

Abstract
Surface states of mesoporous NiO semiconductor films have particular properties differing from the bulk and are able to dramatically influence the interfacial electron transfer and adsorption of chemical species. To achieve a better performance of NiO-based p-type dye-sensitized solar cells (p-DSCs), the function of the surface states has to be understood. In this paper, we applied a modified atomic layer deposition procedure that is able to passivate 72% of the surface states on NiO by depositing a monolayer of Al2O3. This provides us with representative control samples to study the functions of the surface states on NiO films. A main conclusion is that surface states, rather than the bulk, are mainly responsible for the conductivity in mesoporous NiO films. Furthermore, surface states significantly affect dye regeneration (with I–/I3– as redox couple) and hole transport in NiO-based p-DSCs. A new dye regeneration mechanism is proposed in which electrons are transferred from reduced dye molecules to intra-bandgap states, and then to I3– species. The intra-bandgap states here act as catalysts to assist I3– reduction. A more complete mechanism is suggested to understand the particular hole transport behavior in p-DSCs, in which the hole transport time is independent of light intensity. This is ascribed to the percolation hole hopping on the surface states. When the concentration of surface states was significantly reduced, the light-independent charge transport behavior in pristine NiO-based p-DSCs transformed into having an exponential dependence on light intensity, similar to that observed in TiO2-based n-type DSCs. These conclusions on the function of surface states provide new insight into the electronic properties of mesoporous NiO films.
Introduction
NiO crystalline mesoporous films have drawn widespread attention in the past decades,1,2 partly due to NiO being a metal-deficient p-type semiconductor, having valuable electronic,3−5 magnetic,6,7 and catalytic properties.8−10 Another important aspect is that nanocrystalline NiO films combine NiO’s properties into a mesoporous structure which provides a surface area several orders of magnitude larger, compared to the same geometric area in a planar structure.11 This is directly beneficial for electrocatalysis12 or photocatalysis13,14 because of the increased amount of active sites. Another well-known example is p-type dye-sensitized solar cells (p-DSCs), where the large surface area increases dye loading by thousands of times.1 Meanwhile, mesoporous NiO has unique electronic properties, distinguishing it from non-porous bulk NiO. The mesoporous NiO films are generally composed of 10–20-nm-size nanoparticles.11 The size of the particles should be much smaller than the space charge region between non-porous bulk NiO and the electrolyte. This suggests that significant amounts of holes can be stored or transported in the mesoporous NiO films without inducing macroscopic electric fields.15 In addition, electronic properties on NiO surfaces are significantly different from those in the crystalline bulk. Nanoscale NiO particles, allowing for lattice deformations, will significantly increase the occurrence of structural defects. The atomic position disorder will induce energy disorder and then lead to an altered band structure and distribution of the density of states (DOS).16 The high surface area-to-volume ratio even accents the influence of the surface atoms on the electronic properties of mesoporous NiO films. Thus, the electronic properties and functions of surface sites on mesoporous NiO films need to be understood.
The electrochemical17 and electrochromic18 properties of mesoporous NiO films have been studied in some detail. Generally, two pairs of redox peaks are observed by cyclic voltammetry (CV), which are assigned to the two respective transformations between Ni2+ and Ni3+, as well as Ni3+ and Ni4+, on the surface of the NiO films. The surface species seem to be significantly affected by cations adsorbed on the surface. In aqueous solution, the peaks shift by −61 mV per pH unit, suggesting the involvement of protons in the reaction.19 Later researchers suggested that intra-bandgap states of Ni3+ and Ni4+ 20 are trap centers, causing significant charge recombination in p-DSCs.21−23 Very recently, Mayer and co-workers provided a clearer understanding of a proton-coupled electron-transfer (PCET) reaction on a NiO surface.24 However, there is still debate about the true chemical nature of the surface species. Cahoon suggested that the first redox peak should not be Ni2+ to Ni3+ states, but related to oxygen-localized defect states. The redox reaction is not related to the PCET reaction since the oxygen-localized defect states can have similar pH dependence.25,26 Some researchers have also shown indirect evidence that Ni+ is present on NiO surfaces.27 However, there is still a lack of solid evidence for the presence of either Ni+ or Ni4+ on mesoporous NiO films.
