

Abstract
Hydroperoxy-eicosatetraenoyl-phosphatidylethanolamine (HpETE-PE) is a ferroptotic cell death signal. HpETE-PE is produced by the 15-Lipoxygenase (15LOX)/Phosphatidylethanolamine Binding Protein-1 (PEBP1) complex or via an Fe-catalyzed non-enzymatic radical reaction. Ferrostatin-1 (Fer-1), a common ferroptosis inhibitor, is a lipophilic radical scavenger but a poor 15LOX inhibitor arguing against 15LOX having a role in ferroptosis. In the current work, we demonstrate that Fer-1 does not affect 15LOX alone, however, it effectively inhibits HpETE-PE production by the 15LOX/PEBP1 complex. Computational molecular modeling shows that Fer-1 binds to the 15LOX/PEBP1 complex at three sites and could disrupt the catalytically required allosteric motions of the 15LOX/PEBP1 complex. Using nine ferroptosis cell/tissue models, we show that HpETE-PE is produced by the 15LOX/PEBP1 complex and resolve the long-existing Fer-1 anti-ferroptotic paradox.
Keywords: Ferroptosis, Ferrostatin-1, 15-Lipoxygenase, Hydroperoxy-eicosatetraenoyl-phosphatidylethanolamines, Phospholipid peroxidation
Graphical abstract
Highlights
-
•
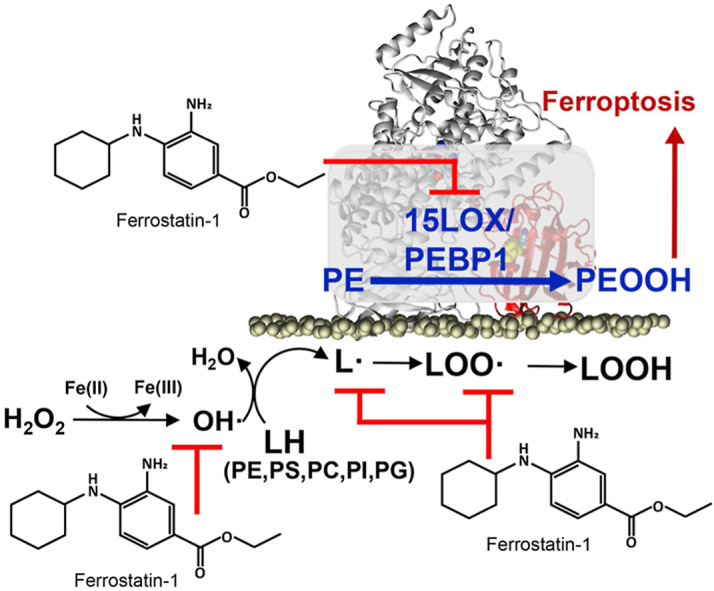
Ferrostatin-1 binds and selectively inhibits arachidonoyl-PE oxidation by 15-LOX/PEBP1.
-
•
Ferrostatin-1 prevention of ferroptosis is mostly due to suppression of 15-LOX/PEBP1.
-
•
Radical scavenging by Ferrostatin-1 in Fe/ascorbate peroxidation is unrelated to ferroptosis.
1. Introduction
Tissue health requires timely removal of harmful or unnecessary cells from the population [1]. This vital function is enacted by several genetically predetermined cell death programs [2]. One of the recently identified programs, ferroptosis, is triggered in cells by the unbalanced redox regulation of three major metabolic pathways related to iron, thiols, and lipids [3]. The role of thiols in the scheme of this irrevocable penalty has been established as the deficiency of the glutathione peroxidase 4 (GPX4) and/or the reduced glutathione (GSH) concentration, which is otherwise uniquely competent in reducing the unstable hydroperoxy-phospholipids (PLOOH) to non-reactive alcohols [4]. While the engagement of iron-catalyzed lipid peroxidation has been strongly implicated, the specific mechanisms are still unclear. Due to the distinctive ability of GPX4 to reduce phospholipid (PL) hydroperoxides, they have been viewed as the most likely death mediators, with our LC-MS studies identifying 15-hydroperoxy-eicosatetraenoyl phosphatidylethanolamine (15-HpETE-PE) as a predictive ferroptosis biomarker [5]. The hypotheses on the types of iron-driven peroxidation processes vary from strictly enzymatic to random free radical reactions [6,7]. One of the major difficulties in identifying the specific mechanism is that in both cases a similar iron-driven catalytic scheme applies: the initial abstraction of bis-allylic hydrogen yielding alkyl radicals that react with molecular oxygen and are oxidized to peroxyl radicals, which further convert to PLOOH as the primary molecular product (Scheme 1).
Scheme 1.

Two alternative oxidation mechanisms: enzymatic (15LOX/PEBP1-driven) and/or non-enzymatic (Fe-driven) reactions of phospholipids (PL) (with the allyl hydrogen shown explicitly as PLH) oxidation yielding hydroperoxy-phospholipids (PLOOH) and their inhibition by ferrostatin-1 (Fer-1).
Among the enzymes involved in the generation of pro-ferroptotic 15-HpETE-PE signals, non-heme iron-containing lipoxygenases (LOXes), including 15LOXes, have been proposed as candidate catalysts [[8], [9], [10]]. However, a number of cell experiments with LOX-knock out cells showed controversial results, only partly confirming the LOX contribution [11,12]. One explanation is that a combination of LOX with yet another factor was necessary for the eicosatetraenoyl-phosphatidylethanolamine (ETE-PE) peroxidation. Indeed, we discovered that a scaffold protein, PEBP1 was required to form the 15LOX/PEBP1 complex which reacts selectively with ETE-PE, one of thousands of oxidizable membrane polyunsaturated fatty acid (PUFA) containing phospholipids (PLs), to generate 15-HpETE-PE, the peroxidation product which induces ferroptotic cell death [13,14].
