

Abstract
Background
Ocular neovascularization is a hallmark of retinal diseases including neovascular age-related macular degeneration and diabetic retinopathy, two leading causes of blindness in adults. Neovascularization is driven by the interaction of soluble vascular endothelial growth factor (VEGF) ligands with transmembrane VEGF receptors (VEGFR), and inhibition of the VEGF pathway has shown tremendous clinical promise. However, anti-VEGF therapies require invasive intravitreal injections at frequent intervals and high doses, and many patients show incomplete responses to current drugs due to the lack of sustained VEGF signaling suppression.
Methods
We synthesized insights from structural biology with molecular engineering technologies to engineer an anti-VEGF antagonist protein. Starting from the clinically approved decoy receptor protein aflibercept, we strategically designed a yeast-displayed mutagenic library of variants and isolated clones with superior VEGF affinity compared to the clinical drug. Our lead engineered protein was expressed in the choroidal space of rat eyes via nonviral gene delivery.
Results
Using a structure-informed directed evolution approach, we identified multiple promising anti-VEGF antagonist proteins with improved target affinity. Improvements were primarily mediated through reduction in dissociation rate, and structurally significant convergent sequence mutations were identified. Nonviral gene transfer of our engineered antagonist protein demonstrated robust and durable expression in the choroid of treated rats one month post-injection.
Conclusions
We engineered a novel anti-VEGF protein as a new weapon against retinal diseases and demonstrated safe and noninvasive ocular delivery in rats. Furthermore, our structure-guided design approach presents a general strategy for discovery of targeted protein drugs for a vast array of applications.
Electronic supplementary material
The online version of this article (10.1007/s12195-020-00641-0) contains supplementary material, which is available to authorized users.
Keywords: Ocular neovascularization, Vascular endothelial growth factor receptor, Directed evolution, Affinity engineering, Nonviral gene therapy
Introduction
Vision loss is a global public health challenge that profoundly impacts patient quality of life, and also imposes an enormous burden on society as a whole.39 Leading causes of vision loss include neovascular age-related macular degeneration (NVAMD), a common cause of vision loss in adults older than 60,23 and diabetic retinopathy (DR), the top cause of blindness in working-age adults.22 NVAMD and DR are both characterized by ocular neovascularization, the growth of abnormal blood vessels with increased vascular permeability and fragility in the choroidal and retinal spaces of the eye. These irregular and leaky vessels interfere with retinal function, leading to vision impairment.19,34
Vascular endothelial growth factor (VEGF) ligands have been identified as major stimulants of choroidal and retinal neovascularization.27,38 The VEGF family of cystine knot growth factors includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factors 1 and 2 (PlGF-1 and PlGF-2).18,47 VEGF family proteins are secreted from producing cells as homodimers, and interact with the VEGF receptor (VEGFR) family of receptor tyrosine kinase (RTK) transmembrane proteins, comprised of VEGFR-1, VEGFR-2, and VEGFR-3.30 VEGF-A, which binds to both VEGFR-1 and VEGFR-2, has been identified as the most prominent regulator of blood vessel formation, playing an active role in regulating angiogenesis, vascular permeability, and inflammation.18
The VEGF/VEGFR network plays an important growth and developmental role in the eye, but also drives progression of NVAMD and DR through its promotion of neovascularization.6,20 The therapeutic benefit of blocking the VEGF/VEGFR pathway to treat retinal diseases was first demonstrated in mouse models27 and later validated in human clinical trials.4,38 Currently, the two most effective classes of clinically approved anti-VEGF drugs are monoclonal antibodies and decoy receptors.52 Antibody drugs include ranibizumab (Lucentis®, Genentech Inc.), a recombinant anti-VEGF antibody fragment that neutralizes all VEGF-A isoforms (approved by the FDA in 2006),4 and bevacizumab (Avastin®, Genentech Inc.), an anti-VEGF-A antibody that was approved in 2004 for cancer treatment but is frequently used off-label for eye diseases.8 There is one FDA approved drug in the decoy receptor category, known as aflibercept (Eylea®, Regeneron Pharmaceuticals Inc., approved in 2011). Aflibercept is a chimeric protein comprised of domain 2 of VEGFR-1 and domain 3 of VEGFR-2 fused to the Fc domain of human immunoglobulin G1 (IgG1).31 Through incorporation of the VEGF-binding domains of VEGFR, aflibercept serves as a receptor mimic with sub-picomolar affinity for all isoforms of VEGF-A, and works by trapping VEGF ligands and preventing their activation of retinal cells.13,31 Notably, the anti-VEGF antibody drugs ranibizumab and bevacizumab bind only VEGF-A isoforms, whereas the decoy receptor drug aflibercept also binds VEGF-B, PlGF-1, and PlGF-2, which may account for its superior outcomes in some patient populations.1,11 In this manuscript, we used VEGF-A165, the most prevalent of the VEGF-A isoforms, as representative of the binding of all VEGF-A isoforms to VEGFR.
Although decoy receptor drugs have greatly advanced retinal disease treatment, anti-VEGF therapies require frequent and invasive intravitreal injections at high doses over long periods of time, which leads to harmful side effects and poor patient compliance.3,14 Indeed, when subjects with NVAMD who received monthly anti-VEGF injections were enrolled in long-term studies with less frequent visits and injections, visual gains were lost.44 In another study, visual outcomes were superior when injections were administered monthly vs. as needed (i.e., only when intraretinal or subretinal fluid was present).8 Moreover, a large observational study of 2227 NVAMD patients treated with anti-VEGF injections in clinical practice found that patients received less frequent injections than in clinical trials, and the resulting outcomes were far inferior.17 Collectively, these data suggest that potent and sustained suppression of VEGF is likely to provide the best long-term outcomes in NVAMD, and that current outcomes are poor because durable suppression is not being achieved. Therefore, aflibercept provides a good starting point as an anti-angiogenic agent, but improvements are needed, such as enhancing the antagonistic potential through increased ligand binding affinity and perpetuating therapeutic persistence through sustained delivery approaches. Gene therapy via intraocular delivery of adeno-associated viral (AAV) or lentiviral (LV) vectors carrying plasmids that encode anti-VEGF proteins is one promising approach that is being explored, including the 2018 FDA approval of a one-time AAV gene therapy treatment (Luxturna®, Spark Therapeutics) for biallelic RPE65 mutation-associated retinal dystrophy.7,15,35 Although the successes of viral gene therapy are notable, there are potential drawbacks to their use. Viral vectors are potent inducers of the innate and adaptive immune system, and inflammation often occurs after intravitreal injection of AAV vectors. This is usually controlled with local or systemic steroids, but sometimes causes complications such as increased intraocular pressure. Neutralizing antibodies are commonly induced after injection of AAV vectors,12,37 and this may preclude repeated injections or treatment of the fellow eye at a later time.
Here, we used an innovative approach that combines structure-based design and protein evolution to discover and characterize novel anti-VEGF proteins based on aflibercept with enhanced affinity compared to the clinical drug. We then delivered the cDNA for the lead anti-VEGF protein into rats using an optimized nonviral gene expression vector and demonstrated strong and persistent ocular expression of the engineered molecule. We anticipate that the new proteins we have designed will serve as the basis for development of superior anti-angiogenic proteins with significant potential to improve patient outcomes in retinal diseases.
Materials and Methods
Protein Expression and Purification
Human VEGF-A Isoform 165 dimers were expressed and purified using a human embryonic kidney cell (HEK) 293F expression system (Thermo Life Technologies). A gene encoding the VEGF-A Isoform 165 (amino acids 27–191) with a C-terminal hexahistidine tag was cloned into the gWiz mammalian expression vector (Genlantis), and constructs were verified by sequence analysis. HEK 293F cells were grown to 1.2 × 106 cells per milliliter and diluted to 1.0 × 106 per milliliter. Midiprepped DNA and polyethyleneimine (Polysciences) were independently diluted to 0.05 and 0.1 mg mL−1 in OptiPro medium (Thermo Life Technologies), respectively, and incubated at room temperature for 15 min. Equal volumes of DNA and polyethyleneimine were mixed and incubated at room temperature for an additional 15 min. Subsequently, the diluted HEK 293F cells and (40 mL L−1) of DNA/polyethyleneimine mixture were added to a shaking flask and incubated at 37 °C and 5% CO2 with rotation at 125 rpm for 5 days. Transfected cells were harvested after 72 h and secreted protein was captured from the supernatant via Ni-NTA (Expedeon) affinity chromatography. Proteins were further purified to > 98% homogeneity with a Superdex 200 sizing column on a fast protein liquid chromatography (FPLC) instrument (GE Healthcare), equilibrated in HEPES-buffered saline (HBS), and purity was confirmed by SDS-PAGE analysis.
