

Abstract
Introduction
Fibroblastic reticular cells (FRCs) support and remodel the lymph node (LN), express and present self-antigens to T cells to promote tolerance. In Type 1 diabetes (T1D), decrease in FRC frequency and in their expression of T1D-related self-antigens may hinder tolerogenic engagement of autoreactive T cells. FRC reticular organization in LNs is critical for adaptive immunity. Thus, we engineered LN-like FRC reticula to determine if FRC reticular properties were altered in T1D and to study engagement of autoreactive T cells in vitro.
Methods
We characterized FRC networks in pancreatic and skin-draining LNs of 4- and 12-week old non-obese diabetic (NOD) and diabetes resistant NOR mice by immunofluorescence. Murine FRCs isolated from NOR, NOD or human pancreatic LNs were cultured in collagen sponges for up to 21 days before immunofluorescence and flow cytometry analysis. NOD FRCs expressing T1D antigens were co-cultured with CellTrace-labeled specific T cells in 2D or in scaffolds. T cell engagement was quantified by CD25 upregulation, CellTrace dilution and by T cell tracking.
Results
FRC networks in both 4- and 12-week old NOD LNs displayed larger reticular pores than NOR controls. NOD FRCs had delayed scaffold remodeling compared to NOR FRCs. Expression of the gp38 FRC marker in NOD FRCs was lower than in NOR but improved in 3D. FRC reticula expressing T1D antigens promoted higher engagement of specific T cells than 2D.
Conclusion
We engineered LN-like FRC reticula that recapitulate FRC organization and phenotype of T1D LNs for studying tolerogenic autoreactive T cell engagement in T1D.
Electronic supplementary material
The online version of this article (10.1007/s12195-020-00627-y) contains supplementary material, which is available to authorized users.
Keywords: Collagen scaffolds, Three-dimensional, Lymph node stromal cells, Peripheral tolerance, Autoimmunity, Human cells, Reticular networks, Secondary lymphoid organs
Introduction
Fibroblastic reticular cells (FRCs) found in lymph nodes (LNs) can be broadly classified as non-hematopoietic (CD45-) stromal cells expressing podoplanin (gp38) and lacking CD31 expression.2,25 As a well-conserved, mucin-type transmembrane protein expressed in multiple tissues playing a critical role in development of the lymphatic system, gp38 is widely used as a marker for lymphatic endothelial cells and FRCs of lymphoid organs and tumor microenvironments. Recent studies showed that gp38 plays crucial roles in the biology of immune cells.2 FRCs have a plethora of functions associated with immunity; they form interconnected reticular networks that provide structural support to LNs, enabling LN contraction and expansion to accommodate T cell proliferation during adaptive immune responses.1 Additionally, FRCs actively participate in the regulation of T cell homeostasis.4,8,39 Through their ability to present peripheral tissue antigens to autoreactive T cells as non-professional antigen-presenting cells (APCs), FRCs may help maintain peripheral tolerance.13,18,24
Peripheral tolerance mechanisms protect against autoimmune diseases like T1D.23 Because some self-antigens are expressed and presented in the periphery rather than in the thymus, both thymic and peripheral tolerance are needed for deletion/inactivation of autoreactive lymphocytes and prevention of autoimmunity.33 T1D results from impairment in both central and peripheral tolerance.21 Numerous alterations in the function of professional APCs6 were described in T1D. Several studies reported FRCs as the source of ectopic expression of insulin, a major T1D antigen, in LNs.5,40,41 Insulin expression was downregulated in pancreatic LNs (PLNs) from 12-week old (late stage T1D) NOD mice and from patients with T1D but not in PLNs from 4-week old (early stage T1D) NOD mice or in PLNs from healthy human donors.41 We recently reported that FRCs themselves are reduced relative to other LN stromal cell subsets in PLNs of both NOD mice and T1D patients.31 Reduced frequency and antigen expression in FRCs from PLNs of 12-week old NOD mice may lead to decreased presentation of β-cell antigens to specific T cells, leading to an impaired peripheral tolerance maintenance to these antigens and contributing to T1D pathogenesis. FRC alterations have been reported in other autoimmune diseases, including in patients suffering from rheumatoid arthritis.17
FRC network integrity in LNs plays a critical role for the activation of adaptive immune responses, and disrupting FRC reticular networks impaired overall immunity.16,29 FRC-FRC contacts are maintained during acute LN expansion, allowing inflamed LNs to maintain conduit size exclusion, to prevent chronic inflammation.26 Acute graft-versus-host disease was recently shown to damage and prevent repair of the FRC network, disabling its capacity to purge autoreactive T cells from the repertoire.9 We hypothesize that in T1D, reticular properties of FRC networks in LNs are altered, which could affect those adaptive immune responses that lead to peripheral tolerance in LNs. Thus, one goal of this report is to characterize the reticular properties of FRC networks in LNs from murine models of T1D and diabetes-resistant controls as Feret diameter of the FRC reticular pores. Feret statistical diameter allows measurement of irregularly shaped particles and pores and could be used to characterize reticular properties of FRC networks in LNs.38
Lack of a physiologically relevant platform to study FRCs in T1D prevents unraveling a definite role of FRCs in the disease pathogenesis to develop innovative therapies for peripheral tolerance induction. Previously tested systems were in vivo based and relied on transgenic mice with artificial antigen expression in FRCs. Traditional two-dimensional (2D) cell culture systems fail to replicate LN FRC reticular organization in vitro and are not clinically translatable. Thus, another goal of this report is to develop FRC reticula that recapitulate T1D LNs through tissue-engineering for in vitro studies to study FRC interactions with T1D antigen-specific T cells.
Traditional systems for studying FRCs do not allow modulation of T1D-relevant antigen expression levels in FRCs and quantification of engagement of specific T cells in vitro. We recently showed that different types of APCs, including immortalized FRC cell lines, can be transduced with a lentiviral construct that allows efficient co-presentation of multiple major β-cell epitopes to both CD4+ and CD8+ diabetogenic T cells following targeting of specific groups of epitopes to appropriate MHC molecules from a single construct.7 Using this system, FRC-mediated engagement of T cells can be quantitatively assessed as T cell proliferation and CD25 upregulation by flow cytometry. Thus, a third goal of this report is to demonstrate that LN-like reticula engineered with T1D-expressing FRCs can promote engagement of specific T cells in vitro.
We report alterations of FRC reticular properties in T1D LNs and demonstrate that our FRC reticula, engineered by culturing LN-derived FRCs in commercially available collagen scaffolds (SpongeCol®), recapitulate FRC reticular alterations in T1D LNs and increase expression of gp38 on FRCs. We also demonstrate that FRC reticula engineered with T1D-antigen expressing NOD FRCs engage autoreactive T cells in vitro more efficiently than in 2D systems.
Research Design and Methods
Mice
All animal procedures were approved by the Institutional Animal Care and Use Committee of University of Miami. NOD/ShiLtJ, NOR/LtJ, NOD.Cg-Tg(TcraTcrbNY8.3)1Pesa/DvsJ (NY8.3) mice were purchased from The Jackson Laboratory (Bar Harbor, Maine).