Acceptance of the enzymatic 15LOX/PEBP1-driven mechanism requires resolution of the conundrum that a variety of radical scavengers are effective in suppressing ferroptosis without displaying sufficient 15LOX inhibitory effects [15,16]. Paradoxically, none of the radical scavengers have been tested as regulators of the formation of 15-HpETE-PE by the 15LOX/PEBP1 complex. The lipophilic radical scavenger, ferrostatin-1 (Fer-1), was identified as an effective inhibitor at the time of the discovery of ferroptosis [17], and since then has become the most popular anti-ferroptotic agent, being used in ~15–20% of the experimental publications on ferroptosis. Fer-1 suppression of cell death has de facto become one of the “functional” tests for ferroptosis vs. other types of cell death. Using a chemical model of azo-initiated peroxidation of arachidonic acid, it was found that the free radical scavenging mechanism of Fer-1 is mainly due to its ortho-amine (-NH) moiety. This moiety can simultaneously interact with lipid radicals forming a planar seven-membered ring in the transition state, which displays a very high scavenging reactivity [18]. In Fe-catalyzed phospholipid peroxidation, the Fer-1 effectiveness was explained by a special mechanism of Fe-chelation by Fer-1, leading to reductive recycling of the compound by alkoxyl radical intermediates [19]. Because Fer-1 acted as a poor 15LOX inhibitor [11,16], it has been argued that chemical, free radical lipid peroxidation rather than enzymatic 15LOX-driven reactions is essential for the execution of the ferroptotic cell death program [11,16]. These contradictory data created a conundrum with several points need to be resolved: i) the high selectivity and specificity of the production of the pro-ferroptotic lipid signals, 15-HpETE-PE from the ETE-PE precursors [5,13], ii) sensitivity of ferroptosis to genetic depletion or chemical poisoning of the enzymes involved in the biosynthesis of ETE-PE, such as acyl-CoA synthase 4 (ACSL4) [20] and lyso-phosphatidylcholine acyl transferase 3 (LPCAT3) [21,22], iii) the ability and sufficiency of 15-HpETE-PE, but not 15-HpETE, for triggering ferroptosis [5], iv) sufficiency of extracellularly added 15LOX activity for the execution of ferroptosis [23].
The effectiveness of Fer-1 as an inhibitor of 15LOX is often tested with free ETE substrate but not with ETE-PE, the precursor of the ferroptotic executor, 15-HpETE-PE. Strikingly, its effectiveness in inhibiting the 15LOX/PEBP1 complex (rather than 15LOX alone) in catalyzing ETE-PE peroxidation has never been tested. Therefore, to test the hypothesis that the 15LOX/PEBP1 complex, rather than 15LOX alone, represents the target for small molecule anti-ferroptotic agents, such as Fer-1, acting via suppression of the enzymatically generated phospholipid death signal, 15-HpETE-PE, in the current work, we analyzed the mechanisms of PL peroxidation in ferroptosis and its inhibition by Fer-1. Using experimental and computational approaches, we demonstrate herein that Fer-1 binds the 15LOX/PEBP1 complex and inhibits its catalytic activity towards ETE-PE peroxidation. We also show that the protective anti-ferroptotic action of Fer-1 is realized predominantly through its ability to inhibit enzymatic conversion of ETE-PE to HpETE-PE.
2. Results
2.1. Ferrrostatin-1 inhibits the production of pro-ferroptotic signals, 15-HpETE-PE
To directly assess the ability of Fer-1 to regulate 15-HpETE-PE production by 15LOX and its complex with PEBP1, we tested Fer-1's effects on oxidation of free ETE and ETE-PE. In accord with published data [12], Fer-1 was ineffective in suppressing free ETE oxidation to HpETE (Fig. 1a) by 15LOX2. However, ETE-PE peroxidation by the 15LOX2/PEBP1 complex revealed two features: i) a 2-fold higher oxidation rate compared to that of 15LOX2 alone (Fig. 1b), and ii) high sensitivity to Fer-1. Notably, Fer-1 decreased the production of 15-HpETE-PE by ~2-fold, vs. the level observed in the absence of PEBP1 (Fig. 1c). The IC50 for Fer-1 inhibition of the 15LOX2/PEBP1 complex was ~10 nM, which is ~an order of magnitude lower than the Fer-1 required for ferroptosis inhibition in cells (above 100 nM) [14].
Fig. 1.

Oxidation of ETE and ETE-PE by 15LOX2 and 15LOX2/PEBP1 complex. a) Effect of Fer-1 on ETE oxidation by 15LOX2 and 15LOX2/PEBP1; b) Oxidation of ETE-PE by 15LOX2 and 15LOX2/PEBP1; c) Inhibition of 15LOX2 and 15LOX2/PEBP1 catalyzed ETE-PE oxidation by Fer-1. The IC50 = 10.1 nM (for 15LOX2/PEBP1; two-way ANOVA with Sidak test).
2.2. Ferrostatin-1 interferes with 15LOX/PEBP1 complex formation
To mechanistically understand this difference in the catalytic properties of 15LOX2 towards ETE-PE in the presence and absence of PEBP1, we analyzed in silico interactions of Fer-1 with 15LOX2, bound or unbound to PEBP1. Docking simulations of Fer-1 binding the 15LOX2/PEBP1 complex revealed three major binding sites (Fig. 2a): Site 1, at the interface between the catalytic and β-barrel domains of 15LOX2; Site 2, at the 15LOX2/PEBP1 interface; and Site 3, at the catalytic site. Simulations repeated for 15LOX2 showed that Site 2 was not a high-affinity site for Fer-1 binding (Supplementary Figure 1a). Statistical analysis of Fer-1 binding poses observed in both sets of simulations demonstrated that the surface areas of the 15LOX2 residues, T178-K198 (α2 helix) at Site 2, is practically unoccupied by Fer-1 in the free 15LOX2 (Fig. 2b, Supplementary Figure 1b) indicating that complexation of 15LOX2 with PEBP1 generates a new druggable site at the interface between the proteins. Next, we compared the fluctuations in the spatial positions of all 15LOX2/PEBP1 amino acid residues predicted by the Gaussian Network Model (GNM) [24] for the Fer-1-free and Fer-1-bound forms of the complex. GNM permits evaluation of sizes and directions of residue fluctuations, as well as collectivity of the modes of motion (see mobility profiles, Fig. 2c). Furthermore, it also predicts residues at hinge regions (Fig. 2c, labeled), which are typically localized at the interface between regions of positive (red shaded region) or negative (blue shade) movements along the global mode axis. Fer-1 induces coupling between the β-barrel of 15LOX2 and PEBP1, restricting anticorrelated motions of PEBP1 with respect to 15LOX2 (Fig. 2c). These motions were previously shown to be essential for insertion of a substrate into the catalytic site in the complex [13]. Fer-1 interacted closely with both T178 on the α2 lid-helix and F14 on the β-barrel loop (Supplementary Figure 2), explaining the constraints of the complex global movements. This new binding site also harbors residues shown to allosterically modulate the catalytic activity of 15LOX2 (Supplementary Fig. 3) [14,23,25].
Fig. 2.

