

Abstract
We assessed the contribution of interleukin-1 (IL1) signaling molecules to malignant tumor growth, using IL1β−/−, IL1α−/−, and IL1R1−/− mice. Tumors grew progressively in IL1R−/− and IL1α−/− mice, but were often absent in IL1β−/− mice. This was observed whether tumors were implanted intradermally or injected intravenously, and was true across multiple distinct tumor lineages. Antibodies to IL1β prevented tumor growth in WT mice but not in IL1R1−/− or IL1α−/− mice. Antibodies to IL1α promoted tumor growth in IL1β−/− mice and reversed the tumor-suppressive effect of anti-IL1β in WT mice. Depletion of CD8+ T cells and blockade of lymphocyte mobilization abrogated the IL1β−/− tumor suppressive effect, as did crossing IL1β−/− mice to SCID or Rag1−/− mice. Finally, blockade of IL1β synergized with blockade of PD-1 to inhibit tumor growth in WT mice. These results suggest that IL1β promotes tumor growth whereas IL1α inhibits tumor growth by enhancing T cell–mediated antitumor immunity.
INTRODUCTION
Both IL1α and IL1β mediate their activities through binding to a single receptor (1). Binding of either IL1α or β to the type I IL1 receptor (IL1R1) activates MyD88 and TRAF6, leading to NF-κB activation and MAP kinase 3 activation with attendant JNK, P38, and ERK activation (2,3). These activities are mediated via a “Toll Interleukin 1 Homology” (TIR) domain which is common to the intracellular domains of both IL1R1 and most TLRs (4). This relationship to Toll-like receptors has led to IL1α and IL1β being considered primarily innate immune cytokines (5), inducing a robust inflammatory response upon binding to IL1R1 on target cells (6–8). Regardless of the focus on innate immunity, IL1 activity was once described as “lymphocyte-” or “thymocyte-activating factor” for its role in costimulating thymocyte cell division (9). Indeed, early assays for IL1 activity involved T-cell proliferation (10,11). More recent studies have shown that IL1 is critical for Th17 cell maturation, is a growth and survival factor for naive T cells, and enhances antigen driven CD8 responses (12–14). Evidence for a role of IL1 in adaptive immune responses involving T cells is abundant, though frequently overlooked.
Although IL1α and IL1β signal through the same receptor, they differ (15). IL1β is secreted from cells as a 17kd molecule cleaved by caspase 1 from an inactive 31 kD precursor (pro- IL1β) through the inflammasome (16,17). In contrast, IL1α is predominantly cell associated (18), and although a 17kd active form of IL1α can be generated by calpain cleavage, its 31kD precursor is also active (19,20). IL1α can also be detected in a cell associated form on the cytoplasmic membrane, where it resides as a biologically active molecule (18). IL1β, but not IL1α, can be detected in circulation in a number of diseases (3,21,22). IL1β has been a therapeutic target: canukinumab (a therapeutic antibody to IL1β) is used to treat a number of inflammatory and rheumatologic disorders (23–25).
The roles of IL1α and IL1β in cancer biology have been studied as well (26,27). Innate immune inflammation is associated with tumor promoting activities (27,28). IL1β is thought to enhance tumor growth by inducing angiogenesis and block antitumor immunity in part by inducing myeloid derived suppressor cells (26,27,29). The role of IL1α in cancer is less clear, with some reports suggesting a pro-tumorigenic role (30) and other reports suggesting tumor inhibition (31). Overexpression of IL1α in fibrosarcoma lines appears to induce antitumor immunity, though the mechanism is thought to involve innate immune cells (32). On the other hand, overexpression of IL1α is a negative prognostic factor in many human cancers (33,34). Blockade of both IL1α and IL1β activity with antagonists or antibodies has been proposed as a general approach to therapy of cancer. (35,36)
To assess the relative roles of IL1α and IL1β in tumor growth, we chose mice deficient in these two molecules as well as mice deficient in the type I IL1R. This allowed us to assess functional deficiency in only IL1α (IL1α−/−), only IL1β (IL1β−/−), or both IL1s (IL1R1−/−). In addition, we used neutralizing antibodies to both IL1α and IL1β. Our results indicate that complete blockade of IL1 activity through deficiency of IL1R1 has no effect on tumor growth; this was true across different cellular lineages and in two different anatomic sites (skin and lung). However, deficiency of IL1β, but not IL1α, reproducibly led to inhibition of tumor growth. This tumor growth inhibition was dependent on an intact adaptive immune system as well as the presence of IL1α. IL1β−/− mice that rejected tumors retained durable antitumor immunity mediated by tumor-specific CD8+ TRM.
Our observations suggest that IL1α may play a larger role than previously considered in adaptive immune antitumor responses, and that altering the balance between IL1α and IL1β may have a role in therapeutic antitumor immunity. Antibodies to IL1α, IL1β, and a soluble form of the receptor antagonist (IL1Ra, which blocks all IL1 signaling at the IL1R1) are already in clinical trials, and our results suggest that globally inhibiting IL1 activity may not be an optimal approach.
MATERIALS AND METHODS
Mice, tumor cells and tumor induction
C57BL/6J mice, IL1R1−/− (Il1r1tm1Imx/J) mice, Rag1−/− (B6.129S7-Rag1tm1Mom/J) mice, B6 SCID (B6.CB17-Prkdcscid/SzJ) mice and Lang-DTR (B6.129S2-Cd207tm3(DTR/GFP)Mal/J) mice were obtained from The Jackson Laboratory. IL1α−/− and IL1β−/− mice were obtained from Dr. Yoichiro Iwakura (Tokyo University of Science, Japan). IL1β−/− mice were crossed with Rag1−/− mice, B6 SCID mice and Lang-DTR mice to generate IL1β−/−Rag1−/− mice, IL1β−/−SCID mice and IL1β−/−Lang DTR mice. Mice were bred in a biosafety level 1 (BL-1) facility at Harvard Medical School (HMS) and Brigham and Women’s Hospital (BWH). All mice were handled in accordance with guidelines set out by the Center for Animal Resources and Comparative Medicine (CCM) at HMS/BWH. All procedures and protocols were advised and approved by an Institutional Animal Care and Use Committee (IACUC) as well as the veterinary staff at the CCM.
EL-4 T-cell lymphoma cells were received in 2014 .B16-F10 melanoma cells were received in 2016. Lewis lung carcinoma cells (LLC) were received in 2011. YUMM1.7 melanoma cells were received in 2015. B16-F10LUC melanoma cells were received in 2017. These cell lines were not authenticated in the past year. Mycoplasma testing was performed in 2019 and was negative for all cell lines. Cell lines used in these experiments were cultured for 7–14 days. EL-4 cells were grown in complete RPMI 1640 medium. B16-F10, YUMM1.7 and Lewis lung carcinoma cells were grown in complete DMEM medium. Tumor cells were injected intradermally (i.d.) into the right flank and tumor development was monitored over time. Tumor volume was calculated as follows: (major circumference × minor circumference2) /2. Mice were euthanized when external necrosis was present or the tumor size reached 2 cm in any direction. For melanoma cell metastasis studies, 2–5 ×105 tumor cells were i.v. injected into the mice. Lung tumor foci were enumerated at different time points. For tumor imaging experiment: 10 days after B16-F10LUC i.v. injection, mice were imaged twice a week for 2–3 weeks with IVIS Lumina III imaging system (Perkin Elmer). 10 μL/g body weight of firefly Luciferin (15 mg/ml) was given to the mice by i.p. injection 10 min before imaging. 7 minutes after Luciferin administration, mice were placed in an induction chamber to be anesthetized with 3% isoflurane. After imaging, mice were allowed to recover in their home cages. Living image 4.2 software (Perkin Elmer Company) was used to optimize image display and analyze images.
In vivo cytokine blocking, T-cell depletion and FTY720 treatment
Mice were injected intraperitoneally (i.p.) with 100 to 400 μg of anti-mIL1α and anti-mIL1β (clones ALF-161 and B122, Biolegend) one day prior to tumor cell injection and then every third day until the end of the experiment. To deplete T cells, mice were injected i.p. with 250 μg of anti-CD4 (clone GK1.5) and (or) anti-CD8(clone 2.43) two days prior to tumor cell injection and then every third day until the end of the experiment. FTY 720 was used to prevent T-cell egress from LNs. Mice were injected i.p. with 1 mg/kg FTY 720 two days prior to tumor cell injection and then every two days until the end of the experiment.
In vivo clodronate treatment and In vivo depletion of LC and Lang+DC
WT and IL1β−/− mice were treated with 200 μL clodronate liposomes (5 mg/ml, Encapsula Nano Sciences, TN, USA) at four sites bordering the tumor implantation site by intradermal injection two days before and 3 times a week after tumor cell inoculation until the end of the experiment. PBS-containing liposomes were used as controls. IL1β−/−Lang DTR mice were treated with 1μg DT at the specified timing of administration to remove LC and Lang+DC.
