

Abstract
Introduction
The intestinal epithelium contains specialized cells including enterocytes, goblet, Paneth, enteroendocrine, and stem cells. Impaired barrier integrity in Inflammatory Bowel Disease is characterized by elevated levels of pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α). Prior studies in immortalized lines such as Caco-2, without native epithelial heterogeneity, demonstrate the amelioration of TNF-α compromised barrier integrity via nicotinic (nAChR) or muscarinic (mAChR) acetylcholine receptor activation.
Methods
A tissue-engineered model of primary human small intestinal epithelium was derived from dissociated organoids cultured on collagen-coated Transwells. Differentiation was accomplished with serum-containing media and compared to Caco-2 and HT-29 regarding alkaline phosphatase expression, transepithelial electrical resistance (TEER), and IL-8 secretion. Inflammation was modeled via basal stimulation with TNF-α (25 ng/mL) with or without nicotine (nAChR agonist) or bethanechol (mAChR agonist). Apoptosis, density (cells/cm2), TEER, lucifer yellow permeability, 70 kDa dextran transport, cell morphology, and IL-8 secretion were characterized.
Results
Primary intestinal epithelium demonstrates significant functional differences compared to immortalized cells, including increased barrier integrity, IL-8 expression, mucus production, and the presence of absorptive and secretory cells. Exposure to TNF-α impaired barrier integrity, increased apoptosis, altered morphology, and increased secretion of IL-8. Stimulation of nAChR with nicotine did not ameliorate TNF-α induced permeability nor alter 70 kDa dextran transport. However, stimulation of mAChR with bethanechol decreased transport of 70 kDa dextran but did not ameliorate TNF-α induced paracellular permeability.
Conclusions
A primary model of intestinal inflammation was evaluated, demonstrating nAChR or mAChR activation does not have the same protective effects compared to immortalized epithelium. Inclusion of other native stromal support cells are underway.
Electronic supplementary material
The online version of this article (10.1007/s12195-020-00633-0) contains supplementary material, which is available to authorized users.
Keywords: Muscarinic, Nicotinic, mAChR, nAChR, Organoid, Inflammation, Tumor necrosis factor
Introduction
A monolayer of epithelial cells forms the main component of the mucosal barrier in the small intestine.24 The epithelium consists of a variety of specialized cells including enterocytes, goblet cells, Paneth cells, enteroendocrine cells, transit-amplifying cells, and stem cells.24 Some cells perform specialized functions to fortify the mucosal barrier, such as mucin or antimicrobial peptide secretion by goblet and Paneth cells, respectively.24 But epithelial permeability is largely regulated by the expression of cohesive junctional complexes, such as tight junctions (TJs), which seal the paracellular space between adjacent epithelial cells.42 Intestinal bowel disease (IBD) patients exhibit increased epithelial permeability and TJ abnormalities.37 Increased epithelial permeability may initiate pathogen translocation from the intestinal lumen to activate the mucosal immune response by underlying immune cells. Thus, IBD patients exhibit increased expression of pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α).50 TNF-α is produced by cells occupying the lamina propria such as macrophages, monocytes, and differentiated Th1 cells.55,61 In return, TNF-α induces epithelial apoptosis,55,74 disrupting the reorganization of TJs, thereby further propagating the inflammatory response.74 Though it is unclear whether increased epithelial permeability precludes pro-inflammatory cytokine production or is a consequence of mucosal immune activation.42 Nevertheless, ameliorating epithelial permeability is critical for IBD treatment.
Previous in-vitro studies demonstrated that cholinergic activation of intestinal epithelial cells (IECs) decreased epithelial permeability following pro-inflammatory challenge.20,31,32,68,77 Acetylcholine (ACh) is a critical IEC regulator, controlling ion transport,26 mucus secretion,5 and cell proliferation.65 In the small intestine, acetylcholine is secreted by enteric neurons of the lamina propria48 and possibly by IECs themselves.64,65 Intestinal epithelial cells express both nicotinic (nAChR) and muscarinic acetylcholine receptors (mAChRs) that interact with ACh.26 However, these studies utilized murine models77 or immortalized epithelial Caco-2142 and HT-2920,32,68 cell lines. Caco-2 and HT-29 cells were originally obtained from human colon adenocarcinoma and spontaneously differentiate after extended culture on permeable membranes.53,54 Differentiated Caco-2 express some morphological and biological characteristics of small intestinal enterocytes such as monolayer formation, polarization, TJ expression, apical microvilli, and small intestinal enzyme activity on the apical cell surface.53,54 The differentiated HT-29 phenotype is similar to small intestinal enterocytes, except HT-29 cells also produce mucin similar to intestinal goblet cells.53 Nevertheless, both Caco-2 and HT-29 are malignant cell types and it is unclear whether these cells faithfully represent native intestinal epithelium. Previous in-vivo studies using murine models also demonstrated cholinergic amelioration of epithelial permeability,77 but differences between human and murine species limit the study relevance.25 Utilizing human small intestinal tissue explants is hindered by tissue availability, particularly for experimental replicates. Furthermore, both in-vivo studies and whole tissue explants complicate mechanistic studies due to multicellular tissue complexity. For example, it is widely accepted that nAChR stimulation of intestinal macrophages reduces TNF-α production.7,41,71 Thus, studying the cholinergic modulation of epithelial permeability using a primary cell-derived intestinal model is a desirable advancement to uncover epithelium-specific processes.
Recently developed intestinal organoids have largely addressed the scalable culture of primary intestinal tissue.38,56 Three dimensional (3D) intestinal organoids are established by embedding biopsy or resection derived LGR5+ intestinal stem cells in a complex extracellular matrix, Matrigel, and supplementing the medium with essential growth factors: Wnt-3A, epidermal growth factor (EGF), noggin, and R-spondin.38 Organoids form cystic structures, contain the intestinal epithelial cell lineages found in the native intestine, and can be passaged to generate enough tissue for experimental replication.58 Though organoids are both physiologically similar to the native intestinal epithelium and scalable, the closed lumen is relatively inaccessible thereby complicating permeability measurements. Two dimensional (2D) monolayers derived from dissociated organoids enable access to both apical and basal media compartments, greatly facilitating epithelial perturbation and analysis.21,30,35,49,69,72
In this study, we describe the development and characterization of a model small intestinal epithelium derived from primary human organoids and its application to investigate cholinergic amelioration of epithelial barrier integrity. This primary cell-derived intestinal model recapitulated the hallmarks of differentiated small intestinal epithelium such as polarization, apical microvilli, TJ formation, small intestinal enzyme expression, but also contained both absorptive enterocytes and mucus-secreting goblet cells which account for the majority of the intestinal epithelium in-vivo.18 Comparison of the primary cell-derived model to Caco-2 and HT-29 monolayers demonstrated increased throughput, barrier integrity, and interleukin-8 production (IL-8), a critical cytokine of the intestinal epithelium in-vivo.36,62 Treatment of primary monolayers with TNF-α altered cell morphology and decreased epithelial barrier integrity (measured by TEER and Lucifer Yellow permeability) through both apoptosis and tight junction rearrangement. Muscarinic stimulation reduced epithelial macromolecule transport of a 70 kDa dextran but neither nAChRs nor mAChRs activation ameliorated TNF-α induced epithelial permeability, potentially highlighting an important functional difference between previously published immortalized cell line models and primary epithelium.
Materials and Methods
Primary Intestinal Organoid Culture and Expansion
De-identified endoscopic tissue biopsies were collected from grossly unaffected (macroscopically normal) areas of the duodenum patients undergoing endoscopy for gastrointestinal complaints. Informed consent and developmentally appropriate assent were obtained at Boston Children’s Hospital from the donors’ guardian and the donor, respectively. All methods were approved and carried out per the Institutional Review Board of Boston Children’s Hospital (Protocol Number IRB-P00000529). Cells were cultured as 3D organoids embedded in 50 μL of Matrigel (cat no. 354230, Corning) on a 24-well plate as previously described.57 Wnt-3A, epidermal growth factor (EGF), noggin, and R-spondin 3 containing organoid expansion medium, termed WENR medium, was prepared from a mixture of Advanced DMEM/F12 medium (cat no. 12634028, Gibco) and 50% LWRN conditioned medium, which was prepared from L-WRN cells, as previously described.44 This cell line produces Wnt-3A, R-spondin 3, and noggin. WENR medium was supplemented with GlutaMAX (1×, cat no. 35050061, Gibco), HEPES (10 mM, cat no. 15630080, Gibco), Primocin (0.1 mg mL−1, Invivogen), B-27 supplement (0.5×, cat no. 12587010, Gibco), N-2 supplement (0.5×, cat no. 17502048, Gibco), nicotinamide (10 mM, cat no. N0636, Sigma-Aldrich), N-acetyl cysteine (0.5 mM, cat no. A7250, Sigma-Aldrich), epidermal growth factor (50 ng mL−1, cat no. 315-09, Peprotech), gastrin (50 nM, cat no. A7250, Sigma-Aldrich), A-83-01 (500 nM, cat no. SML0788, Sigma), prostaglandin E2 (10 nM, cat no. 14010, Cayman Chemical), and SB202190 (10 μM, cat no. S7067, Sigma-Aldrich). Y-27632 ROCK inhibitor (10 μM, cat no. Y0503, Sigma-Aldrich) was added to the organoid medium for the first 48 h following crypt isolation or passage. The culture medium was refreshed every 48 h using 500 μL per well. Cell culture was performed in a humidified, 37 °C, 5% CO2 incubator.
Every 7–10 days, the organoids were passaged to new 24-well plates at a ratio of 1:4–1:8 depending on culture density. Matrigel droplets were scratched off the 24-well plate using a 1000 μL pipette tip and collected into a 15 mL conical tube. The organoids were centrifuged at 500 g for 5 min at room temperature. After aspirating the cell culture medium, the organoids were suspended in 0.5 mM ethylenediaminetetraacetic acid (EDTA, cat no. AM9260G, Gibco) in phosphate-buffered saline (PBS) without Ca2+ or Mg2+ and re-centrifuged at 300 g for 5 min at room temperature. After aspirating the EDTA, the organoids were re-suspended in Trypsin-EDTA and incubated in a 37 °C bath for 2 min. The Trypsin-EDTA was then quenched via a 2:1 dilution with Caco-2 culture medium containing 10% FBS and the organoid suspension was triturated ~10× using a 1000 μL pipette tip to produce single cells and small organoid fragments. The cells were pelleted at 300 g for 5 min at room temperature. The cells were suspended in 4 °C Matrigel and plated on new 24-well plates. The plated Matrigel was incubated at 37 °C for 15 min before adding 500 μL of culture medium containing 10 μM ROCK inhibitor.