In this paper, we define those surface species involved in the two pairs of redox reactions as surface states. A modified atomic layer deposition (ALD) procedure was designed in order to passivate the surface states on NiO by an Al2O3 monolayer. Eventually, we had a NiO film as prepared with rich surface states (named as NiO) and a passivated NiO with less surface states (named as NiO-A). The conductivity on mesoporous NiO films was studied under ambient conditions. The conductivity and the conductivity mechanism on NiO mesoporous films are highly important and determine how charges are transported through the NiO matrix to the external circuit in liquid p-DSCs. We experimentally prove that surface states, not the bulk, dominate the conductivity of NiO mesoporous films. To our knowledge, this has not been proven in previous studies.
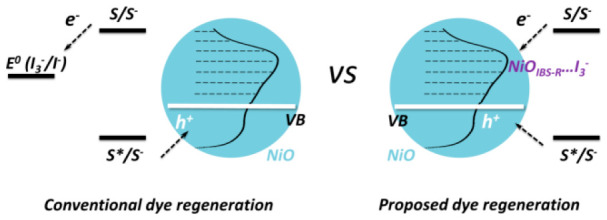
Surface states (with energies located states in the bandgap) on NiO were reported to be the recombination center in liquid p-DSCs.22 In this study, we found that electron injection into intra-bandgap states indeed can decrease the halftime of reduced dye. However, electron injection into intra-bandgap states should not be simply treated as charge recombination, causing detrimental effect in solar-to-electricity conversion efficiency. In NiO-based p-DSCs, electron injection from reduced dye into intra-bandgap states of NiO actually is the step of dye regeneration; namely, an electron will inject into intra-bandgap states of NiO first and then transfer to redox couple in electrolyte. We instead propose a dye regeneration mechanism, in which I3– reduction is mediated by intra-bandgap states. The new proposed dye regeneration diagram is significantly distinguished from the conventional one in which electron transfer is processed from reduced dye into redox couple (e.g., I3– in I–/I3– redox couple). In addition, a particular charge transport has been constantly observed in p-DSCs in which charge transport time is independent of light intensity. Such a light-independent behavior has not been clearly explained yet. Clear evidence in the study herein shows that charge transport is influenced by the density of the surface states. When the surface states on NiO are significantly reduced (from 4 per nm2 to 1 per nm2), the light-independent charge transport can transform into having an exponential dependence on light intensity.
Experimental Section
Mesoporous NiO Films
Mesoporous NiO films were prepared from a reported method.11,21 Briefly, a Ni sol–gel was deposited on a clean fluorine-doped tin oxide (FTO) glass by the doctor-blading method and annealed at 450 °C for 30 min. The whole procedure was repeated twice to obtain a target thickness of ca. 1.3 μm. Details of Ni sol–gel preparation are described in Supporting Information (SI).
Preparation of NiO-A Mesoporous Films by a Modified ALD Procedure
The ALD procedure was modified after considering the particular structure of a mesoporous film. The precursors for ALD of Al2O3 were trimethylaluminum (TMA) and deionized water (H2O) which were stored at 18 °C without additional heating. The temperature in the ALD deposition chamber was set at 70 °C which was stabilized for 60 min before ALD. The pressure inside the deposition chamber was ca. 300 Pa. The pulse time was 0.1 s and purge time of N2 was 8 s for TMA (0.1 s:8 s for TMA:N2 purge). The pulse time was 0.1 s and purge time of N2 was 20 s for H2O (0.1 s:20 s for H2O:N2 purge). The pulse:purge cycle of TMA was solely run 200 cycles before pulsing H2O. Then, the pulse:purge cycle of H2O was run 200 cycles to react with the saturated adsorbed TMA inside the mesoporous NiO films. The conventional ALD procedure is, TMA pulsing:N2 purge:H2O pulsing:N2 purge (called one cycle); the modified ALD procedure is, (TMA pulsing:N2 purge) × 200 times:(H2O pulsing:N2 purge) × 200 times (called one cycle). When the deposition was finished, the sample was taken out from the deposition chamber immediately, and cooled down under the ambient temperature.