For expression of biotinylated VEGF-A, the VEGF-A Isoform 165 (amino acids 27–191) followed by a C-terminal biotin acceptor peptide (BAP)-LNDIFEAQKIEWHE was transiently transfected into HEK 293F cells, as described for non-biotinylated VEGF-A. Secreted VEGF-A was extracted from the cell supernatant via Ni-NTA affinity chromatography and then biotinylated with the soluble BirA ligase enzyme in 0.5 mM Bicine pH 8.3, 100 mM ATP, 100 mM magnesium acetate, and 500 mM biotin (Avidity) for 15 min at room temperature followed by overnight incubation at 4 °C. Excess biotin was removed by size exclusion chromatography on a Superdex 200 column using an FPLC instrument (GE Healthcare), equilibrated in HBS. Complete biotinylation of the VEGF-A protein was verified via SDS-PAGE streptavidin shift assay.
Aflibercept (sequence obtained from drugbank.ca) and engineered variants thereof were expressed through transient transfection of HEK 293F cells, as described for VEGF-A. Specifically, for wild type aflibercept, VEGFR-1 domain 2 (D2) (amino acids 129–231) were fused to VEGFR-2 D3 (amino acids 226–328), followed by the human IgG1 Fc domain. Secreted protein was captured from the supernatant via Protein G agarose (Thermo Scientific) affinity chromatography followed by size exclusion chromatography on a Superdex 200 column using an FPLC instrument (GE Healthcare), equilibrated in HBS. As receptor controls, VEGFR1 domains 2 and 3 (amino acids 106–304) and VEGFR2 domains 2 and 3 (amino acids 120–327) were each fused to human IgG1 Fc at the C-terminus (denoted VEGFR-1-Fc and VEGFR-2-Fc, respectively). Receptor controls were expressed and purified as described for aflibercept. For all Fc-fused constructs, L234A, L235A, and P329G (numbering according to Eu index of Kabat) mutations were introduced into the human IgG1 Fc sequence in order to minimize Fc receptor binding.40
Yeast Surface Binding Studies
General yeast display methodologies were modified from previously described protocols.2,9 The VEGF-A binding domains (D2 of VEGFR-1 and D3 of VEGFR-2) of aflibercept or engineered variants thereof were cloned into the pCT3CBN yeast display vector (a variant of pCT3022,9 with an N-terminal yeast agglutinin protein (Aga2) fusion followed by a 3C protease site, a C-terminal cmyc epitope tag, and BamHI/NotI gene-flanking restriction sites). After induction for 48 h, 1 × 105 yeast cells per well were transferred to a 96-well plate and incubated in PBE buffer containing titrations of biotinylated VEGF-A for 2 h at room temperature. Cells were then washed an incubated for 20 min at 4 °C with 50 nM Alexa647-conjugated streptavidin (SA-647, Thermo Scientific) and a 1:100 v/v ratio of Alexa488-conjugated anti-cmyc epitope monoclonal antibody (Cell Signaling Technologies) diluted in PBE. Expression of the cmyc tag was measured to confirm full-length expression of VEGFR-1 or VEGFR-2. After a final PBE wash, cells were analyzed for binding using a Beckman Coulter CytoFLEX flow cytometer. Binding curves were fitted to a logistic regression model and equilibrium binding constant (KD) values were determined using GraphPad Prism software, assuming first-order binding interactions. Normalized data are presented as mean fluorescence intensity (MFI) for biotinylated VEGF-A binding (as detected by SA-647) divided by MFI for anti-cmyc antibody binding (as detected by Alexa488-conjugated anti-cmyc antibody). Experiments were performed in triplicate.
Yeast Library Design and Generation
PyMOL was used to visualize the binding interface between aflibercept bound to VEGF-A by overlaying the crystallographic structures of VEGF-A bound to VEGFR-1 domain 2 (D2) (PDB ID: 5T89)29 and VEGF-A bound to VEGFR-2 D3 (PDB ID: 3V2A).5 We identified interfacing residues and quantified buried surface area for each residue using the protein interface prediction software PDBePISA.25 Structural visualization of the interface combined with PDBePISA quantitative predictions was used to select eight (aggressive) or seven (conservative) candidate residues to mutate for library generation. The library design strategy is tabulated in Fig. 2, using the numbering from the VEGF-A/VEGFR-1 crystallographic structure (PDB ID: 5T89)29 for D2 residues and the numbering from the VEGF-A/VEGFR-2 crystallographic structure (PDB ID: 3V2A)5 for D3 residues. Mutational predictions were performed using the PyMOL mutagenesis wizard.
Figure 2.

Design of VEGF antagonist libraries. The molecular structure of the interface between VEGF-A and aflibercept was modeled by overlaying the crystallographic structures of VEGF-A bound to VEGFR-1 D2 (PDB ID: 5T89)29 and VEGF-A bound to VEGFR-2 D3 (PDB ID: 3V2A).5 Detailed views of the VEGF-A/VEGFR-1 interface (a) and the VEGF-A/VEGFR-2 interfaces (b) are provided. VEGF-A is colored based on an electrostatic map, and residues that were mutagenized in the library are shown in yellow. The number of potential amino acid outcomes for each mutagenized residue is tabulated at right.
The site-directed mutagenic library used for affinity maturation of aflibercept was constructed via assembly of 16 primers spanning the aflibercept gene. The following degenerate nucleotides were used: I142 (aggressive) = DBW; I145 = NNK; P173 = NNK; Q225 = NNK; L204 = VBB; G255 = NNK; N274 = NNK; and F288 = NNK. The PCR assembly reaction was performed using Pfu Ultra DNA polymerase (Agilent) and the product was further amplified by PCR using primers containing sequence homology to the pCT3CBN yeast display vector for yeast homologous recombination. Aflibercept DNA insert was mixed with linearized vector pCT3CBN backbone and electroporated into EBY100 yeast, as previously described.2,9 Electroporation of the aggressive library yielded 2 × 108 transformants and the conservative library yielded 5 × 107 transformants. The libraries were grown in SDCAA media for 48 h prior to passaging, followed by induction in SGCAA 24 h later at an initial OD of 1. In addition, 106 yeast cells from SDCAA culture was taken to perform yeast plasmid miniprep (Zymogen) according to the manufacturer’s protocol to validate transformed library sequences. Selections on both libraries were initiated 48 h after induction.
Aflibercept Variant Yeast Library Selections
For each round, the initial number of yeast used was chosen to ensure tenfold coverage of the library. Positively selected clones from each round were grown fresh overnight at 30 °C in SDCAA liquid media (pH 4.5) for 2 days, followed by induction in SGCAA liquid media (pH 4.5) for 2 days at 20 °C.
For the first round of selection, the naïve libraries were screened against 50 nM VEGF-A tetramer selection using magnetic-activated cell sorting (MACS) with LS MACS separation columns (Miltenyi Biotec), according to the manufacturer’s protocol. The VEGF-A tetramer was formulated by incubating a 4:1 ratio of biotinylated VEGF-A to SA-647 (Thermo Scientific) for 15 min on ice. Tetramer was incubated with yeast in PBE solution [phosphate-buffered saline pH 7.2, 0.1% bovine serum albumin (BSA), and 1 mM ethylenediaminetetraacetic acid (EDTA)] for 2 h at 4 °C followed by a 15 min incubation at 4 °C in 50 μL anti-Alexa647 microbeads (Miltenyi Biotec) and 950 μL PBE. The second round consisted of a cmyc selection to isolate full-length aflibercept variants. Yeast were incubated with a 1:100 v/v ratio of Alexa647-conjugated anti-cmyc epitope monoclonal antibody (Cell Signaling Technologies) in PBE for 2 h at 4 °C, followed by a 15 min incubation at 4 °C in 50 μL anti-Alexa647 microbeads (Miltenyi Biotec) and 950 μL PBE. Each MACS selection was preceded by a negative selection in which yeast were incubated with anti-Alexa647 microbeads (Miltenyi Biotec) and 950 μL PBE and clones non-specifically binding to the microbeads were discarded.
