

Abstract
SARS‐CoV‐2, a member of the coronavirus family, has caused a global public health emergency. Based on our analysis of hepatitis C virus and coronavirus replication, and the molecular structures and activities of viral inhibitors, we previously reasoned that the FDA‐approved hepatitis C drug EPCLUSA (Sofosbuvir/Velpatasvir) should inhibit coronaviruses, including SARS‐CoV‐2. Here, using model polymerase extension experiments, we demonstrate that the active triphosphate form of Sofosbuvir is incorporated by low‐fidelity polymerases and SARS‐CoV RNA‐dependent RNA polymerase (RdRp), and blocks further incorporation by these polymerases; the active triphosphate form of Sofosbuvir is not incorporated by a host‐like high‐fidelity DNA polymerase. Using the same molecular insight, we selected 3’‐fluoro‐3’‐deoxythymidine triphosphate and 3’‐azido‐3’‐deoxythymidine triphosphate, which are the active forms of two other anti‐viral agents, Alovudine and AZT (an FDA‐approved HIV/AIDS drug) for evaluation as inhibitors of SARS‐CoV RdRp. We demonstrate the ability of two of these HIV reverse transcriptase inhibitors to be incorporated by SARS‐CoV RdRp where they also terminate further polymerase extension. Given the 98% amino acid similarity of the SARS‐CoV and SARS‐CoV‐2 RdRps, we expect these nucleotide analogues would also inhibit the SARS‐CoV‐2 polymerase. These results offer guidance to further modify these nucleotide analogues to generate more potent broad‐spectrum anti‐coronavirus agents.
Keywords: COVID‐19, SARS‐CoV, SARS‐CoV‐2, RNA‐dependent RNA polymerase, nucleotide analogue
The triphosphate form of Sofosbuvir is incorporated by SARS‐CoV polymerase to terminate further primer extension, potentially preventing replication of the virus.
![]()
1. INTRODUCTION
SARS‐CoV‐2, the virus responsible for the COVID‐19 pandemic, is a new member of the subgenus Sarbecovirus, in the Orthocoronavirinae subfamily, but is distinct from MERS‐CoV and SARS‐CoV. 1 The virus has been isolated from the lower respiratory tracts of patients with pneumonia, sequenced and visualized by electron microscopy. 1 Coronaviruses are single‐stranded RNA viruses, sharing properties with other single‐stranded RNA viruses such as hepatitis C virus (HCV), West Nile virus, Marburg virus, HIV virus, Ebola virus, dengue virus, and rhinoviruses. In particular, coronaviruses and HCV are both positive‐sense single‐stranded RNA viruses, 2 , 3 and thus have a similar replication mechanism requiring an RNA‐dependent RNA polymerase (RdRp).
The coronavirus life cycle has been described. 2 Briefly, the virus enters the cell by endocytosis, is uncoated, and ORF1a and ORF1b of the positive‐stranded RNA are translated to produce nonstructural protein precursors, including a cysteine protease and a serine protease; these further cleave the precursors to form mature, functional helicase, and RdRp. A replication‐transcription complex is then formed, which is responsible for making more copies of the RNA genome via a negative‐sense RNA intermediate, as well as the structural and other proteins encoded by the viral genome. The viral RNA is packaged into viral coats in the endoplasmic reticulum‐Golgi intermediate complex, after which exocytosis results in the release of viral particles for subsequent infectious cycles. Potential inhibitors have been designed to target nearly every stage of this process. 2 However, despite decades of research, no effective drug is currently approved to treat serious coronavirus infections such as SARS, MERS, and COVID‐19.
One of the most important druggable targets for coronaviruses is the RdRp. This polymerase is highly conserved at the protein level among different positive‐sense RNA viruses, to which coronaviruses and HCV belong, and shares common structural features in these viruses. 4 Like RdRps in other viruses, the coronavirus enzyme is highly error‐prone, 5 which might increase its ability to accept modified nucleotide analogues as substrates. Nucleotide and nucleoside analogues that inhibit polymerases are an important group of anti‐viral agents. 6 , 7 , 8 , 9
Based on our analysis of hepatitis C virus and coronavirus replication, and the molecular structures and activities of viral inhibitors, we previously reasoned that the FDA‐approved hepatitis C drug EPCLUSA (Sofosbuvir/Velpatasvir) should inhibit coronaviruses, including SARS‐CoV‐2. 10 Sofosbuvir is a pyrimidine nucleotide analogue prodrug with a hydrophobic masked phosphate group enabling it to enter infected eukaryotic cells, and then be converted into its active triphosphate form by cellular enzymes (Fig. 1). In this activated form, it inhibits the HCV RdRp NS5B. 11 , 12 The activated drug (2’‐F,Me‐UTP) binds in the active site of the RdRp, where it is incorporated into RNA, and due to fluoro and methyl modifications at the 2’ position, inhibits further RNA chain extension, thereby halting RNA replication and stopping viral growth. It acts as an RNA polymerase inhibitor by competing with natural ribonucleotides. Velpatasvir inhibits NS5A, a key protein required for HCV replication. NS5A enhances the function of RNA polymerase NS5B during viral RNA synthesis. 13 , 14
FIGURE 1.
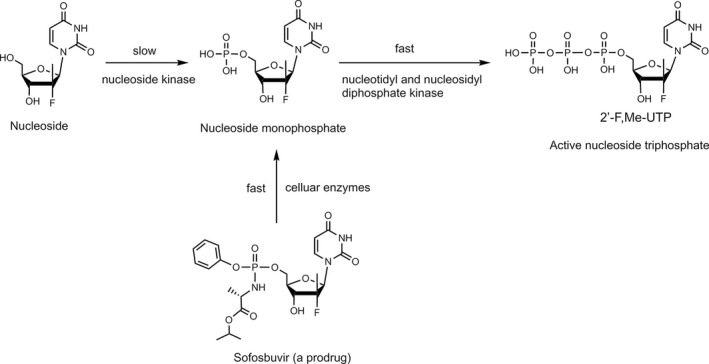
Conversion of Sofosbuvir to active triphosphate (2’‐F,Me‐UTP) in vivo to inhibit viral polymerases. Adapted from 11
There are many other RNA polymerase inhibitors that have been evaluated as antiviral drugs. A related purine nucleotide prodrug, Remdesivir (Fig. 2B), was developed by Gilead to treat Ebola virus infections, though not successfully, and is currently in clinical trials for treating COVID‐19. 15 , 16 In contrast to Sofosbuvir (Fig. 2A), both the 2’‐ and 3’‐OH groups in Remdesivir (Fig. 2B) are unmodified, but a cyano group at the 1’ position serves to inhibit the RdRp in the active triphosphate form. In addition to the use of hydrophobic groups to mask the phosphate in the Protide‐based prodrug strategy, 17 as with Sofosbuvir and Remdesivir, there are other classes of nucleoside prodrugs including those based on ester derivatives of the ribose hydroxyl groups to enhance cellular delivery. 18 , 19
FIGURE 2.