Electrochemical Measurements
Cyclic voltammetry (CV) was performed with a standard three-electrode electrochemical setup inside a glovebox (H2O < 0.1 ppm; O2 < 0.1 ppm). The reference electrode was Ag/AgNO3 soaked in 1.0 mM AgNO3 of acetonitrile (ACN) solution. Platinum (Pt) was used as the counter electrode. 100 mM tetrabutylammonium hexafluorophosphate (TBAPF6) in dry ACN was used as one of the electrolytes. The 5 mM I–/I3– (5 mM I2 and 10 mM LiI) redox couple and 100 mM TBAPF6 in dry ACN were used as the other electrolyte. In this paper, all potentials were referred to Ag/AgNO3 reference electrode (soaked in 1.0 mM AgNO3 of acetonitrile (ACN) solution) unless otherwise stated. For CV characterization of NiO and NiO-A, samples had ca. 80 nm sputtered NiO blocking layer on FTO glasses before doctor-blading mesoporous NiO films, in case of the influence of FTO on the CV characterization.
Conductivity Measurements
Mesoporous NiO films (thickness d = 1.3 μm)
were prepared on a glass with multiple
stripes of FTO on it (Scheme S1). The FTO
layers were rectangles with a width (W) of 5 mm and
a length (L) of 10 mm. Four of the FTO layers were
chosen as the contacting electrodes for the four-probe resistance
measurement (so the contact effect can be ignored). The outer two
electrodes (#1 and #4) were used to force a current, I, while the potentials of the inner two electrodes (#2 and #3), U2
and U3, were monitored at the same time. The resistance of one FTO
stripe can be determined by R = (U2 – U3)/I. The conductance can be described by G = 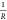 =
= 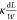 , from which κ (conductivity) can
be calculated. The details can be found in the SI.
, from which κ (conductivity) can
be calculated. The details can be found in the SI.
p-Type Dye-Sensitized Solar Cell (p-DSC) Fabrication
NiO and NiO-A films (5 mm × 5 mm active area) were used to fabricate p-DSCs. The films were sensitized in 0.2 mM PB6 dye34 in ACN:propanol (1:1 in volume) solution overnight. The films were sealed together with a platinized FTO counter electrode by a 25 μm hot-melt film (Surlyn, Solaronix, 6 mm × 6 mm). The electrolyte with 0.05 M I2 and 0.1 M LiI in ACN was filled through a hole pre-drilled in the back of the counter electrode, and the hole was sealed eventually by a glass and Surlyn film.
J–V Measurements
A Newport solar simulator, model 91160, equipped with a Keithley 2400 source meter, was used for J–V measurements. The solar simulator of model 91160A was used with one sun light intensity of 100 mW·cm–2, AM 1.5 G, calibrated by a reference silicon solar cell (Fraunhofer-Institut für Solare Energiesysteme ISE). The photoactive area was about 0.25 cm2 for the solar cell measurement.
Transient Photocurrent Measurements
In transient photocurrent measurements, the light intensity was calibrated by a reference silicon solar cell (Fraunhofer-Institut für Solare Energiesysteme ISE). The target light intensity was achieved by adjusting the applied voltage of the LED light (white light, Luxeon Star 1 W). Light intensity of 15, 36, 64, 100, 140, and 186 mW·cm–2 was obtained with applied voltage at 2.7, 2.8, 2.9, 3.0, 3.1, and 3.2 V, respectively. The transient photocurrent measurement was performed by using of a Dyenamo tool-box DN-AE01 system. A fixed light intensity (15, 36, 64, 100, 140, and 186 mW·cm–2) was applied first, and then a small square-wave light intensity modulation was added upon the fixed light intensity. The corresponding transient current was recorded by a 16-bit resolution data acquisition board (DAQ National Instruments) under the short-circuit condition. The transport time was extracted by fitting the decay of the transient photocurrent.