The third round of selection was performed via fluorescence-activated cell sorting (FACS) using a FACSymphony S6 cell sorter (Becton Dickinson). Yeast libraries were incubated with 1.9 nM biotinylated VEGF-A in PBE for 2 h at 4 °C, followed by 15 min incubation with 50 nM SA-647 (Thermo Scientific) and a 1:100 v/v ratio of Alexa488-conjugated anti-cmyc epitope monoclonal antibody (Cell Signaling Technologies) in PBE at 4 °C. The fourth round of selection was a kinetic sort that was also performed using FACS on a FACSymphony S6 cell sorter (Becton Dickinson). Two tubes of each yeast library were incubated at saturating concentrations of biotinylated VEGF-A (50 nM) in PBE for 2 h at 4 °C. Aflibercept-displaying yeast were also incubated at 50 nM biotinylated VEGF-A to assist in gating for clones with slower dissociation rates relative to the wild type aflibercept. One tube from each yeast cohort was washed and incubated with tenfold excess unbiotinylated VEGF-A (500 nM) in PBE for 2 h at room temperature to allow dissociation and prevent re-binding. The sorting gate was drawn to collect yeast with a slower off-rate in comparison to the wild type aflibercept yeast. Following the fourth round of selection, individual yeast clones were plated and characterized to assess their sequences and VEGF-A binding properties.
Bio-layer Interferometry Binding Measurements
Binding data was obtained using bio-layer interferometry on an OctetRED96 instrument (Molecular Devices). For Fc-fused aflibercept and variants thereof, biotinylated VEGF-A was immobilized to streptavidin (SA)-coated biosensors (Molecular Devices) in 0.45 μm filtered PBSA (phosphate-buffered saline pH 7.2 containing 0.1% BSA). Once baseline measurements were collected in PBSA, binding kinetics were measured by submerging the biosensors in wells containing fivefold serial dilutions of the appropriate analyte for 300 s (association) followed by submerging the biosensor in wells containing only PBSA for 600 s (dissociation). An irrelevant protein was immobilized to a reference SA biosensor for subtraction of non-specific binding. Tips were regenerated in 0.1 M glycine pH 2.7. Curves were fitted using the Octet Data Analysis HT Software version 7.1 (Molecular Devices) assuming a 1:1 binding model to determine the association rates, dissociation rates, and KD values.
Polymer Synthesis and Characterization
Poly(beta-amino ester) (PBAE) was synthesized as previously described46 and as shown in Fig. 6. In a neat solution, 1,4-butanediol diacrylate and 5-amino-1-pentanol were combined at a 1.1:1 molar ratio of acrylate moieties to primary amines. This solution was stirred at 90 °C for 24 h to form the acrylate-terminated base polymer. The base polymer was dissolved in anhydrous tetrahydrofuran (THF) and further reacted by adding the end-capping molecule 1-(3-aminopropyl)-4-methylpiperazine at a final concentration of 200 mg mL−1 polymer and 0.2 M end-cap. This solution was stirred at room temperature for 1 h at 500 rpm. The final end-capped PBAE was precipitated into anhydrous diethyl ether, collected by centrifugation at 3200 rcf for 5 min at 4 °C, and washed twice with anhydrous diethyl ether. The purified PBAE was dried under vacuum for 48 h, then dissolved in anhydrous dimethyl sulfoxide (DMSO) at 100 mg mL−1 and stored at – 20 °C with desiccant in small aliquots to minimize freeze-thaw cycles.
Figure 6.

Polymeric nanoparticle synthesis and characterization. (a) PBAE was synthesized by Michael addition. (b) TEM was used to image nanoparticles after lyophilization. Scale bar: 200 nm. (c) PBAE molecular weight was characterized by GPC. Mn number-average molecular weight, Mw weight-average molecular weight, PDI polydispersity index. Lyophilized nanoparticles were characterized by DLS, NTA, and electrophoretic mobility analysis (zeta potential).
PBAE number- and weight-average molecular weight (Mn and Mw, respectively) and polydispersity index (PDI) were measured. PBAE was dissolved at 5 mg mL−1 in 94% THF, 5% DMSO, and 1% piperidine and filtered through a 0.2-µm polytetrafluoroethylene (PTFE) syringe filter. The polymer was then analyzed by gel permeation chromatography (GPC), and molecular weight was quantified using polystyrene standards.
Nanoparticle Formulation and Characterization
The sequences for Fc-fused wild type aflibercept and mutant clone D4 were inserted into the gWiz mammalian expression vector (Genlantis). PBAE and DNA were individually diluted in 25 mM sodium acetate buffer, pH 5, to 5.55 mg mL−1 and 280 µg mL−1, respectively. The PBAE and DNA solutions were mixed at a 3:2 (v/v) ratio for a 30:1 (w/w) ratio of PBAE to DNA. After 10 min of incubation at room temperature to allow self-assembly, a solution of low-endotoxin (< 50 EU mg−1) sucrose in water was added at a 3:47 (v/v) ratio of sucrose to particles for a final sucrose concentration of 30 mg mL−1. This mixture was immediately frozen at – 80 °C, lyophilized for 24 h, and then stored dry with desiccant at – 20 °C or lower until use. Before injection, nanoparticles were resuspended in water at 0.33 mg mL−1 DNA.
Hydrodynamic diameter was measured by dynamic light scattering (DLS) using a ZetaSizer Pro and nanoparticle tracking analysis (NTA) using a NanoSight NS300 (Malvern Panalytical, United Kingdom). Zeta potential was measured via electrophoretic mobility analysis (ZetaSizer Pro) by diluting nanoparticles in 0.1 × PBS. Transmission electron microscopy (TEM) was used to image nanoparticles. Particles were resuspended in water, then adsorbed to glow-discharged, carbon-coated copper grids for 2 min. Grids were rinsed three times with distilled water, negative-stained with 0.5% uranyl acetate, washed once with distilled water, and then imaged with a Hitachi H7600 TEM. All measurements were made using resuspended lyophilized nanoparticles.
For encapsulation studies, NPs were prepared by diluting DNA in 25 mM NaAc as described above and mixing with diluted PBAE at a range of mass ratios (w/w). After 10 min of incubation for NP formation, sucrose was added as described above, and the NPs were then diluted 1:11 (v/v) in 1xPBS or NaAc. Samples were mixed with 30% glycerol as a loading buffer at a 1:5 ratio (v/v) of loading buffer to NPs, then loaded into a 1% agarose gel (UltraPureTM Agarose, ThermoFisher Scientific) with 1 µg/mL ethidium bromide. Each well contained 110 ng DNA. The gel was run for 30 min at 80 V, then visualized by UV exposure.
Suprachoroidal Injection of Vector in Rats
6–8-week-old Brown Norway rats (Charles River) were anesthetized with ketamine/xylazine, and eyes were visualized with a Zeiss Stereo Dissecting Microscope. A 30-gauge needle on a 1 mL syringe was used to create a partial thickness (four-fifths through sclera) circumferential opening in the sclera 1 mm posterior to the limbus, and a 34-gauge needle with a blunt 45° bevel connected to a 5 μL Hamilton syringe (Hamilton Company) containing vector was inserted into the scleral opening with the bevel facing downward and slowly advanced through the remaining scleral fibers into the suprachoroidal space. The plunger of the syringe was slowly advanced to expand the suprachoroidal space and inject 1 μL of particles containing 1 μg of expression plasmid containing the sequences of Fc-fused wild type or mutant aflibercept and held in place for 30 s, after which the needle was withdrawn while holding a cotton-tipped applicator over the injection site. Visualization of the fundus showed a shallow choroidal detachment on the side of the injection. Antibiotic ointment (Moore Medical LLC) was applied to the ocular surface and rats were returned to their cages.