Tissue Harvest
Pancreatic LNs (PLNs) and/or skin draining LNs (SDLNs, including brachial, axillary and inguinal LNs) were harvested from mice at either 4 or 12 weeks of age (low and high T1D risk, respectively). For LN volume quantification, LNs were photographed after harvesting, LN area was quantified using ImageJ, and the volume calculated using the ellipsoid formula (4/3πa2b). Harvested tissues were either fixed in formalin for histological processing and immunofluorescence or processed fresh for stromal cell isolation. Human PLNs were obtained from the cGMP Facility of the Diabetes Research Institute (DRI, University of Miami, Miller School of Medicine, Miami, FL, USA) before processing the pancreas for islet isolation. The islet isolation protocol, as part of the Clinical Pancreatic Islet Transplantation Study, was approved by the Institutional Review Board (IRB) of the University of Miami and the FDA.
Stromal Cell Isolation
LNs harvested from mice were processed by adapting previously published protocols.12,15 Inguinal, axillary, and brachial SDLNs were pooled (total of 6 LNs per mouse), while PLNs were processed individually (1 PLN per mouse).
For human FRC isolation, freshly isolated PLNs were processed right after harvesting; PLNs were minced to decrease the tissue size before digestion.
Tissues were digested using a DNase I, Dispase II and Collagenase P enzyme mix for no longer than 1 h as previously reported.12 Every 15 min, the supernatant (containing released cells) was transferred to a collection tube containing FACS Buffer (PBS, 2% fetal bovine serum and EDTA). For murine samples, after the tissue was completely digested, the resulting single cell suspension was stained with the following antibodies: gp38-PE, CD31-APC and CD45-PE/Cy7. FRCs were counted and sorted by gating on CD45- CD31- gp38+ using a Beckman Coulter MoFlo AstriosEQ as previously described.12 Alternatively, single cell suspensions obtained after enzymatic digestion were stained with a CD45-Biotin antibody (Biolegend) and Streptavidin Nanobeads (Biolegend). Human samples were only processed using the latter method. Stromal cells were then isolated using the Mojosort magnetic cell separation system (Biolegend) and expanded in traditional 2D culture using tissue culture-treated flasks. After expansion, the cells were harvested by trypsin digestion and stained with our antibody panel for FRC identification (gp38-PE, CD31-APC and CD45-PE/Cy7 for murine cells and gp38-PE, CD31-APC/Cy7 and CD45-Biotin+SA-AF488 for human cells) and CD45− CD31− gp38+ cells were sorted using a Beckman Coulter MoFlo AstriosEQ as previously described.12 Details of antibodies used for flow cytometry on murine and human samples are indicated in Supplementary Table 1.
CD8+ T Cell Isolation
Spleens from NY8.3 transgenic mice were harvested, mechanically disrupted, and CD8+ T cells were isolated using the Mojosort CD8 Mouse Isolation Kit (Biolegend) following the manufacturer’s guidelines.
FRC Cell Culture
After isolation, FRCs were expanded in 2D traditional tissue culture-treated wells or flasks using FRC media, which consisted of RPMI-1640 supplemented with 10% Fetal Bovine Serum, 0.05 mM 2-Mercaptoethanol, 10 mM l-glutamine and 1% antibiotics as we previously reported.35
Collagen Scaffolds
Commercially available Type I collagen sponges were obtained from Advanced Biomatrix (SpongeCol®). Scaffold dimensions: 4 mm diameter, 1.5 mm thickness and 200 µm average pore size (range 100–400 µm, https://advancedbiomatrix.com/spongecol.html).
Cell Seeding in Scaffolds
Experimental methods are indicated in Supplemental Fig. 2. SpongeCol® scaffolds were soaked in FRC media inside an incubator (37 °C) at least overnight. Sponges were then blotted with sterile gauze to remove excess liquid and transferred to a non-tissue culture treated petri dish for cell seeding. FRCs expanded in regular 2D tissue culture-treated plates were trypsinized and resuspended to a final concentration of 3 × 108 cells/mL for murine FRCs or 1 × 108 cells/mL for human FRCs. A total volume of 10 µL cell suspension containing 3 × 105 murine FRCs or 1 × 105 human FRCs was pipetted on top of the sponge and placed inside the incubator. After 30 min, 10 µL fresh media was added to each sponge to prevent the scaffold from drying out. After an additional 30 min (overall incubation of scaffolds with FRCs: 1 h), the sponges were carefully transferred to a sterile non-tissue culture treated 96-well plate, and fresh culture media was added to each well until the scaffolds were completely submerged in media. For the long-term culture studies, media was refreshed every 3 days.
Scaffold Fixing for Histology
At the desired time points after FRC seeding in scaffolds, media was aspirated and the scaffolds were gently washed with 1 × PBS. 4% paraformaldehyde (PFA) was added to the wells until scaffolds were completely submerged and incubated for at least 1 hour at 4 °C. Sponges were then washed with PBS and stored at 4 °C until immunofluorescent processing.
Histological Processing, Immunofluorescence and Imaging
Formalin-fixed paraffin-embedded LN sections (5 µm thick) and PFA-fixed cell-seeded scaffolds were stained with the following antibodies: gp38, CD3, B220, Lyve-1, alpha-smooth muscle actin (αSMA) and F-actin (Phalloidin). Nuclei were counterstained with DAPI. Z-stacks were obtained using a Leica SP5 Inverted Microscope at the Diabetes Research Institute Imaging Core. Details of antibodies used for immunofluorescence are indicated in Supplementary Table 1.
Quantification of FRC Reticular Properties in LNs and Engineered Reticula
Z-stacks obtained from confocal imaging of both LN sections and FRC-seeded scaffolds were analyzed. In single slices, 30 pores per sample were quantified. Images were analyzed using ImageJ (NIH) to calculate the average Feret diameter of FRC reticular pores in each sample. Feret statistical diameter allows measurement of irregularly shaped particles and pores.38 The investigator performing the quantifications was blinded to the treatment.
FRC-T Cell Co-culture to Quantify Engagement of T1D Antigen-Specific T Cells with T1D Antigen-Expressing FRCs
FRCs from PLNs of 12-week old female NOD mice were transduced with lentivectors for expression of GFP and T1D antigens, including the islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) (206-214) peptide that is recognized by CD8+ T cells from NY8.3 TCR-transgenic mice (GFP/Ag). As controls, FRCs were transduced with control lentivectors for expression of GFP only (GFP/No Ag). Collagen scaffolds (SpongeCol®) were placed in 96-well plates; 3 × 105 FRCs in 10 µL media were seeded in the scaffolds (3D) and cultured for 24 h to allow formation of FRC reticula expressing high levels of T1D antigens and GFP (GFP/Ag) or GFP but no antigen controls (GFP/No Ag). As additional controls, 5 × 104 GFP/Ag FRCs or GFP/No Ag FRCs were seeded in tissue culture-treated wells (2D). T1D antigen-specific CD8+ T cells were isolated from NY8.3 NOD mice, labeled with CellTrace Violet and added to the scaffolds 24 h after FRC seeding (3 × 105 T cells/scaffold) or to 2D-cultured FRCs (5 × 104 T cells/well).
T cell tracking Tracking of T1D antigen-specific T cells (CellTrace violet labeled: blue) in the GFP-expressing FRC reticula (green) was performed 24 h after co-culture by confocal imaging through time-lapse z-stacks. Experimental details on confocal time-lapse z-stacks images for cell tracking are indicated in Supplemental Table 2. T cell tracking data were analyzed from confocal time-lapse 3D images using Imaris and custom-made Matlab codes. For each condition, the number of tracked spots per time points and the total number of spots, the mean track speed, the track length, the track displacement length, the mean squared displacement (MSD), and the trajectories were calculated. T cell migration in reticula with GFP/Ag FRCs were compared to migration in GFP/No Ag reticula 24 h after adding T cells and after media addition to the top of the scaffolds.