Femtosecond Transient Absorption Spectroscopy (fs-TAS) Measurements
The detailed experimental procedure for the fs-transient absorption measurements has been described elsewhere.28 Briefly, 800 nm laser pulses from a mode-locked Ti:sapphire laser with temporal widths of roughly 100 fs were amplified by a regenerative amplifier operating at 3 kHz, split, and used to generate pump and probe pulses. Pump pulses were generated by optical parametric amplification. White probe light was generated by focusing the pulse onto a moving CaF2 crystal. The delay between pump and probe pulses was generated by leading the probe pulse through a mechanical delay stage. A chopper, operating at 1.5 kHz, was used to block half of the incoming pump pulses. The difference in intensity of the probe light after the sample with and without pump at wavelengths from roughly 350 to 700 nm was measured with a diode array, and changes in absorbance were calculated. A pump pulse of 300 nJ at wavelength 560 nm was used to excite the PB6 dye molecules. All experiments were done in air at room temperature.
Results and Discussions
A uniform monolayer adsorption of the ALD precursor in the mesoporous films is a crucial factor for achieving the desired configuration. For mesoporous NiO films (1.3 μm thickness), the inner surface area is ca. 100 times higher than its geometric area; this requires a precursor that diffuses windingly inside the mesoporous film, forcing more collisions and longer diffusion time to reach a uniform monolayer adsorption. Thus, achieving a monolayer adsorption of the ALD precursor in mesoporous films should be more difficult than that in planar films. For the work described in this paper, we prepared Al2O3-passivated mesoporous NiO films by one cycle of the modified ALD procedure in order to obtain a monolayer deposition of Al2O3 on the NiO inner surface.
After ALD of Al2O3, NiO films were visibly bleached from a dark brown into a lighter brown color (Figure S1). As is shown in the transmittance spectra (Figure S2), NiO-A has ca. 10% more transmittance compared to NiO films from 550 to 800 nm. Both NiO and NiO-A films show identical X-ray diffraction patterns, corresponding to a typical mesoporous NiO structure29 (Figure S3). Thus, color transformation does not originate from the bulk, but from the surface of NiO particles.
The observed color bleach is consistent with previously published studies25,30 which commonly ascribe it to the change of electronic states localized at the surface of NiO. Here, the surface was characterized by CV, where two clear broad redox peaks were observed, as shown in Figure 1. These redox reactions should occur at the surface of NiO nanoparticles, since the peak current density depends linearly on the scanning rate19 (as reported previously in ref (19) and shown in Figure S8). In contrast to the non-passivated NiO films, the CV of the NiO-A films is significantly altered. The peak around 0.5 V is diminished, and the peak around −0.5 V is not observed. This can be attributed to the loss of surface states or to the surface states being physically covered by Al2O3. Note that only one ALD cycle is performed, indicating a monolayer or sub-monolayer of Al2O3 being present on the NiO films. Such an Al2O3 layer is expected to be virtually transparent to electrons. Additionally, the redox peaks (ca. 0.5 V and ca. −0.5 V in NiO) are asymmetrically diminished after ALD of Al2O3, which cannot be explained by the surface states being physically covered by Al2O3. Since both of the redox reactions are one-electron-transfer processes, the diminished area (proportional to charge) of the peaks should be relatively similar.19 Therefore, the significant change in the CVs after the ALD procedure is ascribed to removal of the surface species active in these reactions.
Figure 1.
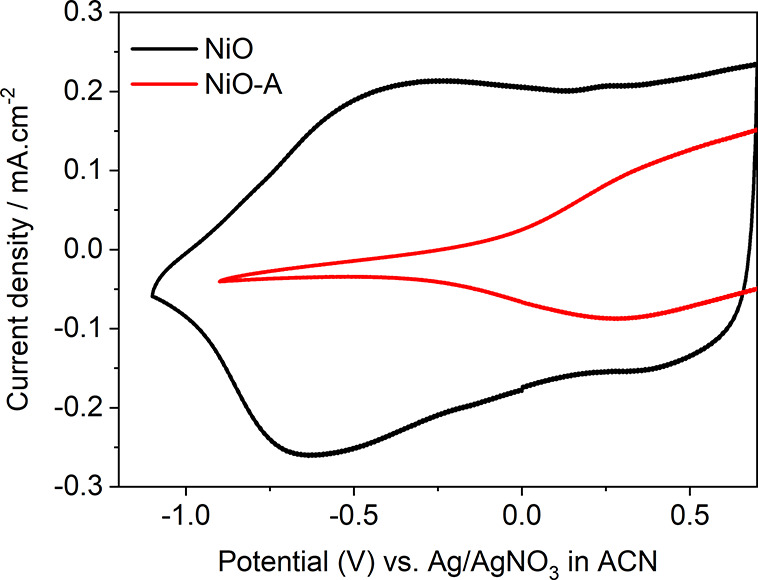
CV of NiO and NiO-A films collected in 100 mM TBAPF6 in ACN solution at a scan rate of 100 mV·s–1. The vertex in the CV of NiO-A is from cutoff of the CV after 0.5 V, and the full CV can be found in Figure S15.