Tissue Harvesting
One month after injection, rats were euthanized and eyes were removed and fixed in 4% paraformaldehyde. Rat choroid samples were isolated under a dissection microscope and put in RIPA buffer (Sigma-Aldrich) containing protease inhibitor (Roche). Samples were sonicated for 4–5 s (Sonic Dismembrator Model 300, Thermo Fisher Scientific), cooled in an ice bath for 5 min, centrifuged for 15 min at 14,000×g, and supernatants were stored at – 80 °C.
Measurement of Human IgG1-Fc Protein in Choroid Homogenates
Total protein concentration was measured using a Bradford CCB-G250 protein-binding assay. Briefly, titrations of BSA (Millipore Sigma) ranging from 200–1400 μg mL−1 were used as standards. A quantity of 25 μL sample or standard was added to duplicate wells of 96-well plates followed by 125 μL Protein Assay Dye (diluted 1:5, Bio-Rad). After a 5-min incubation on a shaker, absorption was measured at 595 nm using a Spectra Max Plus 384 Microplate Reader (Molecular Devices). Human IgG1-Fc protein levels were measured using a sandwich ELISA (NBP2-60068; Novus Biologicals), following the manufacturer’s protocol. Statistical comparisons were performed by unpaired two-tailed Student’s t test using GraphPad Prism software. The VEGF antagonist expression study in rats was performed twice with consistent results, and representative data from one of the studies are presented.
Results
Yeast Surface Display as a Platform for Aflibercept Engineering
Yeast surface display was utilized to engineer variants of the FDA-approved decoy receptor drug aflibercept with higher affinity for VEGF-A. Since all VEGF-A isoforms contain the same binding domains within constitutive exons, the most prevalent isoform (VEGF-A165) was used for experiments. In order to validate the infrastructure for affinity engineering, we first confirmed that the binding domains of aflibercept (VEGFR-1 D2 and VEGFR-2 D3) could be functionally expressed on the surface of yeast. A positive cmyc signal demonstrated full-length expression of the aflibercept binding domains (Fig. 1a). Furthermore, on-yeast surface titration against biotinylated VEGF-A confirmed correct folding of aflibercept on yeast (Fig. 1b), and the affinity (KD = 1.8 nM) closely matched previous reports.51 We further validated our recombinantly-produced aflibercept, Fc-fused VEGFR-1 D2-D3 (denoted VEGFR-1-Fc), and Fc-fused VEGFR-2 D2-D3 (denoted VEGFR-2-Fc) via bio-layer interferometry-based titrations against VEGF-A. (Fig. 1c and Supplementary Fig. 1). Table 1 summarizes the affinity and binding kinetics of each of the three Fc-fused constructs to immobilized VEGF-A, and all measurements closely match literature reports.31 As expected, VEGFR-1-Fc has a higher affinity than VEGFR-2-Fc for VEGF-A due to a significantly slower dissociation rate (koff). Previous reports have demonstrated that aflibercept and VEGF-aflibercept complexes do not interact with VEGFR-expressing cells.28
Figure 1.

Validating the infrastructure for VEGF antagonist engineering. (a) Schematic of the yeast surface display platform for directed evolution of anti-VEGF proteins. The binding domains of aflibercept were expressed as a C-terminal fusion to the yeast agglutinin protein Aga2 for attachment to the yeast surface via disulfide interaction with the yeast cell wall protein Aga1. A C-terminal cmyc epitope tag was included for detection of full-length aflibercept expression using a fluorescent antibody. Binding of the biotinylated target protein (VEGF-A) was detected using fluorescent streptavidin. A representative flow cytometry plot depicting VEGF-A binding vs. expression of yeast surface-displayed aflibercept is shown. (b) Titration of biotinylated soluble VEGF-A on the surface of yeast displaying either aflibercept or a control protein [single-chain variable fragment (scFv) of the antibody nivolumab (Opdivo®, Bristol-Myers Squibb)] to validate proper folding. Binding was quantified by detection of fluorescent streptavidin via flow cytometry, and signal was normalized based on cmyc abundance. Data represent mean ± S.D. (n = 3). (c) Bio-layer interferometry-based analysis of the interaction kinetics of soluble Fc-fused aflibercept, Fc-fused VEGFR-1 domains 2 and 3 (VEGFR-1-Fc), and Fc-fused VEGFR-2 domains 2 and 3 (VEGFR-2-Fc) with immobilized VEGF-A. Aflibercept and VEGFR-1-Fc were used at a concentration of 100 nM and VEGFR-2-Fc was used at a concentration of 50 nM. Response signals were normalized to their respective maximum values.
Table 1.
Bio-layer-interferometry measurements of soluble Fc-fused VEGFR protein interactions with immobilized VEGF-A.
| Construct | KD (pM) | Kon (× 105 Ms−1) | Koff (× 10−4 s−1) |
|---|---|---|---|
| Aflibercept | 330 ± 3.9 | 7.1 ± 0.049 | 2.4 ± 0.023 |
| VEGFR-1-Fc | 140 ± 12 | 2.8 ± 0.023 | 0.39 ± 0.032 |
| VEGFR-2-Fc | 405 ± 11 | 4.2 ± 0.075 | 1.7 ± 0.049 |
Yeast Library Construction and Selection for High Affinity Aflibercept Variants
Aflibercept binds to VEGF-A tightly with a KD of 330 pM (Table 1). In order to improve the strength of an already high-affinity interaction, we applied structural insights from visualization and analysis of the interaction between aflibercept and VEGF-A. To model the aflibercept/VEGF-A interface, we overlaid the molecular structures of VEGF-A bound to VEGFR-1 domain 2 (D2) (PDB ID: 5T89),29 which was determined in 2017, with the structure of VEGF-A bound to VEGFR-2 D3 (PDB ID: 3V2A).5 We identified residues implicated in the VEGF-A interfaces with VEGFR-1 D2 and VEGFR-2 D3 using the protein interface prediction software PDBePISA.25 We then combined PDBePISA predictions of the buried surface area with visual inspection of each interfacial residue to select eight (aggressive) or seven (conservative) residues to mutagenize in aflibercept-based libraries (Fig. 2). Generation of both an aggressive and conservative library allowed us to assess the importance of achieving full empirical coverage of the theoretical library sample space. Our structural analyses revealed that I142, I145, P173, and L204 in VEGFR-1 D2 were key contributors to VEGF-A binding. Electrostatic mapping of VEGF-A also indicated potential electrostatic repulsion between VEGF-A and residue Q225 in VEGFR-1 D2 (Fig. 2a), rationalizing the choice of the five indicated D2 residues. For VEGF-2 D3, we observed that G255 and F288 were integral to the interface with VEGF-A, and we noticed potential electrostatic repulsion between residue N274 and VEGF-A (Fig. 2b), motivating mutagenesis of these three residues.
Following transformation of both the aggressive and conservative libraries, we conducted four rounds of selection against biotinylated VEGF-A. The first two rounds of selection were implemented using MACS, whereas the final two rounds were performed via FACS, using a kinetic sorting strategy in the final round, in which the binding of biotinylated VEGF-A was competed off by non-biotinylated VEGF-A to select for clones that have the slowest dissociation rates.16,48 Comparing round 0 (naive) and post-round 4 staining, we enriched binding of 150 nM VEGF-A by ~ 55-fold (aggressive library) and ~ 25-fold (conservative library) over the course of the selections (Fig. 3a). As anticipated, the naïve conservative library displayed more VEGF-A binding than the naïve aggressive library, although both evolved libraries attained similar levels of target binding. The round 4 kinetic sort enabled us to specifically isolate clones with the slowest dissociation rates. As shown in the pre-sort populations from round 4, both the aggressive and conservative libraries contained substantial populations of clones with slower dissociation rates than wild type aflibercept (Fig. 3b).
Figure 3.