FRC-mediated engagement of T1D antigen-specific T cells was quantified by flow cytometry 3 days after FRC/T cell co-culture. Scaffolds were mechanically processed to isolate T cells, while 2D controls were pipetted vigorously in order to obtain the T cells resting on top of the FRC layer. Isolated cells were stained with antibodies (gp38-PE, CD31-APC, CD45-PE/Cy7 or CD45-AF700, CD8-PerCP/Cy5.5, CD44-PE/CF594, CD25-PE/Cy7) and processed for flow cytometry. CD8+ T cell proliferation (CellTrace dilution) and CD25 upregulation were calculated using Kaluza (Beckman Coulter).
Details of antibodies used for flow cytometry are indicated in Supplementary Table 1.
Statistical Analysis
GraphPad Prism 6 was used for data analysis. Every set of data is presented as mean ± standard deviation. Paired t-test was used in comparative analysis of data sets with two groups. One-way or two-way ANOVA with Tukey post-hoc test for comparison across different time points and Bonferroni for comparison across different experimental groups (NOD vs. NOR) were performed in analysis of data sets with more than two populations. 95% confidence level was considered significant. The investigator carrying out the acquisition and analysis was blinded to the treatment groups.
Results
Reticular Properties of FRC Networks in Lymph Nodes are Altered in T1D Murine Models
FRC network integrity in LNs plays a critical role for the activation of adaptive immune responses and may affect FRC-mediated peripheral tolerance mechanisms. We hypothesize that FRC reticular properties in LNs, in addition to the previously reported decreased frequency and T1D antigen expression levels, are altered in T1D. We characterized the reticular properties of FRC networks in PLNs (Fig. 1) and SDLNs (Fig. S1) of 4-week old female NOD mice as low diabetes risk group (Figs. 1a, S1A) and of 12-week old mice (Figs. 1b, S1B) as high diabetes risk group since diabetes onset in female NOD mice occurs after 12 weeks of age. We euthanized the mice before they turned diabetic, harvested PLNs and SDLNs, measured their volume, and processed them for immunofluorescence to identify FRC reticula in LN sections as gp38+ Lyve-1− networks (to differentiate FRCs from gp38+ Lyve-1+ lymphatic endothelial cells) in T cell areas (identified by staining with antibodies that label T cells and B cells). As controls, we harvested and processed PLNs (Figs. 1c and d) and SDLNs (Fig. S1C, D) from age-matched diabetic-resistant NOR mice. Reticular properties were characterized through quantification of reticular porosity of FRC networks as Feret diameter of the irregularly shaped pores using ImageJ. We found that NOD PLN networks displayed larger reticular pores than NOR PLN controls at both 4 weeks (Feret NOD: 18 ± 6 µm; NOR: 15 ± 4 µm p < 0.01) and 12 weeks of age (Feret NOD: 19 ± 6 µm; NOR: 15 ± 5 µm p < 0.0001), while no age-dependent differences were found for each mouse strain analyzed (Table 1). The volume of freshly harvested PLNs was also calculated to determine if observed alterations were due to inflammation-dependent LN enlargement. We found that volumes were unaffected by either age or strain of the mice. Similar results were also obtained when we quantified reticular properties of FRC networks in SDLNs (Fig. S1).
Figure 1.
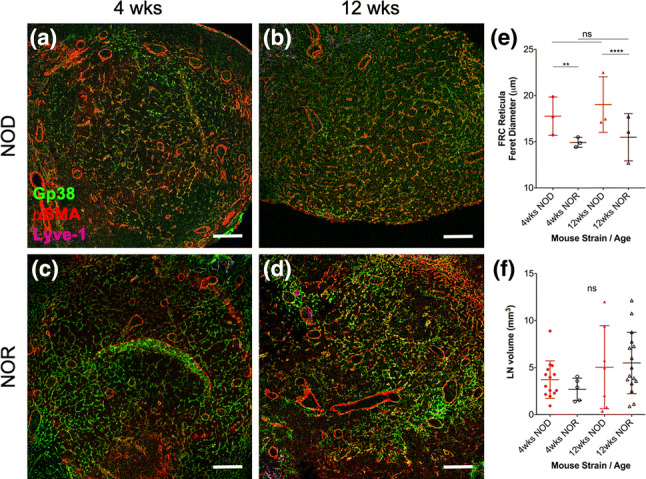
FRC networks in NOD PLNs display larger/more relaxed reticular pores than NOR PLN reticula. (a–d|) PLNs were harvested from either female NOD (a, b) or female NOR (c, d) mice at 4 or 12 weeks of age and their volume was quantified; then, whole LNs were fixed in formalin, embedded in paraffin, sectioned at 5 µm, stained with antibodies against gp38 (identifies both FRCs and lymphatic endothelial cells), Lyve-1 (identifies only lymphatic endothelial cells), and αSMA (identifies myofibroblasts, including FRCs), and imaged using a confocal microscope. Shown are representative images of n = 3 mice. FRC reticula were identified as gp38+ (green), Lyve-1− (cyan) cells. Scale bars: 100 µm. (e, f) Quantification of reticular properties of FRC networks in PLNs as Feret’s diameter of gp38+ Lyve-1− FRC reticular pores (e) and overall PLN volume (f) of NOD PLNs compared to NOR PLNs from either 4-week old or 12-week old mice. Each data point represents the average of 30 pore quantifications per mice; n = 3 mice were analyzed. For quantification of LN volumes from LN photographs, biological replicates (mice) were n = 14 (4 weeks NOD), n = 5 (4 weeks NOR), n = 7 (12 weeks NOD), and n = 10 (12 weeks NOR). ns not significant, **p < 0.01, ****p < 0.0001.
Table 1.
Feret diameter of the irregularly shaped pores of FRC reticula in the paracortex of PLNs and SDLNs from low diabetes risk 4-week old NOD mice and from high diabetes risk 12-week old mice compared to LNs of age-matched diabetes-resistant NOR mice.
| Donor strain (female) | PLNs | SDLNs | ||||||
|---|---|---|---|---|---|---|---|---|
| NOD | NOR | NOD | NOR | NOD | NOR | NOD | NOR | |
| Donor age | 4 weeks | 4 weeks | 12 weeks | 12 weeks | 4 weeks | 4 weeks | 12 weeks | 12 weeks |
| Average diameter (µm) | 18 | 15 | 19 | 15 | 18 | 16 | 19 | 14 |
| Std Dev (µm) | 6 | 4 | 6 | 5 | 5 | 4 | 6 | 3 |
Overall, these results suggest that enlarged reticular pores of FRC networks in PLNs and SDLNs of NOD mice were not due to inflammation-dependent LN expansion. Because FRCs are responsible for building and remodeling their reticular network, we hypothesized that the observed reticular differences in LNs of NOD mice, the main T1D mouse model, were dependent on altered FRC remodeling capabilities. We tissue engineered PLN-like FRC reticula using primary FRCs isolated from PLNs of 12-week old NOD and commercially available collagen sponges to study FRC reticular remodeling in T1D.