Primary Intestinal Monolayer Culture
Polyester Transwell inserts (cat no. 353095, Corning, Corning, NY) in a 24-well plate were coated with 200 μL of a 400 µg mL−1 rat tail type I collagen in Dulbecco’s Modified Eagle Medium (DMEM, cat no. 11995-065, ThermoFisher) for at least 1 h at 37 °C inside a humidified cell culture incubator with 5% CO2. Organoids were harvested for dissociation and monolayer seeding after 7–10 days of culture. Matrigel droplets were harvested and processed in Trypsin-EDTA as described above. The Trypsin-EDTA was then quenched via a 2:1 dilution with Caco-2 culture medium containing 10% FBS and the organoid suspension was triturated ~20× using a 1000 μL pipette tip to produce single cells and small organoid fragments. The cell suspension was filtered through a 40 μm cell strainer (cat no. 22-363-547, Fisher) into a 50 mL conical tube and pelleted at 300 g for 5 min at room temperature. The cells were resuspended in WENR medium with 10 μM ROCK inhibitor. Transwell inserts were seeded using 200 μL of cell suspension (seeding density of 9.09 × 105 cells per cm2) and then 600 μL of media + 10 μM ROCK inhibitor was added to the basolateral compartment. ROCK inhibitor was used for the first 48 h of cell culture and the apical and basal cell culture medium was refreshed every other day. Following 2 days of culture in the EM medium, the apical and basal medium was replaced with differentiation medium (DM) containing Advanced DMEM/F12 + 20% FBS + 4 mM GlutaMAX supplement + 100 U mL−1 Penicillin-Streptomycin. The apical and basal DM medium was replenished every 48 h.
Caco-2 Cell Culture
Caco-2 epithelial cells were obtained from the American Type Culture Collection (ATCC, HTB-37) and cultured in Dulbecco’s Modified Eagle Medium (DMEM, cat no. 11995-065, ThermoFisher) supplemented with 10% fetal bovine serum (FBS, cat no. 35-011-CV, Corning), and 100 U mL−1 Penicillin-Streptomycin (cat no. 15140122, ThermoFisher). Caco-2 cells were cultured on 0.4 μm polyester (cat no. 353095, Corning, Corning, NY) Transwell inserts in a 24 well plate. Before cell seeding, the inserts were coated with 200 μL of a 400 µg mL−1rat tail type I collagen in DMEM for at least 1 h at 37 °C inside a humidified cell culture incubator with 5% CO2. Post at least 1 h, the collagen solution was removed, and Caco-2 cells were seeded on the inserts by adding 200 μL of Caco-2 cell suspension (seeding density of 2.6 × 105 cells per cm2) and then adding 600 μL of media to the basolateral compartment. The apical and basal cell culture medium was refreshed every other day.
HT-29 Cell Culture
HT-29 epithelial cells were obtained from the American Type Culture Collection (ATCC, HTB-38) and cultured in RPMI 1640 Medium with GlutaMAX and HEPES (cat no. 72400047, ThermoFisher) supplemented with 10% fetal bovine serum (FBS, cat no. 35-011-CV, Corning), and 100 U mL−1 Penicillin-Streptomycin (cat no. 15140122, ThermoFisher). HT-29 cells were cultured on 0.4 μm polyester (cat no. 353095, Corning, Corning, NY) Transwell inserts in a 24 well plate. Before cell seeding, the inserts were coated with 200 μL of a 400 µg mL−1 rat tail type I collagen in DMEM for at least 1 h at 37 °C inside a humidified cell culture incubator with 5% CO2. Post at least 1 h, the collagen solution was removed, and HT-29 cells were seeded on the inserts by adding 200 μL of HT-29 cell suspension (seeding density of 2.6 × 105 cells cm2) and then adding 600 μL of media to the basolateral compartment. The apical and basal cell culture medium was refreshed every other day. HT-29 cells were used at passage 69–70.
Tumor Necrosis Factor-α, Nicotinic, and Muscarinic Stimulation of Monolayers
Nicotine (cat no. N3876, Sigma-Aldrich) and bethanechol (cat no. 5057730001, Sigma-Aldrich) stocks were prepared at 50 mM and stored at – 20 °C. Carbachol (cat no. 212385, Sigma-Aldrich) stocks were prepared at 100 mM and stored at – 20 °C. Stocks were thawed only once before use. Nicotine (nAChR agonist), bethanechol (mAChR agonist), or carbachol (nAChR and mAChR agonist) was added to the basal media compartment and incubated at 37 °C and 5% CO2 for 30 min before TNF-α exposure. Nicotine or bethanechol exposure was done using 575 µL of cell culture media in the basal compartment, followed by a 25 µL spike of TNF-α to achieve the desired TNF-α concentration. TNF-α (cat no. 300-01A, PeproTech) was dissolved in deionized water with 0.1% bovine serum albumin at 10,000 µg mL−1 and stored at −80 °C. Stocks were thawed only once before use. TNF-α was spiked in the basal media compartment at the concentration and duration specified in the figure caption following nicotinic or muscarinic stimulation.
Alkaline Phosphatase Measurement
Alkaline phosphatase (AP) expression was measured using a commercial kit (AS-71109, AnaSpec, Fremont, CA). All kit components were prepared as specified by the manufacturer. Cell lysate from Transwell inserts was prepared as follows: medium was removed, and the inserts were washed 2× with PBS in both apical and basal compartments. Next, 200 μL of sterile 10× TrypLE Select (cat no. A1217701, ThermoFisher) was added to the apical side of each insert before incubation at 37 °C for ~15 min. Cells were collected into a centrifuge tube and inserts were washed with 800 μL of PBS and pelleted at 300 g for 5 min at room temperature. The pellet was re-suspended in 150 μL of AP kit supplied buffer, washed and re-suspended in 150 μL of buffer. A 10 μL aliquot of cell suspension was removed to quantify the cell number via hemocytometer. The cells were centrifuged and suspended in 0.2% Triton X-100 (AC327371000, Fisher Scientific). The cells were incubated for 10 min at 4 °C with agitation and then centrifuged at 2500×g for 10 min at 4 °C. The supernatant was used for the AP assay as specified in the kit instructions. The fluorescence was measured via plate reader (EnSightTM, PerkinElmer) using 485 nm and 528 nm emission and excitation wavelengths, respectively. The actual amount of AP was interpolated using a calibration curve generated with the kit supplied AP standard.
Transepithelial Electrical Resistance (TEER) Measurement
To minimize temperature variation of well plates, 24 well plates were allowed to cool to room temperature for 20 min before TEER measurement.6 TEER was measured using an epithelial Volt Ohmmeter (EVOM2, World Precision Instruments, Sarasota, FL) coupled to a chopstick-like electrode (STX2, World Precision Instruments, Sarasota, FL). TEER values (Ω cm2) were determined by subtracting the baseline resistance value measured in the absence of cells and then multiplying the remaining ‘specific’ resistance value (Ω) times the cell culture surface area (cm2).
IL-8 Measurement
IL-8 secretion was measured using an ELISA kit (cat no. 88-8086-22, Thermofisher) according to the manufacturer’s instructions. Note that cell culture supernatant samples were incubated overnight at 4 °C to maximize ELISA sensitivity. Before the ELISA, cell culture supernatants were collected, frozen at − 80 °C, and thawed 1–2 times.
Paracellular Permeability Measurement
Paracellular permeability was measured by measuring the flux of small molecular weight (457 Da) Lucifer Yellow dye (cat no. L0259, Sigma-Aldrich) across primary human intestinal monolayers. Lucifer Yellow salt was dissolved in water at 2.25 mM and stored at − 20 °C. Stocks were thawed and diluted to a working concentration of 100 µM in sterile cell culture medium for the assay. 100 µL of Lucifer Yellow was added to the apical side of monolayers while 600 µL of fresh cell culture medium was added to the basal side of monolayers. After 60–90 min, 50 µL the basal cell culture medium was sampled and the fluorescence intensity of the samples was measured at 360 nm and 528 nm emission and excitation wavelengths, respectively.
70 kDa Dextran Transport Measurement
Transport of a 70 kDa dextran was measured by measuring the flux of large molecular weight (70 kDa) tetramethylrhodamine (TRM) conjugated dextran (cat no. 50-152-6775, Fisher) across primary human intestinal monolayers. 25 mg mL−1 dextran stock was stored at 4 °C. The stock was diluted to a working concentration of 3 mg mL−1 in a sterile cell culture medium for the assay. 100 µL of Lucifer Yellow was added to the apical side of monolayers while 600 µL of fresh cell culture medium was added to the basal side of monolayers. After 60–90 min, 50 µL the basal cell culture medium was sampled and the fluorescence intensity of the samples was measured at 530 and 590 nm emission and excitation wavelengths, respectively.
