RNA by isolated lysosomes
Isolation of lysosomes and uptake assays using isolated lysosomes were performed as previously described [17]. Briefly, lysosomes were isolated from Neuro2a cells using a Lysosome Enrichment Kit for Tissues and Culture Cells (Thermo Fisher Scientific, 89839) (Fig. S1). Isolated lysosomes (5–10 μg of protein) and 5 μg of total RNA or 0.5 μg of plasmid DNA were incubated in 30 μL of 0.3 M sucrose containing 10 mM 3-(N-morpholino)propanesulfonic acid (FUJIFILM Wako, 345–01804) pH 7.0 with or without an energy regenerating system (10 mM ATP, 10 mM MgCl2, 2 mM phosphocreatine [Sigma-Aldrich, P7936] and 50 μg/ml creatine phosphokinase [Sigma-Aldrich, C3755]) at 37°C. After incubation for 3 min, lysosomes were removed by centrifugation and RNA was extracted from the assay solution outside of lysosomes using TRI Reagent. For extraction of DNA, the assay solution outside the lysosomes was incubated for 60 min at 37°C with DNA extraction buffer (see the composition in the affinity-isolation assay section), followed by phenol–chloroform extraction. The levels of RNA and DNA remaining in the assay solution outside the lysosomes were subjected to agarose gel electrophoresis, stained with ethidium bromide (EtBr) (Sigma-Aldrich, E8751), visualized by illumination with ultraviolet light, then quantified using FluorChem 8000 and AlphaView Software. Input indicates 100% input.
Live cell imaging
Neuro2a cells were seeded at 3 × 105 cells/well on 35-mm μ-dishes (ibidi), and grown for 24 h, then cells were transfected with 0.5 μg of each plasmid using 2 μL of 1 μg/μL polyethyleneimine (Polysciences, 24765-2) dissolved in a buffer containing 25 mM 2-(4-[2-hydroxyethyl]piperazin-1-yl)ethanesulfonic acid pH 7 and 150 mM NaCl (1 μg/μL PEI). After 24 h post-transfection, lysosomes were stained with LysoTracker (Thermo Fisher Scientific, L7528) and the nucleus with Hoechst 33342 (Lonza, PA-3014) according to the manufacturers’ instructions. Then cells were rinsed and examined using a FV10i Confocal Laser Scanning Microscope (Olympus, Tokyo, Japan).
Measurement of endogenous RNA degradation
Intracellular RNA degradation was measured according to a previously described method [17], with slight modifications. In brief, Neuro2a cells were seeded at 0.7–1 × 105 cells/well in 24-well culture plates (Corning, CLS3527), grown for 24 h, then co-transfected with 5 ng of each vector together with carrier DNA (pCI-neo vector) up to 0.1 μg DNA/well, using 0.4 μl of 1 μg/μl PEI/well. At 4 or 24 h post-transfection, 0.15 μCi/ml [3H]-uridine (PerkinElmer, NET367001MC or Moravek, MT-602E) was added to label the RNA. At 24 h post-labeling, cells were rinsed, then cultured in culture medium supplemented with 5 mM unlabeled uridine (FUJIFILM Wako, 213–00771). After 0 and 24 h of incubation, cells were trypsinized and acid-insoluble radioactivity was measured using a Tri-Carb 3100TR Low Activity Liquid Scintillation Analyzer (PerkinElmer, MA, USA). RNA degradation levels were calculated by subtracting the relative acid-insoluble radioactivity (percent of 0 h) from 100%.
In vitro transcription
HTTex1 RNA containing 22 or 110 CAG repeats was synthesized using ScriptMAX Thermo T7 Transcription Kit (TOYOBO, TSK-101) according to the manufacture’s standard protocol. pcDNA3-HTTex1 (22 or 110 CAG repeats) plasmids were cut with Eco RV, and 1 μg of each linearized DNA was used as a template. Synthesized RNA was purified using TRI Reagent.