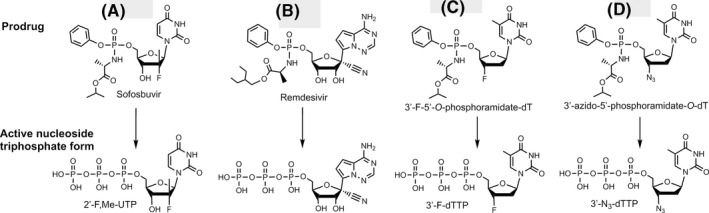
Comparison of structures of prodrug viral inhibitors. Top: Prodrug (phosphoramidate) form; Bottom: Active triphosphate form
The replication cycle of HCV 3 is very similar to that of the coronaviruses. 2 Analyzing the structure of the active triphosphate form of Sofosbuvir (Fig. 2A) compared to that of Remdesivir (Fig. 2B), both of which have already been shown to inhibit the replication of specific RNA viruses (Sofosbuvir for HCV, Remdesivir for SARS‐CoV‐2), we noted in particular that the 2’‐modifications in Sofosbuvir (a fluoro and a methyl group) are substantially smaller than the 1’‐cyano group and the 2’‐OH group in Remdesivir. The bulky cyano group in close proximity to the 2’‐OH may lead to steric hindrance that will impact the polymerase reaction termination efficiency of the activated form of Remdesivir. Interestingly, it was recently reported that, using the MERS‐CoV polymerase, the triphosphate of Remdesivir was preferentially incorporated relative to ATP in solution assays. 20 Nevertheless, it has been shown that the active triphosphate form of Remdesivir does not cause immediate polymerase reaction termination and actually leads to delayed polymerase termination in Ebola virus and respiratory syncytial virus, likely due to its 1’‐cyano group and the free 2’‐OH and 3’‐OH groups. 20 , 21 Compared to the active form of Sofosbuvir (2’‐fluoro‐2’‐methyl‐UTP), two other nucleotide inhibitors with related structures were reviewed as follows: 2’‐fluoro‐UTP is incorporated by polymerase, but RNA synthesis may continue past the incorporated nucleotide analogue; 22 2’‐C‐methyl‐UTP has been shown to terminate the reaction catalyzed by HCV RdRp, 22 but proofreading mechanisms can revert this inhibition in mitochondrial DNA‐dependent RNA polymerases. 23 Additionally, HCV develops resistance to 2’‐C‐methyl‐UTP due to mutations of the RdRp. 24 A computational study published in 2017 considered the ability of various anti‐HCV drugs to dock in the active site of SARS and MERS coronavirus RdRps as potential inhibitors. 25 Recently, Elfiky used a computational approach to predict that Sofosbuvir, IDX‐184, Ribavirin, and Remdesivir might be potent drugs against COVID‐19. 26
Thus, based on our analysis of the biological pathways of hepatitis C and coronaviruses, the molecular structures and activities of viral inhibitors, model polymerase and SARS‐CoV RdRp extension experiments described below, and the efficacy of Sofosbuvir in inhibiting the HCV RdRp, we expect that Sofosbuvir or its modified forms should also inhibit the SARS‐CoV‐2 polymerase. 10
The active triphosphate form of Sofosbuvir (2’‐F,Me‐UTP) was shown to be incorporated by HCV RdRp and prevent any further incorporation by this polymerase. 22 , 27 Other viral polymerases have also been shown to incorporate active forms of various anti‐viral prodrugs to inhibit replication. 28 Since, at the time of the preparation of this manuscript, we did not have access to the RdRp from SARS‐CoV‐2, we first selected two groups of polymerases to test the termination efficiency of the active form of Sofosbuvir, one group with high‐fidelity behavior with regard to incorporation of modified nucleotide analogues, which one would expect for host cell polymerases, the other group with low‐fidelity mimicking viral polymerases, as well as the RdRp from SARS‐CoV, the virus causing the 2003 and subsequent outbreaks of SARS. Our rationale is that the low‐fidelity viral‐like enzymes would incorporate 2’‐F,Me‐UTP and stop further replication, whereas the high‐fidelity polymerases, typical of host cell polymerases, would not. Experimental proof for termination of the SARS‐CoV polymerase reaction would provide further support for this rationale, indicating that Sofosbuvir or its modified forms will inhibit SARS‐CoV‐2.
2. MATERIALS AND METHODS
2.1. Extension reactions with DNA polymerases
Oligonucleotides were purchased from Integrated DNA Technologies (IDT Inc.). The 20 µl extension reactions consisted of 3 µM DNA template and 5 µM DNA primer (sequences shown in Fig. 3), 10 µM 2’‐F,Me‐UTP (Sierra Bioresearch), 1x Thermo Sequenase buffer or 1x ThermoPol buffer (for Therminator enzymes), and either 10 U Thermo Sequenase (GE Healthcare), 4 U Therminator II or 10 U Therminator IX (New England Biolabs). The 1x Thermo Sequenase buffer consists of 26 mM Tris‐HCl, pH 9.5, and 6.5 mM MgCl2. The 1x ThermoPol buffer contains 20 mM Tris‐HCl, pH 8.8, 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgSO4, and 0.1% Triton X‐100. Incubations were performed in a thermal cycler using 15 cycles of 30 sec each at 65°C, 45°C, and 65°C. Following desalting using an Oligo Clean & Concentrator (Zymo Research), the samples were subjected to MALDI‐TOF‐MS (Bruker ultrafleXtreme) analysis, following a previously described method. 29
FIGURE 3.