Fer-1 binds 15LOX2/PEBP1 complex and affects its functional dynamics. a) Fer-1 binding poses (red sticks) cluster in three regions (sites 1–3). b) Histograms of binding population counts (ordinate) and corresponding binding energies (abscissa) for each site. Note the sharp difference between sites 1,3 and 2, where binding occurs to 15LOX2/PEBP1 (red-bars) only. c) Comparison of global dynamics of 15LOX2/PEBP1 complex in absence (red-curve) and presence of Fer-1 (black-curve). d) Final conformers (at100 ns) observed in MD simulations of PEBP1 binding to 15LOX2, with (right) and without (left) Fer-1. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
We further examined the effect of Fer-1 binding to 15LOX2 on the formation of 15LOX2/PEBP1 complex. We carried out a series of independent molecular dynamics (MD) simulations (14 repeats), initiated with three different binding poses of Fer-1 at Site 2 (Supplementary Figure 4a), in addition to 8 runs without Fer-1. In all cases, we placed the PEBP1 molecule 15 Å away (closest atom-atom distance) from 15LOX2 and examined its diffusion and binding to 15LOX2. Six out of eight runs conducted in the absence of Fer-1 led to a 15LOX2/PEBP1 functional complex (Fig. 2d, left panel, Supplementary Fig. 4b) [14,25]; whereas the presence of Fer-1 at Site 2 (Supplementary Figure 4c) caused a variety of non-functional complex conformations (Fig. 2d). We also observed competition between PEBP1 and Fer-1 for binding to the α2 lid-helix (Supplementary Figure 4d), which was another barrier for the complexation of 15LOX2 with PEBP1.
2.3. Ferrostatin-1 inhibits the formation of enzymatically produced PL-OOH in vitro
We further asked whether the inhibitory effect of Fer-1 on the enzymatic phospholipid peroxidation can be realized in cells in which we induced ferroptosis. We employed a model in which cells were co-incubated with supernatants obtained from a Gram-negative bacterium, Pseudomonas aeruginosa (P. aeruginosa), containing a bacteria-specific 15LOX (pLoxA) [23]. The bacterium induces “theft-ferroptosis” in host epithelial cells using this specialized secreted pLoxA that oxidizes the host cell's ETE-PE to produce pro-ferroptotic signals and cause cell death [23]. Structural organization of the pLoxA catalytic site is close to the mammalian 15LOXes [23]. Incubation of human bronchio-epithelial (HBE) cells with pLoxA-containing supernatants resulted in cell death which was completely inhibited by Fer-1 [23]. Cell death was associated with the robust and selective accumulation of HpETE-PE in host cells. The protective effect of Fer-1 was accompanied by a complete suppression of HpETE-PE accumulation (Supplementary Figure 5). Thus pLoxA-generated HpETE-PE is sufficient for executing ferroptosis and Fer-1 blocks this enzymatic peroxidation.
To determine whether PE peroxidation and its suppression by Fer-1 were specific to ferroptosis, we employed a model in which murine lung epithelial (MLE) cells were incubated, in the absence and presence of Fer-1, with either a prototypical Fe-dependent peroxidation system (i.e. Fe/ascorbate) or an inhibitor of GPX4 (i.e. RSL3) [26]. These experiments demonstrated very low selectivity of Fe/ascorbate peroxidation to any particular class of polyunsaturated fatty acyl-phospholipids (PUFA-PL) and revealed a strong correlation between the lipid peroxidation and the abundance of PUFA-PLs (Fig. 3a). The prevalent class of phospholipids, PC (48.8 ± 3.5 mol%), contained most of the peroxidation products followed by PE (25.3 ± 2.3 mol%) and then PS (8.3 ± 1.6 mol%) (Fig. 3a). In contrast, RSL3 induced selective peroxidation whereby 70.5% of the total peroxidized species detected were only PUFA-PE, although the total level of PL peroxidation products was comparable to that generated by Fe/ascorbate (Fig. 3a). Notably, non-selective Fe/ascorbate-driven phospholipid peroxidation did not cause ferroptosis (Fig. 3c). In contrast, RSL3 caused viability loss in ~half of the cells and this toxicity was fully preventable by Fer-1 (Fig. 3c). Notably, the production of pro-ferroptotic PEox species by Fe/ascorbate was significantly lower than by RSL3 (Fig. 3d). In order to assess the specificity of Fer-1 in inhibiting the phospholipid peroxidation, we quantitatively compared the amounts of each of the PLox species inhibited by Fer-1 in both the Fe/ascorbate and the RSL3 systems. It was observed that RSL3-induced peroxidation displayed a markedly greater Fer-1 reduction in the PEox species, including pro-ferroptotic PEox signals, compared to the Fe/ascorbate system. However, peroxidation of PUFA-PC, unrelated to ferroptotic death, was reduced in the Fe/ascorbate system to a greater extent than in cells treated with RSL3.
Fig. 3.

Inhibitory effect of Fer-1 on free radical and enzymatic lipid peroxidation. a) Matrix plot representing the fraction of PEox (red circles), PCox (grey circles), PSox (green circles) and PIox (blue circles) species among the total PLox generated by Fe/ascorbate (top) and RSL3 (bottom) in MLE cells. The total amount of PLox is specified at the bottom of each plot; b) Dot-plot showing amounts of oxidized PL species inhibited by Fer-1 in RSL3 (y-axis) vs. Fe/ascorbate (x-axis). The amount is the difference between RSL3 or Fe/Asc treated cells in the absence and presence of Fer-1 respectively. Larger dot denotes significant differences between the levels in RSL3-Fer-1 and Fe/Asc-Fer-1, smaller dot denotes the non-significant values. Area in dashed rectangle is enlarged as inset, n = 3; c) Cell death induced by Fe/ascorbate (upper panel) and RSL3 (lower panel) in MLE cells. n = 7; d) total PE oxidation in cells exposed to Fe/ascorbate ± Fer-1, RSL3±Fer-1. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.4. Employing effects of Fer-1 on PCox/PEox for the evaluation of enzymatic vs. non-enzymatic peroxidation as the driver of ferroptotic cell death
We further attempted to quantitatively assess the contribution of PCox vs PEox generated via enzymatic of non-enzymatic free radicals ferroptotic mechanisms. To this end, we compared the enzymatic phospholipid peroxidation by pLoxA in HBE cells co-incubated with P. aeruginosa supernatants vs MLE cells treated with Fe/ascorbate. The accumulation of PC and ETE-PE peroxidation products was markedly different between these two systems. Upon non-enzymatic stimulation, the accumulation of PCox (compared to the control) was significantly higher than the accumulation of ETE-PEox, leading to a high ΔPCox/ΔETE-PEox ratio. During enzymatic stimulation, PCox levels decreased below the basal level and PEox levels increased significantly leading to a negative ΔPCox/ΔETE-PEox ratio. Assuming that these ratios characterize the non-enzymatic vs. enzymatic oxidation mechanisms, we used them to assess PL peroxidation in 9 different in vitro and in vivo peroxidation models in which ferroptosis was induced by various agents/conditions (Fig. 4). In all cases, the enzymatic PE peroxidation mechanism dominated over the non-enzymatic free radical PC peroxidation. In all the models, except for HT1080 cells + Erastin the contents of free radical peroxidation products were significantly lower than in Fe/ascorbate system. However, there were no significant RSL3-induced changes between any two ferroptotic systems, except for HT1080+Erastin and BeWo cells. Interestingly, only the enzymatic PE peroxidation products were detected in BeWo cells exposed to RSL3 (Fig. 4).