Localization of 70 kDa dextran in primary human intestinal monolayers was measured by quantifying a 70 kDa TRM conjugated lysine-fixable dextran (cat no. D1818, Thermofisher). Dextran was dissolved in sterile PBS with Ca2+ and Mg2+ at 20 mg mL−1 and stored at − 20 °C. Stocks were thawed and diluted to a working concentration of 3 mg mL−1 in sterile cell culture medium for the assay. Monolayers were incubated in 600 µL of fresh, basal, cell culture medium with or without 50 µM bethanechol for 4 h. The apical medium was not changed. After 4 h, the apical medium was removed, and the 3 mg mL−1 dextran solution was added. After 60–90 min, the apical and basal medium was removed, and the monolayers were washed 5x with PBS with Ca2+ and Mg2+. Then, the monolayers were fixed in 4% formaldehyde (cat no. 28906, ThermoFisher) for 20 min in the dark. Post fixation, the monolayers were washed 2× with PBS and permeabilized in 0.1% Triton X-100 (AC327371000, Fisher Scientific) for 15 min in the dark. Next, the monolayers were washed 2× with PBS and blocked overnight in 2.5% goat serum at 4 °C. The next day, the monolayers were stained with anti-ZO-1 antibody (1:200, 2 h, cat no. 339188, ThermoFisher) at room temperature in the dark. The monolayers were washed 2× with PBS and stained with secondary antibody (1:1000, 1 h, cat no. A-21235, ThermoFisher) in 2.5% goat serum at room temperature in the dark. The monolayers were washed 2x with PBS and the Transwell membranes were isolated and mounted on a standard glass slide and coverslip with Gold Antifade Mountant (cat no. P36931, ThermoFisher). Confocal microscopy was performed on an LSM 710 confocal microscope (Zeiss) equipped with Zen software (Zeiss). Fluorescence microscopy was performed on a Zeiss Axio Observer.Z1 microscope equipped with an ORCA-Flash4.0 camera (cat no. C11440-22CU, Hamamatsu). Images from the Axio Observer were used for quantification of TRM conjugated dextran internalization, rather than confocal microscopy images. The integrated raw fluorescence intensity of the area occupied by the TRM emission was summed for all images of a sample. The summed integrated fluorescence intensity of the TRM emission was then normalized by the total cell area occupied by the DAPI fluorescence. Image processing was performed with CellProfiler11 (www.cellprofiler.org).
Immunostaining and Imaging
For fixation, Transwell monolayers were washed with PBS, fixed for 15–20 min at room temperature with 4% formaldehyde, washed with PBS, permeabilized in 0.1% Triton X-100 for 15–20 min at room temperature, and washed with PBS. Monolayers were blocked with 2.5% goat serum for 1–2 h at room temperature or overnight at 4 °C. Then, primary antibodies were diluted in 2.5% goat serum and applied for 1–2 h at room temperature or overnight at 4 °C. The following primary antibodies were used: anti-ZO-1 (1:200, cat no. 33-9100, Thermofisher), anti-MUC2 (1:200, cat no. PA1-23786, Thermofisher), anti-occludin (1:200, 71-1500, ThermoFisher), anti-Ezrin (1:200, cat no. PA5-17518, Thermofisher), anti-mAChR M3 (1:200, cat no. ab126168, Abcam), and phalloidin (1:500, cat no. A22287, Thermofisher). Transwell membranes were isolated and mounted on a standard glass slide and coverslip with Gold Antifade Mountant (cat no. P36931, ThermoFisher). Secondary antibodies were host species goat Alexa Fluor 488, 546, and 647 conjugates from Thermofisher.
Cellular Morphology Quantification
Cellular morphology was quantified by fixing and immunostaining monolayers for cell nuclei (DAPI) and tight junctions (ZO-1) as described above. Fluorescence microscopy was performed on a Zeiss Axio Observer.Z1 microscope equipped with an ORCA-Flash4.0 camera (cat no. C11440-22CU, Hamamatsu). Image processing was performed with ImageJ60 (https://imagej.nih.gov/ij/). Before cell segmentation, Z-stacks were processed with the Extended Depth of Field plugin23 to obtain an image of tight junctions without interruptions. Cell segmentation was performed with an open-source watershed algorithm (https://biii.eu/tissue-cell-segmentation). The semiautomatic segmentation of each image was manually corrected, and improperly segmented cells were removed.
TNFR1 Measurement
TNFR1 shedding was measured using an ELISA kit (cat no. ab100642, Abcam) according to the manufacturer’s instructions. Note that cell culture supernatant samples were incubated overnight at 4 °C to maximize ELISA sensitivity. Cell culture supernatants were applied to the ELISA kit immediately post experiments without storage and any dilution.
Statistical Analysis
Student’s t tests, ANOVA followed by a Tukey’s posthoc correction, or two-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance as indicated in figure legends. Data are presented as mean ± standard error of the mean (SEM). p values < 0.05 were considered to be significant. Exact p values are indicated in “Results” section.
Results
Human Intestinal Organoids Recapitulate Differentiated Intestinal Epithelium In-Vitro
Despite in-vitro studies demonstrating cholinergic modulation of intestinal epithelial inflammation and barrier injury,20,31,32,68,77 these results have not been demonstrated on a primary human intestinal model. To develop a primary human small intestinal model to study TNF-α induced inflammation and cholinergic modulation, we first established in-vitro organoid cultures via biopsy derived intestinal stem cells (Fig. 1a) based on previously published reports.56,57 Then, we characterized the formation of heterogeneous, polarized, and differentiated epithelial monolayers on commercially available permeable membrane supports (Fig. 1b). Organoids cultured and expanded in 3-dimensional (3D) Matrigel under a Wnt-3A, epidermal growth factor (EGF), noggin, and R-spondin 3 rich media (Fig. 1c), termed WENR media, were dissociated and seeded onto collagen type I coated permeable membrane Transwell supports (Fig. 1d), enabling both apical and basal media sampling, perturbation, and analysis. Several previous studies have demonstrated that culture of organoid or crypt derived monolayers under a WENR media maintained proliferative cells,35,49,67,72,73 most likely LGR5+ stem cells and transit-amplifying cells, which populate the native small intestinal crypt. In contrast, removing WENR and/or adding one of several small molecules resulted in a differentiated monolayer of primarily mucus-secreting goblet cells and absorptive enterocytes,35,49,72,73 which populate the native small intestinal villi. As previous studies of cholinergic modulation of intestinal epithelial inflammation utilized cell lines differentiated on permeable supports,20,31,32,68 primary organoid derived monolayers in this study were cultured in either WENR media (Fig. 1e) or a 20% FBS media (Fig. 1f) and characterized for differentiation. Fluorescence microscopy of monolayers stained for proliferative cell marker EdU and mucin-2 (MUC2) goblet cell marker showed that the culturing monolayers under 20% FBS media rather than WENR Media reduced proliferative cells from 39.6% to 16.6% (p = 0.0063) and increased goblet cells from 0.4 to 12% (p < 0.0001) (Fig. 1g). Immunostaining of cell monolayers revealed that phospho-ezrin expressing enterocytes (Fig. 2c) account for a large proportion of cells when cultured under 20% FBS media. Confocal microscopy of monolayers stained for F-actin (Fig. 2a), and tight junction occludins ZO-1 (Fig. 2b) demonstrated polarized brush border formation and robust tight junction formation. Taken together, these data confirmed a differentiated primary human intestinal model with structural and cellular features of the native small intestinal villi.
Figure 1.

Dissociated human intestinal organoids form differentiated intestinal epithelial monolayers. (a) Schematic representation of the protocol to generate biopsy derived human small intestinal organoids. (b) Schematic representation of a monolayer formed on a permeable insert by seeding organoid derived intestinal cells for 2 days in WENR medium and differentiation media for 5 days. (c) Biopsy derived human small intestinal organoids were expanded in WENR medium and dissociated to establish monolayers (d) on permeable inserts. Scale bars denote 500 and 100 µm, respectively. (e) Representative images of monolayers grown in WENR media or 20% FBS differentiation media (f) for 7 days showed increased proliferation marker Edu+ cells (red) or goblet cell marker MUC2+ cells (green), respectively. Scale bars denote 50 µm. (g) Edu+ cells significantly decreased in 20% FBS cultured monolayers from 39.6 to 16.6% while MUC+ cells significantly increased in 20% FBS from to 0.4 to 12% (*p < 0.05 by student’s t test). Data are presented as mean ± SEM from 3 independent experiments using monolayers generated from one donor.
Figure 2.

Confocal morphological imaging and lineage analysis of primary organoid derived small intestinal epithelium cultured with 20% FBS medium. (a) Orthogonal x–y projection and cross-sectional views of confocal microscopic images showing intestinal epithelium immunostained for F-actin (magenta) and DAPI (blue). (b) Orthogonal x-y projection and cross-sectional views of confocal microscopic images showing intestinal epithelium immunostained for ZO-1 (red) and DAPI (blue). (c) Orthogonal x–y projection and cross-sectional views of confocal microscopic images showing intestinal epithelium immunostained for phospho-ezrin (yellow) and DAPI (blue). (d) Orthogonal x–y projection and cross-sectional views of confocal microscopic images showing intestinal epithelium immunostained for MUC2 (green) and DAPI (blue). Scale bars denote 20 µm.
Given that previous research regarding cholinergic modulation of intestinal inflammation and barrier injury utilized immortalized cell lines,20,31,32,68 the primary intestinal epithelial model was functionally compared to Caco-2 and HT-29 monolayers. To enable the in-vitro study of the intestinal barrier, Caco-2 cells are cultured on permeable supports for the standard 3 weeks, during which the cells differentiate toward an enterocyte phenotype expressing brush border enzymes.53 Brush border enzyme alkaline phosphatase (AP) is used as a differentiation marker22 while transepithelial electrical resistance (TEER) measurements across intestinal cell monolayers indicate barrier integrity. The expression of AP (129.3 pg/105 cells) by a 7-day primary intestinal model (cultured under 20% FBS medium) was equivalent to the AP expression (156 pg/105 cells) by a 21 day Caco-2 intestinal model (p = 0.34) (Fig. 3a). Primary intestinal monolayers exhibited equal to (Donor 1: 818 Ω cm2, p = 0.1270) or greater than TEER (Donor 2: 1008 Ω cm2, p = 0.0001; Donor 3: 974 Ω cm2, p ≤ 0.0001; Donor 4: 1097 Ω cm2, p ≤ 0.0001) as compared to Caco-2 monolayers (Caco-2: 604 Ω cm2) (Fig. 3b). Furthermore, there was a significant variation in the average TEER observed among primary monolayers originating from different donors (Fig. 3b).
Figure 3.