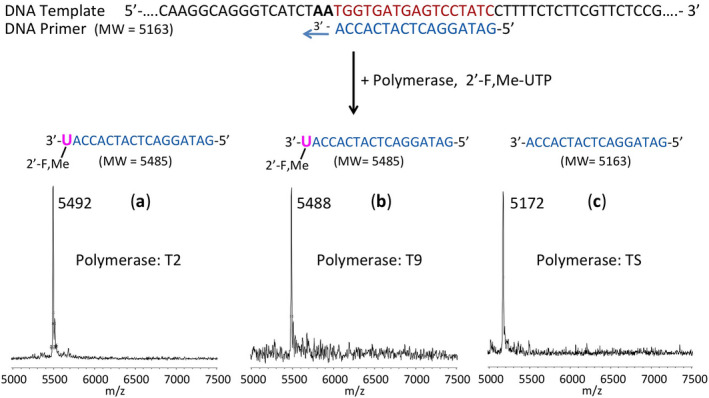
Incorporation of 2’‐F,Me‐UTP as a terminator by two low‐fidelity polymerases but not by a high‐fidelity polymerase. The sequence of the primer and template used for these extension reactions is shown at the top of the figure. Polymerase extension reactions were performed by incubating the primer and template with 2’‐F,Me‐UTP and the appropriate reaction buffer for the specific enzyme, followed by detection of the reaction products by MALDI‐TOF MS. The MS spectra of the extension products generated by Therminator II (T2) in (A) and Therminator IX (T9) in (B) indicate single‐base incorporation and termination, whereas the MS spectrum for Thermo Sequenase (TS) in (C) indicates no incorporation, showing only a primer peak. The accuracy for m/z determination is ± 10 Da
2.2. Extension reactions with SARS‐CoV RNA‐dependent RNA polymerase
Oligonucleotides were purchased from IDT, Inc. Following a published strategy, 41 , 42 the primer and template (sequences are shown in Fig. 4) were annealed by heating to 70°C for 10 min and cooling to room temperature in 1x reaction buffer. The RNA polymerase mixture consisting of 2 µM nsp12 and 6 µM each of cofactors nsp7 and nsp8 was incubated for 15 min at room temperature in a 1:3:3 ratio in 1x reaction buffer. Then 5 µl of the annealed template primer solution containing 2 µM template and 1.7 µM primer in 1x reaction buffer was added to 10 µl of the RNA polymerase mixture and incubated for an additional 10 min at room temperature. Finally, 5 µl of a solution containing either 2 mM 2’‐F,Me‐UTP, 2 mM 3’‐F‐dTTP, 2 mM 3’‐N3‐dTTP or 2 mM UTP in 1x reaction buffer was added, and incubation was carried out for 2 hr at 30°C. The final concentrations of reagents in the 20 µl extension reactions were 1 µM nsp12, 3 µM nsp7, 3 µM nsp8, 425 nM RNA primer, 500 nM RNA template, either 500 µM 2’‐F,Me‐UTP (Sierra Bioresearch), 500 µM 3’‐F‐dTTP (Amersham Life Sciences), or 500 µM 3’‐N3‐dTTP (Amersham Life Sciences), and 1x reaction buffer (10 mM Tris‐HCl pH 8, 10 mM KCl, 2 mM MgCl2 and 1 mM β‐mercaptoethanol). In the experiment with UTP shown in Fig. S2, the final concentrations were 500 nM nsp12, 1.5 µM nsp7, 1.5 µM nsp8, 425 nM RNA primer, 250 nM RNA template, and 500 µM UTP (Fisher) and the reaction time was 1 h at 30°C. Following desalting using an Oligo Clean & Concentrator (Zymo Research), the samples were subjected to MALDI‐TOF‐MS (Bruker ultrafleXtreme) analysis.
FIGURE 4.
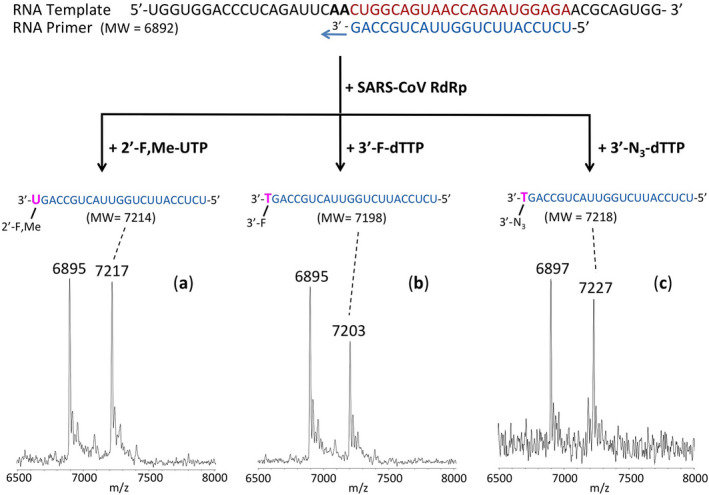
Incorporation of 2’‐F,Me‐UTP, 3’‐F‐dTTP and 3’‐N3‐dTTP by SARS‐CoV RdRp to terminate the polymerase reaction. The sequence of the primer and template used for these extension reactions, which are within the N1 coding sequence of the SARS‐CoV‐2 genome, is shown at the top of the figure. Polymerase extension reactions were performed by incubating (A) 2’‐F,Me‐UTP, (B) 3’‐F‐dTTP, and (C) 3’‐N3‐dTTP with preassembled SARS‐CoV polymerase (nsp12, nsp7, and nsp8), the indicated RNA template and primer, and the appropriate reaction buffer, followed by detection of reaction products by MALDI‐TOF MS. The detailed procedure is shown in the Methods section. For comparison, data for extension with UTP are presented in Fig. S2. The accuracy for m/z determination is ± 10 Da
2.3. Nomenclature of Targets and Ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), 65 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019). 66
3. RESULTS AND DISCUSSION
We first carried out DNA polymerase extension reactions with the active form of Sofosbuvir (2’‐F,Me‐UTP) using Thermo Sequenase as an example of high fidelity, host‐like polymerases, and two mutated DNA polymerases which are known to be more promiscuous in their ability to incorporate modified nucleotides, Therminator II and Therminator IX, as examples of viral‐like low‐fidelity enzymes. A DNA template‐primer complex, in which the next two available bases were A (Fig. 3), was incubated with either 2’‐F,Me‐UTP (structure shown in Fig. 2a), or dTTP as a positive control, in the appropriate polymerase buffer. If the 2’‐F,Me‐UTP is incorporated and inhibits further incorporation, a single‐base primer extension product will be produced. By contrast, dTTP incorporation will result in primer extension by 2 bases. After performing the reactions, we determined the molecular weight of the extension products using MALDI‐TOF‐mass spectrometry (MALDI‐TOF MS).