Tissue and cell preparation, Flow cytometry, Real-Time qPCR
Primary EL-4 tumors and skin draining lymph nodes from the tumor injected site were obtained and digested with collagenase D (Roche, 1108886600, final concentration 400 units/ml) at 37°C for 20–30 min to get single cell suspensions. After cell staining, different subsets of cells were sorted with a FACS Aria III (BD Biosciences) or acquired on a FACS Canto (BD Biosciences) and analyzed using FlowJo software (Version 6.4.7, Tree Star, Ashland, OR). The following mAbs were purchased from Biolegend: FITC-conjugated anti-CD3e (clone 145–2C11/catalog# 100306), anti-MHCII (clone M5/114.15.2/catalog# 107606), anti-Ly6G (clone 1A8/catalog# 127605), PE-conjugated anti-CD11C (clone N418/catalog# 117308), anti-CD103 (clone 2E7/catalog# 121406), anti-CD62L (clone MEL-14/catalog# 104408), anti-CD69 (clone H1.2F3/catalog# 104508), anti-MHCII (clone M5/114.15.2/catalog# 107607), PerCP-conjugated anti-CD44(clone IM7/catalog# 103036), anti-CD11b (clone M1/70catalog# 101230), anti-CD3 (clone 145–2C11/catalog# 100325), anti-CD11c (clone N418/catalog# 117325), APC-conjugated anti-CD8 (clone 53–6.7/catalog# 100712). MHCII (clone M5/114.15.2/catalog# 107613), CD19 (clone 1D3/CD19/catalog# 152409), Ly6C (clone HK1.4/catalog# 128015), PE-Cy7 conjugated CD45 (clone 30F-11/catalog# 103113) and APC-Cy7 conjugated CD8 (clone 53–6.7/catalog# 100714).
For melanoma cell metastasis models, mice were euthanized and lungs were isolated and snap frozen in liquid nitrogen before being stored at −80°C. Total RNA was extracted using RNeasy Fibrous Tissue Mini Kit (Qiagen). For EL-4 i.d. injection models, primary tumors were harvested and total RNA was extracted with RNeasy Mini Kit (Qiagen). RNA Plus mini kit (Qiagen) was used to extract RNA from cultured 3T3, EL-4, B16, LLC cells and sorted cells from tumor and lymph nodes. RNA content in the samples was measured using a Nanodrop. RNA (0.5 μg per sample) was reverse transcribed into cDNA with iScript cDNA Synthesis Kit (Bio-Rad). Triplicate cDNA products were then mixed with Fast SYBR Green Mix and the primers specific for GP100, IL1α, IL1β, IL1R1 or GAPDH. GAPDH was used for normalization. These primers were used: GP100 forward: 5’GAGCTTCCTTCCCGTGCTT3’, GP100 reverse: TGCCTGTTCCAGGTTTTAGTTAC. IL1α forward: 5’CGAAGACTACAGTTCTGCCATT3’. IL1α reverse: 5’GACGTTTCAGAGGTTCTCAGAG3’. IL1β forward: 5’GCAACTGTTCCTGAACTCAACT3’, IL1β reverse: 5’ATCTTTTGGGGTCCGTCAACT3’. IL1R1 forward: 5’GTGCTACTGGGGCTCATTTGT3’, IL1R1 reverse: GGAGTAAGAGGACACTTGCGAAT. GAPDH forward: 5’AGGTCGGTGTGAACGGATTTG’, GAPDH reverse: TGTAGACCATGTAGTTGAGGTCA. The real-time PCR was performed with a StepOne Plus Real-time PCR System (Applied Biosystems, Foster City, CA). The thermal cycle profile was as follows: 95°C for 10 sec, 40X (95°C for 15 sec, 58°C for 30 sec). GP100 gene expression in the lungs from control WT mice was used as a baseline. For qRT-PCR analyses, transcript levels were normalized to GAPDH and represented as 2–ΔCT, where ΔCT is CT(target gene) – CT(GAPDH).
DC, T cells coculture, and cytokine ELISA
Skin draining lymph nodes were obtained from IL1α−/− mice and WT mice, after collagenase D digestion, single cell suspension was prepared and stained with DC markers. After cell sorting, DCs were pulsed with 1μg/ml SIINFEK peptide at 37°C for 2 hours, washed two times and cocultured with OT-1+ T cells with or without exogenous IL1α cytokine (R&D systems). Supernatants were collected 60h after incubation for IFNγ detection with commercial ELISA kits (Biolegend).
Bone marrow (BM) chimeras
IL1 deficient chimeric mice were generated with WT, IL1β−/− and IL1R1−/− mice by irradiating recipient mice at a sub-lethal dose and injecting 5–10 ×106 BM cells intravenously (i.v.) from age matched donor mice. The level of blood chimerism was determined by flow cytometry. 96–98% chimerism was achieved 8 weeks after reconstitution.
Statistics
Unpaired Student t test or repeated-measures two-way ANOVA followed by Bonferroni correction were performed with Prism software. P values < 0.05 were considered significant.
RESULTS
Tumor growth reduced in IL1β−/− mice across many tumor types and tissues
To examine the role of the IL1 family in tumor immunity, we implanted EL-4 lymphoma cells intradermally in C57BL/6 (WT), IL1R1−/− mice, IL1α−/− mice and IL1β−/− mice. Our initial results demonstrated that tumors grew rapidly and with similar kinetics in WT, IL1R1−/− and IL1α−/− mice. These mice had to be sacrificed between day 18–20 because of increasing tumor burden. However, EL-4 tumor growth was significantly reduced in IL1β−/− mice. Many IL1β−/− mice developed small tumors that subsequently disappeared, suggesting tumor rejection. More than 75% of IL1β−/− mice eventually cleared tumor and became long-term survivors (Fig. 1A). To ensure that this was not a phenomenon unique to EL4 lymphoma cells, similar experiments were done with malignant tumors derived from different cellular lineages: YUMM1.7 melanoma cells (derived from a genetically defined BRAF600EPten−/− melanoma), B16-F10 melanoma cells and Lewis Lung carcinoma (LLC) cells (Fig. 1B, 1C, 1D). In all cases, WT, IL1R1−/− and IL1α−/− mice developed large tumors with similar kinetics, whereas tumor growth in IL1β−/− mice was reduced. In many cases the tumors grew transiently and then regressed in IL1β−/− mice, suggesting active rejection.
Figure 1. IL1β−/− mice, but not IL1α−/− or IL1R1−/− mice demonstrated reduced tumor growth in skin and tumor metastasis in the lung.
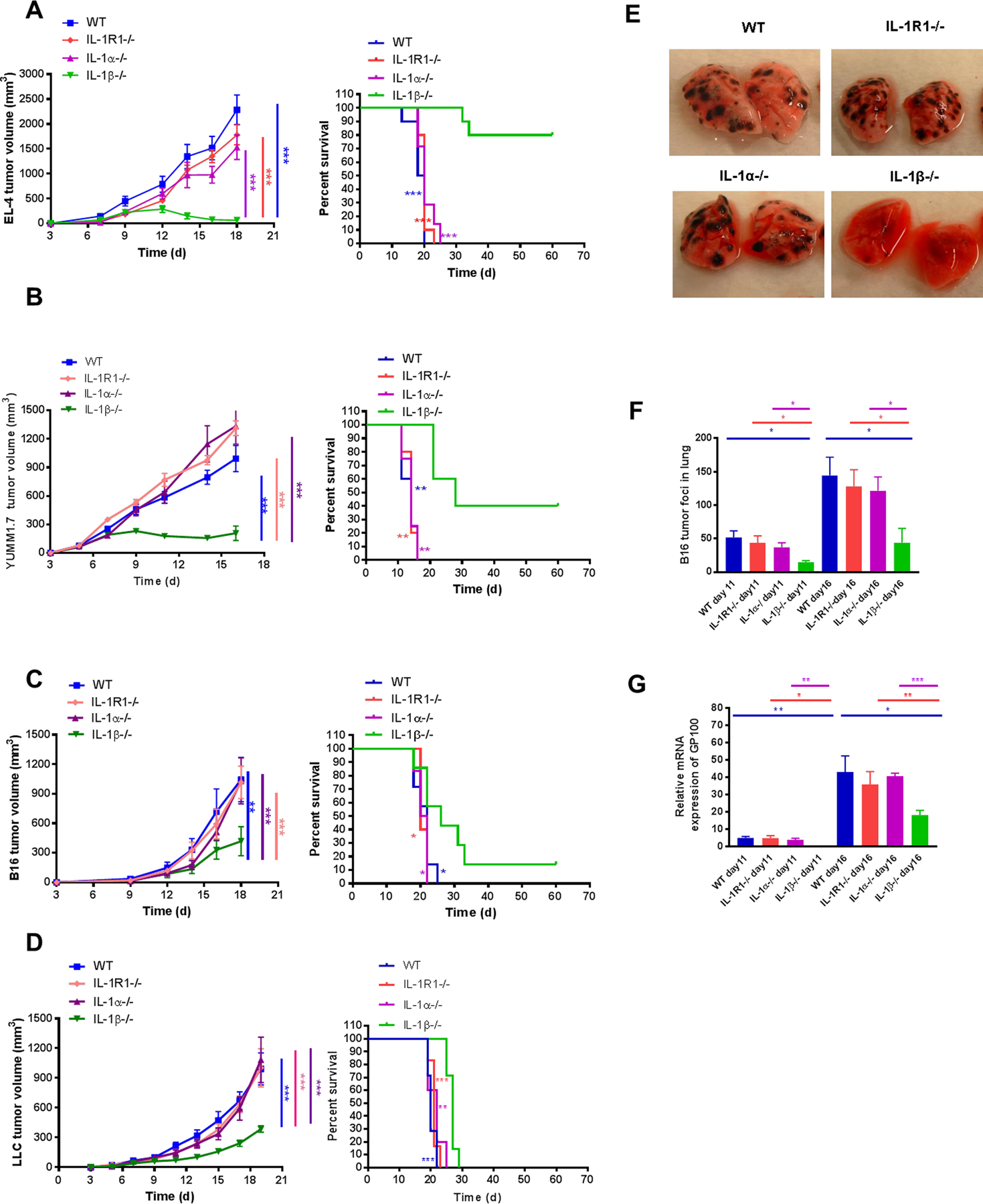
(A) EL4 lymphoma (2×105), (B) YUMM1.7 melanoma (3×106), (C) B16-F10 melanoma (3×105), and (D) LLC (2×105) cells were injected i.d. into WT and IL1-deficient mice. Tumor growth (left) and survival (right) were recorded every 2–3 days and statistics generated as compared to IL1β−/− mice (n=5–10 mice per group). B16F10 melanoma cells (5×105) were injected i.v. into WT and IL1-deficient mice. Tumor foci in the lungs were (E) photographed at day 16 and (F) counted at day 11 and 16 (n=4–5 mice per group). (G) Expression of GP100 in lungs of WT and IL1-deficient mice was assessed using qPCR. GP100 was quantified relative to the housekeeping gene GAPDH. Relative mRNA expression of GP100 was normalized to control WT mice set at 1. Graphs show mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. NS=not significant.