Fig. 4.

Proportion of enzymatic and non-enzymatic PL oxidation during ferroptosis. Ratio of changes in free radical induced non-specific PCox products (amount of PCox in oxidation induced condition – amount of PCox in basal condition) to specific enzymatic ETE-PE oxidation products (amount of ETE-PEox in oxidation induced condition – amount of ETE-PEox in basal condition) (primary y-axis) for two systems HBE+ΔwspF and MLE + Fe/ascorbate defines the complete enzymatic and free radical oxidation, respectively. Using these values, the percentage of free radical oxidation (secondary y-axis) was calculated for various ferroptotic conditions. *P < 0.05 vs MLE + Fe/Asc, One way ANOVA.
3. Discussion
One of the major enigmatic issues in understanding the ferroptotic mechanism is the production of death signals, hydroperoxy phospholipids. Our direct LC-MS redox lipidomics analysis showed that accumulation of PEox, (ETE-PE oxidation products), represents the dominant specific response vs. formation of peroxidation products in other classes of phospholipids. As Fe is believed to play a leading role in ferroptosis [3], the major question is whether enzymatic Fe of LOXes or non-enzymatic Fe acts as the catalyst of ferroptosis-associated phospholipid peroxidation. Redox-cycling of Fe and the production of ROS, particularly of HO• radicals, react aggressively and indiscriminatively towards biomolecular targets [7], and cannot selectively peroxidize ETE-PE species, leaving other more abundant polyunsaturated fatty acids (PUFA) phospholipids non-oxidized [7]. Indeed, the Fe/ascorbate catalyzed reaction caused robust peroxidation of the more abundant PUFA-PC. This peroxidation, however, did not trigger ferroptotic cell death thus emphasizing the necessity of PEox for ferroptosis.
Normally, cells may contain phospholipid peroxidation products generated by both enzymatic and non-enzymatic pathways. The distinction between the contribution of the two types of mechanisms to the overall peroxy-phospholipidome has not been yet established. Based on the available redox lipidomics data it is likely that enzymatic mechanisms are phospholipid class-specific while non-enzymatic reactions will lead to profiles dependent on the levels of poly-unsaturation rather than the structure of the phospholipid polar head-groups. Indeed, in model systems, free radical azo-initiators oxidize various phospholipids classes independently of their headgroup [27]. Free radical peroxidation of a mixture of sn-1-stearoyl, sn-2-linoleoyl PC and PE initiated by Fe2+/ascorbate yielded at least 13 species of oxidized PLS (including hydroperoxy-products), and oxidatively truncated PC and PE species [19]. 7-(diethylamino)coumarin-3-carbohydrazide initiated free radical oxidation resulted in the formation of 31 oxidized species of PG, 23 species of PC-ox, 34 species of PE-ox, 7 species of PS-ox, 17 species of PA-ox and 6 species of oxidized phosphatidylinositiol phosphate (PIox) species [27]. In line with this, our experiments on Fe/ascorbate induced peroxidation in MLE cells revealed increased levels of 76 individual PLox including 24 PCox species, 33 PE-ox species, 10, PIox species and 9 PSox species. In sharp contrast, our analysis revealed that 81 out of 120 PLox species induced in 9 different pro-ferroptotic systems were represented by PEox class compared to only 20 PCox species and 19 PIox species thus emphasizing selective PE peroxidation in ferroptosis [5,13,14,28]. Redox lipidomics assessments of PL oxidation during the execution of several death programs such as apoptosis, pyroptosis and necroptosis demonstrated that PEox was by far the most dominant peroxidation product accountable for ~ half of all PLox both in terms of the number of oxidized individual species and their abundance. This characteristic of ferroptosis PE peroxidation did not occur during the execution of other examined cell death programs [29].
The conundrum still remains as how do the diverse free radical scavengers – which are not effective LOX inhibitors, act as anti-ferroptotic agents. Given that the 15LOX/PEBP1 complex, rather than LOX alone, is engaged in the generation of pro-ferroptotic PEox signals, we examined the suppressive potency of Fer-1 on 15-HpETE-PE production by 15LOX2/PEBP1. Importantly, we found that, in spite of the Fer-1's broad-range radical scavenging activity, its anti-ferroptotic protection is largely due to the inhibition of the 15LOX2/PEBP1-catalyzed formation of pro-ferroptotic HpETE-PE. Notably, Fer-1's IC50 against the 15LOX2/PEBP1 complex is an order of magnitude lower than its half-maximal ferroptosis inhibitory concentration. Mechanistically, our computer model predicts that Fer-1 binds at the 15LOX2 and PEBP1 interaction sites and disrupts the allosteric movement of 15LOX's lid helix that facilitates the entry/binding of ETE-PE to the catalytic site as well as the formation of the productive 15LOX2/PEBP1 complex. Thus it is likely that the ability of Fer-1 to prevent the formation of 15LOX2/PEBP1 complexes and to interfere with the required allosteric 15LOX motions in the complex are the major contributors to the observed 50% decrease in 15LOX2/PEBP1's activity by Fer-1 observed in our experiments in model biochemical systems.
It should be noted that the ultimate executioners of ferroptotic death have not yet been identified. It is possible that after the initial instigation of ferroptosis by the 15LOX2/PEBP1 complex, 15-HpETE-PE undergoes oxidative truncation, forming electrophilic products which attack specific proteins in a PE-targeted process. If so, then free radical scavengers may exert their anti-ferroptotic action at a later stage of the ferroptotic process, after the 15LOX2/PEBP1 complex catalyzed reaction. Alternatively, the random radical scavenging activity of Fer-1 may be related to non-specific side effects of Fer-1 that limit its therapeutic potential.
Overall, our results suggest that oxidation of ETE-PE to HpETE-PE is a highly regulated process, in line with the concept of death program. This is emphasized by two major features of peroxidation: i) remarkable selectivity towards ETE-PE, which is a minor lipid out of hundreds of alternative PUFA-PLs, and ii) high specificity of Fer-1 selectively eliminating the pro-ferroptotic HpETE-PE oxidation products. The ability of Fer-1 to suppress the production of 15-HpETE-PE by inhibiting the 15LOX2/PEBP1 complex but not by inhibiting 15LOX2 alone resolves a long-standing conundrum related to an apparent ineffectiveness of Fer-1 as a LOX inhibitor. This suggests that the effects of other anti-ferroptotic agents should be tested in models utilizing 15LOX/PEBP1 (rather than 15LOX alone) in the reactions catalyzing the production of 15-HpETE-PE. Furthermore, the discovery of selective inhibitors of ferroptosis that suppress the activity of the 15LOX/PEBP1 complex without affecting the catalytic potency of 15LOX alone may lower ferroptosis, while maintaining the production of vital regulators of inflammatory response.