As seen in Figure 3A and B, when the primer‐template complex (sequences shown at top of Figure 3) and 2’‐F,Me‐UTP were incubated with the low‐fidelity 9oN polymerase mutants, 29 , 30 , 31 Therminator II (T2) and Therminator IX (T9), we observed single‐product peaks with molecular weights of 5492 Da and 5488 Da, indicating single base extension in the polymerase reaction. Thus 2’‐F,Me‐UTP was able to be incorporated and block further nucleotide incorporation. In contrast, when the extension reactions were carried out with high‐fidelity Thermo Sequenase DNA polymerase (TS), 32 there was no incorporation, as evidenced by a single primer peak at 5172 Da (Figure 3C). This supports our rationale that Thermo Sequenase, a high‐fidelity enzyme originally designed for accurate Sanger sequencing, will not incorporate 2’‐F,Me‐UTP, whereas a low‐fidelity polymerase, such as T2 or T9, will incorporate 2’‐F,Me‐UTP and stop further nucleotide incorporation. When dTTP was used as a positive control with these three enzymes, incorporation continued past the first A in the template, resulting in a higher molecular weight peak.
These results demonstrate that lower fidelity polymerases will have a high likelihood of incorporating 2’‐F,Me‐UTP and inhibit viral RNA replication, whereas high‐fidelity enzymes, more typical of the host DNA and RNA polymerases, will have a low likelihood of being inhibited by 2’‐F,Me‐UTP. Anti‐viral drug design based on this principle may lead to potent viral polymerase inhibitors with fewer side effects. To provide further proof that SARS‐CoV‐2 RdRp might be inhibited by 2’‐F,Me‐UTP, we next tested the ability of this molecule to be incorporated into an RNA primer to terminate the reaction catalyzed by the RdRp from SARS‐CoV, using an RNA template. As shown in Figure 4A, the active triphosphate form of the drug Sofosbuvir not only was incorporated by the RdRp, but prevented further incorporation, behaving as a terminator in the polymerase reaction.
Based on our similar insight related to their molecular structures and previous antiviral activity studies, in comparison with Sofosbuvir, we selected the triphosphate forms of Alovudine (3’‐deoxy‐3’‐fluorothymidine) and azidothymidine (AZT, the first FDA‐approved drug for HIV/AIDS) for evaluation as inhibitors of the SARS‐CoV RdRp. These two molecules share a similar backbone structure (base and ribose) with Sofosbuvir, but have only one modification group at the 3’ carbon of the deoxyribose. Furthermore, because these modifications on Alovudine and AZT are on the 3’ carbon in place of the OH group, they directly prevent further incorporation of nucleotides leading to permanent termination of RNA synthesis and replication of the virus. Both Alovudine and AZT are deoxythymidine analogues. However, because their size, structure and base‐pairing properties are similar to uridine and the SARS‐CoV RdRp has low fidelity, the triphosphate forms of these two analogues might still be substrates of the viral polymerase.
Alovudine is one of the most potent inhibitors of HIV reverse transcriptase and HIV‐1 replication. 33 This promising drug was discontinued after a Phase II trial due to its hematological toxicity. However, subsequent in vitro studies showed Alovudine was very effective at suppressing several nucleoside/nucleotide reverse transcriptase inhibitor (NRTI)‐resistant HIV‐1 mutants. 34 New clinical studies were then carried out in which low doses of Alovudine were given as supplements to patients showing evidence of infection by NRTI resistant HIV strains and not responding well to their current drug regimen. A 4‐week course of 2 mg/day Alovudine reduced viral load significantly and was relatively well tolerated with no unexpected adverse events. 35
AZT is another antiretroviral medication that has long been used to prevent and treat AIDS. 36 , 37 , 38 Upon entry into the infected cells, similar to Alovudine, cellular enzymes convert AZT into the effective 5'‐triphosphate form (3’‐N3‐dTTP, structure shown in Figure 2D), which competes with dTTP for incorporation into DNA by HIV‐reverse transcriptase resulting in termination of HIV's DNA synthesis. 39 Since the side effects and toxicity of AZT are well understood, novel methodologies have been directed at enhancing AZT plasma levels and its bioavailability in all human organs in order to improve its therapeutic efficacy. Among these possibilities, an AZT prodrug strategy was proposed. 40
We thus assessed the ability of 3’‐N3‐dTTP and 3’‐F‐dTTP, the active triphosphate forms of AZT and Alovudine, along with 2’‐F,Me‐UTP, to be incorporated by SARS‐CoV RdRp into an RNA primer and terminate the polymerase reaction.
The RdRp of SARS‐CoV, referred to as nsp12, and its two protein cofactors, nsp7 and nsp8, shown to be required for the processive polymerase activity of nsp12, were cloned and purified as described. 41 , 42 These three viral gene products have high homology (e.g., 96% identity and 98% similarity for nsp12, with similar homology levels at the amino acid level for nsp7 and nsp8) to the equivalent gene products from SARS‐CoV‐2, the causative agent of COVID‐19. A detailed description of the homologies of nsp7, nsp8, and nsp12 is included in Fig. S1 which highlights key functional motifs in nsp12 described by Kirchdoerfer and Ward. 42 Of these, Motifs A, B, E, F, and G are identical in SARS‐CoV and SARS‐CoV‐2 at the amino acid level, and Motifs C and D display only conservative substitutions.