Enrichment of enhanced-affinity aflibercept variants by directed evolution. (a) Flow cytometry plots depicting binding of 150 nM VEGF-A to the yeast-displayed aggressive and conservative aflibercept libraries at round 0 vs. post-round 4 of selections. Binding of the biotinylated target protein (VEGF-A) was detected using fluorescent streptavidin and full-length protein expression was detected using a fluorescent anti-cmyc antibody. (b) Flow cytometry plots showing the gating strategy for round 4 FACS kinetic selections. Each library was incubated with saturating concentrations of biotinylated VEGF-A (50 nM) and then washed and incubated with an excess of 500 nM non-biotinylated VEGF-A to isolate clones with slower dissociation rates compared to aflibercept. Biotinylated VEGF-A binding was detected using fluorescent streptavidin and full-length protein expression was detected using a fluorescent anti-cmyc antibody.
Characterization of Enhanced Affinity Aflibercept Variants
Following selections, we characterized the binding properties and sequences of individual clones from each library and identified three mutants that exhibited improved affinity due to an apparent reduction in koff. Two of the selected mutants (C5 and D4) came from the aggressive library, whereas one mutant (F3) came from the conservative library. Bio-layer interferometry-based binding studies were performed against immobilized VEGF-A to compare Fc-fused aflibercept to the three Fc-fused aflibercept variants (Fig. 4 and Supplementary Fig. 2). As shown, all three aflibercept variants exhibited slower dissociation rates and tighter KD values compared to wild type aflibercept (Table 2).
Figure 4.
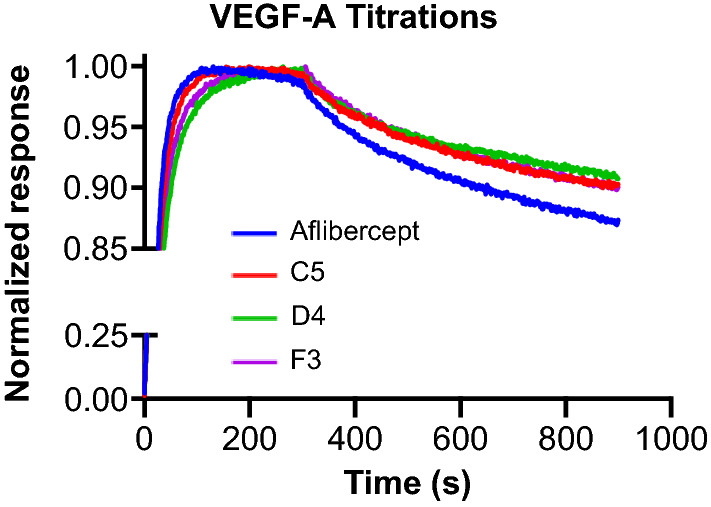
Enhanced VEGF-A binding affinity of engineered aflibercept variants. Bio-layer interferometry-based measurements of the interaction kinetics of soluble Fc-fused aflibercept and Fc-fused aflibercept variants (C5, D4, and F3) with immobilized VEGF-A. All soluble proteins were used at a concentration of 100 nM. Response signals were normalized to their respective maximum values.
Table 2.
Bio-layer-interferometry measurements of soluble Fc-fused aflibercept variant protein interactions with immobilized VEGF-A.
| Construct | KD (pM) | Kon (× 105 Ms−1) | Koff (× 10−4 s−1) |
|---|---|---|---|
| Aflibercept | 330 ± 3.9 | 7.1 ± 0.049 | 2.4 ± 0.023 |
| C5 | 270 ± 28 | 6.6 ± 0.032 | 1.8 ± 0.016 |
| D4 | 250 ± 34 | 5.7 ± 0.028 | 1.4 ± 0.018 |
| F3 | 260 ± 28 | 6.4 ± 0.030 | 1.7 ± 0.016 |
To determine the molecular changes responsible for the affinity improvements, we analyzed the sequences of our engineered aflibercept variants. In VEGFR-1 D2, positions 173 and 204 showed strong preference for the wild type residues (proline and leucine, respectively), and position 142 favored hydrophobic residues although side chain size was flexible (allowing isoleucine, valine, and phenylalanine) (Fig. 5a). Note that clone F3 was constrained to have isoleucine at position 142 since it originated from the conservative library. Position 225 was surprisingly tolerant of mutation to chemically diverse amino acids and did not show evidence of convergence (Fig. 5a). Interestingly, position 145 was mutated from isoleucine to leucine in all three affinity-matured aflibercept variants. As depicted in Fig. 5a, the predicted conformation for the mutant leucine has enhanced shape complementarity with VEGF-A due to the expected positioning of the methyl groups. In VEGFR-2 D3, position 255 exhibited a convergent mutation from the flexible glycine with the highly rigid proline, which enforces a sharp bend that could complement the concave interface of VEGF-A (Fig. 5b). The predicted potential electrostatic repulsion by the asparagine at position 277 appears to have been relieved through mutation to a nonpolar residue, although size and shape do not appear to be important at this position (as phenylalanine, leucine, and tryptophan are all allowed) (Fig. 5b). Finally, there appeared to be a preference for a less bulky residue in place of phenylalanine at position 288, as the enhanced-affinity aflibercept variants substituted either threonine or valine. (Fig. 5b).
Figure 5.

Sequence convergence of enhanced-affinity aflibercept variants. The molecular structure of the complex between VEGF-A and aflibercept was modeled by overlaying the crystallographic structures of VEGF-A bound to VEGFR-1 D2 (PDB ID: 5T89)29 and VEGF-A bound to VEGFR-2 D3 (PDB ID: 3V2A).5 Enlarged views of the VEGF-A/VEGFR-1 interface (a) and the VEGF-A/VEGFR-2 interfaces (b) are shown. VEGF-A is colored based on an electrostatic map, and residues that were mutagenized in the library are shown in yellow. Predicted orientations of convergently mutated residues are shown in red. Sequences of the engineered aflibercept variants at each mutated position are presented in the table at right. Mutations from the wild type aflibercept residue are indicated with red text and convergent substitutions are indicated with red shading.
Expression of Aflibercept Variants in Rat Choroid via Nonviral Gene Delivery
Current clinical regimens for aflibercept mandate frequent and damaging intravitreal injections over long time periods.3,14,44 As an alternative, there has been growing interest in gene delivery approaches to achieve robust and durable expression of the drug directly from retinal pigmented epithelial cells, in order to perpetuate high ocular levels of the therapy. Although viral gene delivery methods for ocular diseases have shown potential and have been safe and well-tolerated in clinical trials to date, 15 these methods can be limited by pre-existing immunity, developed immunity following vector delivery,24 or, in some cases, vector-induced ocular toxicity due to AAV cis-regulatory sequences.50 To circumvent these issues, we utilized a nonviral gene transfer approach using polymer-based nanoparticles to deliver our engineered aflibercept variants. Moreover, treatments were administered into the suprachoroidal space, which is actually a potential space that is expanded when fluid is injected just internal to the sclera, resulting in a new route of delivery to the retina.32,33 Suprachoroidal injections have several advantages over intravitreal injections, as they can be done in an outpatient clinic setting, are safer, and result in gene transfer to the entire retina.
Based on prior results showing robust transfection of human retinal endothelial and pigment epithelial cells,42 we selected the PBAE shown in Fig. 6a as a delivery vector and synthesized the polymer using the chemical scheme shown. PBAE was chosen to deliver the DNA encoding our engineered VEGF antagonists due to its demonstrated efficacy and low toxicity in targeted transfection of retinal pigment epithelial (RPE) cells in vitro and in vivo,45 including when delivered via suprachoroidal injection.41 PBAE forms nanoparticles with anionic nucleic acids, such as DNA, and is hydrolytically degradable, simultaneously facilitating intracellular release of the plasmid from the particles and minimizing potential toxicity. We mixed this polymer with mammalian expression vectors encoding the gene for either Fc-fused wild type aflibercept or Fc-fused aflibercept mutant clone D4 to allow self-assembly of nanoparticles. Complete DNA encapsulation within the nanoparticle was observed for polymer-to-DNA mass ratios of > 2 in pH 5 sodium acetate buffer and ratios of > 5 in PBS (Supplementary Fig. 3). The resulting particles were imaged via transmission electron microscopy (TEM) (Fig. 6b). Polymer length was characterized by gel permeation chromatography (GPC) and nanoparticle size and charge properties were assessed by dynamic light scattering (DLS), nanoparticle tracking analysis (NTA), and electrophoretic mobility analysis (Fig. 6c). Particles were then administered to Brown Norway rats via suprachoroidal injection, which was previously demonstrated to elicit significantly higher levels of gene-delivered protein expression compared to intravitreal injections.41 One month after injection, previously found to be the peak time point for expression,41 we detected both wild type aflibercept and aflibercept variant D4 (5–10 μg mg−1 total protein) in the rat choroid (Fig. 7). Protein concentrations were similar between rats injected with vectors containing wild type and mutant aflibercept, and significantly above choroidal background, demonstrating proof of concept for the delivery of potent anti-angiogenic proteins to treat retinal diseases.