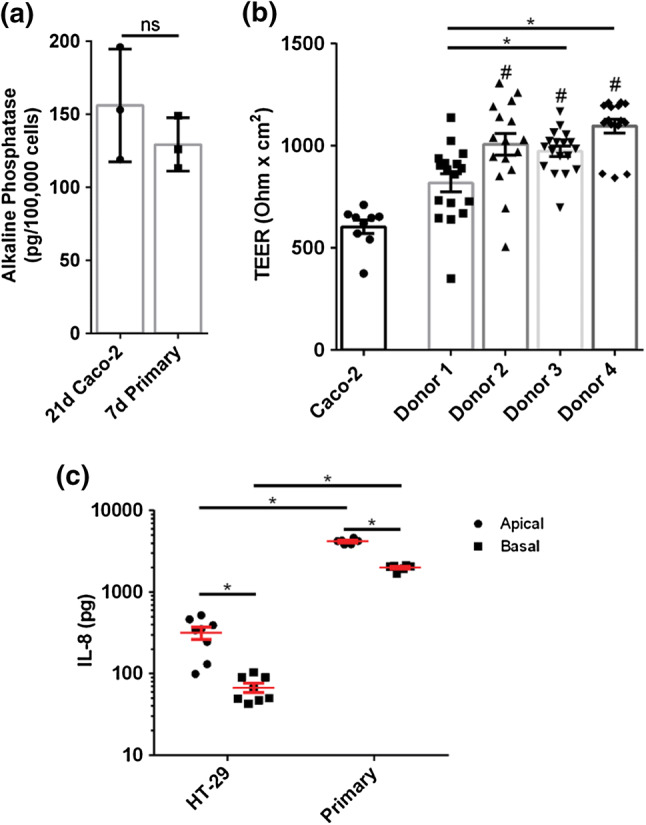
Functional comparison of organoid derived primary human intestinal monolayers (cultured in 20% FBS) and immortalized Caco-2 and HT-29 cell derived human intestinal monolayers. (a) Primary human monolayers cultured for 5 days in 20% FBS comparably expressed alkaline phosphatase as Caco-2 monolayers cultured for 21 days (p > 0.05 by student’s t test). (b) Primary human monolayers cultured for 5 days in 20% FBS showed comparable or greater TEER as Caco-2 monolayers cultured for 5 days (* and # p < 0.05 by ANOVA followed by Tukey’s HSD test, # denotes comparison to Caco-2 monolayers). (c) HT-29 monolayers and primary human monolayers cultured for 7 days both showed significantly increased apical IL-8 secretion compared to basal secretion. Primary human monolayers showed significantly increased apical and basal IL-8 secretion compared to HT-29 monolayers (*p < 0.05 by two-way ANOVA with Sidak’s multiple comparisons test). Seeding densities were: Caco-2: 2.5E5 cells/cm2, HT-29 cells: 2.6E5 cells/cm2, and smaller primary cells: 9.09E5 cells/cm2. All data are presented as mean ± SEM from at least 3 independent experiments from one donor unless specified as in panel b.
Interleukin-8 (IL-8) is a major intestinal pro-inflammatory molecule that is released upon intestinal infection or injury, resulting in neutrophil recruitment and a further proinflammatory cascade.36,52 Thus, IL-8 may serve as an immunological marker for in-vitro intestinal epithelial inflammation or barrier injury.72 IL-8 was secreted in a polarized manner for both HT-29 and primary intestinal epithelium (cultured under 20% FBS medium): primary and HT-29 monolayers secreted 2.1× (p < 0.0001) and 4.7× (p = 0.0494) more IL-8, respectively, in their apical compartments as compared to basal compartments (Fig. 3c). However, primary intestinal epithelium secreted more IL-8 than HT-29 monolayers in both the apical (Mean diff = 3873 pg, p < 0.0001) and basal compartment (Mean diff = 1928 pg, p < 0.0001) (Fig. 3c). These results indicated that while Caco-2 and HT-29 monolayers bear some similarity to primary monolayers (AP expression and polarized IL-8 secretion, respectively), primary monolayers exhibited increased IL-8 secretion, increased barrier integrity, reduced differentiation time as measured by AP expression, and both absorptive and secretory cell types.
Tumor Necrosis Factor-Α Alters Cell Morphology and Decreases Barrier Integrity Through Apoptosis and Tight Junction Disorder
To study cholinergic modulation of inflammation and barrier injury using the primary intestinal epithelial model, we characterized barrier disruption induced by tumor necrosis factor-alpha (TNF-α). TNF-α is a pro-inflammatory cytokine upregulated in inflammatory bowel disease (IBD)55 and was previously used to investigate cholinergic modulation of inflammation in endothelial39 and epithelial HT-2932,68 monolayers. Primary monolayers were cultured for 7 days under 20% FBS medium as described, and then varying doses (specified in figures and figure captions) of TNF-α were applied to the basal media compartment. Phase-contrast microscopy of control (Fig. 4a) and TNF-α treated monolayers (Fig. 4b) revealed a higher number of dead cells floating above the monolayer, particularly at doses of 25 and 50 ng mL−1. Furthermore, multicellular structures (Fig. 4c), likely remnant from seeding undissociated organoid fragments, were particularly susceptible to TNF-α induced cell death (Fig. 4d). Apoptotic cells, identified by nuclear fragmentation and blebbing,2,4,76 were quantified for control and TNF-α treated monolayers. The percentage of apoptotic cells increased (Fig. 4e) from 1.6% (control monolayers) to 3.2% (p 0.6180), 5.6% (p = 0.0292), and 5.1% (p = 0.0656) for 4, 25, and 50 ng mL−1 TNF-α, respectively. The monolayer density was quantified and while the cell density decreased from 173,208 cells per cm2 to 151,557, 143,718, and 148,197 cells per cm2 for 4, 25, and 50 ng mL−1 TNF-α, respectively, the results were insignificant (Fig. 4f).
Figure 4.

The effect of cytokine TNF-α on organoid derived primary human intestinal monolayers. (a) Representative phase contrast image of a monolayer cultured for 5 days in 20% FBS. (b) Monolayer exposure to 50 ng mL−1 TNF-α in the basal compartment for 48 h increased cell death and decreased visibility of cell-cell junctions. Panels a and b: scale bars denote 100 µm. (c) Representative phase contrast image of a monolayers with multicellular morphology. (d) Monolayer exposure to 50 ng mL−1 TNF-α in the basal compartment for 48 h eliminated multicellular morphology. Panels c and d: scale bars denote 1 mm. (e) Monolayer exposure to varying doses of TNF-α in the basal compartment for 48 h increased the number of apoptotic cells. (f) Monolayers exposure to varying doses of TNF-α in the basal compartment for 48 h decreased monolayer density. (g) TEER decreased upon exposure to varying doses of TNF-α in the basal compartment for 48 h. All panels: * and # p < 0.05 by ANOVA followed by Tukey’s HSD test, # denotes comparison to control. All data are presented as mean ± SEM from at least 3 independent experiments from one donor.
To quantify barrier injury induced by TNF-α, TEER was measured before and after TNF-α exposure and the final TEER value was normalized by the initial TEER value. Normalized TEER values significantly decreased from 1.62 to 1.29 (p = 0.0021), 0.74 (p < 0.0001), and 0.64 (p < 0.0001) for 4, 25, and 50 ng mL−1 TNF-α, respectively (Fig. 4g). Furthermore, the differences in TEER between a low dose of 4 ng mL−1 TNF-α and higher doses of 25 and 50 ng mL−1 TNF-α were statistically different (p ≤ 0.0001). These data demonstrate that even a low dose of 4 ng mL−1 TNF-α decreases primary intestinal epithelial barrier integrity, at least partly due to cell apoptosis. However, given that apoptotic cells and cell density were non-significantly altered following a 4 ng mL−1 TNF-α dose exposure, the primary intestinal epithelial barrier may also be compromised by TJ alterations.
Immunofluorescent labeling of control and TNF-α treated monolayers for TJ protein ZO-1 revealed alterations in both cell morphology and TJ structure (Figs. 5a–5d). Qualitatively, TNF-α treated cells appeared both more elongated and larger as compared to non-treated cells. To quantitatively analyze TNF-α induced cell morphology alterations, immunofluorescent images of tight junction protein ZO-1 were processed using a segmentation algorithm implemented in ImageJ (Fig. 5e). Following cell segmentation, all of the segmented cells were manually verified for accuracy. Cell elongation was quantified via the parameter ‘Aspect Ratio’, which was calculated by first fitting an ellipse to the segmented cell and calculating the ratio of the major and minor ellipse axes. Only exposure to 50 ng mL−1 TNF-α resulted in significant (p < 0.0001) cell elongation in comparison to control (Fig. 5f). Cell size was quantified via the parameter ‘Area’ which was simply the area of the segmented cell. Both 4 and 50 ng mL−1 TNF-α exposures elicited significant cell size increase (p < 0.0001 and p = 0.0013, respectively) in comparison to control while 25 ng mL−1 TNF-α did not (p = 0.1018) (Fig. 5g). Interestingly, cell size increased significantly more following a 4 ng mL−1 TNF-α exposure in comparison to a 50 ng mL−1 TNF-α exposure. Qualitatively, tight junction appeared rippled and/or kinked following TNF-α exposure (Figs. 5a–5d). Tight junction rippling was quantified via the difference between the ‘Convex Area’ and ‘Area’; kinked or rippled tight junctions increase the convex hull selection among a set of points, thereby increasing this difference (See Supplementary Fig. S1). Tight junction rippling significantly increased following TNF-α exposure to 4 (p < 0.0001), 25 (p = 0.0388), and 50 ng mL−1 (p < 0.0001). Taken together, these data demonstrate that TNF-α induces a leaky primary intestinal epithelium through both apoptosis, and cellular or tight junction reorganization.
Figure 5.

Quantitative morphological analysis of primary human intestinal monolayers exposed to cytokine TNF-α. (a) The morphology of monolayers cultured for 5 days in 20% FBS was visualized by immunostaining and fluorescent microscopy of tight junctions (ZO-1, green) and cell nuclei (blue). Scale bar denotes 50 µm. (b–d) Representative images of monolayers exposed to varying doses of TNF-α in the basal compartment for 48 h demonstrated morphological changes such as elongation and rippled or kinked tight junctions. Scale bars denote 50 µm. (e) Schematic representation of monolayer segmentation of the image in panel a, using tight junction protein ZO-1 for cell border detection. (f) The aspect ratio of cells significantly increased following exposure to 50 ng mL−1 TNF-α in the basal compartment for 48 h. (g) Cell size significantly increased following exposure to 4 and 50 ng mL−1 TNF-α in the basal compartment for 48 h. (h) The difference between the convex area and the cell area significantly increased following exposure to 4, 25, and 50 ng mL−1 TNF-α in the basal compartment for 48 h. All panels: * and # p < 0.05 by ANOVA followed by Tukey’s HSD test, # denotes comparison to control. All data are presented as mean ± SEM from at least 3 independent experiments from one donor.