To determine whether reduced tumor growth in IL1β−/− mice was specific to skin, B16-F10 melanoma cells were injected intravenously into WT, IL1R1−/−, IL1α−/− and IL1β−/− mice to simulate metastatic tumor spread. At day 11 and 16, metastatic tumor development was assessed in the lungs. Visual inspection showed diminished tumor growth in the IL1β−/− lungs compared to WT, IL1R1−/−, and IL1α−/− lungs (Fig. 1E). The number of B16-F10 tumor foci was significantly decreased in the lungs of IL1β−/− mice (Fig. 1F). When lungs were assayed for melanoma-specific GP100 by RT-PCR, there was a significant reduction of GP100 mRNA in the lungs of IL1β−/− mice compared to WT, IL1R1−/− and IL1α−/− mice (Fig. 1G).
We further explored whether EL4 or B16-F10 cells produced IL1α or β in vitro. IL1α, IL1β, and IL1R1 mRNA was undetectable in these cell lines. Furthermore, EL-4 tumors growing in IL1α−/− mice did not express IL1α, tumors growing in IL1β−/− mice did not express IL1β and tumors growing in IL1R1−/− mice did not express IL1R1 (Supplementary Fig. S1).
IL1α inhibits tumor progression whereas IL1β promotes tumor growth.
IL1α and IL1β both signal through the IL1R1, yet only deficiency of IL1β inhibited tumor growth. This suggested a role for IL1α in contributing to the tumor suppressive phenotype seen in IL1β−/− mice. To test this possibility, WT mice were treated with neutralizing antibodies to IL1α or IL1β. Blocking antibody directed at IL1β inhibited EL-4 tumor growth reproducibly, whereas neutralizing antibody to IL1α had no effect, indicating that IL1β blockade could partially recapitulate the IL1β−/− phenotype. Combining neutralizing antibodies to IL1α and IL1β in WT mice abrogated the effect of anti-IL1β alone, suggesting a role for IL1α in promoting tumor inhibition (Fig. 2A). In contrast to WT mice, neutralizing antibody to IL1β did not reduce tumor growth in IL1R1−/− or in IL1α−/− mice, suggesting that IL1α:IL1R1 interactions are critical for tumor inhibition (Fig. 2B). The requirement for intact IL1α signaling was highlighted when anti-IL1α blocking antibody partially reversed the tumor suppression phenotype of IL1β−/− mice. More than 80% of IL1β−/− mice cleared EL-4 tumors and became long-term survivors, whereas all of the WT mice were sacrificed for progressive tumor growth by day 20. However, treatment of IL1β−/− mice with neutralizing antibody to IL1α restored tumor growth; only 30% of these IL1β−/− mice were able to permanently clear EL-4 tumors (Fig. 2C). These data suggested that the presence of both an intact IL1 signaling pathway (IL1R1) and IL1α were critical for the IL1β−/− tumor suppressive phenotype.
Figure 2. Effects of blocking IL1α and/or IL1β on tumor growth.
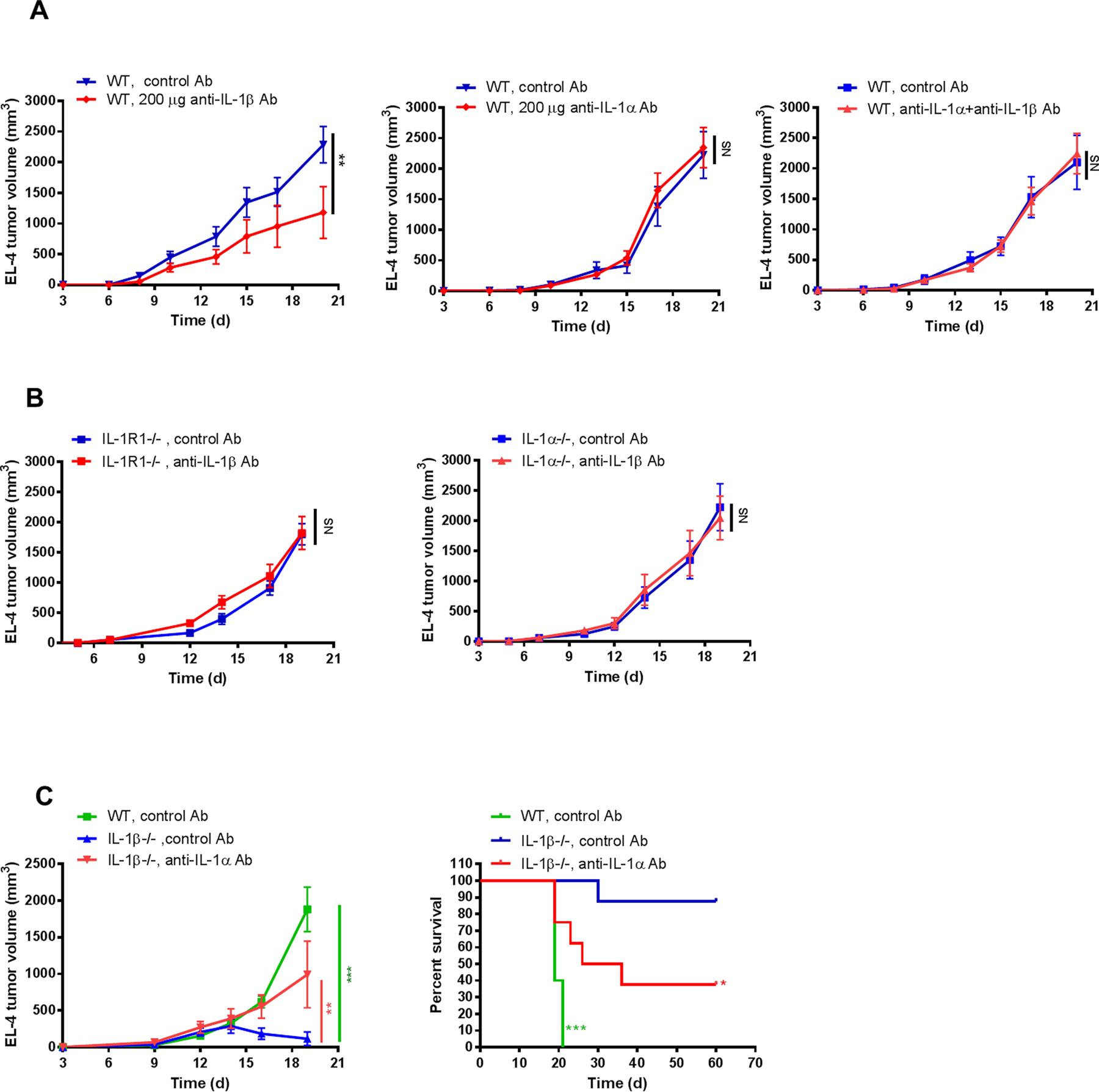
2×105 EL-4 cells were injected i.d. into mice. (A) WT mice were treated with anti-IL1β, anti-IL1α or both 2 days before and then every 2–3 days after tumor cell injection. Mice injected with control Ab were used as controls (n=5–6 mice per group). (B) IL1R1−/− mice and IL1α−/− mice were treated with anti-IL1β or control Ab 2 days before and then every 2–3 days after tumor cell injection (n=5–8 mice per group). (C) IL1β−/− mice were treated with anti-IL1α or control Ab. Tumor size and survival was recorded every 2–3 days and plotted (n=5–8 mice per group). Statistics were generated compared to IL1β−/− mice with control Ab. Graphs show mean ± SEM. *P<0.05, **P<0.01, ***P<0.001. NS=not significant.
The tumor suppressive phenotype in IL1β−/− mice is mediated by T cells.
The observations that tumors initially grew and then subsequently regressed in IL1β−/− mice led us to suspect a process mediated by adaptive immunity. To investigate further, we treated IL1β−/− and WT mice with either control antibodies or antibodies to both CD4 and CD8 to deplete T cells from the circulation prior to intradermal injection of EL-4 cells. As shown in Fig. 3A, tumors grew in WT mice, whether treated with control antibody or antibodies to CD4 and CD8. In IL1β−/− mice treated with the control antibody, tumors grew briefly and then regressed, consistent with previous observations. This phenotype was reversed in IL1β−/− mice treated with antibodies to CD4 and CD8: in such mice, tumors grew as rapidly as in WT mice without evidence of rejection. Similar results were obtained in mice implanted with B16-F10 melanoma cells (Supplementary Fig. S2).