4. Materials and methods
Materials. 1-Octadecanoyl-2-(5Z,8Z,11Z,14Z-eicosatetraenoyl)-sn-glycero-3-phosphatidylethanolamine (ETE-PE), 1-Steraoyl-2-15(S)-hydroperoxy-5Z,8Z,11Z,13E-eicosatetraenoyl-sn-glycero-3-phosphatidyl-ethanolamine (15-HpETE-PE), eicosatetraenoic acid (ETE), and 15(S)-hydroperoxy-5Z,8Z,11Z,13E-eicosatetraenoic acid (15-HpETE) were purchased from Cayman Chemical (Ann Arbor, MI). Unless otherwise stated, all other reagents were purchased from Fisher Scientific (Waltham, MA).
Cell and cell treatments. To induce ferroptosis, BeWo cells, primary human kidney proximal tubule epithelial cells (PHKCs) were incubated with RAS-selective lethal compound 3 (RSL3, Selleck Chemicals, Houston, TX) (100 nM) for 12 h RSL3 (750 nM) for 5 h, respectively in the absence or in the presence of ferrostatin-1 (Fer-1, 0.4 μM) [5]. Human fibrosarcoma cells (HT-1080) were treated with erastin (10 μM) for 24 h or FINO2 (10 μM) for 24 h in the absence or in the presence of Fer-1 (2 μM) as previously described [30]. Human bronchial epithelial cells (HBE, a cell line originally established by Dieter Gruenert) were treated for 20 h with supernatants from P. aeruginosa hyper-biofilm mutant (ΔwspF) strains in the absence and in the presence of Fer-1 (0.2 μM) as previously described [23]. P. aeruginosa hyper-biofilm mutant (ΔwspF) strains were obtained from the P. aeruginosa transposon mutant library, University of Washington (Seattle, Washington, USA) [31]. Mouse embryonic fibroblasts (Pfa1) WT and ACSL4 KO cells were incubated RSL3 (100 nM) for 6 h as described before [5]. Conditional knockout (GPX4 KO) Pfa1 treated with 4-hydroxytamoxifen. MLE cells were seeded (1.5 × 106 cells/dish), and incubated with 800 nM of RSL3 in the presence or absence of FER-1 (0.4 μM) for 6 h. For Fe/Ascorbate experiment, MLE cells were treated with Fe2SO4 (10 μM) plus ascorbic acid (500 μM) for 45 min in the presence or absence of Fer-1 (0.4 μM). Cell death was determined by measuring released lactate dehydrogenase (LDH) activity at different time-points. LDH activity was quantified using the Cytotoxicity Detection Kit (LDH) according to the manufacturer's instructions (Promega Corporation, Madison, WI).
Acute kidney injury model of ferroptosis (AKI). AKI data were obtained from the previously published study [14]. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Protein expression and purification. PEBP1 protein was expressed in the Escherichia coli strain BL21 (DE3) Codon+ (Agilent, Santa Clara, CA) harboring a pET21-derived (EMD Millipore, Billerica, MA) bacterial expression plasmid modified to express PEBP1 with N-terminal His10 and mRuby2 tags. Following initial growth to OD600 of 0.6, cells were induced using 0.2 mM isopropylthio-beta-galactosidase (IPTG) and cultured overnight at room temperature. Cells were harvested by centrifugation and lysed in a buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl, 5% glycerol, 5 mM imidazole, 1 mM β-mercaptoethanol, and protease inhibitors. Lysates were cleared by centrifugation at 29,000×g. Protein was purified by nickel affinity chromatography (Qiagen, Hilden, Germany) and the N-terminal, His10-mRuby2 tag was subsequently removed by overnight digestion with TEV protease. After digestion, PEBP1 was isolated via a second round of nickel affinity chromatography. The protein was then dialyzed overnight at 4 °C into a buffer containing 20 mM sodium acetate (pH 5.5) before cation exchange chromatography was performed in sodium acetate (pH 5.5) using a HiTrap SP column (GE Healthcare, Little Chalfont, UK). The resulting fractions were further refined by size exclusion chromatography in 20 mM HEPES (pH 8.0), 500 mM NaCl buffer using a Sephacryl S-200 column (GE Healthcare). Before performing experiments, PEBP1 was desalted into 5 mM Bis-Tris (pH 6.5), 25 mM NaCl [14].
15-LOX2 was expressed as N-terminal His6-tagged protein and was purified via immobilized metal affinity chromatography using a Ni-NTA resin [32,33]. The protein purity was evaluated by SDS-PAGE analysis and was found to be greater than 90%. Metal content was assessed on a Finnigan inductively coupled plasma-mass spectrometer (ICP-MS) via comparison with an iron standard solution. Cobalt-EDTA was used as an internal standard.
Oxidation of ETE-PE by 15LOX in model system. Liposomes (50 μl) containing ETE-PE (5 nmols) and di-oleoyl-phosphatidylcholine (DOPC) (1 nmol) were incubated with recombinant human15LOX2 (3 pmol) [32] in the presence and in the absence of recombinant PEBP1 [14] and Fer-1 at 37 °C. For reactions with the 15LOX2/PEBP1 complex, equal quantity of PEBP1 and 15LOX2 (1:1 ratio) were added into the reaction system. The mixture was incubated at 37 °C for 20 min using a shaker mixer. The reaction was stopped with the addition of 9 vol of 100% acetonitrile and the samples were centrifuged at 10,000 g for 15 min at 4 °C. The supernatant was analyzed by LC-MS/MS system.
Oxidation of free ETE by 15LOX2 in model system. 15LOX2 (0.06 μM) catalyzed oxidation of ETE (arachidonic acid, 10 μM) was performed in the absence and in the presence of different concentrations of Fer-1 (0.1–4.0 μM) in 20 mM HEPES buffer pH 7.4, containing 100 μM DTPA, 0.6 μM 15-HpETE at 37 °C for 10 min.