Muscarinic Receptor Activation Decreases Primary Intestinal Epithelial Transport of Large Molecular Weight (70 kDa) Dextran But Does Not Ameliorate TNF-α Induced Paracellular Leakiness or IL-8 Production
Lastly, we used the primary human intestinal epithelial model to investigate the cholinergic amelioration of TNF-α induced epithelial permeability. The enteric nervous system resides adjacent to the intestinal epithelium and releases the neurotransmitter acetylcholine.48 Acetylcholine interacts with intestinal epithelial cells via nicotinic (nAChR) or muscarinic choline receptors (mAChR) to regulate epithelial fluid and electrolyte transport across the intestinal lumen.26 Previously published data demonstrated protective effects on intestinal barrier integrity via cholinergic activation.20,31,32,39,68,77 However, all previous evidence was acquired using murine models and immortalized cell line models of the intestinal epithelium.
Primary monolayers were cultured for 7 days as previously described, and then 25 ng mL−1 TNF-α was applied to the basal media compartment following a 30 min exposure to 50 µM nicotine in the basal media compartment. After TNF-α exposure, TEER significantly decreased compared to control, regardless if cell monolayers were pretreated with nicotine (p < 0.0001) or not (p = 0.0006) (Fig. 6a). Nicotine treatment alone did not affect TEER (p = 1.000). To complement paracellular permeability measurement via TEER, the flux of Lucifer Yellow (457 Da) added to the apical compartment was measured by sampling the basal compartment. Corroborating TEER measurements, Lucifer Yellow flux significantly increased compared to control, regardless if cell monolayers were pretreated with nicotine (p < 0.0001) or not (p < 0.0001) (Fig. 6b). Nicotine treatment alone did not affect Lucifer Yellow flux (p = 0.9669). To investigate larger molecular weight transport, the flux of a 70 kDa tritcmethylrhodamine (TRM) conjugated dextran added to the apical compartment was measured by sampling the basal compartment. The transport of 70 kDa dextran did not significantly change following under any treatment (Fig. 6c). Together, these results indicated that nAChR activation did not alter primary intestinal epithelial barrier integrity under healthy or TNF-α induced inflammatory conditions.
Figure 6.

Primary human intestinal epithelial barrier integrity after exposure to nicotine and/or TNF-α. (a) TEER significantly decreased following exposure to 25 ng mL−1 TNF-α in the basal compartment for 4 h. TEER did not change following exposure to 50 µM nicotine in the basal compartment for 4 h. Pre-incubation with 50 µM nicotine in the basal compartment for 30 min prior to TNF-α did not eliminate TNF-α induced TEER decrease. (b) Lucifer yellow flux significantly increased following exposure to 25 ng mL−1 TNF-α in the basal compartment for 4 h. Lucifer yellow flux did not change following exposure to 50 µM nicotine in the basal compartment for 4 h. Pre-incubation with 50 µM nicotine in the basal compartment for 30 min prior to TNF-α did not eliminate TNF-α increased lucifer yellow permeability. (c) 70 kDa dextran flux did not change following exposure to 25 ng mL−1 TNF-α or 50 µM nicotine in the basal compartment for 4 h. All panels: * and # p < 0.05 by ANOVA followed by Tukey’s HSD test, # denotes comparison to control. All data are presented as mean ± SEM from at least 3 independent experiments from one donor.
To investigate mAChR activation, 25 ng mL−1 TNF-α was applied to the basal media compartment following a 30 min exposure to 50 µM muscarinic receptor agonist bethanechol in the basal media compartment. Similar to nAChR experiments, TEER significantly decreased after TNF-α as compared to control, regardless if cell monolayers were pretreated with bethanechol (p = 0.0002) or not (p = 0.0002) (Fig. 7a). Bethanechol treatment alone did not affect TEER (p = 0.7256). Similarly, Lucifer Yellow flux corroborated TEER measurements; Lucifer Yellow flux significantly increased compared to control, regardless if cell monolayers were pretreated with bethanechol (p = 0.0002) or not (p < 0.0001) (Fig. 7b). Bethanechol treatment alone did not affect Lucifer Yellow flux (p = 0.0904). Though neither nAChR or mAChR activation with nicotine or bethanechol, respectively, altered paracellular barrier integrity, bethanechol treatment of monolayers significantly decreased transport of a 70 kDa dextran under healthy (p = 0.0002) and TNF-α induced inflammatory conditions (p < 0.0001) (Fig. 7c).
Figure 7.

Primary human intestinal epithelial barrier integrity after exposure to bethanechol and/or TNF-α. (a) TEER significantly decreased following exposure to 25 ng mL−1 TNF-α in the basal compartment for 4 h. TEER did not change following exposure to 50 µM bethanechol in the basal compartment for 4 h. Pre-incubation with 50 µM bethanechol in the basal compartment for 30 min prior to TNF-α did not eliminate TNF-α induced TEER decrease. (b) Lucifer yellow flux significantly increased following exposure to 25 ng mL−1 TNF-α in the basal compartment for 4 h. Lucifer yellow flux did not change following exposure to 50 µM bethanechol in the basal compartment for 4 h. Pre-incubation with 50 µM bethanechol in the basal compartment for 30 min prior to TNF-α did not eliminate TNF-α increased lucifer yellow permeability. (c) 70 kDa dextran flux did not change following exposure to 25 ng mL−1 TNF-α in the basal compartment for 4 h. 70 kDa dextran flux significantly decreased following exposure to 50 µM bethanechol with or without TNF-α co-incubation in the basal compartment for 4 h. Panels a–c: * and # p < 0.05 by ANOVA followed by Tukey’s HSD test, # denotes comparison to control. Panels a–c: data is presented as mean ± SEM from at least 3 independent experiments from one donor. (d) A representative image of a monolayer showing internalization of an apically introduced lysine fixable TMR conjugated 70 kDa dextran. (e) A representative image of a monolayer incubated with 50 µM bethanechol for 4 showing a lack of internalization of an apically introduced lysine fixable TMR conjugated 70 kDa dextran. (f) The integrated raw fluorescence intensity due to internalized TRM conjugated dextran normalized by the nuclei area occupied by DAPI fluorescence was lower for monolayers exposed to 50 µM bethanechol in the basal compartment for 4 h. Data are presented as mean ± SEM from 2 independent experiments from one donor.
To further investigate mAChR dependent macromolecule transport, we designed a second assay allowing visualization and quantification of macromolecule localization. The primary epithelium was exposed to a 70 kDa TRM-conjugated ‘lysine fixable’ dextran in the presence or absence of 50 µM bethanechol. Monolayers were extensively washed, fixed, stained for tight junction protein ZO-1 and cell nuclei, and imaged via fluorescence microscopy, allowing for direct visualization of internalized 70 kDa dextran. Fluorescence microscopy revealed bethanechol treated monolayers (Fig. 7e) with qualitatively less dextran localization as compared to non-treated monolayers (Fig. 7d). The normalized fluorescent intensity revealed 2.7× higher localization for un-treated monolayers as compared to bethanechol treated monolayers (Fig. 7f).
TNF-α results in pro-inflammatory IL-8 production by intestinal epithelial cells.72 Correspondingly, IBD patients typically exhibit elevated TNF-α and IL-8 levels.16 Previous results demonstrated significant IL-8 suppression by pre-treatment of HT-29/B6 monolayers, with carbachol, a cholinergic agonist stimulating both nAChRs and mAChRs, before TNF-α.32 Furthermore, pre-treatment of HT-29/B6 monolayers with atropine, a muscarinic antagonist, negated the effects of carbachol.32 Mechanistically, these results were attributed to subtype M3 mAChR induced shedding of tumor necrosis factor receptor 1 (TNFR1) which reduced the density of cell surface TNF-α receptors and neutralized TNF-α in the cell culture medium.32,68 Immunofluorescent staining against subtype M3 mAChR revealed that primary human intestinal monolayers express the implicated muscarinic receptor (See Supplementary Fig. S2). Given that bethanechol reduced macromolecular transport in the primary human epithelium (Figs. 7c–7f), we analyzed basal IL-8 secretion after 4 or 8 h post-TNF-α and/or bethanechol treatment. TNF-α exposure for 8 h significantly increased basal IL-8 production compared to control, regardless if cell monolayers were pretreated with bethanechol (p < 0.0001) or not (p < 0.0001) (Fig. 8a). Pretreatment with bethanechol before TNF-α did not suppress basal IL-8 production (p = 0.9991). Similarly, TNF-α exposure for 4 h significantly increased basal IL-8 production compared to control, regardless if cell monolayers were pretreated with bethanechol (p < 0.0001) or not (p < 0.0001) (Fig. 8b). Pretreatment with bethanechol before TNF-α did not suppress basal IL-8 production (p = 0.9999). Furthermore, both temporally resolved (Fig. 9a) experiments and a 30-min endpoint experiment (Fig. 9b) demonstrated that mAChR activation via carbachol did not induce TNFR1 shedding in either the apical nor basal compartments.
Figure 8.

IL-8 secretion by organoid derived human intestinal epithelial barrier integrity after exposure to bethanechol and/or TNF-α. (a) Exposure to 25 ng/mL TNF-α in the basal compartment for 8 h significantly increased basal IL-8 secretion. Exposure to 50 µM bethanechol in the basal compartment for 8 h did not alter IL-8 secretion. Pre-incubation with 50 µM bethanechol in the basal compartment for 30 min prior to 25 ng/mL TNF-α did not eliminate TNF-α increased basal IL-8 secretion. (b) The same experiment was conducted after a 4-h exposure to TNF-α, with identical results as an 8 h exposure. (* and # p < 0.05 by ANOVA followed by Tukey’s HSD test, # denotes comparison to control). Data are presented as mean + SEM from at least three independent experiments from one donor.
Figure 9.