The density of states (DOS) in the pristine mesoporous NiO films prepared by the well-known sol–gel method has been characterized.23 In this study, the DOS as a function of potential was derived from the cathodic scanning curve of CV (see SI for the detailed calculations).25,31 The DOS obtained is similar to that reported in a previous study, where the DOS of surface states was extracted from the impedance spectra in an identically prepared NiO films.23 Therefore, we conclude that the capacitance of NiO films is mainly caused by chemical capacitance from redox reactions of the surface states, while the influence of the double-layer capacitance on the measured capacitance can be ignored.
In pristine NiO sample, DOS is about 2.9 × 1020 cm–3, integrated from 0.6 to −0.9 V, and the DOS is ca. 8.1 × 1019 cm–3 in NiO-A from the identical potential range. 72% of the surface states were removed in NiO-A with insertion of Al2O3. The potential of valence band maximum (VBM) in crystalline NiO is labeled at ca. 0.02 V in Figure 2. Above the VB, there is supposed to be a wide bandgap range (3.5 eV) until the conduction band minimum (3.52 V). We found a significant amount of surface states above the VB in NiO, with the DOS of ca. 1.95 × 1020 cm–3 integrated from 0.02 to −0.9 V. We named those surface states in the bandgap of NiO as the intra-bandgap states. The DOS value is consistent with other reported values of mesoporous NiO films.23,25 However, the DOS decreased to ca. 3.68 × 1019 cm–3 in same potential range (0.02 to −0.9 V) in NiO-A films. This means that 80% of those intra-bandgap states have been removed after ALD of Al2O3. Similar passivation in mesoporous films has been reported, indicating that ALD of Al2O3 is able to passivate the surface states in NiO25 or SnO233 with a single ALD cycle. In addition, the VBM of NiO-A shows a positive shift of 0.23 V compared to that in NiO, measured by X-ray photoelectric spectroscopy (XPS). The quasi-Fermi level in NiO-A shows a similar positive shift of 0.27 V, confirmed by charge extraction under open-circuit conditions (this measurement was done in an intact p-DSCs based on NiO and NiO-A; more information will be given below). Both of those positive shifts result from the loss of the intra-bandgap states (Figures S4 and S5).
Figure 2.
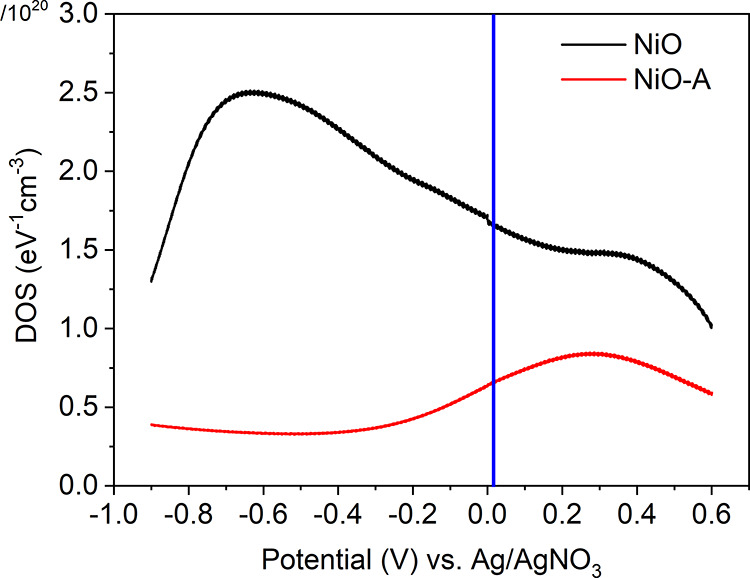
DOS of NiO and NiO-A films at different potentials, calculated from the cathodic CV in Figure 1. Details can be found in the SI. The valence band position is labeled with a blue line based on refs (17 and 32).