Figure 3. EL-4 tumor growth and B16 melanoma metastasis were restored in IL1β−/− mice after T-cell depletion.
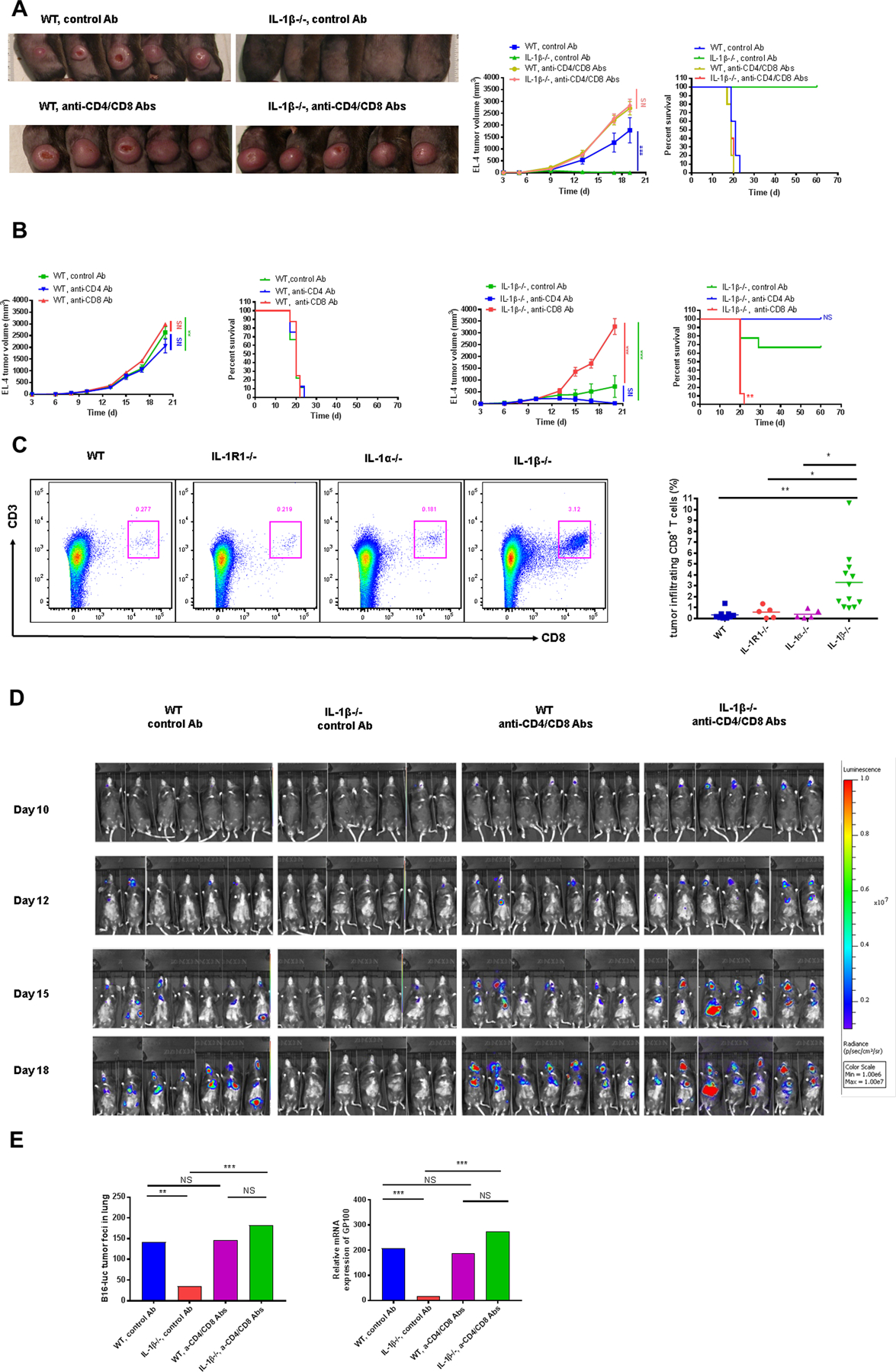
(A) 2×105 EL-4 cells were injected i.d. into WT and IL1β−/− mice treated with anti-CD4/CD8 or control Abs 2 days before and then every 2–3 days after tumor cell injection. Tumors were photographed at day 17 after EL-4 injection (left) and tumor volumes (middle) were plotted. Survival was also recorded (right) (n=5 mice per group). (B) 2×105 EL-4 cells were injected i.d. into IL1β−/− and WT mice treated with either anti-CD4, anti-CD8 or control Ab. Tumor growth and survival was recorded every 2–3 days and plotted. (n=8–9 mice per group). (C) 2×105 EL-4 cells were injected i.d. into WT and IL1 deficient mice. Representative dot plots were shown and percentage of tumor infiltrating CD8+CD3+ T cells were measured at day 14 after EL-4 cell injection (n=5–12 mice per group). (D) 5×105 B16-F10 cells expressing luciferase (B16-Luc) were injected i.v. into WT and IL1β−/− mice treated with either control Ab or antibodies to both CD4 and CD8 2 days before and then every 2–3 days after tumor cell injection. Bioluminescence imaging was performed at different time points (n=7 mice per group). (E) Tumor foci in the lungs were numerated and the expression of GP100 was measured at day 19 after B16-Luc i.v. injection (n=7 mice per group).
To further elucidate the roles of CD4+ and CD8+ T cells in this T cell-mediated tumor regression in IL1β−/− mice, we treated IL1β−/− and WT mice with either anti-CD4 or anti-CD8. Anti-CD4 –treated IL1β−/− mice could still clear EL-4 tumors and became long-term survivors, similar to IL1β−/− mice treated with control Ab. In contrast, EL-4 tumor growth was restored in anti-CD8–treated IL1β−/− mice (Fig. 3B). We also compared tumor infiltrating CD8+ T cells among WT and IL1 deficient mice. We found that tumors from IL1β−/− mice contained significantly more CD8+ T cells compared to tumors from WT, IL1α−/− and IL1R1−/− mice assessed at the same time point (Fig. 3C).
To explore this observation in the B16-F10 lung metastasis model, we treated IL1β−/− and WT mice with either control antibodies or antibodies to both CD4 and CD8 prior to i.v. injection of B16-F10luc cells. Diminished tumor growth by bioluminescence imaging was observed in IL1β−/− mice treated with control antibody at different time points. In contrast, after T-cell depletion, B16 melanoma tumor growth was restored in IL1β−/− mice (Fig. 3D). Lungs were harvested from these mice at day 18 for tumor foci numeration and GP100 analysis. Consistent with the imaging results, control IL1β−/− mice manifested fewer tumor foci and decreased expression of GP100 compared to control WT mice whereas T-cell depleted IL1β−/−mice demonstrated comparable numbers of tumor foci and more GP100 expression in the lungs compared to T cell–depleted WT mice (Fig. 3E).
Tumor resistance and antitumor memory require adaptive immunity in IL1β−/− mice
To further investigate the contribution of the adaptive immune system to tumor immunity in IL1β−/− mice, we bred these mice with immunodeficient Rag 1−/− and SCID backgrounds. After the requisite number of backcrosses, we subjected IL1β−/−Rag 1−/− and IL1β−/−SCID mice to intradermal EL-4 tumor implantation. Although IL1β−/− mice showed tumor resistance, both IL1β−/−Rag 1−/− and IL1β−/−SCID mice showed tumor growth indistinguishable from WT mice (Fig. 4A). Thus, in the absence of adaptive immune cells, the IL1β−/− tumor suppressive phenotype is abolished.
Figure 4. IL1β−/− mice acquire long-lasting antitumor immune memory after primary tumor clearance.