Kinetic and endpoint analysis of TNFR1 shedding by organoid derived human intestinal epithelium after basal exposure to carbachol. (a) Exposure to 100 µM carbachol in the basal compartment did not increase the apical or basal TNFR1 concentration for up to 2 h. Data are presented as mean ± SD from 1 independent experiment with three independent monolayers. (b) The experiment was repeated as an endpoint analysis after a 30 min exposure to 100 µM carbachol in the basal compartment. Exposure to 100 µM carbachol in the basal compartment for 30 min did not increase the apical or basal TNFR1 concentration. Data are presented as mean ± SEM from at least three independent experiments from one donor.
Discussion
In this study, we described an in-vitro human small intestinal model featuring a primary intestinal epithelium on permeable membrane supports (Figs. 1b and 1d). Derived from primary 3D intestinal organoids (Figs. 1a and 1c), this system recapitulates small intestinal functionality and diversity more accurately compared to highly prevalent immortalized cell line models. Using the in-vitro human small intestinal model, we simulated an inflamed intestine by treating the primary epithelium with pro-inflammatory cytokine TNF-α. The in-vitro primary human intestinal model was used, for the first time, to investigate amelioration of TNF-α impaired barrier integrity via nicotinic and muscarinic receptor activation.
The in-vitro human small intestinal model was established using solely primary human cells by obtaining and expanding LGR5+ stem cells in 3D Matrigel culture as previously described (Fig. 1a).56,57 Following differentiation, monolayers exhibited cellular heterogeneity, containing both mucus-secreting goblet cells (Fig. 2d) and absorptive enterocytes (Fig. 2c), thereby mimicking the cellular phenotype of the native small intestinal villi. In-vivo, mucus-producing goblet cells constitute 4–12% of the epithelium.59 MUC2+ goblet cells constituted 12% of the differentiated primary monolayers presented here (Fig. 1g). Furthermore, differentiated monolayers exhibited an F-actin rich brush border in the apical plasma membrane (Fig. 2a), indicating a highly polarized epithelium as found on native small intestinal villi. As previously demonstrated, maintenance of the primary epithelium in WENR media (Fig. 1e) mimicked the cellular phenotype of the native small intestinal crypts.35,49,67,72,73 Namely, monolayers remained highly proliferative with practically zero goblet cells present (Fig. 1g). Therefore, the protocol described herein may be adapted to model distinct compartments of the native intestinal crypt-villus axis, depending on the biological phenomena under investigation.
Differentiated monolayers exhibited robust tight junction formation (Fig. 2b), which led to TEER values higher than similarly formed Caco-2 monolayers (Fig. 3b). The epithelial barrier integrity was comparable across monolayers derived via biopsy samples from multiple human donors, indicating high reproducibility (Fig. 3b). Caco-2 monolayers differentiate in approximately 3 weeks to represent intestinal enterocytes.53 Compared to Caco-2, the in-vitro human small intestinal model differentiated approximately 3× faster to equally express differentiation marker alkaline phosphatase22 (Fig. 3a), thereby significantly increasing experimental throughput while simultaneously recapitulating the native small intestine with higher fidelity. Indeed, we demonstrated significantly higher IL-8 (Fig. 3c) by primary intestinal epithelium compared to similarly formed HT-29 monolayers, highlighting the functional differences between primary and immortalized cell-derived intestinal models. While all of the monolayers described herein were derived from small intestinal duodenal segments, it was demonstrated that LGR5+ stem cells recapitulate the cellular phenotype of the intestinal segments from their origin.43 Thus, the protocol described herein could be adapted to model other regions of the human intestine.
Intestinal cell-derived monolayers cultured on permeable membrane supports are frequently used to study the epithelial barrier in response to pro-inflammatory cytokine,1 viral,21,79 or bacterial27,49 challenge. In the present study, we exposed the basal side of the primary epithelium to pro-inflammatory cytokine TNF-α to simulate an inflamed intestine. TNF-α is a key mediator of inflammatory bowel disease, including both ulcerative colitis and Crohn’s disease.55 While the concentrations of TNF-α utilized here (4-50 ng mL−1) are significantly higher compared to serum TNF-α concentrations (7.8 pg mL−1) of IBD patients,34 the concentrations are comparable to those previously used to study TNF-α and the intestinal epithelium in-vitro.9,29,40,63 The barrier integrity of the epithelial monolayer significantly decreased following exposure to TNF-α (Fig. 4g). Analysis of TNF-α exposed monolayers with reduced barrier integrity revealed significantly increased apoptosis (Fig. 4e) and decreased epithelial monolayer density (Fig. 4f), indicative of cell shedding. Indeed, TNF-α is a potent stimulus of IEC shedding.75 Interestingly, basal exposure to TNF-α strongly targeted cells growing as multicellular clumps on the apical cell surface (Figs. 4c and 4d). This may suggest that “villus-like” cells on primary monolayers are more susceptible to TNF-α induced apoptosis. In accordance, previous in-vivo studies demonstrated that acute TNF-α exposure significantly blunts villi.8 Though the mechanisms of IEC apoptosis are still under debate, IECs show differential susceptibility to apoptosis depending on IEC differentiation3 or location.19 Recently, researchers cultured Caco-2 cells with 3D “villus-like” morphology on microfluidic organ chips and used villi height as an assay of injury.28,33 Thus, it is plausible that multicellular growths on primary monolayers can be leveraged to model the TNF-α induced villus blunting observed in-vivo.
While apoptosis was induced by TNF-α, immunostaining of monolayers for TJ protein ZO-1 and nuclei revealed that confluent monolayers with intact tight junctions spanned the permeable supports (Figs. 5a–5d). Using image segmentation (Fig. 5e), we quantitatively analyzed the cell morphology of > 103 cells in response to basal TNF-α exposure. Cells exposed to both low (4 ng mL−1) and high (50 ng mL−1) doses of TNF-α exhibited significant enlargement (Fig. 5g). However, only a high TNF-α dose (50 ng mL−1) resulted in a significant aspect ratio increase (Fig. 5f). Regardless of TNF-α dose, cells exhibited rippled tight junctions (Figs. 5b–5d). This TJ alteration was quantitatively analyzed using a new parameter, the convex hull area—cell area (Fig. S1), which demonstrated significant tight junction rippling in response to TNF-α at all doses (Fig. 5h). In vivo, IECs undergoing apoptosis alter TJs and integrins to detach from the basal membrane and the adjacent IECs extend their cytoplasm underneath the shedding IEC to create new junctions that maintain an intact epithelial barrier.19 The observed TJ rippling (Fig. 5h) indicates that TJ remodeling is necessary at all TNF-α doses to shed apoptotic IECs and reseal the epithelium. At a high (50 ng mL−1) TNF-α dose, increased apoptosis (Fig. 4e) and decreased monolayer density (Fig. 4f) was more pronounced as compared to a low (4 ng mL−1) TNF-α dose. The increased cell aspect ratio at only a high TNF-α dose (Fig. 5f) may indicate that epithelial cell migration is required for more extensive cellular reorganization, as compared to a low TNF-α dose where epithelial cells may only be required to extend their cytoplasm to maintain epithelial contiguity. Together, these data suggest that TNF-α impairs epithelial barrier integrity through apoptosis and TJ remodeling while epithelial cells migrate and/or adopt morphological changes to maintain an intact barrier, depending on the extent of epithelial cell loss. This theory is supported by several in-vivo studies. For example, the “zipper model” theorizes that neighboring cells extend cytoplasmic processes underneath the shedding cell as it leaves the monolayer to reform TJs and maintain an intact barrier under physiological conditions.75 In diseased intestinal states, cell shedding is markedly increased and epithelial cells undergo flattening and attenuation to cover the exposed basement membrane.45
Intestinal cell monolayers derived from immortalized cell lines were used to study nAChR and mAChR activation for the amelioration of intestinal epithelial permeability.20,31,32,68 The present study is the first utility of a primary human intestinal epithelium towards said application. Nicotinic stimulation of primary epithelium did not alter barrier integrity or macromolecule transport, nor did it rescue TNF-α induced barrier impairment (Figs. 6a–6c). Similarly, a previous study demonstrated that nicotine did not impact interleukin 1 beta (IL-1β) induced epithelial permeability of Caco-2 monolayers.20 However, a plethora of in-vivo studies demonstrated amelioration of epithelial injury via nAChR activation by nicotine,66 alternative nicotinic agonists,66 or vagal nerve stimulation.14,15,17,41,71,78 Thus, nAChR activation most likely does not target epithelial cells directly, but rather exerts anti-inflammatory effects via another cell type found in the intestinal mucosa. Indeed, these studies demonstrated that nicotinic agonists reduced TNF-α production by macrophages71 and activated enteric glial cells (EGCs).14 Further supporting this hypothesis, in-vitro studies utilizing co-cultures of the intestinal epithelium and EGCs demonstrated that nAChR activation targeted EGCs to exert protective effects on the intestinal epithelium.12,13,15
Similarly, mAChR activation with bethanechol did not ameliorate TNF-α barrier impairment (Figs. 7a–7b). However, mAChR activation significantly decreased the transport of a 70 kDa dextran in both a healthy state and post-TNF-α treatment (Fig. 7c). To further verify the effect of mAChR activation on macromolecule transport, we visualized a ‘lysine-fixable’ 70 kDa dextran (Figs. 7d and 7e) and quantified the results via image processing (Fig. 7f), demonstrating that mAChR activation with bethanechol decreased 70 kDa dextran internalization . Though mAChR stimulation did not ameliorate TNF-α barrier impairment, we tested whether the decreased macromolecule transport also decreased basal IL-8 secretion following TNF-α exposure, which would minimize the immune response in-vivo. Muscarinic activation 30 min before TNF-α treatment did not inhibit basal IL-8 secretion after 8 h (Fig. 8a) or 4 h (Fig. 8b) of TNF-α treatment, suggesting that intestinal epithelial macromolecule transport and IL-8 secretion are dependent on different cellular pathways. These results are interesting as they highlight functional differences between in-vitro experiments utilizing immortalized human cell lines, primary human cells, and ex-vivo experiments using whole human mucosa. For example, in contrast to our results, one study demonstrated that activation of subtype M3 mAChR with bethanechol increased macromolecular permeability of horseradish peroxidase (HRP) in ex-vivo human mucosa and T84 cell monolayers,10 a human colorectal derived epithelial cell line. However, a contradicting study demonstrated that carbachol, a mAChR, and nAChR agonist, did not alter HRP permeability of T84 cell monolayers.70 Contrary to our results, mAChR activation of Caco-2 monolayers via bethanechol ameliorated IL-1β induced barrier injury.20 However, mAChR activation did not decrease IL-1β induced IL-8 production.20 Khan et al. demonstrated that mAChR activation of HT-29 monolayers with carbachol ameliorated TNF-α induced barrier damage, IL-8 production, and NF-κB activation. It was determined that M3 mAChR activation with carbachol rapidly (within 5 min) augments the shedding of TNFR1 to attenuate TNF-α signaling.32,68 Given that these two studies also investigated TNF-α induced barrier damage, we conducted experiments to determine that mAChR activation of primary monolayers with carbachol did not augment TNFR1 shedding (Fig. 9). The discrepancies in the literature and our study may be due to differential mAChR expression between immortalized cells, primary cells, and ex-vivo tissue, which is frequently unreported, or assessed using functional tests with antagonists rather than direct observation at the mRNA or protein level. Furthermore, it was reported that mAChR subtypes are differentially expressed across the crypt-villus axis in mouse small intestine.46,47 Thus, it may be plausible that even a single immortalized cell line may exhibit differential mAChR expression dependent on tissue culture protocols and differentiation.