Development and Characterization of a Tissue-Engineered FRC Reticula that Recapitulates FRC Reticular Alterations of PLNs from High T1D Risk NOD Mice
As cell source for tissue-engineering the FRC reticula, we isolated FRCs from 4-week old or from 12-week old NOD or NOR mice by FACS sorting of gp38+ CD31− (FRC markers) LN cells after CD45+ cell depletion as previously described12 (Fig. 2a). Expression of the main FRC marker, gp38 as mean fluorescence intensity (MFI) in freshly sorted FRCs was comparable between age and strains of donor mice (Fig. 2b). We previously reported that in PLNs, the frequency of FRCs from 12-week old NOD mice is reduced compared to 4-week old NOD and to age-matched NOR mice. Analyzing FRCs in SDLNs, we found that the total number of FRCs from 4-week old but not 12-week old NOD mice was lower than from NOR controls (Fig. 2c). However, no differences in the total number of SDLN FRCs was observed in 4-week old compared to 12-week old donor mice for any of the strains analyzed. When we compared the frequency of FRCs in SDLN cells, we found that this was reduced in 12-week old NOD compared to 4-week old NOD mice while it was comparable between NOD and NOR at both 4 weeks and 12 weeks of age (Fig. 2d). Similarly, when we compared the frequency of FRCs in SDLN stromal (CD45−) cells, we found that those were also comparable between age-matched NOD and NOR and it was reduced in 12-week old NOR compared to 4-week old NOR mice (Fig. 2e).
Figure 2.

Characterization of FRCs freshly isolated from PLNs and SDLNs of NOD and NOR mice and of FRCs after culture in 2D substrates. FRCs were isolated from PLNs and SDLNs of 4-week old and 12-week old NOD and NOR mice by fluorescence activated cell sorting (FACS) of gp38+ CD31− LN cells after CD45+ cell depletion as previously reported25. (a, b) FACS dot plots (a) and gp38 mean fluorescence intensity (MFI) (b) of freshly sorted FRCs. Biological replicates (mice) were n = 6 (4 weeks NOD), n = 35 (12 weeks NOD), n = 5 (4weeks NOR), n = 5 (12 weeks NOR). (c–e) Quantification of total number (c), frequency relative to total live LN cells (d) and of total CD45− LN stromal cells (e) FRCs isolated from single SDLNs. (f, g) Phase contrast images (d), FACS dot plot (d) and gp38 MFI (g) of cultured (standard 2D tissue-treated plates) FRCs from PLNs and SDLNs of 12-week old NOR and NOD. Data are representatives of cells at passages 17–21. ns not significant, *p < 0.05, **p < 0.01.
Sorted FRCs were expanded in 2D cultures using tissue culture-treated plates and analyzed for gp38 expression (the main FRC marker) after expansion (Fig. 2f). We found that NOD-derived FRCs expressed lower levels of gp38, as gp38 MFI, than NOR LN-derived FRCs after 2D culture (Fig. 2g).
FRCs in LNs wrap around collagen fibers that are mainly composed of Type I collagen.34 In order to tissue engineer the FRC reticula, we decided to use commercially available collagen Type I sponges because of the higher clinical applicability. To exclude that scaffold processing could affect its porosity, we characterized the porosity of acellular collagen scaffolds before and after wetting the scaffolds with culture media, and after scaffold fixation with PFA (Fig. S2A). We found that the collagen scaffolds did not change their pore dimension after either hydration or fixation (Fig. S2B).
We decided to focus on PLN reticula because of the critical importance of PLNs in T1D pathogenesis, as the site of activation of β-cell autoreactive T cells. We also decided to focus on FRCs derived from 12-week old NOD mice because of the high risk of diabetes development in mice of this age.
We seeded 3 × 105 FRCs from 12-week old NOD or NOR PLNs in 10 µL media in scaffolds pre-wet with culture media and cultured them for at least 24 h (Fig. S2C).
We cultured the FRC-seeded scaffolds for up to 21 days (Fig. 3a) and determined FRC remodeling of their reticular networks in the scaffolds by quantifying the Feret diameter of reticular pores. Quantification of reticula engineered using NOD PLN FRCs demonstrated that up to 3 days after culture in collagen sponges, reticular pore sizes were larger than reticula of NOR PLN FRCs (Fig. 3b and Table 2). Compared to acellular scaffolds, the reticular pores of NOR-derived FRCs cultured in the scaffolds for 3 days was reduced by 70 ± 4%, while the reticula of NOD-derived FRCs was reduced only by 57 ± 3%.
Fig. 3.

Long-term culture of PLN FRCs in collagen scaffolds recapitulates LN-like reticula structure of donor mice and reticular alterations of T1D PLNs. (a) Representative maximum projection images of confocal z-stacks taken 1, 7, 14 or 21 days after seeding of 12-week old NOD (top row) or NOR (bottom row) PLN FRCs in collagen sponges with protocols shown in Supplemental Fig. 2. FRCs are identified by gp38 (green) and F-actin (red); nuclei are counterstained with DAPI (blue). Scalebars = 100 µm. (b) FRC remodeling of their reticular networks in 3D scaffolds as reticular pore size (Feret diameter) as a function of culture time (3–21 days) and of type of FRCs seeded (derived from NOD: red vs. NOR: black). For each condition, n = 30 pores per scaffolds were quantified blindly and n = 3 independent scaffolds were measured. (c) FRC proliferation in 3D scaffolds as total FRC numbers retrieved from the scaffolds at each time point per treatment (NOD: red vs. NOR: black derived FRCs). (d) Expression of the important FRC marker gp38, as mean fluorescence intensity (MFI) of NOD (red) and NOR (black) cells isolated from scaffolds 7–21 days after seeding and normalized to the MFI of cells at same passage but cultured in traditional 2D tissue culture flasks. Biological replicates were n = 3 scaffolds. Both the effects of culture time and of FRC origin (NOD vs. NOR) were evaluated. ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Table 2.
Feret diameter of the irregularly shaped pores of FRC reticula engineered by seeding FRCs from PLNs of high diabetes risk 12-week old NOD mice compared to age-matched diabetes-resistant NOR mice cultured up to 21 days.
| Donor strain (female) | Engineered reticula | |||||
|---|---|---|---|---|---|---|
| NOD d1 | NOR d1 | NOD d7 | NOR d7 | NOD d21 | NOR d21 | |
| Donor age | 12 weeks | 12 weeks | 12 weeks | 12 weeks | 12 weeks | 12 weeks |
| Average diameter (µm) | 111 | 81 | 107 | 83 | 72 | 79 |
| Std Dev (µm) | 24 | 30 | 30 | 27 | 17 | 21 |
Also, while reticular pores of NOR-derived FRCs cultured for 3–21 days were comparable, suggesting that scaffold remodeling occurred within three days after seeding, NOD-derived FRCs at 21 days were smaller (70 ± 2% reduction on pore diameter compared to acellular scaffolds), than day 3 and day 7, suggesting that scaffold remodeling took up to 21 days after seeding to occur (Fig. 3b and Table 2). The total number of NOD PLN FRCs retrieved per scaffold at day 21 was comparable to day 7 and day 14 cultures suggesting that NOD-derived FRCs did not proliferate in the scaffolds (Fig. 3c). Thus, NOD-derived FRCs remodeled their reticular network by tightening their reticular pores, likely by cell contraction, rather than proliferation. While NOD-derived FRCs displayed progressive remodeling between 3 and 21 days after culture in the scaffold, NOR-derived FRCs did not further reduce their reticular pore sizes after seeding in the scaffold.
When we compared the number of NOD FRCs recovered from the scaffolds to the number of NOR FRCs at each time point, we found that this was not significantly different at day 7 and 14. Thus, the observed alterations in reticular properties of NOD FRC networks 3 days after FRC seeding in scaffolds were likely due to FRC delayed remodeling rather than FRC reduced proliferation compared to NOR FRCs at those early time points after FRC seeding in the scaffolds. These results suggest that our tissue-engineered FRC reticula recapitulates alterations of FRC reticular properties observed in LNs of T1D murine models, validating the physiological significance of our model for study FRCs in T1D.