Figure 7.
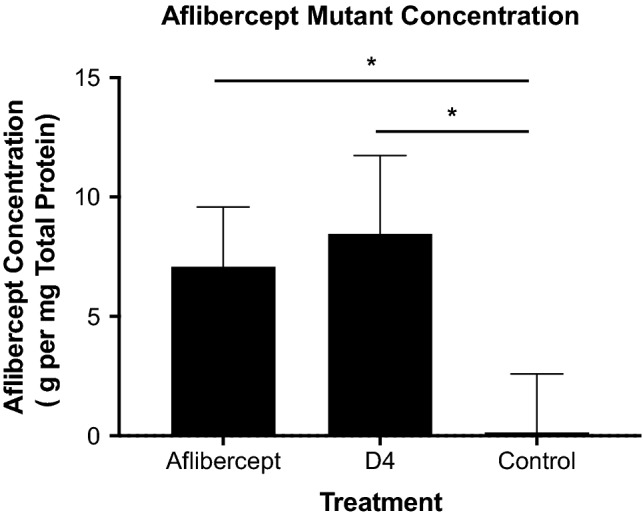
Aflibercept variant expression in rat choroid. Rats were injected suprachoroidally with polymeric nanoparticles loaded with DNA plasmids containing the genes for either Fc-fused aflibercept (n = 6) or the Fc-fused aflibercept mutant D4 (n = 6). One month after injection, expression of aflibercept or D4 was quantified via human IgG-Fc sandwich ELISA. Untreated control eyes (n = 5) were analyzed for comparison. IgG-Fc expression was normalized to 1 mg of total choroid protein content. Data represent mean ± S.D. Statistical analysis was performed via unpaired two-tailed Student’s t test. *p < 0.01.
Discussion
NVAMD and DR are debilitating diseases that represent two leading causes of blindness in adults.21,23 Despite significant advances in treating retinal diseases through inhibition of neovascularization via VEGF signaling blockade, several factors including potency, safety, and tolerability of treatments remain to be addressed. Thus, there is an urgent need for new approaches to enhance the efficacy and durability of anti-angiogenic therapies. In this study, we built off of a clinically approved decoy receptor drug, aflibercept, to create a higher affinity VEGF antagonist. Furthermore, a minimally invasive suprachoroidal ocular gene delivery mechanism was used to successfully induce sustained ocular expression of our new anti-VEGF protein in rats. The durability of expression evident after one month in our gene transfer study (Fig. 7) is a remarkable result for nonviral and non-integrating plasmid delivery vectors, and this finding demonstrates the potential for our new strategy to reduce the frequency and invasiveness of retinal disease therapies. Validation of this novel approach for engineered protein design and delivery presents an exciting opportunity for combatting choroidal and retinal neovascularization, and the anti-angiogenic proteins we developed could have additional applications in other diseases such as cancer.
One serious complication of intravitreal injections of anti-VEGF agents for treatment of neovascular eye diseases is endophthalmitis, which can potentially lead to blindness. The incidence of endophthalmitis per intravitreal injection is about 0.02% and the risk is compounded by the need for repeated therapeutic injections, often for the remainder of the patient’s life.10 Since our gene therapy approach will require only one or at most a few injections, the risk of endophthalmitis will be dramatically decreased. In addition, sustained suppression of VEGF will reduce treatment burden and reduce the likelihood of recurrent collections of fluid within and under the retina that can cause permanent vision impairment.
In addition to the promising clinical implications of our study, this work offers important insights into protein engineering and directed evolution approaches. Further improvement of aflibercept’s picomolar affinity for VEGF-A posed a formidable challenge, but integration of recently reported crystallographic data29 enabled deployment of a structure-informed design strategy, allowing us to construct focused libraries for the isolation of clones with enhanced target affinity and slower dissociation rates. Importantly, structural and computational predictions were not sufficient to design an enhanced-affinity version of aflibercept, but coupling the focused library approach with the yeast surface display platform for directed evolution enabled discovery of aflibercept variants with slower dissociation rates through surprising biochemical changes. In one case, a subtle isoleucine to leucine substitution (position 145 in VEGFR-1 D2) was significantly favored in variants with improved affinity, and modeling of the amino acid side chain rotamer provided rationale for this mutational preference (Fig. 5a). In an even more striking observation, a highly disruptive mutation from a flexible glycine residue to a rigid proline residue (position 255 in VEGFR-2 D3) was conserved amongst all enhanced-affinity aflibercept variants, stabilizing a bend of the protein backbone (Fig. 5b). Although it would be formally possible to observe these conserved changes using a random error-prone mutagenesis methodology, the likelihood of identifying clones that simultaneously incorporate these two changes at once would be greatly diminished due to the much larger sample space and the variability in error rate. By sampling all possibilities for this limited interface library, we were able to more thoroughly explore the co-variation between key residues contributing to VEGF-A binding energetics. In addition to the information obtained regarding favorable substitutions, the use of focused libraries also allowed us to identify an amino acid (residue 173 in VEGFR-1 D2) that was constrained to its wild type residue of proline for all three enhanced-affinity aflibercept variants (Fig. 5a). This result emphasizes the importance of including the possibility of wild type residues at each mutagenized position, and also provides key feedback for future library design. Equally important information was observed from position 225 in VEGFR-1 D2, which tolerated side chains with varying polarity and size, suggesting that mutations at this position had less impact on binding energetics (Fig. 5a). An exciting advantage of this strategy is that the results should apply to all VEGF-A isoforms since they contain the same VEGFR binding domains as VEGF-A165, the prototypical isoform used in this manuscript. Taken together, these findings illustrate the wealth of information gained from our structure-guided design approach. This approach is readily adaptable to a variety of physiological systems with known structural information, and it is particularly suitable for targeted enhancement of high-affinity interfaces.
Implementation of both an aggressive library and a conservative library, with a tenfold difference in theoretical diversity between the two, allowed us to interrogate the effects of sample size in our focused libraries. As expected, the conservative library contained a higher percentage of binders prior to selection (Fig. 3a), but both evolved libraries achieved similar target affinities (Fig. 3b) and converged on similar sequences (Fig. 5). This finding suggests that the tenfold difference in theoretical library diversity may not have played a significant role in the evolutionary outcome, making a case for use of slightly larger libraries to investigate more protein variants without enduring a significant change in library selection results. As the molecular engineering field continues to integrate more information from structural and computational studies into the design process,26,36,43,49 it will be interesting to see what role theoretical library size plays in the success of various evolutionary strategies.
Although we were able to enhance the affinity of aflibercept, the improvement was modest, and further library generations will be required to further potentiate antagonistic activity. Feedback from the library designed herein will provide valuable insight into which positions hold the most promise for mutagenesis in future library generations. Additionally, further studies will be required to explore the therapeutic potential of our engineered aflibercept variants in animal models of ocular neovascularization. Despite these limitations, our multi-faceted approach to anti-VEGF protein engineering and delivery promises to inspire meaningful advances that will improve treatment options for ocular diseases.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure 1: VEGF-A affinity characterization of receptor variants. Bio-layer interferometry-based kinetic titrations of soluble Fc-fused aflibercept (a), Fc-fused VEGFR-1 domains 2 and 3 (VEGFR-1-Fc) (b), and Fc-fused VEGFR-2 domains 2 and 3 (VEGFR-2-Fc) (c) against immobilized VEGF-A. Fivefold titrations of soluble protein were used starting at 100 nM for aflibercept and VEGFR-1-Fc and starting at 250 nM for VEGFR-2-Fc. Binding constants were determined using Octet Data Analysis HT Software and are presented in Table 1.