We performed polymerase extension assays with 2’‐F,Me‐UTP, 3’‐F‐dTTP, 3’‐N3‐dTTP, or UTP following the addition of a preannealed RNA template and primer to a preassembled mixture of the RdRp (nsp12) and two cofactor proteins (nsp7 and nsp8). The extended primer products from the reaction were subjected to MALDI‐TOF‐MS analysis. The RNA template and primer, corresponding to the N1 epitope region of the N protein of the SARS‐CoV‐2 virus, were used for the polymerase assay, and their sequences are indicated at the top of Figure 4. Because there are two As in a row in the next available positions of the template for RNA polymerase extension downstream of the priming site, if 2’‐F,Me‐UTP, 3’‐F‐dTTP, or 3’‐N3‐dTTP are incorporated by the viral RdRp, the nucleotide analogue will be added to the 3’‐end of the primer strand. If they are indeed inhibitors of the polymerase, the extension should stop after this incorporation; further 3’‐extension should be prevented. In the case of the UTP control reaction, two UTPs should be incorporated. As shown in Figure 4 and Fig. S2, this is exactly what we observed. In the MALDI‐TOF MS trace in Figure 4a, a peak indicative of the molecular weight of a primer extension product terminated with one 2’‐F,Me‐UTP was obtained (7217 Da observed, 7214 Da expected). Similarly, in the trace in Figure 4b a single extension peak indicative of a single‐base extension product terminated by 3’‐F‐dTTP is revealed (7203 Da observed, 7198 Da expected), with no further incorporation. And in the trace in Figure 4c, a single extension peak indicative of a single‐base extension by 3’‐N3‐dTTP is seen (7227 Da observed, 7218 Da expected), with no evidence of further incorporation. As a positive control, primer extension by 2 UTPs occurred (7506 Da observed, 7504 Da expected) as shown in the MALDI‐TOF MS trace in Fig. S2.
In summary, these results demonstrate that the nucleotide analogues 2’‐F,Me‐UTP, 3’‐F‐dTTP, and 3’‐N3‐dTTP, are permanent terminators for the SARS‐CoV RdRp. Their prodrug versions (Sofosbuvir, 3’‐F‐5’‐O‐phosphoramidate dT nucleoside and 3’‐N3‐5’‐O‐phosphoramidate dT nucleoside) can be readily synthesized using the ProTide prodrug approach, as shown in Figure 2A, C and D, and can be evaluated as potential therapeutics for both SARS and COVID‐19.
One factor that has confounded the development of RdRp inhibitors in coronaviruses is the presence of a 3’‐exonuclease‐based proofreading activity such as that associated with nsp14, a key component of the replication‐transcription complex in SARS‐CoV, 43 , 44 and also encoded in SARS‐CoV‐2. This exonuclease activity can be overcome with the use of 2’‐O‐methylated nucleotides. 43 Importantly, since both Sofosbuvir and AZT are FDA‐approved drugs for the treatment of other viral infections and their toxicity profiles are well‐established, their ability to inhibit coronaviruses can be evaluated quickly in laboratory and clinical settings.
A preprint of this manuscript was posted on bioRxiv on March 14, 2020. 45 In our two recent publications, we used the polymerase extension and MS detection approach described in this manuscript to evaluate a larger library of nucleotide analogues as SARS‐CoV‐2 inhibitors, demonstrating that the approach is robust. 46 , 47 We confirmed our prediction that the three molecules (2’‐F,Me‐UTP, 3’‐F‐dTTP, 3’‐N3‐dTTP) reported in this paper as well as four others also inhibited SARS‐CoV‐2 polymerase to varying degrees. 46 In addition, we analyzed a library of 11 additional nucleoside triphosphates as inhibitors of SARS‐CoV and SARS‐CoV‐2. 47
The field of COVID‐19 therapeutics development is moving rapidly, and while this manuscript was under review and revision, numerous publications and preprints have appeared. Structural studies have indicated possible binding sites in the SARS‐CoV‐2 RdRp for potential polymerase inhibitors. 48 , 49 , 50 , 51 , 52 Given the high homology of the SARS‐CoV and SARS‐CoV‐2 RdRp active site domains, it is likely that they will bind nucleotide analogues such as Sofosbuvir in a similar way, as we have recently reported. 46 The structures of the SARS‐CoV‐2 RNA‐dependent RNA polymerase nsp12 and its complex with nsp7 and nsp8 have been determined by cryo‐EM, 49 , 50 and these structures were compared with those of other RdRps including the SARS‐CoV RdRp and HCV NS5B. These investigators performed docking studies to reveal likely binding sites for potential inhibitors and natural nucleotides. Gao et al. modeled Remdesivir diphosphate binding to SARS‐CoV‐2 nsp12 based on superposition with Sofosbuvir diphosphate bound to HCV NS5B, and found that the nsp12 of SARS‐CoV‐2 has the highest similarity with the Apo state of NS5B. 49 Yin et al. indicated that the orientations of the template‐primer RNA in the active site of SARS‐CoV‐2 and hepatitis C virus NS5B are similar, and the amino acid residues involved in RNA binding and those making up the active site are highly conserved. 50
Several investigators have recently recommended Sofosbuvir as a possible antiviral for COVID‐19, based on structural studies and multiple alignment analysis. 48 By comparing the positive‐stranded RNA genomes of HCV and SARS‐CoV‐2, Buonaguro et al. postulated that Sofosbuvir might be an optimal nucleotide analogue to repurpose for COVID‐19 treatment. 53 Gordon et al. performed a detailed kinetic study including Km values of triphosphates of Remdesivir, Sofosbuvir, and other nucleotide analogues using gel electrophoresis, indicating that Sofosbuvir triphosphate has an apparent lower efficiency than the natural nucleotide. 54 Sofosbuvir was recently shown to inhibit SARS‐CoV‐2 replication in human hepatoma‐derived (Huh‐2) and Type II pneumocyte‐derived (Calu‐3) cells with EC50 values of 6.2 and 9.5 µM, respectively. 55 Sofosbuvir was also reported to protect human brain organoids from SARS‐CoV‐2 infection. 56
After considering the potential advantages of Sofosbuvir including its low toxicity, its ability to be rapidly activated to the triphosphate form by cellular enzymes, and the high intracellular stability of this active molecule, COVID‐19 clinical trials with EPCLUSA (a combination of Sofosbuvir and Velpatasvir) 57 and with Sofosbuvir plus Daclatasvir 58 have been initiated in several countries. Recently, Sadeghi et al. reported promising results in a clinical trial using the combination drug Sofosbuvir (SOF) and Daclatasvir (DCV) to treat moderate or severe COVID‐19 patients. 59 These investigators showed that SOF/DCV treatment increased 14‐day clinical recovery rates and reduced hospital stays. Two similar SOF/DCV clinical trials were also performed and provided evidence that this drug combination may have some benefit; 60 , 61 the authors recommended that larger well‐controlled randomized trials are necessary to confirm their results. Indeed, a network of larger COVID‐19 clinical trials has been established in Brazil, Egypt, Iran, and South Africa.