The total number of NOR PLN FRCs recovered from the scaffolds at day 14 and 21 was higher than day 7 but comparable to day 21 suggesting FRC proliferation in the scaffolds occurs between day 7 and 14 (Fig. 3c). When we compared the number of NOD FRCs in the scaffold to the number of NOR FRCs at day 21, we found that number of NOD FRCs was lower than NOR FRCs suggesting also reduced proliferation of NOD FRCs in the scaffold at day 21.
Reduced remodeling capabilities of NOD FRCs may result from reduced contractility of NOD compared to NOR FRCs and may have important implications for adaptive immunity. Expression of the main FRC surface marker gp38 has been linked to FRC remodeling capability, as gp38 expression correlates with FRC contractility.3 Culture in the collagen scaffolds for 14 days increased expression of gp38 in both NOD and NOR-derived FRCs cultured in scaffolds and normalized to culture in 2D (Fig. 3d). Normalization of 3D cultured FRCs to same passage FRCs cultured in 2D was necessary because we found that 2D-cultured FRCs tended to decrease their gp38 expression (Fig. S3A). When comparing gp38 expression of 3D cultured FRCs not normalized to 2D cultures, we found that gp38 expression of 3D cultured NOD FRCs was lower than 3D cultured NOR FRCs at all time points analyzed (Fig. S3B).
These results suggest that 3D culture in collagen sponges improves FRC phenotype as gp38 expression and may help rescue the reticular remodeling capabilities of NOD FRCs by enhancing their contractility. While 14 days culture in 3D collagen sponges is sufficient to increase gp38 expression in NOD FRCs, culture for 21 days is necessary to allow NOD-derived FRCs to remodel their scaffold to the same levels as FRCs from diabetes-resistant NOR mice.
Applicability of the Tissue-Engineered FRC Reticula to Recapitulate FRC Networks in Human PLNs
For clinical translatability, we characterized FRC reticular networks in PLNs from healthy human subjects and tested whether reticula engineered with human PLN FRCs could recapitulate reticular networks of human PLNs.
We characterized FRC networks in human PLNs from healthy donors by immunofluorescence (Fig. 4a). Reticular pore sizes as Feret pore diameters, were overall smaller than murine NOR PLN reticular pores (Fig. 4b). To validate our tissue-engineered FRC reticula with human cells, we isolated human CD45− gp38+ CD31− FRCs from PLNs applying the same protocol we used for isolation of murine FRCs by FACS (Fig. 4c). After culture in 2D tissue-culture treated dishes, we seeded human FRCs into collagen scaffolds using protocols established for murine FRCs (Fig. S2C). Similar to murine FRCs, human PLN FRCs formed a LN-like reticular network (Fig. 4d). Culture of human PLN FRCs in collagen scaffolds resulted in scaffold remodeling and average reduction of the reticula pore size of 73 ± 6% after 1-day culture in the scaffolds and resulting average Feret pore diameters of 171 ± 13 µm.
Fig. 4.

Applicability of the tissue-engineered model to human FRCs: Characterization of PLN FRCs from healthy human donors in LNs and of sorted FRCs cultured in 3D collagen scaffolds. (a) Representative maximum projection image of confocal z-stacks of human PLNs from a healthy donor. FRCs are identified by the FRC marker gp38 (green). Scalebar = 50 µm. (b) Reticular pore size (Feret diameter) of human FRC networks in PLNs compared to PLNs from 12-week old NOR mice (n = 3 human PLN donors; n = 3 NOR PLN donors). For each PLN, n = 30 pores were quantified (c) Gating strategy for FACS sorting of gp38+ CD31− LN cells after depletion of CD45+ cells isolated from enzyme-based digestion of PLNs as previously reported25. (d) Representative maximum projection images of confocal z-stacks taken 1–5 days after seeding 1 × 105 human PLN FRCs in collagen sponges. FRCs are identified by gp38 (green), and F-actin (red); nuclei are counterstained with DAPI (blue). Scalebars = 100 µm. *p < 0.05
We concluded that FRC engineered reticula can also be used to study human PLN FRCs in vitro in a physiomimetic environment.
FRCs in Engineered Reticula Expressing T1D Antigens Engaged T1D Autoreactive T Cells More Effectively than in 2D Culture Systems
Finally, we aimed at validating the applicability of our engineered FRC reticula to quantify engagement of T1D antigen-specific T cells in vitro using T1D antigen-expressing FRCs compared to traditional 2D culture systems.
We transduced FRCs from 12-week old NOD PLNs to express multiple T1D-relevant epitopes,7 including a natural peptide from IGRP recognized by NY8.3 CD8+ T cells in the context of H2-Kd MHC class I and GFP to allow T cell tracking on FRC reticula through confocal microscopy. To determine if FRC reticula engineered with T1D antigen-expressing FRCs could engage T1D autoreactive T cells, we co-cultured antigen-expressing FRCs (GFP/Ag) with CellTrace-labeled IGRP-reactive CD8+ T cells (isolated from NY8.3 mice) in 3D reticula and compared T cell engagement to 2D cultures and to FRCs transduced with control lentivectors to express GFP but not the antigens (GFP/No Ag). Three days after co-culture, we harvested the T cells and analyzed them by flow cytometry to determine T cell engagement through proliferation by CellTrace dilution and by upregulation of CD25 (Fig. 5a). Autoreactive T cells cultured within 3D antigen-expressing FRC reticula (GFP/Ag) lead to increased T cell engagement indicated by increased frequency of proliferated (Fig. S4A) and CD25+ (Fig. S4B) antigen-specific CD8+ T cells relative to 2D cultures and to non-antigen expressing FRCs (GFP/no Ag). Interestingly, we noted that T cell engagement by FRCs resulted in CD25 upregulation mainly in unproliferated cells (Fig. 5c). Thus, 3D culture of FRCs improves interactions with T cells resulting in more productive engagement of antigen-specific T cells.
Fig. 5.