Quantitative PCR
cDNA was synthesized using a PrimeScript RT reagent Kit with gDNA Eraser (Perfect Real Time) (Takara, RR047A) according to the manufacturer’s protocol. HTTex1 and Actb mRNA were quantified by qPCR using a CFX96TM Real-Time System (BIO-RAD, CA, USA) with SYBR Premix Ex TaqTM II (Tli RNaseH Plus) (Takara, RR0820A). In the affinity-isolation assays, the following primers were used: HTT forward primer, 5ʹ- GGGAGACCCAAGCTTGGTACC-3ʹ; HTT reverse primer, 5ʹ- GAAGGACTTGAGGGACTCGAAG-3ʹ. To quantify the HTTex1-EGFP mRNA levels, the following primers were used: EGFP forward primer, 5ʹ- GTAAACGGCCACAAGTTCAGCGTG-3ʹ; EGFP reverse primer, 5ʹ- AAGTCGTGCTGCTTCATGTGGTCG-3ʹ; mo-use Actb forward primer, 5ʹ-CGTGCGTGACATCAAAGAGAA-3ʹ; mouse Actb reverse primer, 5ʹ-CAATAGTGATGACCTGGCCGT-3ʹ. The levels of HTTex1-EGFP were normalized by Actb levels.
HTT mRNA degradation assay
Neuro2a cells were seeded at 2 × 105 cells/well in 12-well culture plates (Corning, CLS3513), grown for 24 h, then transfected with 0.28 μg pTet-Off (Clontech, 631017), 30 ng pTRE-Tight-HTTex1-EGFP and 40 ng pCI-neo (Promega, E1841) (control) or pCI-neo-SIDT2 together with carrier DNA (pCI-neo vector) up to 0.6 μg DNA/well using 1.8 μl of 1 μg/μl PEI/well. At 48 h post-transfection, 1 μg/ml doxycycline (Clontech, 631311) was added to suppress the transcription of HTTex1-EGFP gene and after 1 h cells were harvested, and total RNA was extracted using TRI Reagent. Expression levels of HTTex1-EGFP mRNA were quantified by qPCR. Results are shown as percent of RNA degradation, which were calculated by subtracting the relative mRNA levels (percent of 0 h) from 100%.
To investigate the effect of SIDT2 overexpression on HTTex1-EGFP mRNA levels, Neuro2a cells were harvested 48 h post-transfection, RNA was extracted and HTTex1-EGFP mRNA levels were quantified by qPCR. Results are expressed as percentage of control.
Filter-trap assay
Neuro2a cells were seeded at 1.0 × 105 cells/well in 24-well culture plates. After 24 h, cells were co-transfected with 20 ng of pTRE-Tight-HTTex1-CAG-22-EGFP or HTTex1-CAG-145-EGFP) and 20 ng of pCI-neo or pCI-neo-SIDT2 together with carrier DNA (pCI-neo vector) up to 0.3 μg DNA/well, using 0.9 μl of 1 μg/μl PEI/well. After 48 h, cells were harvested with lysis buffer, then genomic DNA was cut using a syringe. A cellulose acetate membrane (GE Healthcare, 10404131) was immersed in a dilution buffer containing 1% SDS in Dulbecco’s PBS (dilution buffer), then placed on top of a Filter Paper 60 (BIO-RAD, 1620161) which was previously set in the Bio-Dot Microfiltration Apparatus (BIO-RAD, CA, USA). Samples diluted with dilution buffer were applied to wells and filtered using an aspirator. After filtration, 200 μL of Dulbecco’s PBS containing 0.1% SDS was applied, and the wells were washed by aspiration. This operation was performed twice. Next, the membrane was incubated with dilution buffer for 5 min, then subjected to western blotting analysis.
Statistical analysis
Student’s t-test was used for the comparison of two sets of data. Analysis of variance with Tukey-Kramer test was used for comparisons of more than 2 sets of data. Mean values are shown with SEM.