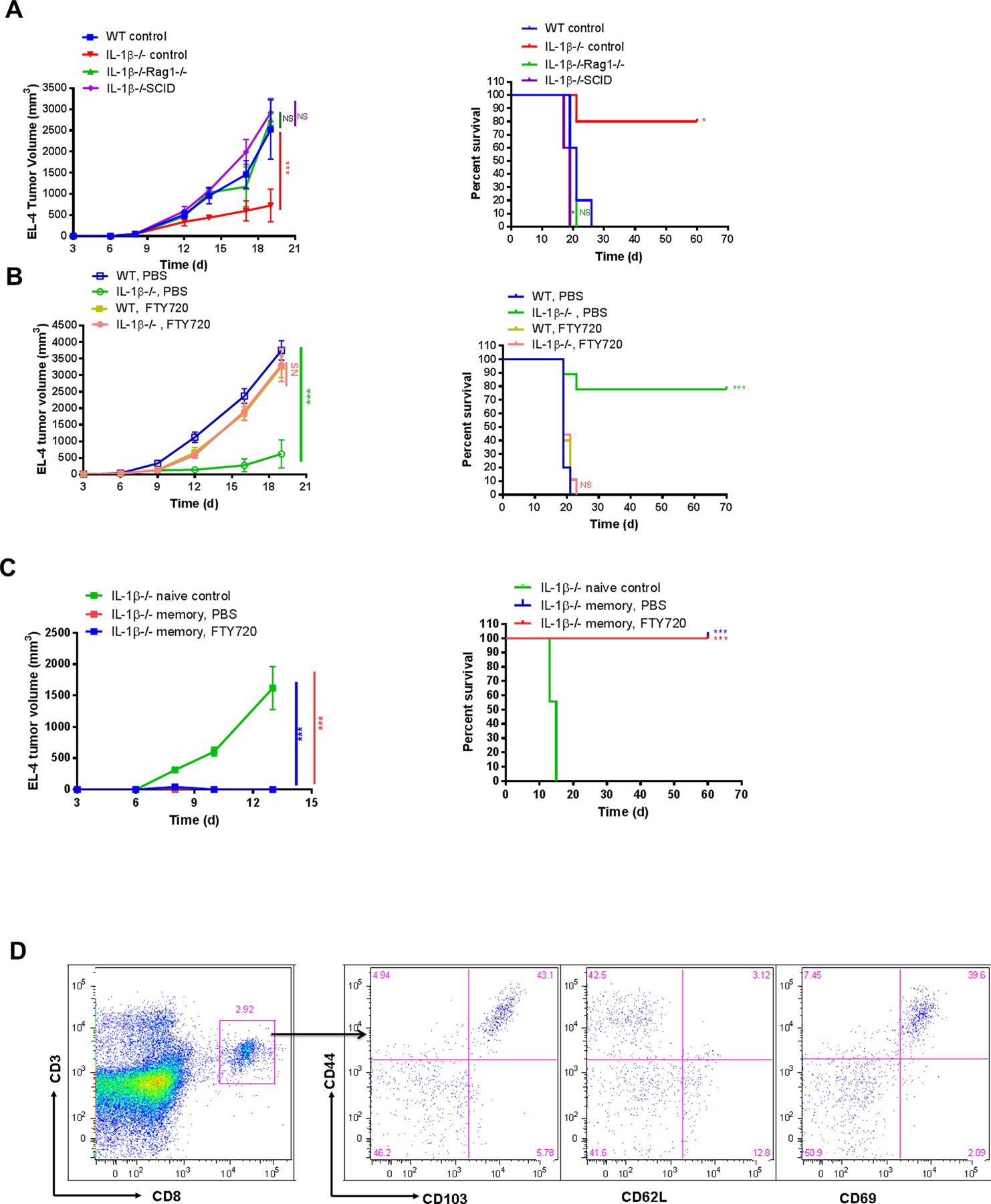
(A) 2×105 EL-4 cells were injected i.d. into WT, IL1β−/−, IL1β−/− Rag1−/− and IL1β−/− SCID mice. Tumor growth (left) and survival (right) was recorded every 2–3 days and plotted. Statistics were generated compared to WT control mice (n=5 mice per group). (B) 2×105 EL-4 cells were injected i.d. into WT and IL1β−/− mice treated with PBS or FTY720. Tumor growth (left) and survival (right) was recorded every 2–3 days and plotted. Statistics were generated by comparing WT PBS vs IL1β−/− PBS and then WT FTY720 vs IL1β−/− FTY720 (n=9–10 mice per group). (C) 5×106 EL-4 cells were injected i.d. into naive and memory IL1β−/− mice treated with or without FTY720. Tumor growth (left) and survival (right) was recorded every 2–3 days and plotted. Statistics were generated compared to IL1β−/− naive control mice (n=7–9 mice per group). (D) Distant skin was harvested from EL-4 tumor survivor memory IL1β−/− mice and phenotype of TRM was examined by flow cytometry.
A primary adaptive immune response to tumor involves recognition of tumor antigen in draining lymph nodes, followed by proliferation of tumor specific T cells and migration of effector T cells from lymph node to tumor-bearing tissue. Effector T-cell migration out of lymph nodes requires downregulation of sphinogosine-1-phosphate receptor activity and can be blocked by FTY720, an S1PR1 agonist that traps T cells within lymph nodes. Treatment of mice with FTY720 allowed for rapid tumor growth IL1β−/− mice, indicating that exit of T cells from the LN and their recruitment to tumors was required to mediate protective immunity in the IL1β−/− setting (Fig. 4B).
To investigate whether the IL1β−/− mice that previously cleared EL-4 tumor developed long-lasting antitumor immune memory, we injected EL4 cells (5×106) into these IL1β−/− tumor survivor “memory” mice. Age-matched naive IL1β−/− mice grew tumors with this EL4 cell inoculum. However, memory IL1β−/− mice that had previously rejected lower dose intradermal implantation of EL4 cells showed no tumor growth, suggesting durable immune memory. In contrast to naive IL1β−/− mice, FTY720-treated memory IL1β−/− mice did not develop tumors, indicating that mobilization of T cells from lymph nodes was not required, consistent with a protective role mediated by tissue resident memory T cells (TRM) (Fig. 4C). Naive IL1β−/− mice had few skin CD8+ T cells, but a distinct population of CD103+CD62L−CD69+ CD8+ T cells could be found in tumor survivor memory mice, in both tumor-exposed and distant skin, consistent with the presence of TRM cells (Fig. 4D). Finally, we asked whether myeloid derived suppressor cells could be identified in IL1β−/− mice. We found that both monocyte MDSC (M-MDSC) and polymorphonuclear MDSC (PMN-MDSC) were decreased in numbers in IL1β−/− mice in tumors, blood and lymphoid tissue (Supplementary Fig. S3).
Depletion of antigen presenting cells abrogates IL1β−/− tumor immunity
To determine the contribution of antigen presenting cells in the antitumor immune response observed in IL1β−/− mice, IL1β−/− and WT control mice were injected at the site of tumor implantation with either PBS or clodronate-encapsulated liposomes 2 days before EL-4 cell intradermal injection and then every 4 days until the end of the experiment. Intradermal injection of clodronate liposomes at the site of tumor implantation in WT mice had little effect. However, clodronate liposome injection in IL1β−/− mice reversed the tumor suppressive phenotype and tumors grew progressively. Control liposome injection had no effect on tumor growth or survival in either IL1β−/− or WT mice (Fig. 5A). The efficacy of the clodronate mediated depletion of antigen presenting cells was confirmed by significant reduction of migratory DCs (mDC, MHCIIhigh CD11c+ cells) and partial depletion of MHCII+CD11b+ cells in the skin draining lymph nodes of treated mice (Fig. 5B).
Figure 5. Depletion of antigen presenting cells reverses IL1β−/− mouse tumor immunity.
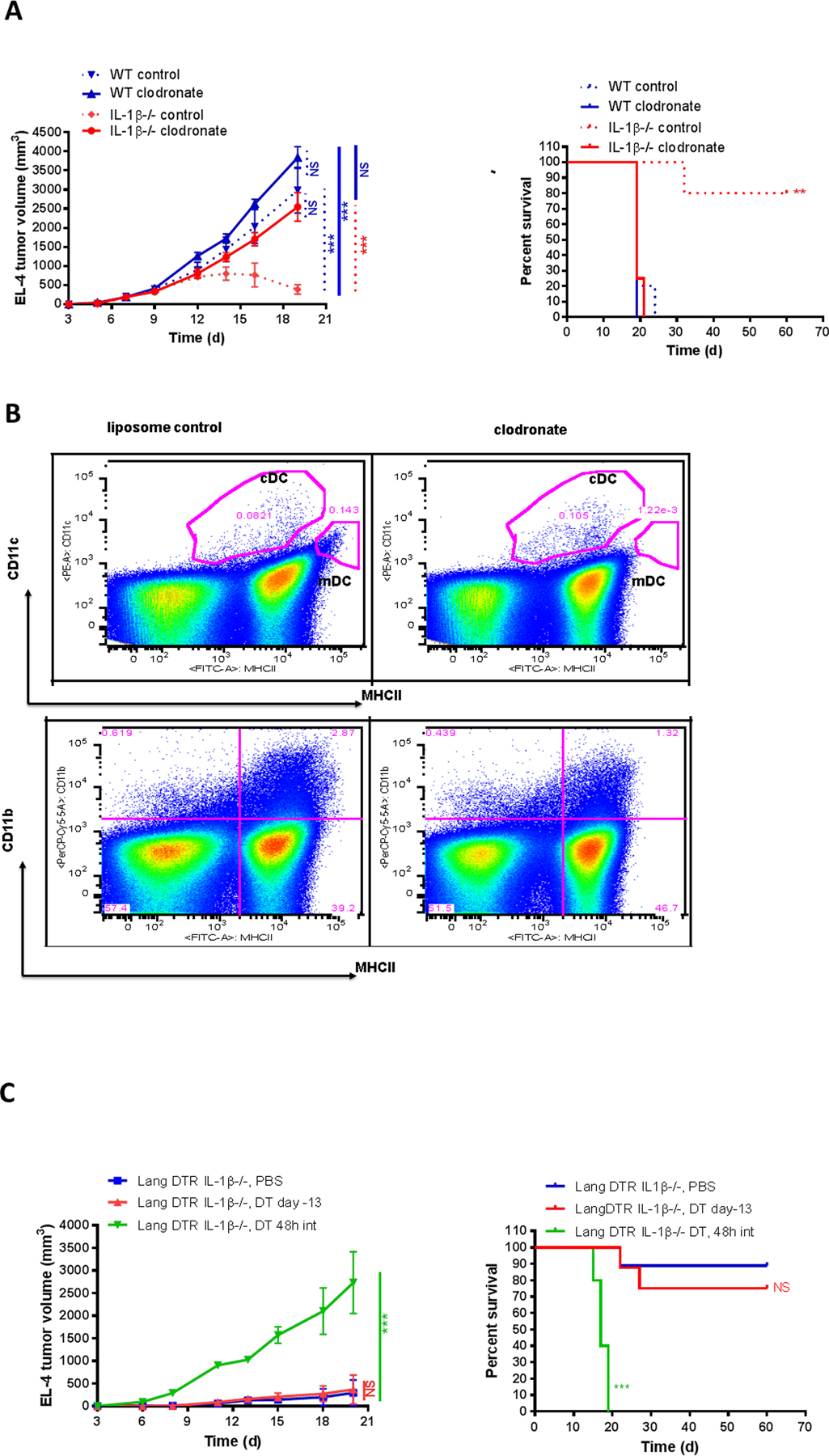
(A) 2×105 EL-4 cells were injected i.d. into WT and IL1β−/− mice treated with clodronate liposomes two days before and 3 times a week after tumor cell inoculation. PBS-containing liposomes were used as controls. EL-4 tumor growth (left) and survival (right) were recorded every 2–3 days (n=4–5 mice per group). (B) WT and IL1β−/− mice were treated with clodronate liposomes; PBS-containing liposomes were used as controls. Skin draining lymph nodes were harvested 3 days after clodronate treatment. Classical DC (cDC) and migratory DC (mDC) were examined by flow cytometry. Representative dot plots and gating strategy were shown. (C) 2×105 EL-4 cells were injected i.d. into Lang DTR IL1β−/−mice that were injected i.p. with 1μg DT at the specified time. EL-4 tumor growth (left) and survival (right) were recorded every 2–3 days (n=5–9 mice per group).