Applicability of 3D FRC reticula engineered with T1D antigen-expressing FRCs for studying FRC interactions with specific T cells in vitro in a LN-like environment. FRCs from 12-week old NOD PLNs were transduced with a lentiviral vector expressing GFP and T1D antigens, including the IGRP(206-214) peptide that is recognized by CD8+ T cells from NY8.3 TCR-transgenic mice (GFP/Ag) or with control lentivectors (GFP/No Ag). (a) Schematic of experimental methods for quantification of T cell engagement in FRC reticula. Collagen scaffolds (SpongeCol®) were placed in 96-well plates; 3 × 105 FRCs were seeded in the scaffolds (3D) and cultured for 24 h to allow formation of reticula expressing T1D antigens and GFP (GFP/Ag) or GFP but no antigen controls (GFP/No Ag). As additional controls, FRCs were also seeded in tissue culture-treated wells (2D). T1D antigen-specific CD8+ T cells were isolated from NY8.3 NOD mice, labeled with CellTrace (red or blue) and added to the scaffolds 24 hours after FRC seeding or to 2D-cultured FRC wells. Tracking of T1D antigen-specific T cells (violet CellTrace labeled: blue) in the GFP-expressing reticula (green) was performed 24 hours after co-culture by confocal imaging through time-lapse z-stacks and analyzed using Imaris and Matlab. FRC-mediated engagement of T1D antigen-specific T cells was quantified by flow cytometry 3 days after co-culture. To isolate T cells, scaffolds were mechanically processed, whereas cells were released from 2D control wells through vigorous pipetting/mixing. Isolated cells were stained with antibodies and processed for flow cytometry. CD8+ T cell proliferation (CellTrace dilution) and T cell receptor engagement (CD25 upregulation) were calculated using Kaluza. (b, c) Representative flow cytometry bivariate plots (b) and quantification (c) of FRC-mediated engagement of T1D antigen-specific CD8+ T cells as proliferation (CellTrace dilution) and T cell receptor engagement (CD25 upregulation) 3 days after co-culture in tissue culture-treated plates (2D) or in collagen sponges (3D). Engagement of specific T cells by T1D antigen-expressing FRCs (GFP/Ag) in 3D scaffolds is compared to control FRCs expressing GFP but no antigen (GFP/No Ag) in 3D scaffolds and to 2D cultures. (d, e) Tracking of CellTrace-labeled (blue) T1D antigen-specific T cells in GFP-expressing 3D FRC reticula (green). Trajectory of specific T cells in T1D antigen-expressing FRC reticula (GFP/Ag, red) is compared to control FRCs reticula expressing GFP but no antigen (GFP/No Ag, black). For GFP/No Ag 403 ± 18 spots per time points and 590 tracks were quantified through Imaris. For GFP/Ag 982 ± 66 spots per time points and 1090 tracks were quantified through Imaris
Through intravital microscopy it was previously shown that T cells crawl along FRC fibers in the LN paracortex. Thus, to further characterize FRC-T cell interactions in the reticula, we tracked in real time x, y and z migration of T1D antigen-specific T cells within the antigen-expressing reticula (GFP/Ag) compared to non-antigen expressing FRCs (GFP/No Ag) 24 h after adding T cells to the FRC reticula. We found that the mean track speed, the track displacement and the mean squared displacement but not the total track length of T cells migrating on top of FRCs in the antigen-expressing reticula (GFP/Ag) was higher than in control non antigen-expressing reticula (GFP/No Ag) (Fig. 5d and Supplemental Videos 1, 2). We also performed T cell tracking on the reticula after adding media containing T cells to the scaffolds to mimic T cell entry in the LN-like environment and found similar results (Supplemental Fig. 4c and Supplemental Videos 3, 4). Thus, we confirmed that engineered FRC reticula can be used for studying interactions of autoreactive T cells with FRCs in vitro in a physiomimetic environment of the LN paracortex.
Discussion
Autoimmune diseases like T1D result from impairment in both central and peripheral tolerance.21 So far, alterations in the function of the APCs, as well as the phenotype and the suppressive capabilities of regulatory T cells6,19 were described in T1D and could be linked to T1D pathogenesis. Here, we focus on another non-professional APC that has been recently identified as modulator of peripheral tolerance. We recently showed that FRC abundance and antigen expression levels31,41 are affected in T1D. While the role of FRCs as non-professional APCs that could modulate antigen-specific T cells has been previously established, these studies have relied on transgenic mice for overexpression of artificial (not disease relevant) antigens in FRCs. Whether FRCs expressing T1D antigens can present them to specific T cells establishing a definitive role for FRCs in peripheral tolerance still needs to be demonstrated for developing new FRC-centered therapies for autoimmune diseases like T1D.
The purpose of this report was (i) to characterize reticular properties of FRC networks in murine models of T1D to determine if these FRC properties are also affected in the disease, and (ii) to report the development of a novel tissue-engineered FRC reticula that recapitulates structural observations from LNs in disease (NOD) and control (NOR) models, applicability to human cells, and potential for studying engagement of T1D antigen-specific T cells with FRCs in a physiomimetic environment reflective of the LN paracortex. We report the development of an innovative tissue-engineered FRC reticula using collagen scaffolds and FRCs from diabetes-prone NOD mice or from diabetes-resistant NOR mice that recapitulates reticular alterations of FRCs observed in T1D murine models. We also show that reticula engineered with T1D antigen-expressing FRCs allow quantifying engagement of antigen-specific T cells in vitro and this was more effectively achieved than with 2D culture systems. This was made possible by genetically engineering LN-derived FRCs to express T1D antigenic epitopes.
FRCs (gp38+ CD31−) provide structural integrity to LNs14 and their network integrity in LNs plays a critical role in adaptive immunity, including tolerance induction.9,16,26,29 The myofibroblastic phenotype (αSMA+) of FRCs allows them to modulate their contractility3 and to accommodate LN expansion during an acute immune response. We found that reticular organization of NOD-derived FRCs in LNs is altered compared to NOR controls where NOD FRC networks have larger reticular pores than NOR controls. Though alterations of FRC reticular networks in NOD mice compared to NOR are minimal (yet statistically different), these quantifications have been made on steady-state LNs, while in vivo LN overall size can vary dramatically depending on the status of the host (e.g. during inflammation). Thus, small size differences in steady-state NOD reticula compared to NOR controls may result in larger differences in LNs in vivo during inflammatory processes. Importantly, we show that those differences in reticular properties of FRC reticula in T1D models are found not only in PLNs, but also in the SDLNs.
It was previously reported that FRC contractility is controlled by their gp38 expression and their interaction with dendritic cells.3 CLEC2 upregulation by both LN-resident and infiltrating migratory dendritic cells in response to inflammatory cues inhibits gp38-driven actomyosin contractility of FRCs, resulting in FRC elongation and reduced LN tension, which enables rapid LN expansion in the absence of stromal cell proliferation.3 FRC reticular alterations in T1D were consistently observed in both low (4-week old) and high (12-week old) T1D risk mice and in both PLNs and SDLNs, and was not associated with a change in overall LN volume or cellularity, at least at 12 weeks of age. This was somewhat unexpected given the uniqueness of PLNs as a more “inflamed” site where β-cell antigens from the pancreas drain to and promote the activation of T1D antigen-reactive T cells. Thus, alteration in FRC reticular properties are unlikely to be a consequence of the inflammatory process in T1D leading to dendritic cell hyperactivation and CLEC2-mediated inhibition of gp38. Long-term culture of isolated and expanded FRCs in collagen scaffolds further supports this hypothesis because similar reticular alterations were observed in engineered reticula with NOD FRCs compared to NOR FRCs. In those studies, delayed scaffold remodeling of NOD FRCs was not associated with decreased cell proliferation in the scaffold at least during the first 14 days of culture in the scaffolds, suggesting that larger-pore/looser reticula might result from more relaxed FRCs. Culture in 3D scaffolds for at least 21 days was able to rescue the remodeling capability of NOD PLN-derived FRCs. Another phenotypical alteration found in NOD FRCs compared to NOR controls was the overall reduced gp38 expression during ex vivo culture, despite the fact that gp38 expression on NOD FRCs was comparable to NOR cells in freshly sorted FRCs. Interestingly, NOD FRC remodeling, which occurred 21 days after culture in the scaffolds unlike NOR cells that started the remodeling process by day 1, was associated with increased expression of gp38, supporting the hypothesis that 3D culture rescues NOD-derived FRC remodeling capability, potentially through gp38 upregulation. Future studies will aim at unraveling the mechanisms for NOD FRC reduced remodeling capacity and contractility by identifying potential genetic or post-translational modifications that could be targeted for therapeutic treatments in T1D.