Supplementary Material
Acknowledgments
We thank Saito Ikawa, Yoshiko Hara and Dr. Yosif El-Darawish for technical support.
Funding Statement
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science [16H05146, 16H01211 and 19H05710 to T.K. and 17K07124 to K.W.], Grants-in-Aid for JSPS Research Fellows [15J06868 to K.H. and 15J06173 to V.R.C], a Grant-in-Aid for JSPS International Research Fellow [18F18384 to V.R.C.], research grants from Takeda Science Foundation (to T.K. and M.T.) and Intramural Research Grants for Neurological and Psychiatric Disorders (30-5 and 30-9 to T.K. and 27-9 to K.W.) from the National Center of Neurology and Psychiatry (Japan).
Disclosure statement
The authors declare no competing financial interests.
Supplemental Material
Supplemental data for this article can be accessed here.
References
- [1].Fujiwara Y, Furuta A, Kikuchi H, et al. Discovery of a novel type of autophagy targeting RNA. Autophagy. 2013;9:403–409. PMID:23291500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fujiwara Y, Kikuchi H, Aizawa S, et al. Direct uptake and degradation of DNA by lysosomes. Autophagy. 2013;9:1167–1171. PMID:23839276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. PMID:26799652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schroder B, Wrocklage C, Pan C, et al. Integral and associated lysosomal membrane proteins. Traffic. 2007;8:1676–1686. PMID:17897319. [DOI] [PubMed] [Google Scholar]
- [5].Chapel A, Kieffer-Jaquinod S, Sagne C, et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol Cell Proteomics. 2013;12:1572–1588. PMID:23436907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jialin G, Xuefan G, Huiwen Z.. SID1 transmembrane family, member 2 (Sidt2): a novel lysosomal membrane protein. Biochem Biophys Res Commun. 2010;402:588–594. PMID:20965152. [DOI] [PubMed] [Google Scholar]
- [7].Eskelinen EL, Cuervo AM, Taylor MR, et al. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic. 2005;6:1058–1061. PMID:16190986. [DOI] [PubMed] [Google Scholar]
- [8].Aizawa S, Fujiwara Y, Contu VR, et al. Lysosomal putative RNA transporter SIDT2 mediates direct uptake of RNA by lysosomes. Autophagy. 2016;12:565–578. PMID:27046251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aizawa S, Contu VR, Fujiwara Y, et al. Lysosomal membrane protein SIDT2 mediates the direct uptake of DNA by lysosomes. Autophagy. 2017;13:218–222. PMID:27846365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fujiwara Y, Wada K, Kabuta T.. Lysosomal degradation of intracellular nucleic acids-multiple autophagic pathways. J Biochem. 2017;161:145–154. PMID:28039390. [DOI] [PubMed] [Google Scholar]
- [11].Gough NR, Hatem CL, Fambrough DM.. The family of LAMP-2 proteins arises by alternative splicing from a single gene: characterization of the avian LAMP-2 gene and identification of mammalian homologs of LAMP-2b and LAMP-2c. DNA Cell Biol. 1995;14:863–867. PMID:7546292. [DOI] [PubMed] [Google Scholar]
- [12].Hatem CL, Gough NR, Fambrough DM. Multiple mRNAs encode the avian lysosomal membrane protein LAMP-2, resulting in alternative transmembrane and cytoplasmic domains. J Cell Sci. 1995;108(Pt 5):2093–2100. PMID:7657727. [DOI] [PubMed] [Google Scholar]
- [13].Fukuda M, Viitala J, Matteson J, et al. Cloning of cDNAs encoding human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. Comparison of their deduced amino acid sequences. J Biol Chem. 1988;263:18920–18928. PMID:3198605. [PubMed] [Google Scholar]