The DC dark conductivity of both films was measured under ambient conditions (temperature of ca. 21 °C, humidity of ca. 42%) with four-probe resistance measurements (see details in the SI). The conductivity measurements were performed on an identical NiO film before/after ALD Al2O3. The whole measurement avoids any direct connection with the top surface of the NiO and NiO-A films. The resultant conductivity values of NiO and NiO-A films are (1.7 ± 0.25) × 10–4 and (2.4 ± 0.27) × 10–6 Ω–1 cm–1, respectively, as shown in Figure 3. This corresponds to a decrease in conductivity of the NiO-A film by a factor of 70 after ALD of Al2O3, indicating that the conductivity of the as-prepared NiO films is mainly from the surface states, not the bulk. The dominant conductivity by surface states suggests that hopping conductivity should be the conductivity mechanism in the mesoporous NiO films. As a control experiment, we also prepared 140 nm non-porous NiO films by sputtering, and an identical ALD of Al2O3 was performed. The conductivity of the NiO films before/after deposition of Al2O3 does not change (see SI, p S15). This also supports the unique electronic properties of surface states on mesoporous NiO films.
Figure 3.

Effect of current density
on applied potential in a dark DC conductivity
measurement of NiO (black squares) and NiO-A (red circles) films.
Conductivities are extracted by linear fits to the function 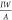 =
= 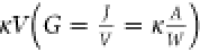 , where I is current, A is the
surface area of the cross section (L ×
d), W is the distance current passes, and
κ is the conductivity.
, where I is current, A is the
surface area of the cross section (L ×
d), W is the distance current passes, and
κ is the conductivity.
The influence of dye adsorption on the conductivity of NiO and NiO-A was studied as well. NiO-PB6 and NiO-A-PB6 films were obtained after sensitization with the previously reported PB6 dye.34 The conductivity of NiO decreases by a factor of 4, being (4.1 ± 0.46) × 10–5 Ω–1cm–1 in NiO-PB6. A conductivity loss of only a factor of 2 is observed on NiO-A (1.0 ± 0.11) × 10–6 Ω–1cm–1) after the dye sensitization. The dye molecules themselves may act as conductors on the films, and therefore an increase in conductivity may be expected. The observed loss in conductivity suggests that sensitization decreases the density of surface states. The chemical nature of these surface states may be transformed by covalent band formation. The effect being smaller for the passivated NiO-A-PB6 films can be explained by their lower density of surface states, leading to a lower probability for occupation of surface states.
Intra-bandgap states are often assigned as the main reason for the significant charge recombination and poor energy conversion efficiency in p-DSCs.21,22,35 Here, 80% of these intra-bandgap states are removed through surface passivation (Figure 2), making the NiO-A film a seemingly more “ideal” semiconductor. This encouraged us to investigate the effect of removal of NiO surface states on the performance of p-DSCs. The two types of films, NiO and NiO-A, were therefore employed to fabricate p-DSCs with PB6 dye as the photosensitizer and I–/I3– as the redox couple. J–V curves of the solar cells are shown in Figure 4. The NiO-A-based device shows twice the open-circuit voltage (Voc = 0.20 ± 0.009 V) as that in NiO-based p-DSCs (0.105 V). The improved Voc is ascribed to suppression of charge recombination and the fact that the quasi-Fermi level of NiO is positively shifted after the removal of surface states from the bandgap. Nevertheless, the short-circuit current (Jsc) of NiO-A-based solar cells is surprisingly low, 0.022 ± 0.002 mA·cm–2. This is 2 orders of magnitude lower than that of the NiO-based solar cells, 3.32 ± 0.14 mA·cm–2 (Figure 4 and Table S2).