Using human FRCs isolated from PLNs of healthy donors and collagen scaffolds, we demonstrated that similarly to murine FRCs, we can recapitulate the FRC reticular organization of human PLNs, increasing the translational potential of our tissue-engineering approach. Human FRCs from healthy donors were able to remodel their reticula network leading to a more contracted reticula after short-term 3D culture in the scaffolds, which resembled the case of FRCs from diabetes-resistant NOR mice. Future studies will aim at studying the reticula formation capabilities of FRCs obtained from T1D patients.
Even after long-term culture of FRCs in collagen scaffolds, reticula pore size was larger than what we observed in LN reticula, but we are attributing that difference to the initial dimensions and properties of the collagen scaffold we used for this study. Future studies will assess whether modifying the scaffold structure and/or composition to decrease scaffold pore size, will promote formation of engineered FRC reticula with LN-relevant pore sizes.
LN stromal cells including FRCs, express self-antigens, as well as T1D antigens.10,11,22,41 Using transgenic mice expressing non-disease relevant model antigens in FRCs, it was demonstrated that FRCs are able to present endogenously expressed antigens via MHC class I to autoreactive CD8+ T cells without co-stimulatory signals, leading to initial proliferation followed by deletion.11 We previously reported that FRC frequency in native LNs and T1D antigen expression levels is decreased in T1D.31,41 These alterations in FRC tolerogenic properties could impair tolerogenic engagement of T1D autoreactive T cells. Here, we show that reticula with FRCs genetically engineered to express T1D allow quantification of engagement of T1D antigen-specific T cells, and T cell engagement in 3D reticula was more efficient than in 2D cultures. We also show that FRCs can remodel their reticula even when they are not in contact with dendritic cells. Thus, FRCs could modulate their interactions with autoreactive T cells by remodeling their reticular network in LNs. By decreasing their contractility through gp38 downregulation, FRCs can loosen/relax their network, potentially decreasing FRC tolerogenic interactions with autoreactive T cells and promoting their escape from peripheral regulation in LNs. Our model could be applied in the future to determine how reticular properties of LN FRCs orchestrate immune responses in the LN27,28 and whether reduced antigen presentation in LN FRCs decreases their peripheral regulation of autoreactive T cells. This work contributes to generate a new field of research for understanding the role of FRCs in peripheral tolerance and in autoimmune diseases like T1D.
We also demonstrated that our tissue engineered FRC reticula can be employed for tracking T cell movements, including migration within the reticula and interactions with FRCs. Potentially, our model can be used for characterizing the immunological synapse that forms between T1D antigen-expressing FRCs and specific T cells, which occurs at earlier time points than what was tested in this report.
Promoting peripheral expression in professional APCs and presentation of self-antigens related to specific autoimmune diseases to autoreactive T cells without co-stimulation or in combination with immunosuppressive agents have shown promising results in preventing or attenuating autoimmune responses in murine models of multiple sclerosis and T1D.20,30,32,36,37 While professional APCs can switch from a tolerogenic to an immunogenic function in the context of inflammation by upregulating co-stimulatory molecules and pro-inflammatory cytokines thereby raising some concerns on in vivo stability of these approaches, other non-professional APCs, like FRCs, lack co-stimulatory molecules, are more stably tolerogenic by nature and may represent a safer option for tolerance induction. We recently showed that promoting extra-LN formation of FRC-like reticula expressing T1D antigens is sufficient to induce tolerance to T1D antigens and prevent T1D in NOD mice without affecting overall immunity.15 Thus, in future work, we can test our engineered reticula with T1D antigen-expressing FRCs in therapeutic applications to determine whether, after implantation, they can attract and delete autoreactive T cells from the systemic circulation to ameliorate T1D.
In conclusion, we engineered a physiologically relevant model of the LN FRC reticula that could recapitulate FRC organization and phenotype in LNs of T1D-prone mice, including reduced remodeling capabilities, and in human LNs. When engineered to express and present T1D antigens to antigen-reactive T cells, FRCs seeded on 3D collagen scaffold can engage T1D antigen-specific T cells more efficiently than 2D systems. Future applications of our model will help understand the role of FRCs in tolerance and T1D and how reticular organization and antigen expression levels affect FRC-T cell interactions and T1D development and lead to the development of novel therapeutics for inducing tolerance in autoimmune diseases like T1D.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors are grateful to the personnel of the DRI Preclinical Cell Processing and Translational Models Core for their help with the management of mice colonies, the DRI Imaging Core lead by Dr. Maria Boulina for providing expertise on confocal imaging, the DRI Histology core headed by Kevin Johnson for his help with histological processing of all samples and the DRI Flow Cytometry core led by Dr. Oliver Umland for his help and expertise in cell sorting and flow cytometry data analysis. Funding was provided by philanthropic funds from the Diabetes Research Institute Foundation, a grant from the Juvenile Diabetes Research Foundation (Grant # 2-SRA-2016-316-S-B), a grant from the National Institute of Health (Grant # DK109929), Dr. Tomei’s 2017 provost’s research award, and Dr. Tomei’s BME start-up package. Dr. Postigo-Fernandez was supported by a Postdoctoral Fellowship from the American Diabetes Association (1-18-PDF-151).
Author Contributions
FGB, FZT, SW, MS, ML and LH generated data; JPF prepared cells and tissues for some of the experiments; FGB, FZT, RJC and AAT reviewed/edited the manuscript and contributed to discussion. AAT and RJC designed the research and AAT wrote the manuscript.
Conflict of interest
Freddy Gonzalez Badillo, Flavia Zisi Tegou, Riccardo Masina, Shane Wright, Mackenzie Scully, Laura Harwell, Michael Lupp, Jorge Postigo-Fernandez, Remi J. Creusot, and Alice Tomei declare that they have no conflicts of interest.
Ethical Standards
No human studies were carried out by the authors for this article. All animal studies were carried out in accordance with established guidelines for animal care and were approved by the Institutional Animal Care and Use Committee of University of Miami.