Sofosbuvir and Velpatasvir together form the combination drug EPCLUSA, which is widely used for the treatment of HCV. Velpatasvir inhibits the viral replication protein NS5A in HCV; 13 , 14 Daclatasvir also inhibits this protein. 62 Sacramento et al. reported that Daclatasvir was able to reduce SARS‐CoV‐2‐induced enhancement of TNF‐α and IL‐6, which are key contributors to the cytokine storm. 55 Because Velpatasvir and Daclatasvir have strong structural similarity and target the same NS5A protein in HCV, and Daclatasvir has also been shown to inhibit SARS‐CoV‐2 replication 55 and is currently in COVID‐19 clinical trial, 58 it is plausible that Velpatasvir will display similar inhibitory activity for SARS‐CoV‐2. Finally, Remdesivir has been approved under FDA emergency use authorization, 63 and is currently being tested for its safety and effectiveness in various COVID‐19 clinical trials; in contrast, Sofosbuvir is an FDA‐approved hepatitis C drug with wide availability and a well‐characterized safety and clinical profile.
We recently demonstrated that Sofosbuvir terminated RNA is more resistant to the SARS‐CoV‐2 proofreading exonuclease than RNAs terminated by Remdesivir and natural nucleotides. 64 The higher resistance to exonuclease of Sofosbuvir‐RNA relative to the RNAs containing the natural nucleotide or Remdesivir can compensate for the apparently lower SARS‐CoV‐2 RdRp incorporation efficiency of Sofosbuvir triphosphate. Therefore, in view of the fact that Sofosbuvir triphosphate inhibits the SARS‐CoV and SARS‐CoV‐2 RdRps and has better resistance to the exonuclease than the natural nucleotide or Remdesivir, it is likely that Sofosbuvir will inhibit replication of SARS‐CoV‐2. These results provide a molecular basis supporting the current use of Sofosbuvir in combination with other drugs in COVID‐19 clinical trials.
4. CONCLUSION
We demonstrated the capability of low‐fidelity DNA polymerases, as well as SARS‐CoV RNA‐dependent RNA polymerase, which is nearly identical to the SARS‐CoV‐2 RdRp responsible for COVID‐19, to incorporate 2’‐F,Me‐UTP, the active form of Sofosbuvir, where it serves to terminate the polymerase reaction. We also showed two other nucleoside triphosphates, 3’‐F‐dTTP, the active form of Alovudine, and 3’‐N3‐dTTP, the active form of AZT, can be incorporated and terminate further nucleotide extension by the RdRp in the polymerase reaction, potentially preventing further replication of the virus. If prodrugs of these nucleotide analogues display efficacy in inhibiting SARS‐CoV‐2 replication in cell culture, as recently demonstrated for Sofosbuvir in virus‐infected cells, 55 they can be potential candidates for clinical trials for the treatment and prevention of COVID‐19.
DATA SHARING STATEMENT
All relevant data are in the paper.
DISCLOSURES
The authors declare no competing interests.
AUTHORS’ CONTRIBUTIONS
JJ conceived and directed the project; the approaches and assays were designed and conducted by JJ, XL, SK, SJ, JJR, MC, and CT, comparative sequence analysis was performed by IM and SK, and SARS‐CoV polymerase and associated proteins nsp12, 7, and 8 were cloned and purified by RNK. Data were analyzed by all authors. All authors wrote and reviewed the manuscript.
Supporting information
Supplementary Material
Acknowledgments
This research is supported by Columbia University, a grant from the Jack Ma Foundation, a generous gift from the Columbia Engineering Member of the Board of Visitors Dr. Bing Zhao, and Fast Grants to J.J. and a National Institute of Allergy and Infectious Disease grant AI123498 to R.N.K.
Ju J, Li X, Kumar S, et al. Nucleotide analogues as inhibitors of SARS‐CoV Polymerase. Pharmacol Res Perspect. 2020;8:e00674 10.1002/prp2.674
REFERENCES
- 1. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Eng J Med. 2020;382:727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zumla A, Chan JFW, Azhar EI, Hui DSC, Yuen K‐Y. Coronaviruses – drug discovery and therapeutic options. Nat Rev Drug Discovery. 2016;15:327–347. 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dustin LB, Bartolini B, Capobianchi MR, Pistello M. Hepatitis C virus: life cycle in cells, infection and host response, and analysis of molecular markers influencing the outcome of infection and response to therapy. Clin Microbiol Infect. 2016;22:826–832. 10.1016/j.cmi.2016.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. te Velthuis AJW. Common and unique features of viral RNA‐dependent polymerases. Cell Mol Life Sci. 2014;71:4403–4420. 10.1007/s00018-014-1695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Selisko B, Papageorgiou N, Ferron F, Canard B. Structural and functional basis of the fidelity of nucleotide selection by Flavivirus RNA‐dependent RNA polymerases. Viruses. 2018;10:59 10.3390/v10020059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKenna CE, Levy JN, Khawli LA, et al. Inhibitors of viral nucleic acid polymerases. Pyrophosphate analogues. ACS Symposium Series. 1989;401:1–16. 10.1021/bk-1989-0401.ch001. [DOI] [Google Scholar]
- 7. Öberg B. Rational design of polymerase inhibitors as antiviral drugs. Antiviral Res. 2006;71:90–95. 10.1016/j.antiviral.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 8. Eltahla AA, Luciani F, White PA, Lloyd AR, Bull RA. Inhibitors of the hepatitis C virus polymerase; mode of action and resistance. Viruses. 2015;7:5206–5224. 10.3390/v7102868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Clercq E, Li G. Approved antiviral drugs over the past 50 years. Clin Microbiol Rev. 2016;29:695–747. 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ju J, Kumar S, Li X, Jockusch S, Russo JJ. Nucleotide analogues as inhibitors of viral polymerases. bioRxiv. 2020;. 10.1101/2020.01.30.927574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kayali Z, Schmidt WN. Finally sofosbuvir: an oral anti‐HCV drug with wide performance capability. Pharmgenomics Pers Med. 2014;7:387–398. 10.2147/PGPM.S52629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sofia MJ, Bao D, Chang W, et al. Discovery of a β‐D‐2′‐Deoxy‐2′‐α‐fluoro‐2′‐β‐C‐methyluridine nucleotide prodrug (PSI‐7977) for the treatment of hepatitis C virus. J Med Chem. 2010;53:7202–7218. 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 13. Gitto S, Gamal N, Andreone P. NS5A inhibitors for the treatment of hepatitis C infection. J Viral Hepatitis. 2017;24:180–186. 10.1111/jvh.12657. [DOI] [PubMed] [Google Scholar]