Supplementary Figure 2: VEGF-A affinity characterization of enhanced-affinity aflibercept variants. Bio-layer interferometry-based kinetic titrations of soluble Fc-fused aflibercept variants C5 (a), D4 (b), and F3 (c) against immobilized VEGF-A. Fivefold dilutions of soluble protein were used starting at 100 nM for all aflibercept variants. Binding constants were measured using Octet Data Analysis HT Software and are presented in Table 2.
Supplementary Figure 3: DNA encapsulation in NPs. PBAE/DNA NPs were made by diluting DNA in 25 mM sodium acetate buffer at pH 5 (NaAc), and mixing with diluted PBAE at increasing polymer-to-DNA mass ratios (w/w). After 10 min of incubation for NP formation, sucrose was added, and the NPs were then diluted 1:11 (v/v) in additional NaAc (a) or PBS (b). Samples were mixed with 30% glycerol as a loading buffer at a 1:5 ratio (v/v) of loading buffer to NPs, then loaded into a 1% agarose gel with 1 µg/mL ethidium bromide. Each well contained 110 ng DNA. The gel was run for 30 min under 80 V, then visualized by UV exposure. DNA was completely bound in the NPs at 2 w/w or higher at pH 5, and even after dilution in PBS, DNA was completely bound at 5 w/w or higher.
Acknowledgments
The authors thank Patrick James Krohl for technical assistance with the project. The authors also acknowledge the NIH (R01EY031097, R01CA228133), the E. Matilda Ziegler Foundation for the Blind, the Louis B. Thalheimer Translational Fund, and Research to Prevent Blindness (Dr. H. James and Carole Free Catalyst Award and unrestricted grant) for support.
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical Approval
All animals were treated in accordance with the Association for Research in Vision and Ophthalmology Statement for Use of Animals in Ophthalmic and Vision Research, and protocols were reviewed and approved by the Johns Hopkins University Animal Care and Use Committee. No human studies were carried out by the authors for this article.
Abbreviations
- NVAMD
Neovascular age-related macular degeneration
- DR
Diabetic retinopathy
- VEGF
Vascular endothelial growth factor
- PDGF
Platelet derived growth factor
- VEGFR
Vascular endothelial growth factor receptor
- RTK
Receptor tyrosine kinase
- IgG1
Immunoglobulin G1
- AAV
Adeno-associated virus
- LV
Lentivirus
- HEK
Human embryonic kidney
- FPLC
Fast protein liquid chromatography
- HBS
HEPES-buffered saline
- BAP
Biotin-acceptor protein
- PDBePISA
Protein data bank in Europe proteins, interfaces, structures, and assemblies
- MACS
Magnetic-activated cell sorting
- SA
Streptavidin
- BSA
Bovine serum albumin
- EDTA
Ethylenediaminetetraacetic acid
- FACS
Fluorescence-activated cell sorting
- PBAE
Poly(beta-amino ester)
- THF
Tetrahydrofuran
- DMSO
Dimethyl sulfoxide
- PDI
Polydispersity index
- PTFE
Polytetrafluoroethylene
- GPC
Gel permeation chromatography
- DLS
Dynamic light scattering
- NTA
Nanoparticle tracking analysis
- PBS
Phosphate-buffered saline
- TEM
Transmission electron microscopy
- BLI
Bio-layer interferometry
- scFv
Single-chain variable fragment
Author Contributions
RK, AZ, JS, SYT, LRA, and PRS designed, conducted, and analyzed experiments. PAC, JJG, and JBS conceptualized the study, supervised all research, interpreted data, and provided funding. RK, PRS, and JBS wrote the manuscript with input from co-authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Avery RL, et al. Systemic pharmacokinetics and pharmacodynamics of intravitreal aflibercept, bevacizumab, and ranibizumab. Retina. 2017;37:1847–1858. doi: 10.1097/IAE.0000000000001493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 3.Boyer DS, Heier JS, Brown DM, Francom SF, Ianchulev T, Rubio RG. A phase IIIb study to evaluate the safety of ranibizumab in subjects with neovascular age-related macular degeneration. Ophthalmology. 2009;116:1731–1739. doi: 10.1016/j.ophtha.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 4.Brown DM, et al. Ranibizumab vs. verteporfin for neovascular age-related macular degeneration. N. Engl. J. Med. 2006;355:1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- 5.Brozzo MS, et al. Thermodynamic and structural description of allosterically regulated VEGFR-2 dimerization. Blood. 2012;119:1781–1788. doi: 10.1182/blood-2011-11-390922. [DOI] [PubMed] [Google Scholar]
- 6.Campochiaro PA. Ocular neovascularization. J. Mol. Med. 2013;91:311–321. doi: 10.1007/s00109-013-0993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campochiaro PA, et al. Lentiviral vector gene transfer of endostatin/angiostatin for macular degeneration (GEM) study. Hum. Gene Ther. 2017;28:99–111. doi: 10.1089/hum.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.CATT Research Group Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011;364:1897–1908. doi: 10.1056/NEJMoa1102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 10.Daien V, et al. Incidence and outcomes of infectious and noninfectious endophthalmitis after intravitreal injections for age-related macular degeneration. Ophthalmology. 2018;125:66–74. doi: 10.1016/j.ophtha.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Diabetic Retinopathy Clinical Research Network Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N. Engl. J. Med. 2015;372:1193–1203. doi: 10.1056/NEJMoa1414264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding K, et al. AAV8-vectored suprachoroidal gene transfer produces widespread ocular transgene expression. J. Clin. Invest. 2019;129:4901–4911. doi: 10.1172/JCI129085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dixon JA, Oliver SCN, Olson JL, Mandava N. VEGF trap-eye for the treatment of neovascular age-related macular degeneration. Expert Opin. Investig. Drugs. 2009;18:1573–1580. doi: 10.1517/13543780903201684. [DOI] [PubMed] [Google Scholar]
- 14.Falavarjani KG, Nguyen QD. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: a review of literature. Eye. 2013;27:787–794. doi: 10.1038/eye.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heier JS, et al. Intravitreous injection of AAV2-sFLT01 in patients with advanced neovascular age-related macular degeneration: a phase 1, open-label trial. Lancet. 2017;390:50–61. doi: 10.1016/S0140-6736(17)30979-0. [DOI] [PubMed] [Google Scholar]
- 16.Ho CCM, et al. “Velcro” engineering of high affinity CD47 ectodomain as signal regulatory protein α (SIRPα) antagonists that enhance antibody-dependent cellular phagocytosis. J. Biol. Chem. 2015;290:12650–12663. doi: 10.1074/jbc.M115.648220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holz FG, et al. Multi-country real-life experience of anti-vascular endothelial growth factor therapy for wet age-related macular degeneration. Br. J. Ophthalmol. 2015;99:220–226. doi: 10.1136/bjophthalmol-2014-305327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holz FG, Schmitz-Valckenberg S, Fleckenstein M. Recent developments in the treatment of age-related macular degeneration. J. Clin. Invest. 2014;124:1430–1438. doi: 10.1172/JCI71029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N. Engl. J. Med. 2008;358:2606–2617. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- 20.Kieran MW, Kalluri R, Cho Y-J. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb. Perspect. Med. 2012;2:1–17. doi: 10.1101/cshperspect.a006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein R, et al. Prevalence of age-related macular degeneration in 4 racial/ethnic groups in the multi-ethnic study of atherosclerosis. Ophthalmology. 2006;113:373–380. doi: 10.1016/j.ophtha.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. II. Prevalence and risk of diabetic retinopathy when age at diagnosis is less than 30 years. Arch. Ophthalmol. 1984;102:520–526. doi: 10.1001/archopht.1984.01040030398010. [DOI] [PubMed] [Google Scholar]
- 23.Klein R, Klein BEK, Tomany SC, Meuer SM, Huang G-H. Ten-year incidence and progression of age-related maculopathy: the Beaver Dam eye study. Ophthalmology. 2002;109:1767–1779. doi: 10.1016/s0161-6420(02)01146-6. [DOI] [PubMed] [Google Scholar]