Because macrophages are also depleted by clodronate, we investigated the role of epidermal Langerhans cells and dermal CD207+ dendritic cells in the tumor suppressive response by crossing IL1β−/− mice with Lang-DTR transgenic mice expressing the diphtheria toxin receptor under the control of the murine CD207 promoter (37). Administration of a single dose of DT can deplete both LC and CD207+dDC within 48h. Epidermal LC repopulation is slow and does not reach completion for 8 weeks, whereas CD207+dDC repopulate dermis within a few days. Systemic DT injections 13 days before tumor implantation allow repopulation of CD207+dDC by the first week of tumor growth, while systemic DT injections every 48 hours throughout the time course of the experiment would deplete both LC and CD207+dDC. Most of the PBS-treated Lang DTR IL1β−/− mice cleared EL-4 tumor and became long-term survivors, as observed in the IL1β−/− mice. Similar results were found in Lang DTR IL1β−/− mice that received systemic treatment with DT 13 days before tumor injection (Lang DTR IL1β−/−, DT day −13). In contrast, all Lang DTR IL1β−/− mice treated with systemic DT 1 day before EL-4 tumor cell injection and then every 48 hours (Lang DTR IL1β−/− mice, DT 48h int) developed large tumors and had to be sacrificed at day 20 due to high tumor burden (Fig. 5C). Thus, repetitive DT treatment during the tumor growth phase restored tumor growth in Lang DTR IL1β−/− mice although a single dose prior to tumor implantation did not. These results suggest that CD207+dDC are required to cross-present tumor antigen to T cells in IL1β−/− mice and inhibit tumor growth. We next asked if antigen presenting cells (APCs) expressed IL1α and β. We determined that both MHCII+CD11c+ DCs and MHCII+CD11b+CD11c– myeloid APCs expressed IL1α, β, and IL1R1. Moreover, IL1α−/− DC were less effective at stimulating CD8+T cells to produce IFNγ, whereas exogenous IL1α enhanced IFNγ production (Supplementary Fig. S4).
Expression of IL1R1 on bone marrow derived cells is required for tumor inhibition.
We next subjected mice to lethal irradiation and reconstitution with bone marrow to prepare chimeric mice. When WT mice were irradiated and reconstituted with IL1β−/− bone marrow, tumor growth was rapid, even more rapid than when WT bone marrow was used for reconstitution. In contrast, when IL1β−/− mice were irradiated and reconstituted with either IL1β−/− or WT bone marrow, significant tumor suppression was observed, recapitulating the IL1β−/− tumor suppressive phenotype (Fig. 6, left). However, this protection against tumor growth was abrogated if bone marrow from IL1R−/− mice was transferred to IL1β−/− mice (Fig. 6, right), suggesting that IL1R1 expression on effector T cells was required for the tumor suppressive effect.
Figure 6. EL-4 tumor growth in IL1 deficient chimeric mice.
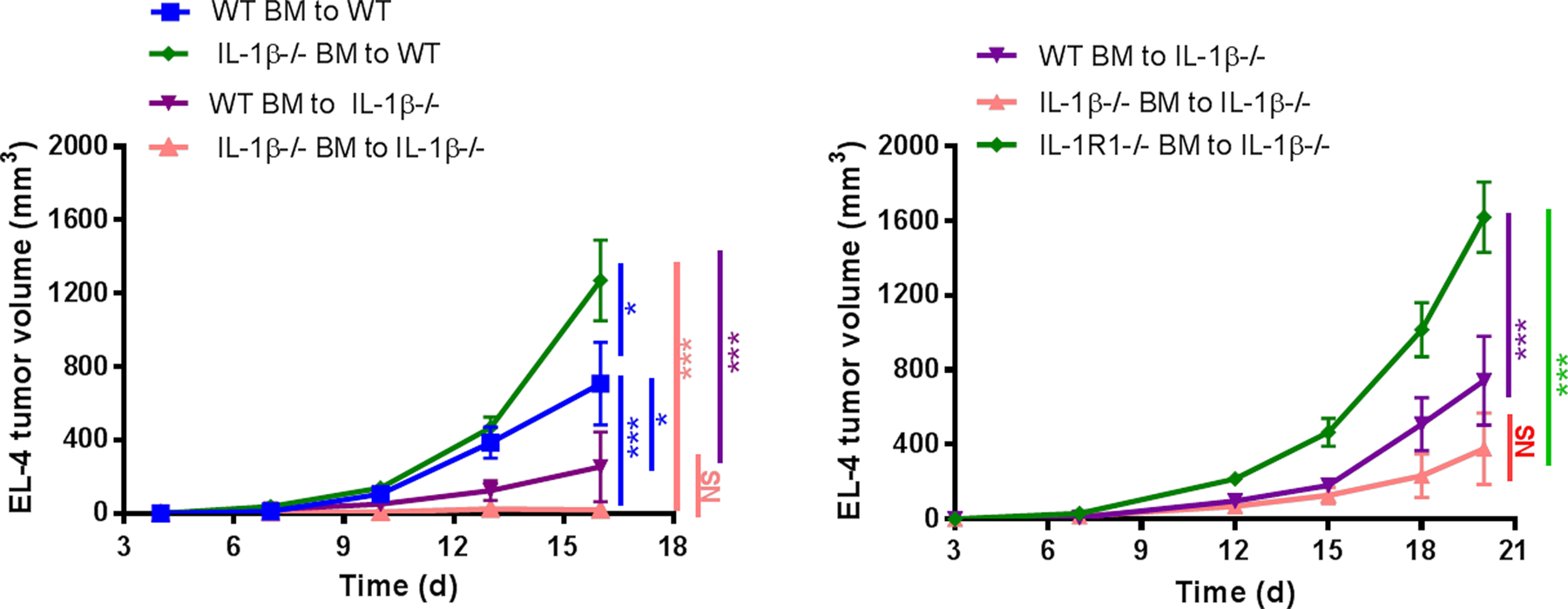
IL1 deficient chimeric mice were generated using WT, IL1R1−/− and IL1β−/− mice. 2×105 EL-4 cells were injected i.d. into IL1 deficient chimeric mice. Tumor growth was recorded 2 to 3 times per week and plotted (n=7–9 mice per group). Graphs show mean ± SEM. *P<0.05, **P<0.01, ***P<0.001. NS=not significant.
IL1β blockade recapitulates IL1β−/− tumor resistance independently of PD-1 blockade
Our previous data (Fig. 2A) indicates that administration of blocking antibodies specific for IL1β can suppress EL-4 T-cell lymphoma tumor growth in WT mice and partially mimic the antitumor phenotype seen in IL1β−/− mice. These findings prompted us to test the effect of anti-IL1β on tumor growth in lung using our model. B16-F10LUC melanoma cells were injected intravenously in WT mice and tumor growth was compared using bioluminescent imaging. Anti-IL1β significantly slowed growth of melanoma cell tumors in lung, as compared to isotype control (Fig. 7A). Lungs were harvested from control Ab–treated and anti-IL1β –treated mice at day 10 and day 19 for tumor foci numeration. Consistent with the imaging results, anti-IL1β –treated mice manifested fewer tumor foci compared with control Ab–treated mice (Fig. 7B).
Figure 7. Anti-IL1β alone or in combination with anti-PD-1 inhibits tumor growth and metastasis.