LC-MS/MS analysis of oxygenated PL. Oxygenated PE species formed in 15LOX2 catalyzed reaction were analyzed by an LC-MS/MS method using a Dionex Ultimate 3000 RSLCnano System coupled online to a Q-Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific, San Jose, CA) using a hydrophilic interaction liquid chromatography (HILIC) column (BEH HILIC Column, 100 Å, 1.7 μm, 1.0 mm × 50 mm (Waters, Milford, MA)). A gradient solvent system that consisted of solvent A (acetonitrile:water, 90:10 (v:v)) and solvent B (acetonitrile:water, 50:50 (v:v)), each containing 5 mM ammonium acetate, was used as follows: 0–3 min of 100% A at 50 μl/min flow rate, 3–8 min linear gradient of 0%–50% B at 50 μl/min flow rate, 8–9 min linear gradient of 50%–0% B at 50 μl/min flow rate, 9–12 min re-equilibration of 100% A at 50 μl/min flow rate. All solvents were LC/MS-grade. MS analysis was performed in negative ion mode at a resolution of 140,000 full width at half maximum (FWHM) for the full MS scan in a data-dependent mode. Minimum three technical replicates for each sample were run to increase the reproducibility.
LC-MS/MS analysis of oxygenated ETE. Oxygenated ETE was analyzed by LC/MS using a Dionex Ultimate™ 3000 HPLC system coupled on-line to Q-Exactive hybrid quadrupole-orbitrap mass spectrometer (ThermoFisher Scientific, San Jose, CA) as previously described [34]. Briefly, arachidonic and its oxidative derivatives were separated by a C18 column (Acclaim PepMap RSLC, 300 μm 15 cm, Thermo Scientific) using a gradient solvents (A: Methanol (20%)/Water (80%) (v/v) and B: Methanol (90%)/Water (10%) (v/v) both containing 5 mM ammonium acetate. The column was eluted at a flow rate of 12 μL/min using a linear gradient from 30% solvent B to 95% solvent B over 70 min, held at 95%B from 70 to 80 min followed by a return to initial conditions by 83 min and re-equilibration for an additional 7 min. Spectra were acquired in negative ion mode. Minimum three technical replicates for each sample were run to increase the reproducibility.
LC-MS/MS analysis of cell and tissue samples. Non-targeted lipidomics analysis of cell and tissue samples are performed as described previously [14]. Briefly, lipids from the samples were extracted using the Folch procedure, and phosphorus was determined by a micro-method as described previously [5]. Phospholipids were analyzed by LC/MS using a Dionex Ultimate 3000 HPLC system coupled on-line to a Q-Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher) using a normal phase column (Luna 3 μm Silica (2) 100 Å, 150 × 2.0 mm, (Phenomenex)). The analytes were eluted using gradient solvents (A and B) at a flow rate of 0.2 ml/min. Solvent A is isopropanol/hexane/water (285:215:5, v/v/v), and solvent B is isopropanol/hexane/water (285:215:40, v/v/v) while both solvent contained 10 mM ammonium formate. The following gradient was used for the elution: 0-23 min-linear gradient from 10% to 32% B; 23-32 min-linear gradient of 32%–65% B; 32–35 min - linear gradient of 65%–100% B; 35–62 min held at 100% B; 62-64 min-linear gradient from 100% to 10% B; followed by an equilibration from 64 to 80 min at 10% B. Analysis was performed in negative ion mode at a resolution of 140,000 for the full MS scan in a data-dependent mode. The scan range for MS analysis was m/z 400–1800 with an isolation window of 1.0 Da was set for the MS2 scans.
Mass spectrometry data analysis. MS data were analyzed using Compound Discoverer™ 2.0 (Thermo scientific) as described previously [14]. Briefly, aligned peaks with 5000 intensity and signal to noise ratio more than 5 were identified. The identity of the peaks was assigned based on the exact mass (±7 ppm) and retention time. The retention times of the PLox species were fixed as 5 min within the PL class internal standard. The PLox species were confirmed with representative fragmentation analysis of selected m/z. The amounts of each lipid species were calculated using calibration data obtained with reference standards. Supplementary table 1 details the list of reference and internal standards used in the study. Representative MS data from pfa-1 cells treated with RSL3 are shown in supplementary table 2.
Molecular Simulations. Target conformers were generated by molecular dynamics (MD) simulations in explicit water (TIP3P), following earlier work [[35], [36], [37]]. The initial structures used in simulations were the human 15LOX2 resolved with a substrate mimic [38] (PDB: 4nre) where the substrate was replaced by ETE-PE, and the 15LOX2/PEBP1 complex reported in our previous work [14]. For each system, we performed three MD runs of 100 ns each, using NAMD [39] with CHARMM27 force field. Force field parameters for Fer-1 were generated using SwissParam server [40]. Preparatory simulations used the following protocol: 0.2 ns of water equilibration, 10,000 steps of minimization, 0.35 ns of heating from 0 to 300 K, and 0.15 ns equilibration of the whole system before initiating the production MD run. A cutoff of 12 Å for non-bonded interactions was applied. Langevin dynamics and the Langevin piston algorithm were used to maintain the temperature at 300 K and the pressure at 1 atm. Snapshots at 100 ps intervals were analyzed to identify clusters of conformations using ensemble analysis tool in ProDy [41], which led to 13 clusters for 15LOX2, and 13 for 15LOX2/PEBP1 complex. A representative member from each cluster was used in Fer-1 docking simulations using Smina [45]. Five runs were performed for each conformer, each yielding 14 high-scoring poses (with Fer-1 within 4 Å interatomic distance from 15LOX2) such that 13 x 5 x 14 = 910 poses were generated for each target, 15LOX2 and for 15LOX2/PEBP1. Also, a series of independent MD simulations of 15LOX2/ETE-PE complex, 14 runs with three different binding poses of Fer-1 and 8 runs without Fer-1 (each 100 ns), were performed with PEBP1 (1beh) [42] placed 15 Å away (supplementary Fig. 4a). VMD [43], Pymol [44] and ProDy package were used for analysis and visualization. The collective dynamics of 15LOX2/PEBP1 was analyzed before and after Fer-1 binding using the Gaussian network model implemented in ProDy [35].
Acknowledgement
This work was supported by NIH grants: AI145406, CA165065, AI068021, GM113908, HL114453, AI156924, NS076511, AI156923, NS061817; National Science Centre, Poland Grant (2019/35/D/ST4/02203), Natural Science Foundation of China (81873209, 81622050, 81903821); 111 Project of Chinese MoE (B13038); the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y036), and GDUPS (2019).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101744.
Contributor Information
Hülya Bayır, Email: bayihx@ccm.upmc.edu.
Valerian E. Kagan, Email: kagan@pitt.edu.