Figure 4.
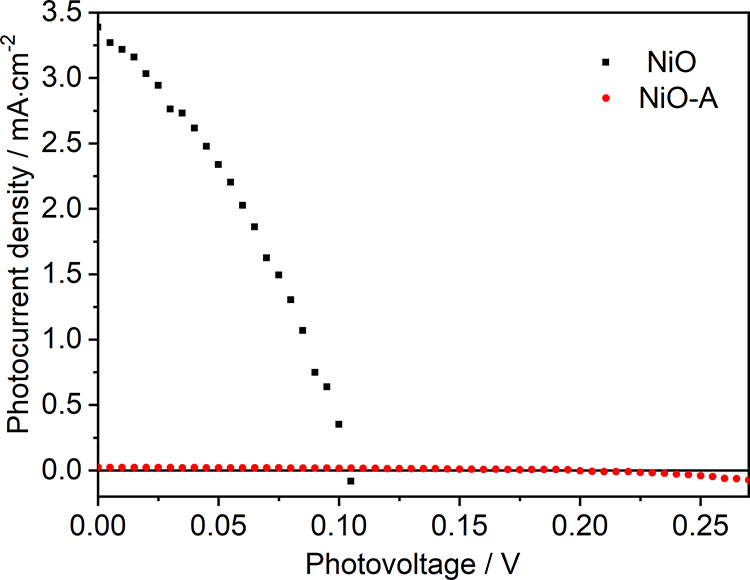
J–V curves of NiO- and NiO-A-based p-DSCs under one sun illumination. The electrolyte is 0.05 M I2 and 0.1 M LiI in ACN.
In addition, photoelectrochemical properties of NiO-PB6 and NiO-A-PB6 photocathodes were characterized with linear sweep voltammetry under dark, light, and chopped light conditions (see Figure S7). Dark current and photocurrent are clearly distinguishable for each of photocathodes, indicating that the light absorbed by the PB6 dye makes a contribution to the photocurrent. The photocurrent density in NiO-PB6 photocathode is ca. 150 times larger than that in the NiO-A-PB6 photocathodes, similar to the trend of Jsc in solar cells mentioned above.
To understand the low photocurrent observed, CV measurements were performed with NiO and NiO-A as working electrodes and in I–/I3– redox electrolytes (see Figure 5). Platinum, glassy carbon, FTO, and TiO2 were also chosen as working electrodes to perform control experiments. With Pt, the CV shows two pairs of redox peaks corresponding to the oxidation of 3I– – 2e– → I3– at −0.02 V (Ox1, the peak potential) and the reduction of I3– + 2e–→ 3I– at −0.35 V (Re1), and the oxidation of 2I3– – 2e– → 3I2, 0.42 V (Ox2) and the reduction of 3I2 + 2e– → 2I3– at 0.28 V (Re2) (Figure S9). The results are consistent with those reported in the literature.36,37 [The redox reaction of I–/I3– is rather complex since many one-electron transfer processes could potentially happen.38] The Re2 peak is missing on the glassy carbon electrode, and both reduction peaks (Re1 and Re2) essentially disappear with the FTO as the working electrode. This indicates that both glassy carbon and FTO are not suitable catalysts for these reactions (see Figures S10 and S11). Interestingly, two redox peaks are clearly observed with NiO as the working electrode, giving Ox1 at 0.20 V, Re1 at −0.55 V, Ox2 at 0.70 V, and Re2 at 0.13 V. These redox reactions require a larger overpotential (0.25 V more) for each oxidation or reduction reaction with respect to that from Pt electrode. But without doubt, NiO possesses catalytic ability in the reduction of I2 and I3–.
Figure 5.