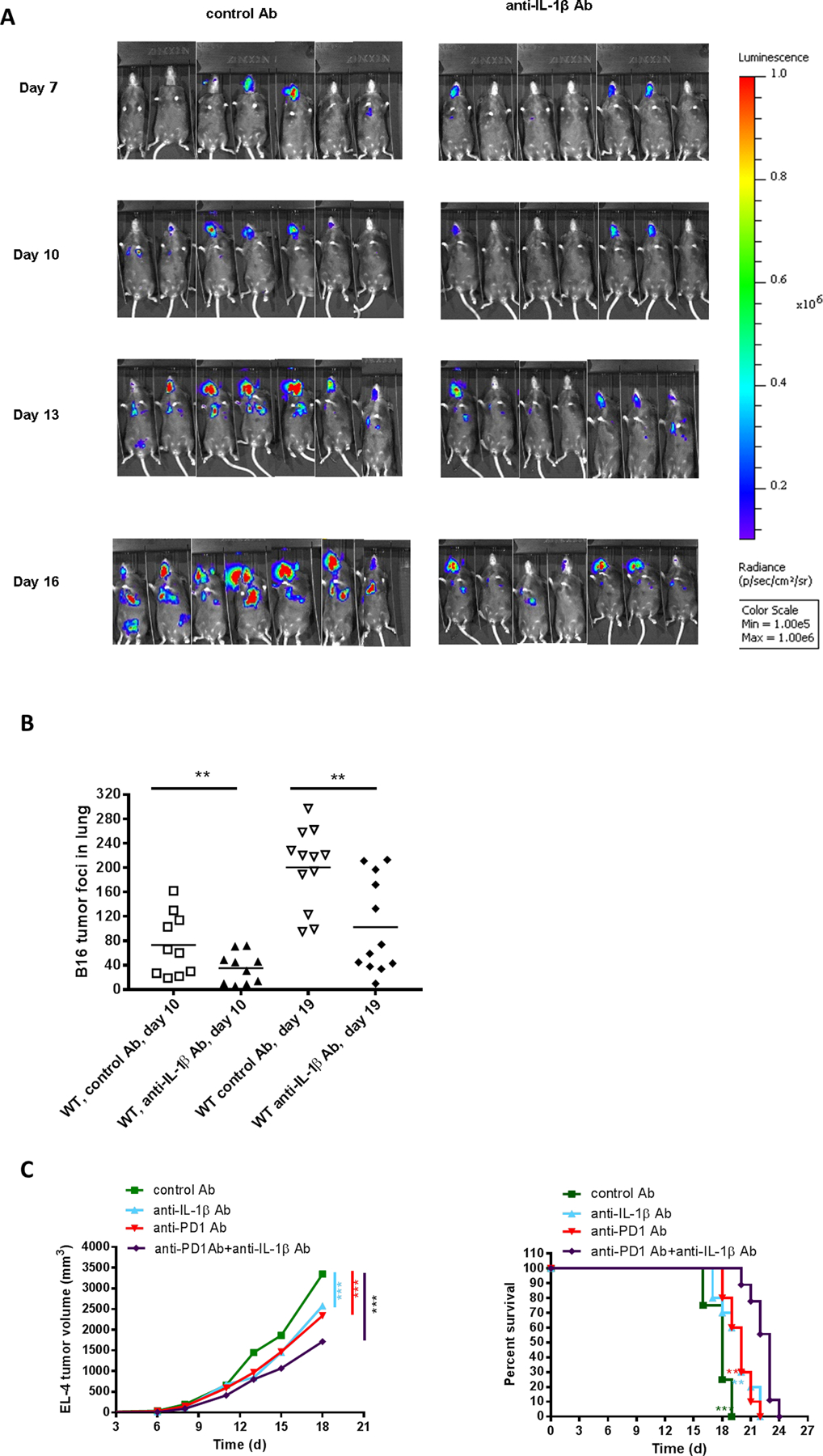
(A) 5×105 B16-Luc cells were injected i.v. into WT mice treated with control Ab or anti-IL1β. Bioluminescence imaging was performed at different time points (n=7 mice per group). (B) 5×105 B16-F10 cells were injected i.v. into WT mice treated with control Ab or anti-IL1β. Tumor foci in the lungs were numerated at day 10 and day 19 after tumor cell injection (n=10–12 mice per group). (C) 2×105 EL-4 cells were injected i.d. into WT mice treated with anti-IL1β, anti-PD-1 or both before and after tumor cell injection. Tumor growth (left) and survival (right) were recorded every 2–3 days (n=8–10 mice per group). Graphs show mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, NS=not significant.
Since our experiments showed that the tumor resistance of IL1β−/− mice is dependent on CD8+ T cell–mediated tumor rejection, we hypothesized that anti-IL1β might synergize with immune checkpoint inhibitor antibodies directed at PD-1. In mice treated with anti-IL1β combined with anti-PD-1, T lymphoma tumors in the skin of WT mice grew at a slower rate and to a smaller final volume than with either antibody treatment alone (Fig. 7C). In conclusion, neutralizing antibodies to IL1β, alone or in combination with anti-PD-1, show promise for cancer immunotherapy.
DISCUSSION
IL1α and IL1β arose from a gene duplication event less than a million years ago and have only 25% homology at the amino acid level (1,38,39). Although IL1β is found in all vertebrate species studied, IL1α is only found in mammals (39). Both ligands, however, signal exclusively through the type I IL1 receptor (IL1R1) (1). Although IL1α and IL1β do bind equally to IL1R1, there is evidence that the soluble “decoy” IL1R2 binds preferentially to IL1β (40). The relative effect of this phenomenon on tumor growth in IL1β−/− and IL1α−/− mice was not explored in this study but may be worth investigating. We have demonstrated that IL1α and IL1β have distinct roles in tumor biology. Deficiency in IL1β, whether caused by genetics or by antibody blockade, leads to inhibition of tumor growth and often immune-mediated tumor rejection. However, complete blockade of IL1 signaling in IL1R−/− mice abrogates this tumor suppressive effect, suggesting a possible antitumor role for IL1α. This activity of IL1α was further demonstrated by the use of IL1α neutralizing antibodies, which blocked the immune mediated rejection of tumors in IL1β−/− mice. In addition, the slower tumor growth in WT mice induced by anti-IL1β is reversed by the simultaneous administration of anti-IL1α. Further indirect evidence for an immunostimulatory role of IL1α comes from the lack of a tumor suppressive phenotype when anti-IL1β was given to either IL1α−/− or IL1R1−/− mice.
The evidence for adaptive immune mediated tumor protection in IL1β−/− mice is compelling. First, tumors grew initially in most animals, then subsequently diminished in size and became undetectable. These resolving tumors contained abundant CD8+ T cells, in contrast to tumors from WT, IL1α−/−, and IL1R1−/− mice. Second, depletion of CD8+ T cells from tumor bearing mice reversed the suppressive phenotype conferred by the IL1β−/− background. Depletion of CD4+ T cells had little effect. Third, crossing IL1β−/− mice to immunodeficient mice, SCID and Rag1−/−, also abrogated the tumor suppressive phenotype. Fourth, blocking the migration of effector T cells from lymph node to peripheral tissue with FTY720 also blocked tumor immunity in IL1β−/− mice. Fifth, depletion of CD207+ dermal dendritic cells in IL1β−/− mice abrogated the tumor suppressive effect, suggesting that these cross-presenting DC interacted with CD8+ T cells to mediate tumor immunity. Finally, the >70% of IL1β−/− mice that reject tumors develop durable immunity and are resistant to subsequent implantation of high numbers of tumor cells. This immunity could not be abrogated by FTY720, indicating that it resided within skin. When skin was examined for the presence of tissue resident memory T cells (TRM), CD8+ TRM that also expressed CD69 and CD103 could be identified.
We demonstrated the IL1β−/− antitumor immune effect in mice bearing tumors of multiple cellular lineages: EL4 lymphoma cells, B16-F10 and Yumm1.7 melanoma cells, and Lewis lung carcinoma cells. The effect was reproducible whether the cells were injected intradermally or intravenously. Thus, tumor growth in both the skin and the lung was reduced in IL1β−/− mice. These data suggest that this observation is not an artifact of a single tumor model system but is more broadly applicable. Our bone marrow chimera experiments demonstrate that radioresistant cells from the IL1β−/− host are critical for the antitumor immune effect. The effect was strongest when IL1β−/− bone marrow was transferred, but was still evident when WT bone marrow was transferred. However, the effect was abrogated when IL1R1−/− bone marrow was transferred, suggesting that that the putative immune effector cell requires intact IL1 signaling (Fig. 6). We speculate that T cells activated by IL1α (e.g., on the cell surface of antigen presenting cells) are responsible for the observed antitumor immunity.
Our study suggests that inhibiting IL1 signaling indiscriminately (e.g., with anakinra or rilanocept) may not be an optimal approach in cancer immunotherapy, as the beneficial effects of IL1α signaling on antitumor immunity would be lost. In contrast, blockade of IL1β appears to induce antitumor immunity, provided IL1α and IL1R1 are present. Whether it is blockade of angiogenesis or inhibition of the generation of MDSC’s (or both) that are responsible for the tumor promoting effects of IL1β is unclear. Other variables in the tumor microenvironment may also be at play. We also showed an additive, possibly synergistic, effect of IL1β antibody blockade and PD-1 blockade. Three therapeutic antibodies against PD-1 and one against IL1β (canakinumab) are FDA approved. The clinical trial that used canakinumab to prevent complications of atherosclerotic disease had the unexpected effect of reducing the incidence of lung cancer in recipients. This circumstantial evidence is consistent with our results and suggests that clinical trials aimed at blocking IL1β activity in cancer are warranted.