Conflict of interest
B.R.S. is an inventor on patents and patent applications involving ferroptosis and co-founded and serves as a consultant to Inzen Therapeutics and Nevrox. The other authors declare no competing financial interests.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Danial N.N., Korsmeyer S.J. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/S0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 2.Tang D., Kang R., Vanden Berghe T., Vandenabeele P., Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bayır H., Anthonymuthu T.S., Tyurina Y.Y., Patel S.J., Amoscato A.A., Lamade A.M., Yang Q., Vladimirov G.K., Philpott C.C., Kagan V.E. Achieving life through death: redox biology of lipid peroxidation in ferroptosis. Cell Chem. Biol. 2020;27:387–408. doi: 10.1016/j.chembiol.2020.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cozza G., Rossetto M., Bosello-Travain V., Maiorino M., Roveri A., Toppo S., Zaccarin M., Zennaro L., Ursini F. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: the polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic. Biol. Med. 2017;112:1–11. doi: 10.1016/j.freeradbiomed.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Kagan V.E., Mao G., Qu F., Angeli J.P., Doll S., Croix C.S., Dar H.H., Liu B., Tyurin V.A., Ritov V.B., Kapralov A.A., Amoscato A.A., Jiang J., Anthonymuthu T., Mohammadyani D., Yang Q., Proneth B., Klein-Seetharaman J., Watkins S., Bahar I., Greenberger J., Mallampalli R.K., Stockwell B.R., Tyurina Y.Y., Conrad M., Bayir H. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinman A., Holst C.R., Latham J.C., Bruegger J.J., Ulas G., McCusker K.P., Amagata A., Davis D., Hoff K.G., Kahn-Kirby A.H., Kim V., Kosaka Y., Lee E., Malone S.A., Mei J.J., Richards S.J., Rivera V., Miller G., Trimmer J.K., Shrader W.D. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PloS One. 2018;13 doi: 10.1371/journal.pone.0201369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stoyanovsky D.A., Tyurina Y.Y., Shrivastava I., Bahar I., Tyurin V.A., Protchenko O., Jadhav S., Bolevich S.B., Kozlov A.V., Vladimirov Y.A., Shvedova A.A., Philpott C.C., Bayir H., Kagan V.E. Iron catalysis of lipid peroxidation in ferroptosis: regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019;133:153–161. doi: 10.1016/j.freeradbiomed.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaitidis P., Schewe T., Sutherland M., Kuhn H., Nigam S. 15-Lipoxygenation of phospholipids may precede the sn-2 cleavage by phospholipases A2: reaction specificities of secretory and cytosolic phospholipases A2 towards native and 15-lipoxygenated arachidonoyl phospholipids. FEBS Lett. 1998;434:437–441. doi: 10.1016/s0014-5793(98)01024-2. [DOI] [PubMed] [Google Scholar]
- 9.Maskrey B.H., Bermudez-Fajardo A., Morgan A.H., Stewart-Jones E., Dioszeghy V., Taylor G.W., Baker P.R., Coles B., Coffey M.J., Kuhn H., O'Donnell V.B. Activated platelets and monocytes generate four hydroxy phosphatidylethanolamines via lipoxygenase. J. Biol. Chem. 2007;282:20151–20163. doi: 10.1074/jbc.M611776200. [DOI] [PubMed] [Google Scholar]
- 10.Shintoku R., Takigawa Y., Yamada K., Kubota C., Yoshimoto Y., Takeuchi T., Koshiishi I., Torii S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Canc. Sci. 2017;108:2187–2194. doi: 10.1111/cas.13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zilka O., Shah R., Li B., Friedmann Angeli J.P., Griesser M., Conrad M., Pratt D.A. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 2017;3:232–243. doi: 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah R., Shchepinov M.S., Pratt D.A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 2018;4:387–396. doi: 10.1021/acscentsci.7b00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anthonymuthu T.S., Kenny E.M., Shrivastava I., Tyurina Y.Y., Hier Z.E., Ting H.C., Dar H.H., Tyurin V.A., Nesterova A., Amoscato A.A., Mikulska-Ruminska K., Rosenbaum J.C., Mao G., Zhao J., Conrad M., Kellum J.A., Wenzel S.E., VanDemark A.P., Bahar I., Kagan V.E., Bayir H. Empowerment of 15-lipoxygenase catalytic competence in selective oxidation of membrane ETE-PE to ferroptotic death signals, HpETE-PE. J. Am. Chem. Soc. 2018;140:17835–17839. doi: 10.1021/jacs.8b09913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wenzel S.E., Tyurina Y.Y., Zhao J., St Croix C.M., Dar H.H., Mao G., Tyurin V.A., Anthonymuthu T.S., Kapralov A.A., Amoscato A.A., Mikulska-Ruminska K., Shrivastava I.H., Kenny E.M., Yang Q., Rosenbaum J.C., Sparvero L.J., Emlet D.R., Wen X., Minami Y., Qu F., Watkins S.C., Holman T.R., VanDemark A.P., Kellum J.A., Bahar I., Bayir H., Kagan V.E. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171:628–641. doi: 10.1016/j.cell.2017.09.044. e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griesser M., Shah R., Van Kessel A.T., Zilka O., Haidasz E.A., Pratt D.A. The catalytic reaction of nitroxides with peroxyl radicals and its relevance to their cytoprotective properties. J. Am. Chem. Soc. 2018;140:3798–3808. doi: 10.1021/jacs.8b00998. [DOI] [PubMed] [Google Scholar]
- 16.Shah R., Margison K., Pratt D.A. The potency of diarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem. Biol. 2017;12:2538–2545. doi: 10.1021/acschembio.7b00730. [DOI] [PubMed] [Google Scholar]
- 17.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., Morrison B., 3rd, Stockwell B.R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheng X.H., Cui C.C., Shan C., Li Y.Z., Sheng D.H., Sun B., Chen D.Z. O-Phenylenediamine: a privileged pharmacophore of ferrostatins for radical-trapping reactivity in blocking ferroptosis. Org. Biomol. Chem. 2018;16:3952–3960. doi: 10.1039/c8ob00546j. [DOI] [PubMed] [Google Scholar]