Abbreviations
- FRCs
Fibroblastic reticular cells
- T1D
Type-1 diabetes
- LN
Lymph node
- PLN
Pancreatic LN
- SDLN
Skin-draining LN
- gp38
Podoplanin
- αSMA
Alpha-smooth muscle actin
- NOD
Non-obese diabetic mice
- NOR
Diabetes-resistant NOD mice
- APCs
Antigen-presenting cells
- SpongeCol®
Commercially available collagen-based scaffold
- 2D
Two-dimensional
- 3D
Three-dimensional
- MFI
Mean fluorescence intensity
- IGRP
Islet-specific glucose-6-phosphatase catalytic subunit-related protein
- MHC
Major histocompatibility complex
- MSD
Mean squared displacement
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Acton SE, et al. Dendritic cells control fibroblastic reticular network tension and lymph node expansion. Nature. 2014;514:498–502. doi: 10.1038/nature13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Astarita JL, Acton SE, Turley SJ. Podoplanin: emerging functions in development, the immune system, and cancer. Front. Immunol. 2012;3:283. doi: 10.3389/fimmu.2012.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Astarita JL, et al. The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nat. Immunol. 2015;16:75–84. doi: 10.1038/ni.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown FD, Turley SJ. Fibroblastic reticular cells: organization and regulation of the T lymphocyte life cycle. J. Immunol. 2015;194:1389–1394. doi: 10.4049/jimmunol.1402520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen JN, et al. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J. Exp. Med. 2010;207:681–688. doi: 10.1084/jem.20092465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Creusot RJ, Postigo-Fernandez J, Teteloshvili N. Altered function of antigen-presenting cells in type 1 diabetes: a challenge for antigen-specific immunotherapy? Diabetes. 2018;67:1481–1494. doi: 10.2337/db17-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dastagir SR, et al. Efficient presentation of multiple endogenous epitopes to both CD4(+) and CD8(+) diabetogenic T cells for tolerance. Mol Ther. 2017;4:27–38. doi: 10.1016/j.omtm.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denton AE, Roberts EW, Linterman MA, Fearon DT. Fibroblastic reticular cells of the lymph node are required for retention of resting but not activated CD8+ T cells. Proc. Natl. Acad. Sci. U.S.A. 2014;111:12139–12144. doi: 10.1073/pnas.1412910111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dertschnig S, et al. Graft-versus-host disease reduces lymph node display of tissue-restricted self-antigens and promotes autoimmunity. J. Clin. Investig. 2020 doi: 10.1172/jci133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fletcher AL, Malhotra D, Turley SJ. Lymph node stroma broaden the peripheral tolerance paradigm. Trends Immunol. 2011;32:12–18. doi: 10.1016/j.it.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fletcher AL, et al. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J. Exp. Med. 2010;207:689–697. doi: 10.1084/jem.20092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fletcher AL, et al. Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells. Front. Immunol. 2011;2:35. doi: 10.3389/fimmu.2011.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuhlbrigge R, Yip L. Self-antigen expression in the peripheral immune system: roles in self-tolerance and type 1 diabetes pathogenesis. Curr. Diab. Rep. 2014;14:525. doi: 10.1007/s11892-014-0525-x. [DOI] [PubMed] [Google Scholar]
- 14.Gardner JM, et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science. 2008;321:843–847. doi: 10.1126/science.1159407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.GonzalezBadillo FE, et al. CCL21 expression in beta-cells induces antigen-expressing stromal cell networks in the pancreas and prevents autoimmune diabetes in mice. Diabetes. 2019;68:1990–2003. doi: 10.2337/db19-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graw F, Regoes RR. Influence of the fibroblastic reticular network on cell-cell interactions in lymphoid organs. PLoS Comput. Biol. 2012;8:e1002436. doi: 10.1371/journal.pcbi.1002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hahnlein JS, et al. Impaired lymph node stromal cell function during the earliest phases of rheumatoid arthritis. Arthritis Res. Ther. 2018;20:35. doi: 10.1186/s13075-018-1529-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirosue S, Dubrot J. Modes of antigen presentation by lymph node stromal cells and their immunological implications. Front. Immunol. 2015;6:446. doi: 10.3389/fimmu.2015.00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hull CM, Peakman M, Tree TIM. Regulatory T cell dysfunction in type 1 diabetes: what’s broken and how can we fix it? Diabetologia. 2017;60:1839–1850. doi: 10.1007/s00125-017-4377-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter Z, et al. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano. 2014;8:2148–2160. doi: 10.1021/nn405033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kont V, et al. Modulation of Aire regulates the expression of tissue-restricted antigens. Mol. Immunol. 2008;45:25–33. doi: 10.1016/j.molimm.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JW, et al. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat. Immunol. 2007;8:181–190. doi: 10.1038/ni1427. [DOI] [PubMed] [Google Scholar]
- 23.Lu P, Wang YL, Linsley PS. Regulation of self-tolerance by CD80/CD86 interactions. Curr. Opin. Immunol. 1997;9:858–862. doi: 10.1016/S0952-7915(97)80190-2. [DOI] [PubMed] [Google Scholar]
- 24.Magnusson FC, et al. Direct presentation of antigen by lymph node stromal cells protects against CD8 T-cell-mediated intestinal autoimmunity. Gastroenterology. 2008;134:1028–1037. doi: 10.1053/j.gastro.2008.01.070. [DOI] [PubMed] [Google Scholar]
- 25.Malhotra D, et al. Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat. Immunol. 2012;13:499–510. doi: 10.1038/ni.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez VG, et al. Fibroblastic reticular cells control conduit matrix deposition during lymph node expansion. Cell Rep. 2019;29:2810–2822. doi: 10.1016/j.celrep.2019.10.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novkovic M, Onder L, Bocharov G, Ludewig B. Topological structure and robustness of the lymph node conduit system. Cell Rep. 2020;30:893–904.e896. doi: 10.1016/j.celrep.2019.12.070. [DOI] [PubMed] [Google Scholar]
- 28.Novkovic M, Onder L, Cheng HW, Bocharov G, Ludewig B. Integrative computational modeling of the lymph node stromal cell landscape. Front. Immunol. 2018;9:2428. doi: 10.3389/fimmu.2018.02428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novkovic M, et al. Topological small-world organization of the fibroblastic reticular cell network determines lymph node functionality. PLoS Biol. 2016;14:e1002515. doi: 10.1371/journal.pbio.1002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearson RM, et al. Overcoming challenges in treating autoimmuntity: development of tolerogenic immune-modifying nanoparticles. Nanomedicine. 2019;18:282–291. doi: 10.1016/j.nano.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Postigo-Fernandez J, Farber DL, Creusot RJ. Phenotypic alterations in pancreatic lymph node stromal cells from human donors with type 1 diabetes and NOD mice. Diabetologia. 2019;62:2040–2051. doi: 10.1007/s00125-019-04984-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prasad S, et al. Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells. J. Autoimmun. 2018;89:112–124. doi: 10.1016/j.jaut.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pugliese A, et al. Self-antigen-presenting cells expressing diabetes-associated autoantigens exist in both thymus and peripheral lymphoid organs. J. Clin. Investig. 2001;107:555–564. doi: 10.1172/jci10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sobocinski GP, et al. Ultrastructural localization of extracellular matrix proteins of the lymph node cortex: evidence supporting the reticular network as a pathway for lymphocyte migration. BMC Immunol. 2010;11:42. doi: 10.1186/1471-2172-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomei AA, Siegert S, Britschgi MR, Luther SA, Swartz MA. Fluid flow regulates stromal cell organization and CCL21 expression in a tissue-engineered lymph node microenvironment. J. Immunol. 2009;183:4273–4283. doi: 10.4049/jimmunol.0900835. [DOI] [PubMed] [Google Scholar]
- 36.Tostanoski LH, Gosselin EA, Jewell CM. Engineering tolerance using biomaterials to target and control antigen presenting cells. Discov. Med. 2016;21:403–410. [PubMed] [Google Scholar]
- 37.Tostanoski LH, et al. Reprogramming the local lymph node microenvironment promotes tolerance that is systemic and antigen specific. Cell Rep. 2016;16:2940–2952. doi: 10.1016/j.celrep.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walton WH. Feret’s statistical diameter as a measure of particle size. Nature. 1948;162(4113):329–330. doi: 10.1038/162329b0. [DOI] [Google Scholar]
- 39.Wendland M, et al. Lymph node T cell homeostasis relies on steady state homing of dendritic cells. Immunity. 2011;35:945–957. doi: 10.1016/j.immuni.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 40.Yip L, Creusot RJ, Pager CT, Sarnow P, Fathman CG. Reduced DEAF1 function during type 1 diabetes inhibits translation in lymph node stromal cells by suppressing Eif4g3. J. Mol. Cell Biol. 2013;5:99–110. doi: 10.1093/jmcb/mjs052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yip L, et al. Deaf1 isoforms control the expression of genes encoding peripheral tissue antigens in the pancreatic lymph nodes during type 1 diabetes. Nat. Immunol. 2009;10:1026–1033. doi: 10.1038/ni.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.