Supplementary Material
Acknowledgments & Author Contributions
T.T., T.S.K. searched the literature and designed the study, performed the data analysis and interpretation, and wrote the manuscript. T.T., S.L., Y.P., C.A.S., J.W.M., K.L., T.P., J.Z. performed the animal experiments and in vitro experiments. R.L., K.C. assisted with tumor imaging. S.K. assisted with reviewing and editing the manuscript. H.W. generated the B16-F10LUC cells for the experiments. R.F. assisted designing the experiments. This work was funded in part by grants from the National Institutes of Health (R01 AI127654 to TSK).
Footnotes
Competing Interests
The authors declare no competing interests.
Publisher's Disclaimer: Disclaimers
Publisher's Disclaimer: The authors do not have any disclaimers.
REFERENCES
- 1.Dower SK, Qwarnstrom EE, Page RC, Blanton RA, Kupper TS, Raines E, et al. Biology of the interleukin-1 receptor. J Invest Dermatol 1990;94(6 Suppl):68S–73S. [DOI] [PubMed] [Google Scholar]
- 2.Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol 2010;10(2):89–102 doi 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Dinarello CA, Molgora M, Garlanda C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019;50(4):778–95 doi 10.1016/j.immuni.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boraschi D, Italiani P, Weil S, Martin MU. The family of the interleukin-1 receptors. Immunol Rev 2018;281(1):197–232 doi 10.1111/imr.12606. [DOI] [PubMed] [Google Scholar]
- 5.Moorlag S, Roring RJ, Joosten LAB, Netea MG. The role of the interleukin-1 family in trained immunity. Immunol Rev 2018;281(1):28–39 doi 10.1111/imr.12617. [DOI] [PubMed] [Google Scholar]
- 6.Keyel PA. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine 2014;69(1):136–45 doi 10.1016/j.cyto.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Rubartelli A Autoinflammatory diseases. Immunol Lett 2014;161(2):226–30 doi 10.1016/j.imlet.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Schett G, Dayer JM, Manger B. Interleukin-1 function and role in rheumatic disease. Nat Rev Rheumatol 2016;12(1):14–24 doi 10.1038/nrrheum.2016.166. [DOI] [PubMed] [Google Scholar]
- 9.Dinarello CA. Studies on the biological properties of purified and recombinant human interleukin-1. Methods Find Exp Clin Pharmacol 1986;8(2):57–61. [PubMed] [Google Scholar]
- 10.Kupper T, Horowitz M, Lee F, Robb R, Flood PM. Autocrine growth of T cells independent of interleukin 2: identification of interleukin 4 (IL 4, BSF-1) as an autocrine growth factor for a cloned antigen-specific helper T cell. J Immunol 1987;138(12):4280–7. [PubMed] [Google Scholar]
- 11.Koide S, Steinman RM. Induction of murine interleukin 1: stimuli and responsive primary cells. Proc Natl Acad Sci U S A 1987;84(11):3802–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Sasson SZ, Caucheteux S, Crank M, Hu-Li J, Paul WE. IL-1 acts on T cells to enhance the magnitude of in vivo immune responses. Cytokine 2011;56(1):122–5 doi 10.1016/j.cyto.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8+ T cells. J Exp Med 2013;210(3):491–502 doi 10.1084/jem.20122006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeda S, Saijo S, Murayama MA, Shimizu K, Akitsu A, Iwakura Y. Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGF-beta-induced Foxp3 expression. J Immunol 2014;192(4):1449–58 doi 10.4049/jimmunol.1300387. [DOI] [PubMed] [Google Scholar]
- 15.Krumm B, Xiang Y, Deng J. Structural biology of the IL-1 superfamily: key cytokines in the regulation of immune and inflammatory responses. Protein Sci 2014;23(5):526–38 doi 10.1002/pro.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu A, Wu H. Structural mechanisms of inflammasome assembly. FEBS J 2015;282(3):435–44 doi 10.1111/febs.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eder C Mechanisms of interleukin-1beta release. Immunobiology 2009;214(7):543–53 doi 10.1016/j.imbio.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Kurt-Jones EA, Beller DI, Mizel SB, Unanue ER. Identification of a membrane-associated interleukin 1 in macrophages. Proc Natl Acad Sci U S A 1985;82(4):1204–8 doi 10.1073/pnas.82.4.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carruth LM, Demczuk S, Mizel SB. Involvement of a calpain-like protease in the processing of the murine interleukin 1 alpha precursor. J Biol Chem 1991;266(19):12162–7. [PubMed] [Google Scholar]
- 20.Lomedico PT, Gubler U, Hellmann CP, Dukovich M, Giri JG, Pan YC, et al. Cloning and expression of murine interleukin-1 cDNA in Escherichia coli. Nature 1984;312(5993):458–62. [DOI] [PubMed] [Google Scholar]
- 21.Simon AD, Yazdani S, Wang W, Schwartz A, Rabbani LE. Circulating levels of IL-1beta, a prothrombotic cytokine, are elevated in unstable angina versus stable angina. J Thromb Thrombolysis 2000;9(3):217–22. [DOI] [PubMed] [Google Scholar]
- 22.Pountain G, Hazleman B, Cawston TE. Circulating levels of IL-1beta, IL-6 and soluble IL-2 receptor in polymyalgia rheumatica and giant cell arteritis and rheumatoid arthritis. Br J Rheumatol 1998;37(7):797–8. [DOI] [PubMed] [Google Scholar]
- 23.Torene R, Nirmala N, Obici L, Cattalini M, Tormey V, Caorsi R, et al. Canakinumab reverses overexpression of inflammatory response genes in tumour necrosis factor receptor-associated periodic syndrome. Ann Rheum Dis 2017;76(1):303–9 doi 10.1136/annrheumdis-2016-209335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N Engl J Med 2018;378(20):1908–19 doi 10.1056/NEJMoa1706314. [DOI] [PubMed] [Google Scholar]
- 25.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017;377(12):1119–31 doi 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 26.Voronov E, Carmi Y, Apte RN. The role IL-1 in tumor-mediated angiogenesis. Front Physiol 2014;5:114 doi 10.3389/fphys.2014.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mantovani A, Barajon I, Garlanda C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol Rev 2018;281(1):57–61 doi 10.1111/imr.12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamura K, Smyth MJ. Targeting cancer-related inflammation in the era of immunotherapy. Immunol Cell Biol 2017;95(4):325–32 doi 10.1038/icb.2016.126. [DOI] [PubMed] [Google Scholar]
- 29.Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, Baron R, et al. The role of IL-1beta in the early tumor cell-induced angiogenic response. J Immunol 2013;190(7):3500–9 doi 10.4049/jimmunol.1202769. [DOI] [PubMed] [Google Scholar]
- 30.Kumar S, Kishimoto H, Chua HL, Badve S, Miller KD, Bigsby RM, et al. Interleukin-1 alpha promotes tumor growth and cachexia in MCF-7 xenograft model of breast cancer. Am J Pathol 2003;163(6):2531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voronov E, Dinarello CA, Apte RN. Interleukin-1alpha as an intracellular alarmin in cancer biology. Semin Immunol 2018;38:3–14 doi 10.1016/j.smim.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Voronov E, Dotan S, Krelin Y, Song X, Elkabets M, Carmi Y, et al. Unique Versus Redundant Functions of IL-1alpha and IL-1beta in the Tumor Microenvironment. Front Immunol 2013;4:177 doi 10.3389/fimmu.2013.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xuan Y, Wang YN. Hypoxia/IL-1alpha axis promotes gastric cancer progression and drug resistance. J Dig Dis 2017;18(9):511–20 doi 10.1111/1751-2980.12496. [DOI] [PubMed] [Google Scholar]
- 34.Sorrentino R, Terlizzi M, Di Crescenzo VG, Popolo A, Pecoraro M, Perillo G, et al. Human lung cancer-derived immunosuppressive plasmacytoid dendritic cells release IL-1alpha in an AIM2 inflammasome-dependent manner. Am J Pathol 2015;185(11):3115–24 doi 10.1016/j.ajpath.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev 2010;29(2):317–29 doi 10.1007/s10555-010-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dinarello CA. An expanding role for interleukin-1 blockade from gout to cancer. Mol Med 2014;20 Suppl 1:S43–58 doi 10.2119/molmed.2014.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett CL, van Rijn E, Jung S, Inaba K, Steinman RM, Kapsenberg ML, et al. Inducible ablation of mouse Langerhans cells diminishes but fails to abrogate contact hypersensitivity. J Cell Biol 2005;169(4):569–76 doi 10.1083/jcb.200501071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Copeland NG, Silan CM, Kingsley DM, Jenkins NA, Cannizzaro LA, Croce CM, et al. Chromosomal location of murine and human IL-1 receptor genes. Genomics 1991;9(1):44–50. [DOI] [PubMed] [Google Scholar]
- 39.Rivers-Auty J, Daniels MJD, Colliver I, Robertson DL, Brough D. Redefining the ancestral origins of the interleukin-1 superfamily. Nat Commun 2018;9(1):1156 doi 10.1038/s41467-018-03362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burger D, Chicheportiche R, Giri JG, Dayer JM. The inhibitory activity of human interleukin-1 receptor antagonist is enhanced by type II interleukin-1 soluble receptor and hindered by type I interleukin-1 soluble receptor. J Clin Invest 1995;96(1):38–41 doi 10.1172/JCI118045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.