- [14].Fujiwara Y, Hase K, Wada K, et al. An RNautophagy/DNautophagy receptor, LAMP2C, possesses an arginine-rich motif that mediates RNA/DNA-binding. Biochem Biophys Res Commun. 2015;460:281–286. PMID:23839276. [DOI] [PubMed] [Google Scholar]
- [15].Hase K, Fujiwara Y, Kikuchi H, et al. RNautophagy/DNautophagy possesses selectivity for RNA/DNA substrates. Nucleic Acids Res. 2015;43:6439–6449. PMID:26038313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shih JD, Hunter CP. SID-1 is a dsRNA-selective dsRNA-gated channel. RNA. 2011;17:1057–1065. PMID:21474576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Contu VR, Hase K, Kozuka-Hata H, et al. Lysosomal targeting of SIDT2 via multiple YxxPhi motifs is required for SIDT2 function in the process of RNautophagy. J Cell Sci. 2017;130:2843–2853. PMID:28724756. [DOI] [PubMed] [Google Scholar]
- [18].Weiss MA, Narayana N. RNA recognition by arginine-rich peptide motifs. Biopolymers. 1998;48:167–180. PMID:10333744. [DOI] [PubMed] [Google Scholar]
- [19].Frankel AD. Fitting peptides into the RNA world. Curr Opin Struct Biol. 2000;10:332–340. PMID:10851193. [DOI] [PubMed] [Google Scholar]
- [20].Li W, Koutmou KS, Leahy DJ, et al. Systemic RNA interference deficiency-1 (SID-1) extracellular domain selectively binds long double-stranded RNA and is required for RNA transport by SID-1. J Biol Chem. 2015;290:18904–18913. PMID:26067272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. PMID:17417937. [DOI] [PubMed] [Google Scholar]
- [22].Shao J, Diamond MI. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet. 2007;16(2):R115–23. PMID:17911155. [DOI] [PubMed] [Google Scholar]
- [23].Mangiarini L, Sathasivam K, Seller M, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. PMID:8898202. [DOI] [PubMed] [Google Scholar]
- [24].Fan HC, Ho LI, Chi CS, et al. Polyglutamine (PolyQ) diseases: genetics to treatments. Cell Transplant. 2014;23:441–458. PMID:24816443. [DOI] [PubMed] [Google Scholar]
- [25].Cooper JK, Schilling G, Peters MF, et al. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998;7:783–790. PMID:9536081. [DOI] [PubMed] [Google Scholar]
- [26].Nagai Y, Inui T, Popiel HA, et al. A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol. 2007;14:332–340. PMID:17369839. [DOI] [PubMed] [Google Scholar]
- [27].Takahashi M, Contu VR, Kabuta C, et al. SIDT2 mediates gymnosis, the uptake of naked single-stranded oligonucleotides into living cells. RNA Biol. 2017;14:1534–1543. PMID:28277980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Quick WM editor. Transmembrane Transporters. Cham: John Wiley & Sons, Inc.; 2002. DOI: 10.1002/0471434043 [DOI] [Google Scholar]
- [29].Deng D, Sun P, Yan C, et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature. 2015;526:391–396. PMID:26176916 [DOI] [PubMed] [Google Scholar]
- [30].Yu J, Ge J, Heuveling J, et al. Structural basis for substrate specificity of an amino acid ABC transporter. Proc Natl Acad Sci U S A. 2015;112:5243–5248. PMID:25848002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bayer TS, Booth LN, Knudsen SM, et al. Arginine-rich motifs present multiple interfaces for specific binding by RNA. RNA. 2005;11:1848–1857. PMID:16314457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kabuta T, Suzuki Y, Wada K. Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem. 2006;281:30524–30533. PMID:16920710. [DOI] [PubMed] [Google Scholar]
- [33].Liu W, Goto J, Wang YL, et al. Specific inhibition of Huntington’s disease gene expression by siRNAs in cultured cells. Proc Jpn Acad. 2003;79:293–298. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.