While this study did not demonstrate cholinergic regulation of the primary intestinal epithelial barrier, several recently published studies utilizing intact organoids demonstrated cholinergic control of intestinal epithelial proliferation and differentiation. For example, it was shown that mAChR antagonists enhanced organoid growth and increased the number of LGR5+ stem cells.65 It was also demonstrated that activation of Paneth cell α2 and β4 nAChRs induced Wnt5a expression which promoted organoid proliferating and LGR5+ stem cell differentiation.64 Two years prior, Pastula et al. demonstrated that co-cultured murine myenteric neurons were able to replace exogenous Wnt necessary for organoid growth.51 It may very well be that neuron derived ACh stimulated Paneth cell nAChRs to increase Wnt5a production. Perhaps a 2D intestinal epithelium that features both Paneth cells and proliferative cells would enable cholinergic regulation of the epithelial barrier. Further, since TNF-α is also modulated by stromal cells in vivo,55,61 this platform could be used as a co-culture model beyond examining epithelial response alone in future studies. Given these findings and the rapid advancement of primary human intestinal organoids, the 2D intestinal epithelium described herein is a powerful tool to study cholinergic regulation of intestinal epithelial barrier integrity with higher phenotype fidelity. The use of monolayers as opposed to organoids greatly facilitates studying epithelial permeability which is critical in the context of IBD.
This study demonstrated a primary cell-derived in-vitro model of the human small intestinal epithelium, which was functionally different compared to common immortalized cell-derived models. The results provide insight into TNF-α induced disruption of the intestinal epithelium and suggest that cholinergic activation may have significantly different effects on intestinal epithelium derived from immortalized cells as compared to primary cells. These results warrant further investigation, including a characterization of mAChR expression. The intestinal model described here was reproducible and practical concerning setup, throughput, and ease of perturbing and analyzing the intestinal epithelial barrier.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
S.H., and A.K. conceived the study. S.H., A.K., and S.M. provided experimental design input. S.H., and W.L. conducted the experiments. S.H. analyzed the data and was the primary manuscript author. S.H., W.L., and E.S., prepared and maintained human organoid cultures. E.S., and D.B. supplied human intestinal tissue for organoid establishment and WRN conditioned medium. All authors provided input towards the manuscript. The authors thank funding support from the National Institute of Health award numbers R21EB025395 Trailblazer (AK and RK), R01EB021908 BRP (AK and DB), R01DK084056, P30HD18655 and P30DK034854 (DTB) and the Department of Chemical Engineering at Northeastern University for start-up funding (AK and RK).
Conflict of interest
The authors Sanjin Hosic, Will Lake, Eric Stas, Ryan Koppes, David T. Breault, Shashi K. Murthy, Abigail N. Koppes do not declare any competing interests or conflicts.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sanjin Hosic, Email: hosic.s@gmail.com.
Will Lake, Email: williamericlake@gmail.com.
Eric Stas, Email: Eric.Stas@childrens.harvard.edu.
Ryan Koppes, Email: r.koppes@northeastern.edu.
David T. Breault, Email: david.breault@childrens.harvard.edu
Shashi K. Murthy, Email: s.murthy@northeastern.edu
Abigail N. Koppes, Email: a.koppes@northeastern.edu
References
- 1.Andrews C, McLean MH, Durum SK. Cytokine tuning of intestinal epithelial function. Front. Immunol. 2018;9:1270. doi: 10.3389/fimmu.2018.01270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkin-Smith GK, Poon IKH. Disassembly of the dying: mechanisms and functions. Trends Cell Biol. 2017;27:151–162. doi: 10.1016/j.tcb.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Beausejour M, et al. Suppression of anoikis in human intestinal epithelial cells: differentiation state-selective roles of alpha2beta1, alpha3beta1, alpha5beta1, and alpha6beta4 integrins. BMC Cell Biol. 2013;14:53. doi: 10.1186/1471-2121-14-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharyya A, et al. Apoptogenic effects of black tea on Ehrlich’s ascites carcinoma cell. Carcinogenesis. 2003;24:75–80. doi: 10.1093/carcin/24.1.75. [DOI] [PubMed] [Google Scholar]
- 5.Birchenough GM, Johansson ME, Gustafsson JK, Bergstrom JH, Hansson GC. New developments in goblet cell mucus secretion and function. Mucosal. Immunol. 2015;8:712–719. doi: 10.1038/mi.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blume L-F, Denker M, Gieseler F, Kunze T. Temperature corrected transepithelial electrical resistance (TEER) measurement to quantify rapid changes in paracellular permeability. Die Pharmazie. 2010;65:19–24. [PubMed] [Google Scholar]
- 7.Borovikova LV, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 8.Brown KS, et al. Tumor necrosis factor induces developmental stage-dependent structural changes in the immature small intestine. Mediators Inflamm. 2014;2014:852378. doi: 10.1155/2014/852378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruno MEC, Kaetzel CS. Long-term exposure of the HT-29 human intestinal epithelial cell line to TNF causes sustained up-regulation of the polymeric Ig receptor and proinflammatory genes through transcriptional and posttranscriptional mechanisms. J. Immunol. 2005;174:7278–7284. doi: 10.4049/jimmunol.174.11.7278. [DOI] [PubMed] [Google Scholar]
- 10.Cameron HL, Perdue MH. Muscarinic acetylcholine receptor activation increases transcellular transport of macromolecules across mouse and human intestinal epithelium in vitro. Neurogastroenterol. Motil. 2007;19:47–56. doi: 10.1111/j.1365-2982.2006.00845.x. [DOI] [PubMed] [Google Scholar]
- 11.Carpenter AE, et al. Cell profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheadle GA, Costantini TW, Bansal V, Eliceiri BP, Coimbra R. Cholinergic signaling in the gut: a novel mechanism of barrier protection through activation of enteric glia cells. Surg. Infect. 2014;15:387–393. doi: 10.1089/sur.2013.103. [DOI] [PubMed] [Google Scholar]
- 13.Cheadle GA, et al. Enteric glia cells attenuate cytomix-induced intestinal epithelial barrier breakdown. PLoS ONE. 2013;8:e69042. doi: 10.1371/journal.pone.0069042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costantini TW, et al. Vagal nerve stimulation protects against burn-induced intestinal injury through activation of enteric glia cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;299:G1308–1318. doi: 10.1152/ajpgi.00156.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costantini TW, et al. Targeting alpha-7 nicotinic acetylcholine receptor in the enteric nervous system: a cholinergic agonist prevents gut barrier failure after severe burn injury. Am. J. Pathol. 2012;181:478–486. doi: 10.1016/j.ajpath.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Daig R, et al. Increased interleukin 8 expression in the colon mucosa of patients with inflammatory bowel disease. Gut. 1996;38:216–222. doi: 10.1136/gut.38.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Jonge WJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 2005;6:844–851. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- 18.de Santa Barbara P, van den Brink GR, Roberts DJ. Development and differentiation of the intestinal epithelium. Cell Mol. Life Sci. 2003;60:1322–1332. doi: 10.1007/s00018-003-2289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delgado ME, Grabinger T, Brunner T. Cell death at the intestinal epithelial front line. FEBS J. 2016;283:2701–2719. doi: 10.1111/febs.13575. [DOI] [PubMed] [Google Scholar]
- 20.Dhawan S, et al. Cholinergic receptor activation on epithelia protects against cytokine-induced barrier dysfunction. Acta Physiol. (Oxf) 2015;213:846–859. doi: 10.1111/apha.12469. [DOI] [PubMed] [Google Scholar]
- 21.Ettayebi K, et al. Replication of human noroviruses in stem cell-derived human enteroids. Science. 2016;353:1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferruzza S, Rossi C, Scarino ML, Sambuy Y. A protocol for differentiation of human intestinal Caco-2 cells in asymmetric serum-containing medium. Toxicol. In Vitro. 2012;26:1252–1255. doi: 10.1016/j.tiv.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 23.Forster B, Van De Ville D, Berent J, Sage D, Unser M. Complex wavelets for extended depth-of-field: a new method for the fusion of multichannel microscopy images. Microsc. Res. Tech. 2004;65:33–42. doi: 10.1002/jemt.20092. [DOI] [PubMed] [Google Scholar]
- 24.Gerbe F, Legraverend C, Jay P. The intestinal epithelium tuft cells: specification and function. Cell. Mol. Life Sci. 2012;69:2907–2917. doi: 10.1007/s00018-012-0984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibbons DL, Spencer J. Mouse and human intestinal immunity: same ballpark, different players; different rules, same score. Mucosal. Immunol. 2011;4:148–157. doi: 10.1038/mi.2010.85. [DOI] [PubMed] [Google Scholar]
- 26.Hirota CL, McKay DM. Cholinergic regulation of epithelial ion transport in the mammalian intestine. Br. J. Pharmacol. 2006;149:463–479. doi: 10.1038/sj.bjp.0706889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.In J, et al. Enterohemorrhagic Escherichia coli reduces mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell. Mol. Gastroenterol. Hepatol. 2016;2:48–62.e43. doi: 10.1016/j.jcmgh.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jalili-Firoozinezhad S, et al. Modeling radiation injury-induced cell death and countermeasure drug responses in a human Gut-on-a-Chip. Cell Death Dis. 2018;9:223. doi: 10.1038/s41419-018-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janes KA, et al. The response of human epithelial cells to TNF involves an inducible autocrine cascade. Cell. 2006;124:1225–1239. doi: 10.1016/j.cell.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 30.Kauffman AL, et al. Alternative functional in vitro models of human intestinal epithelia. Front Pharmacol. 2013;4:79. doi: 10.3389/fphar.2013.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan RI, et al. Activation of focal adhesion kinase via M1 muscarinic acetylcholine receptor is required in restitution of intestinal barrier function after epithelial injury. Biochim. Biophys. Acta. 1842;635–645:2014. doi: 10.1016/j.bbadis.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 32.Khan MR, et al. Activation of muscarinic cholinoceptor ameliorates tumor necrosis factor-alpha-induced barrier dysfunction in intestinal epithelial cells. FEBS Lett. 2015;589:3640–3647. doi: 10.1016/j.febslet.2015.10.029. [DOI] [PubMed] [Google Scholar]