- 14. Quezada EM, Kane CM. The hepatitis C virus NS5A stimulates NS5B during in vitro RNA synthesis in a template specific manner. Open Biochem J. 2009;3:39–48. 10.2174/1874091X00903010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holshue ML, DeBolt C, Lindquist S, et al. First case of 2019 novel coronavirus in the United States. N Eng J Med. 2020;382:929–936. 10.1056/NEJMoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 2020;30:269–271. 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alanazi AS, James E, Mehellou Y. The ProTide prodrug technology: where next? ACS Med Chem Lett. 2019;10:2–5. 10.1021/acsmedchemlett.8b00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Clercq E, Field HJ. Antiviral prodrugs – the development of successful prodrug strategies for antiviral chemotherapy. Br J Pharmacol. 2006;147:1–11. 10.1038/sj.bjp.0706446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roberts SK, Cooksley G, Dore GJ, et al. Robust antiviral activity of R1626, a novel nucleoside analog: A randomized, placebo‐controlled study in patients with chronic hepatitis C. Hepatol. 2008;48:398–406. 10.1002/hep.22321. [DOI] [PubMed] [Google Scholar]
- 20. Gordon CJ, Tchesnokov EP, Feng JY, Porter DP, Götte M. The antiviral compound remdesivir potently inhibits RNA‐dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J Biol Chem. 2020;295:4773–4779. 10.1074/jbc.AC120.013056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tchesnokov EP, Feng JY, Porter DP, Götte M. Mechanism of inhibition of Ebola virus RNA‐dependent RNA polymerase by Remdesivir. Viruses. 2019;11:326 10.3390/v11040326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fung A, Jin Z, Dyatkina N, Wang G, Beigelman L, Deval J. Efficiency of incorporation and chain termination determines the inhibition potency of 2’‐modified nucleotide analogs against hepatitis C virus polymerase. Antimicrob Agents Chemother. 2014;58:3636–3645. 10.1128/AAC.02666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arnold JJ, Sharma SD, Feng JY, et al. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLoS Pathog. 2012;8:e1003030 10.1371/journal.ppat.1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dutartre H, Bussetta C, Boretto J, Canard B. General catalytic deficiency of hepatitis C virus RNA polymerase with an S282T mutation and mutually exclusive resistance towards 2’‐modified nucleotide analogues. Antimicrob Agents Chemother. 2006;50:4161–4169. 10.1128/AAC.00433-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elfiky AA, Mahdy SM, Elshemey WM. Quantitative structure‐activity relationship and molecular docking revealed a potency of anti‐hepatitis C virus drugs against human corona viruses. J Med Virol. 2017;89:1040–1047. 10.1002/jmv.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elfiky AA. Anti‐HCV, nucleotide inhibitors, repurposing against COVID‐19. Life Sci. 2020;248:117477 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deval J, Symons JA, Beigelman L. Inhibition of viral RNA polymerases by nucleoside and nucleotide analogs: therapeutic applications against positive‐strand RNA viruses beyond hepatitis C virus. Curr Opin Virol. 2014;9:1–7. 10.1016/j.coviro.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fearns R, Deval J. New antiviral approaches for respiratory syncytial virus and other mononegaviruses: Inhibiting the RNA polymerase. Antiviral Res. 2016;134:63–76. 10.1016/j.antiviral.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 29. Ju J, Kim DH, Bi L, et al. Four‐color DNA sequencing by synthesis using cleavable fluorescent nucleotide reversible terminators. Proc Natl Acad Sci USA. 2006;103:19635–19640. 10.1073/pnas.0609513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo J, Xu N, Li Z, et al. Four‐color DNA sequencing with 3’‐O‐modified nucleotide reversible terminators and chemically cleavable fluorescent dideoxynucleotides. Proc Natl Acad Sci USA. 2008;105:9145–9150. 10.1073/pnas.0804023105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ruparel H, Bi L, Li Z, et al. Design and synthesis of a 3’‐O‐allyl photocleavable fluorescent nucleotide as a reversible terminator for DNA sequencing by synthesis. Proc Natl Acad Sci USA. 2005;102:5932–5937. 10.1073/pnas.0501962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tabor S, Richardson CC. A single residue in DNA polymerases of Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy‐ and dideoxynucleotides. Proc Natl Acad Sci USA. 1995;92:6339–6343. 10.1073/pnas.92.14.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Camerman N, Mastropaolo D, Camerman A. Structure of the anti‐human immunodeficiency virus agent 3’‐fluoro‐3’‐deoxythymidine and electronic charge calculations for 3’‐deoxythymidines. Proc Natl Acad Sci USA. 1990;87:3534–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim E‐Y, Vrang L, Öberg B, Merigan TC. Anti‐HIV type 1 activity of 3′‐fluoro‐3′‐deoxythymidine for several different multidrug‐resistant mutants. AIDS Res Human Retroviruses. 2001;17:401–407. 10.1089/088922201750102445. [DOI] [PubMed] [Google Scholar]
- 35. Ghosn J, Quinson A‐M, Sabo ND, et al. Antiviral activity of low‐dose alovudine in antiretroviral‐experienced patients: results from a 4‐week randomized, double‐blind, placebo‐controlled dose‐ranging trial. HIV Med. 2007;8:142–147. 10.1111/j.1468-1293.2007.00444.x. [DOI] [PubMed] [Google Scholar]
- 36. Mitsuya H, Weinhold KJ, Furman PA, et al. 3'‐Azido‐3'‐deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T‐lymphotropic virus type III/lymphadenopathy‐associated virus in vitro. Proc Natl Acad Sci USA. 1985;82:7096–7100. 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yarchoan R, Weinhold KJ, Lyerly HK, et al. Administration of 3'‐azido‐3'‐deoxythymidine, an inhibitor of HTLV‐III/LAV replication, to patients with AIDS or AIDS‐related complex. Lancet. 1986;327:575–580. 10.1016/S0140-6736(86)92808-4. [DOI] [PubMed] [Google Scholar]
- 38. Mitsuya H, Yarchoan R, Broder S. Molecular targets for AIDS therapy. Science. 1990;249:1533–1544. 10.1126/science.1699273. [DOI] [PubMed] [Google Scholar]