- 24.Kotterman MA, Yin L, Strazzeri JM, Flannery JG, Merigan WH, Schaffer DV. Antibody neutralization poses a barrier to intravitreal adeno-associated viral vector gene delivery to non-human primates. Gene Ther. 2015;22:116–126. doi: 10.1038/gt.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Krohl PJ, Ludwig SD, Spangler JB. Emerging technologies in protein interface engineering for biomedical applications. Curr. Opin. Biotech. 2019;60:82–88. doi: 10.1016/j.copbio.2019.01.017. [DOI] [PubMed] [Google Scholar]
- 27.Kwak N, Okamoto N, Wood JM, Campochiaro PA. VEGF is major stimulator in model of choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2000;41:3158–3164. [PubMed] [Google Scholar]
- 28.MacDonald DA, et al. Aflibercept exhibits VEGF binding stoichiometry distinct from bevacizumab and does not support formation of immune-like complexes. Angiogenesis. 2016;19:389–406. doi: 10.1007/s10456-016-9515-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markovic-Mueller S, et al. Structure of the full-length VEGFR-1 extracellular domain in complex with VEGF-A. Structure. 2017;25:341–352. doi: 10.1016/j.str.2016.12.012. [DOI] [PubMed] [Google Scholar]
- 30.Pandey Arvind K, et al. Mechanisms of VEGF (Vascular Endothelial Growth Factor) inhibitor-associated hypertension and vascular disease. Hypertension. 2018;71:e1–e8. doi: 10.1161/HYPERTENSIONAHA.117.10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papadopoulos N, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15:171–185. doi: 10.1007/s10456-011-9249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel SR, Berezovsky DE, McCarey BE, Zarnitsyn V, Edelhauser HF, Prausnitz MR. Targeted administration into the suprachoroidal space using a microneedle for drug delivery to the posterior segment of the eye. Invest. Ophthalmol. Vis. Sci. 2012;53:4433–4441. doi: 10.1167/iovs.12-9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel SR, Lin ASP, Edelhauser HF, Prausnitz MR. Suprachoroidal drug delivery to the back of the eye using hollow microneedles. Pharm. Res. 2011;28:166–176. doi: 10.1007/s11095-010-0271-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pennington KL, DeAngelis MM. Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. (Lond) 2016;3:1–20. doi: 10.1186/s40662-016-0063-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rakoczy EP, et al. Gene therapy with recombinant adeno-associated vectors for neovascular age-related macular degeneration: 1 year follow-up of a phase 1 randomised clinical trial. Lancet. 2015;386:2395–2403. doi: 10.1016/S0140-6736(15)00345-1. [DOI] [PubMed] [Google Scholar]
- 36.Raman S, et al. Structure-guided design fine-tunes pharmacokinetics, tolerability, and antitumor profile of multispecific frizzled antibodies. Proc. Natl. Acad. Sci. USA. 2019;116:6812–6817. doi: 10.1073/pnas.1817246116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reichel FF, et al. AAV8 can induce innate and adaptive immune response in the primate eye. Mol. Ther. 2017;25:2648–2660. doi: 10.1016/j.ymthe.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenfeld PJ, et al. Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2006;355:1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- 39.Saaddine JB, Narayan KMV, Vinicor F. Vision loss: a public health problem? Ophthalmology. 2003;110:253–254. doi: 10.1016/s0161-6420(02)01839-0. [DOI] [PubMed] [Google Scholar]
- 40.Schlothauer T, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 2016;29:457–466. doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
- 41.Shen J, et al. Suprachoroidal gene transfer with nonviral nanoparticles. Sci. Adv. 2020;6:1–10. doi: 10.1126/sciadv.aba1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shmueli RB, Sunshine JC, Xu Z, Duh EJ, Green JJ. Gene delivery nanoparticles specific for human microvasculature and macrovasculature. Nanomedicine. 2012;8:1200–1207. doi: 10.1016/j.nano.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silva D-A, et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature. 2019;565:186–191. doi: 10.1038/s41586-018-0830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singer MA, et al. HORIZON: an open-label extension trial of ranibizumab for choroidal neovascularization secondary to age-related macular degeneration. Ophthalmology. 2012;119:1175–1183. doi: 10.1016/j.ophtha.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 45.Sunshine JC, Sunshine SB, Bhutto I, Handa JT, Green JJ. Poly(β-Amino Ester)-nanoparticle mediated transfection of retinal pigment epithelial cells in vitro and in vivo. PLoS ONE. 2012;7:e37543. doi: 10.1371/journal.pone.0037543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tzeng SY, Higgins LJ, Pomper MG, Green JJ. Biomaterial-mediated cancer-specific DNA delivery to liver cell cultures using synthetic poly(beta-amino ester)s. J. Biomed. Mater. Res. 2013;101A:1837–1845. doi: 10.1002/jbm.a.34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vitt UA, Hsu SY, Hsueh AJW. Evolution and classification of cystine knot-containing hormones and related extracellular signaling molecules. Mol. Endocrinol. 2001;15:681–694. doi: 10.1210/mend.15.5.0639. [DOI] [PubMed] [Google Scholar]
- 48.Weiskopf K, et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88–91. doi: 10.1126/science.1238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wrenbeck EE, Faber MS, Whitehead TA. Deep sequencing methods for protein engineering and design. Curr. Opin. Struct. Biol. 2017;45:36–44. doi: 10.1016/j.sbi.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiong W, et al. AAV cis-regulatory sequences are correlated with ocular toxicity. Proc. Natl. Acad. Sci. USA. 2019;116:5785–5794. doi: 10.1073/pnas.1821000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, et al. Comparison of binding characteristics and in vitro activities of three inhibitors of vascular endothelial growth factor A. Mol. Pharm. 2014;11:3421–3430. doi: 10.1021/mp500160v. [DOI] [PubMed] [Google Scholar]
- 52.Yorston D. Anti-VEGF drugs in the prevention of blindness. Community Eye Health. 2014;27:44–46. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: VEGF-A affinity characterization of receptor variants. Bio-layer interferometry-based kinetic titrations of soluble Fc-fused aflibercept (a), Fc-fused VEGFR-1 domains 2 and 3 (VEGFR-1-Fc) (b), and Fc-fused VEGFR-2 domains 2 and 3 (VEGFR-2-Fc) (c) against immobilized VEGF-A. Fivefold titrations of soluble protein were used starting at 100 nM for aflibercept and VEGFR-1-Fc and starting at 250 nM for VEGFR-2-Fc. Binding constants were determined using Octet Data Analysis HT Software and are presented in Table 1.
Supplementary Figure 2: VEGF-A affinity characterization of enhanced-affinity aflibercept variants. Bio-layer interferometry-based kinetic titrations of soluble Fc-fused aflibercept variants C5 (a), D4 (b), and F3 (c) against immobilized VEGF-A. Fivefold dilutions of soluble protein were used starting at 100 nM for all aflibercept variants. Binding constants were measured using Octet Data Analysis HT Software and are presented in Table 2.
Supplementary Figure 3: DNA encapsulation in NPs. PBAE/DNA NPs were made by diluting DNA in 25 mM sodium acetate buffer at pH 5 (NaAc), and mixing with diluted PBAE at increasing polymer-to-DNA mass ratios (w/w). After 10 min of incubation for NP formation, sucrose was added, and the NPs were then diluted 1:11 (v/v) in additional NaAc (a) or PBS (b). Samples were mixed with 30% glycerol as a loading buffer at a 1:5 ratio (v/v) of loading buffer to NPs, then loaded into a 1% agarose gel with 1 µg/mL ethidium bromide. Each well contained 110 ng DNA. The gel was run for 30 min under 80 V, then visualized by UV exposure. DNA was completely bound in the NPs at 2 w/w or higher at pH 5, and even after dilution in PBS, DNA was completely bound at 5 w/w or higher.