- 33.Kim HJ, Li H, Collins JJ, Ingber DE. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. U S A. 2016;113:E7–15. doi: 10.1073/pnas.1522193112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komatsu M, et al. Tumor necrosis factor-α in serum of patients with inflammatory bowel disease as measured by a highly sensitive immuno-PCR. Clin. Chem. 2001;47:1297–1301. doi: 10.1093/clinchem/47.7.1297. [DOI] [PubMed] [Google Scholar]
- 35.Kozuka K, et al. Development and characterization of a human and mouse intestinal epithelial cell monolayer platform. Stem Cell Rep. 2017;9:1976–1990. doi: 10.1016/j.stemcr.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kucharzik T, et al. Acute induction of human IL-8 production by intestinal epithelium triggers neutrophil infiltration without mucosal injury. Gut. 2005;54:1565–1572. doi: 10.1136/gut.2004.061168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landy J, et al. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 2016;22:3117–3126. doi: 10.3748/wjg.v22.i11.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leushacke M, Barker N. Ex vivo culture of the intestinal epithelium: strategies and applications. Gut. 2014;63:1345–1354. doi: 10.1136/gutjnl-2014-307204. [DOI] [PubMed] [Google Scholar]
- 39.Li Y-Z, Liu X-H, Rong F, Hu S, Sheng Z-Y. Carbachol inhibits TNF-α-induced endothelial barrier dysfunction through alpha 7 nicotinic receptors. Acta Pharmacol. Sin. 2010;31:1389–1394. doi: 10.1038/aps.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;288:G422–430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 41.Matteoli G, et al. A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut. 2014;63:938–948. doi: 10.1136/gutjnl-2013-304676. [DOI] [PubMed] [Google Scholar]
- 42.Michielan A, D’Inca R. Intestinal permeability in inflammatory bowel disease: pathogenesis, clinical evaluation, and therapy of leaky gut. Mediators Inflamm. 2015;2015:628157. doi: 10.1155/2015/628157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Middendorp S, et al. Adult stem cells in the small intestine are intrinsically programmed with their location-specific function. Stem Cells. 2014;32:1083–1091. doi: 10.1002/stem.1655. [DOI] [PubMed] [Google Scholar]
- 44.Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013;8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore R, Carlson S, Madara JL. Villus contraction aids repair of intestinal epithelium after injury. Am. J. Physiol. 1989;257:G274–283. doi: 10.1152/ajpgi.1989.257.2.G274. [DOI] [PubMed] [Google Scholar]
- 46.Muise ED, Gandotra N, Tackett JJ, Bamdad MC, Cowles RA. Localization of muscarinic acetylcholine receptor 2 to the intestinal crypt stem cell compartment. Data Brief. 2017;10:482–486. doi: 10.1016/j.dib.2016.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muise ED, Gandotra N, Tackett JJ, Bamdad MC, Cowles RA. Distribution of muscarinic acetylcholine receptor subtypes in the murine small intestine. Life Sci. 2017;169:6–10. doi: 10.1016/j.lfs.2016.10.030. [DOI] [PubMed] [Google Scholar]
- 48.Nezami BG, Srinivasan S. Enteric nervous system in the small intestine: pathophysiology and clinical implications. Curr. Gastroenterol. Rep. 2010;12:358–365. doi: 10.1007/s11894-010-0129-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noel G, et al. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep. 2017;7:45270. doi: 10.1038/srep45270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olsen T, et al. Tissue levels of tumor necrosis factor-alpha correlates with grade of inflammation in untreated ulcerative colitis. Scand. J. Gastroenterol. 2007;42:1312–1320. doi: 10.1080/00365520701409035. [DOI] [PubMed] [Google Scholar]
- 51.Pastula A, et al. Three-dimensional gastrointestinal organoid culture in combination with nerves or fibroblasts: a method to characterize the gastrointestinal stem cell niche. Stem Cells Int. 2016;2016:3710836. doi: 10.1155/2016/3710836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qazi BS, Tang K, Qazi A. Recent advances in underlying pathologies provide insight into interleukin-8 expression-mediated inflammation and angiogenesis. Int. J. Inflamm. 2011 doi: 10.4061/2011/908468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rousset M. The human colon carcinoma cell lines HT-29 and Caco-2: two in vitro models for the study of intestinal differentiation. Biochimie. 1986;68:1035–1040. doi: 10.1016/S0300-9084(86)80177-8. [DOI] [PubMed] [Google Scholar]
- 54.Sambuy Y, et al. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005;21:1–26. doi: 10.1007/s10565-005-0085-6. [DOI] [PubMed] [Google Scholar]
- 55.Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 2008;14:4280–4288. doi: 10.3748/wjg.14.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340:1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- 57.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 58.Sato T, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 59.Schneider H, Pelaseyed T, Svensson F, Johansson MEV. Study of mucin turnover in the small intestine by in vivo labeling. Sci. Rep. 2018;8:5760. doi: 10.1038/s41598-018-24148-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Silva FA, Rodrigues BL, Ayrizono ML, Leal RF. The immunological basis of inflammatory bowel disease. Gastroenterol. Res. Pract. 2016;2016:2097274. doi: 10.1155/2016/2097274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smythies LE, et al. Mucosal IL-8 and TGF-β recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J. Leukoc. Biol. 2006;80:492–499. doi: 10.1189/jlb.1005566. [DOI] [PubMed] [Google Scholar]
- 63.Sonnier DI, Bailey SR, Schuster RM, Lentsch AB, Pritts TA. TNF-alpha induces vectorial secretion of IL-8 in Caco-2 cells. J. Gastrointest. Surg. 2010;14:1592–1599. doi: 10.1007/s11605-010-1321-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takahashi T, Shiraishi A, Murata J. The coordinated activities of nAChR and Wnt signaling regulate intestinal stem cell function in mice. Int. J. Mol. Sci. 2018 doi: 10.3390/ijms19030738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takahashi T, et al. Non-neuronal acetylcholine as an endogenous regulator of proliferation and differentiation of Lgr5-positive stem cells in mice. FEBS J. 2014;281:4672–4690. doi: 10.1111/febs.12974. [DOI] [PubMed] [Google Scholar]
- 66.The FO, et al. Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology. 2007;133:1219–1228. doi: 10.1053/j.gastro.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 67.Thorne CA, et al. Enteroid monolayers reveal an autonomous WNT and BMP circuit controlling intestinal epithelial growth and organization. Dev. Cell. 2018;44:624–633. doi: 10.1016/j.devcel.2018.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uwada J, et al. Activation of muscarinic receptors prevents TNF-alpha-mediated intestinal epithelial barrier disruption through p38 MAPK. Cell. Signal. 2017;35:188–196. doi: 10.1016/j.cellsig.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 69.VanDussen KL, et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut. 2015;64:911–920. doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wallon C, et al. Eosinophils express muscarinic receptors and corticotropin-releasing factor to disrupt the mucosal barrier in ulcerative colitis. Gastroenterology. 2011;140:1597–1607. doi: 10.1053/j.gastro.2011.01.042. [DOI] [PubMed] [Google Scholar]
- 71.Wang H, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 72.Wang Y, et al. Analysis of interleukin 8 secretion by a stem-cell-derived human-intestinal-epithelial-monolayer platform. Anal. Chem. 2018;90:11523–11530. doi: 10.1021/acs.analchem.8b02835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, et al. Formation of human colonic crypt array by application of chemical gradients across a shaped epithelial monolayer. Cell. Mol. Gastroenterol. Hepatol. 2018;5:113–130. doi: 10.1016/j.jcmgh.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watson AJM, Hughes KR. TNF-α–induced intestinal epithelial cell shedding: implications for intestinal barrier function. Ann. N. Y. Acad. Sci. 2012;1258:1–8. doi: 10.1111/j.1749-6632.2012.06523.x. [DOI] [PubMed] [Google Scholar]
- 75.Williams JM, et al. Epithelial cell shedding and barrier function: a matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015;52:445–455. doi: 10.1177/0300985814559404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu XF, et al. Ardipusilloside I induces apoptosis by regulating Bcl-2 family proteins in human mucoepidermoid carcinoma Mc3 cells. BMC Compl. Altern. Med. 2013;13:322. doi: 10.1186/1472-6882-13-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y, Li J. Carbachol ameliorates lipopolysaccharide-induced intestinal epithelial tight junction damage by down-regulating NF-kappabeta and myosin light-chain kinase pathways. Biochem. Biophys. Res. Commun. 2012;428:321–326. doi: 10.1016/j.bbrc.2012.10.056. [DOI] [PubMed] [Google Scholar]
- 78.Zhou H, et al. Vagus nerve stimulation attenuates intestinal epithelial tight junctions disruption in endotoxemic mice through alpha7 nicotinic acetylcholine receptors. Shock. 2013;40:144–151. doi: 10.1097/SHK.0b013e318299e9c0. [DOI] [PubMed] [Google Scholar]
- 79.Zou WY, et al. Human intestinal enteroids: new models to study gastrointestinal virus infections. Methods Mol. Biol. 2017 doi: 10.1007/7651_2017_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.