- 39. Furman PA, Fyfe JA, St Clair MH, et al. Phosphorylation of 3’‐azido‐3’‐deoxythymidine and selective interaction of the 5’‐triphosphate with human immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci USA. 1986;83:8333–8337. 10.1073/pnas.83.21.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. D’Andrea G, Brisdelli F, Bozzi A. AZT: An old drug with new perspectives. Curr Clin Pharmacol. 2008;3:20–37. 10.2174/157488408783329913. [DOI] [PubMed] [Google Scholar]
- 41. Subissi L, Posthuma CC, Collet A, et al. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc Natl Acad Sci USA. 2014;111:E3900–E3909. 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kirchdoerfer RN, Ward AB. Structure of the SARS‐CoV nsp12 polymerase bound to nsp7 and nsp8 co‐factors. Nature Commun. 2019;10:2342 10.1038/s41467-019-10280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Minskaia E, Hertzig T, Gorbalenya AE, et al. Discovery of an RNA virus 3’‐5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc Natl Acad Sci USA. 2006;103:5108–5113. 10.1073/pnas.0508200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Agostini ML, Andres EL, Sims AC, et al. Coronavirus susceptibility to the antiviral Remdesivir (GS‐5734) is mediated by the viral polymerase and the proofreading exonuclease. mBio. 2018;9:e00221–e00218. 10.1128/mBio.00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ju J, Li X, Kumar S, et al. Nucleotide analogues as inhibitors of SARS‐CoV polymerase. bioRxiv. 2020;. 10.1101/2020.03.12.989186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chien M, Anderson TK, Jockusch S, et al. Nucleotide analogues as inhibitors of SARS‐CoV‐2 polymerase, a Key Drug Target for COVID‐19. J Proteome Res. 2020;. 10.1021/acs.jproteome.0c00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jockusch S, Tao C, Li X, et al. A library of nucleotide analogues terminate RNA synthesis catalyzed by polymerases of coronaviruses that cause SARS and COVID‐19. Antiviral Res. 2020;180:104857 10.1016/j.antiviral.2020.104857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jácome R, Campillo‐Balderas JA, Ponce de León S, Becerra A, Lazcano A. Sofosbuvir as a potential alternative to treat the SARS‐CoV‐2 epidemic. Sci Rep. 2020;10:9294. 10.1038/s41598-020-66440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gao Y, Yan L, Huang Y, et al. Structure of the RNA‐dependent RNA polymerase from COVID‐19 virus. Science. 2020;368:779–782. 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA‐dependent RNA polymerase from SARS‐CoV‐2 by remdesivir. Science. 2020;368:1499–1504. 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hillen HS, Koci G, Farnung L, Dienemann C, Tegunov D, Cramer P. Structure of replicating SARS‐CoV‐2 polymerase. Nature. 2020;584:154–156. 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
- 52. Elfiky AA. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS‐CoV‐2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020;253:117592 10.1016/j.lfs.2020.117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Buonaguro L, Tagliamonte M, Tornesello ML, Buonaguro FM. SARS‐CoV‐2 RNA polymerase as target for antiviral therapy. J Transl Med. 2020;18:185 10.1186/s12967-020-02355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gordon CJ, Tchesnokov EP, Woolner E, et al. Remdesivir is a direct‐acting antiviral that inhibits RNA‐dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J Biol Chem. 2020;295:6785–6797. 10.1074/jbc.RA120.013679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sacramento CQ, Fintelman‐Rodrigues N, Temerozo JR, et al. The in vitro antiviral activity of the anti‐hepatitis C virus (HCV) drugs daclatasvir and sofosbuvir against SARS‐CoV‐2. bioRxiv 2020. 10.1101/2020.06.15.153411. [DOI] [PMC free article] [PubMed]
- 56. Mesci P, Macia A, Saleh A, et al. Sofosbuvir protects human brain organoids against SARS‐CoV‐2. bioRxiv. 2020. 10.1101/2020.05.30.125856. [DOI] [Google Scholar]
- 57. Sayad B, Sobhani M, Khodarahmi R. Sofosbuvir as repurposed antiviral drug against COVID‐19: Why were we convinced to evaluate the drug in a registered/approved clinical trial? Arch Med Res. 2020;51:577–581. 10.1016/j.arcmed.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. World Hepatitis Alliance press release: Hepatitis C drugs may offer an inexpensive treatment option for COVID‐19 . https://www.worldhepatitisalliance.org/latest‐news/infohep/3548907/hepatitis‐c‐drugs‐may‐offer‐inexpensive‐treatment‐option‐covid‐19.
- 59. Sadeghi A, Asgari AA, Norouzi A, et al. Sofosbuvir and daclatasvir compared with standard of care in the treatment of patients admitted to hospital with moderate or severe coronavirus infection (COVID‐19): a randomized controlled trial. J Antimicrob Chemother. 2020;. 10.1093/jac/dkaa334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eslami G, Mousaviasl S, Radmanesh E, et al. The impact of sofosbuvir/daclatasvir or ribavirin in patients with severe COVID‐19. J Antimicrob Chemother. 2020;. 10.1093/jac/dkaa331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kasgari HA, Moradi S, Shabani AM, et al. Evaluation of the efficacy of sofosbuvir plus daclatasvir in combination with ribavirin for hospitalized COVID‐19 patients with moderate disease compared with standard care: a single‐centre, randomized controlled trial. J Antimicrob Chemother. 2020;. 10.1093/jac/dkaa332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smith MA, Regal RE, Mohammad RA. Daclatasvir: A NS5A replication complex inhibitor for hepatitis C infection. Ann Pharmacother. 2016;50:39–46. 10.1177/1060028015610342. [DOI] [PubMed] [Google Scholar]
- 63. Eastman RT, Roth JS, Brimacombe KR, et al. Remdesivir: A review of its discovery and development leading to emergency use authorization for treatment of COVID‐19. ACS Cent Sci. 2020;6:672–683. 10.1021/acscentsci.0c00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jockusch S, Tao C, Li X, et al. Sofosbuvir terminated RNA is more resistant to SARS‐CoV‐2 proofreader than RNA terminated by Remdesivir. Sci Rep. 2020;10:16577 10.1038/s41598-020-73641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46:D1091–D1106. 10.1093/nar/gkx1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol. 2019;176:S297–S396. 10.1111/bph.14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material