- 19.Miotto G., Rossetto M., Di Paolo M.L., Orian L., Venerando R., Roveri A., Vuckovic A.M., Bosello Travain V., Zaccarin M., Zennaro L., Maiorino M., Toppo S., Ursini F., Cozza G. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28:101328. doi: 10.1016/j.redox.2019.101328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doll S., Proneth B., Tyurina Y.Y., Panzilius E., Kobayashi S., Ingold I., Irmler M., Beckers J., Aichler M., Walch A., Prokisch H., Trümbach D., Mao G., Qu F., Bayir H., Füllekrug J., Scheel C.H., Wurst W., Schick J.A., Kagan V.E., Angeli J.P.F., Conrad M. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017;13:91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon S.J., Winter G.E., Musavi L.S., Lee E.D., Snijder B., Rebsamen M., Superti-Furga G., Stockwell B.R. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 2015;10:1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., Fulda S., Gascón S., Hatzios S.K., Kagan V.E., Noel K., Jiang X., Linkermann A., Murphy M.E., Overholtzer M., Oyagi A., Pagnussat G.C., Park J., Ran Q., Rosenfeld C.S., Salnikow K., Tang D., Torti F.M., Torti S.V., Toyokuni S., Woerpel K.A., Zhang D.D. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dar H.H., Tyurina Y.Y., Mikulska-Ruminska K., Shrivastava I., Ting H.C., Tyurin V.A., Krieger J., St Croix C.M., Watkins S., Bayir E., Mao G., Armbruster C.R., Kapralov A., Wang H., Parsek M.R., Anthonymuthu T.S., Ogunsola A.F., Flitter B.A., Freedman C.J., Gaston J.R., Holman T.R., Pilewski J.M., Greenberger J.S., Mallampalli R.K., Doi Y., Lee J.S., Bahar I., Bomberger J.M., Bayir H., Kagan V.E. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Invest. 2018;128:4639–4653. doi: 10.1172/JCI99490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Chang Y.-Y., Yang L.-W., Bahar I. iGNM 2.0: the Gaussian network model database for biomolecular structural dynamics. Nucleic Acids Res. 2016;44:D415–D422. doi: 10.1093/nar/gkv1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mikulska-Ruminska K., Shrivastava I., Krieger J., Zhang S., Li H., Bayır H., Wenzel S.E., VanDemark A.P., Kagan V.E., Bahar I. Characterization of differential dynamics, specificity, and allostery of lipoxygenase family members. J. Chem. Inf. Model. 2019;59:2496–2508. doi: 10.1021/acs.jcim.9b00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang W.S., SriRamaratnam R., Welsch M.E., Shimada K., Skouta R., Viswanathan V.S., Cheah J.H., Clemons P.A., Shamji A.F., Clish C.B., Brown L.M., Girotti A.W., Cornish V.W., Schreiber S.L., Stockwell B.R. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni Z., Milic I., Fedorova M. Identification of carbonylated lipids from different phospholipid classes by shotgun and LC-MS lipidomics. Anal. Bioanal. Chem. 2015;407:5161–5173. doi: 10.1007/s00216-015-8536-2. [DOI] [PubMed] [Google Scholar]
- 28.Doll S., Freitas F.P., Shah R., Aldrovandi M., da Silva M.C., Ingold I., Goya Grocin A., Xavier da Silva T.N., Panzilius E., Scheel C.H., Mourão A., Buday K., Sato M., Wanninger J., Vignane T., Mohana V., Rehberg M., Flatley A., Schepers A., Kurz A., White D., Sauer M., Sattler M., Tate E.W., Schmitz W., Schulze A., O'Donnell V., Proneth B., Popowicz G.M., Pratt D.A., Angeli J.P.F., Conrad M. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 29.Wiernicki B., Dubois H., Tyurina Y.Y., Hassannia B., Van Coillie S., Bayir H., Kagan V., Vandenabeele P., Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis, Cell Death Dis.. (n.d.). [DOI] [PMC free article] [PubMed]
- 30.Gaschler M.M., Andia A.A., Liu H., Csuka J.M., Hurlocker B., Vaiana C.A., Heindel D.W., Zuckerman D.S., Bos P.H., Reznik E., Ye L.F., Tyurina Y.Y., Lin A.J., Shchepinov M.S., Chan A.Y., Peguero-Pereira E., Fomich M.A., Daniels J.D., Bekish A.V., Shmanai V.V., Kagan V.E., Mahal L.K., Woerpel K.A., Stockwell B.R. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018;14:507–515. doi: 10.1038/s41589-018-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobs M.A., Alwood A., Thaipisuttikul I., Spencer D., Haugen E., Ernst S., Will O., Kaul R., Raymond C., Levy R., Chun-Rong L., Guenthner D., Bovee D., Olson M.V., Manoil C. Comprehensive transposon mutant library of <em>Pseudomonas aeruginosa</em>. Proc. Natl. Acad. Sci. Unit. States Am. 2003;100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jameson J.B., Kenyon V., Holman T.R. A high-throughput mass spectrometric assay for discovery of human lipoxygenase inhibitors and allosteric effectors. Anal. Biochem. 2015;476:45–50. doi: 10.1016/j.ab.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amagata T., Whitman S., Johnson T.A., Stessman C.C., Loo C.P., Lobkovsky E., Clardy J., Crews P., Holman T.R. Exploring sponge-derived terpenoids for their potency and selectivity against 12-human, 15-human, and 15-soybean lipoxygenases. J. Nat. Prod. 2003;66:230–235. doi: 10.1021/np020462l. [DOI] [PubMed] [Google Scholar]
- 34.Tyurina Y.Y., Poloyac S.M., Tyurin V.A., Kapralov A.A., Jiang J., Anthonymuthu T.S., Kapralova V.I., Vikulina A.S., Jung M.Y., Epperly M.W., Mohammadyani D., Klein-Seetharaman J., Jackson T.C., Kochanek P.M., Pitt B.R., Greenberger J.S., Vladimirov Y.A., Bayir H., Kagan V.E. A mitochondrial pathway for biosynthesis of lipid mediators. Nat. Chem. 2014;6:542–552. doi: 10.1038/nchem.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H., Chang Y.-Y., Lee J.Y., Bahar I., Yang L.-W. DynOmics: dynamics of structural proteome and beyond. Nucleic Acids Res. 2017;45:W374–W380. doi: 10.1093/nar/gkx385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin J., Perryman A.L., Schames J.R., McCammon J.A. The relaxed complex method: accommodating receptor flexibility for drug design with an improved scoring scheme. Biopolym. Orig. Res. Biomol. 2003;68:47–62. doi: 10.1002/bip.10218. [DOI] [PubMed] [Google Scholar]
- 37.Lin J.-H., Perryman A.L., Schames J.R., McCammon J.A. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J. Am. Chem. Soc. 2002;124:5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- 38.Kobe M.J., Neau D.B., Mitchell C.E., Bartlett S.G., Newcomer M.E. The structure of human 15-lipoxygenase-2 with a substrate mimic. J. Biol. Chem. 2014;289:8562–8569. doi: 10.1074/jbc.M113.543777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zoete V., Cuendet M.A., Grosdidier A., Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011;32:2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 41.Bakan A., Meireles L.M., Bahar I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27:1575–1577. doi: 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banfield M.J., Barker J.J., Perry A.C., Brady R.L. Function from structure? The crystal structure of human phosphatidylethanolamine-binding protein suggests a role in membrane signal transduction. Structure. 1998;6:1245–1254. doi: 10.1016/s0969-2126(98)00125-7. [DOI] [PubMed] [Google Scholar]
- 43.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 44.DeLano W.L. Pymol: an open-source molecular graphics tool CCP4. Newsl. Protein Crystallogr. 2002;40:82–92. [Google Scholar]
- 45.Koes D.R., Baumgartner M.P., Camacho C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. Journal of Chemical Information and Modeling. 2013;53(8):1893–1904. doi: 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.