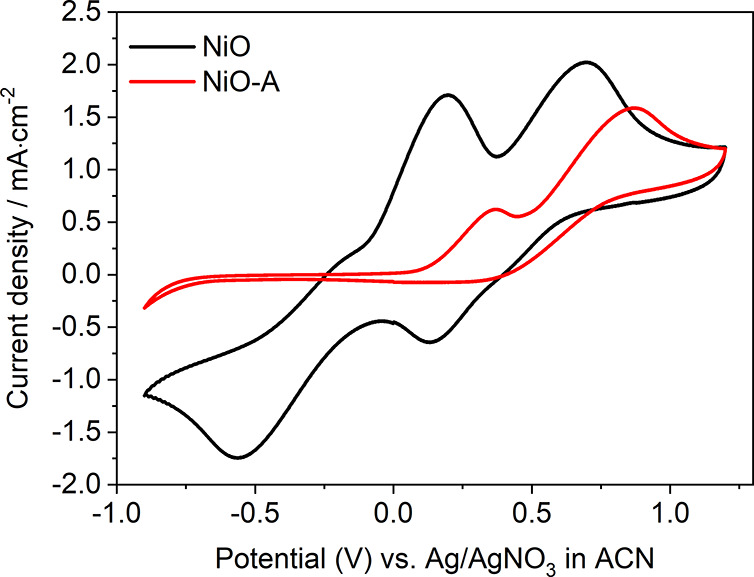
CV of NiO and NiO-A films in 5 mM I–/I3– (5 mM I2 and 10 mM LiI) and 100 mM TBAPF6 in ACN at a scan rate of 100 mV·s–1.
After removal of surface states, NiO-A shows a completely different CV in the presence of the I–/I3– redox couple. Two oxidation peaks are observed at 0.37 V of Ox1 and at 0.87 V of Ox2. However, no reduction peaks are detected. This could be due to the surface states acting as the catalyst for reduction of I3–. The removal of the surface states certainly passivates the catalytic process. In other words, those active surface atoms which were involved in I3– reduction on NiO have changed their chemical properties after ALD Al2O3. Another possibility is that loss of these surface states causes loss of conductivity, resulting in electrons not being able to access the catalytic sites. Note that the DOS in the bandgap of the NiO-A films still is ca. 4 × 1019 cm–3 eV–1, which is comparable to the DOS of bandgap states in ZnO and TiO2 mesoporous semiconductor films.39,40 [The concentration of the bandgap states is 3.68 × 1019 cm–3 in NiO-A, similar as that of TiO2 mesoporous films31,41 (e.g., 2.5 × 1019 cm–3 31).] This indicates that the conductivity should not be the limiting factor preventing the charge accessibility to these remaining intra-bandgap states in NiO, in consideration of the trapping/detrapping charge transport mechanism in n-DSCs (both ZnO and TiO2-based n-DSCs). The charges should be able to access remaining intra-bandgap states in the NiO-A. The removed surface states from the intra-bandgap states, named as NiOIBS-R, determine the catalytic reduction of I3– + 2e– → 3I– at −0.35 V (Re1).
Annealing has been reported to change the color of NiO films and reduce the surface states.30 Here, we annealed NiO films at 200 °C under an inert Ar atmosphere. We found that the Re1 peak still cannot be detected in CV, although a high current density (Jsc = 0.3 mA·cm–2 at least) flows through the NiO films (Figure S16). This further confirms that the conductivity should not be the limiting factor and that the inactivation of Re1 results from the loss of catalytic sites after the removal of intra-bandgap states (Figures S15 and S16). The passivation by annealing is not permanent, and the states can recover during the scanning process of CV, as shown by the recovery of the R1 peak in Figure S17.
Based on the discussion above, to complete the Re1 reaction, a suitable catalyst is needed. The low photocurrent observed in the NiO-A-based solar cells indicates that the loss of surface states can affect the dye regeneration in p-DSCs with I–/I3– as the redox couple. More specifically, intra-bandgap states of NiOIBS-R are involved in the dye regeneration. The low photocurrent in the NiO-A-based p-DSCs derives from the inefficient dye regeneration. The dye regeneration efficiency has been estimated to be 50% in NiO-PB6-based p-DSCs,42 while the halftime of charge recombination on NiO-dye is ca. 5 ps.34 This suggests that the dye regeneration halftime should be ca. 5 ps in order to achieve the estimated dye regeneration efficiency. The required dye regeneration rate is, however, far beyond the diffusion limitation of I3– in the electrolyte: 50 mM I3– gives an upper-limit pseudo-first-order diffusion rate constant of 5 × 108 s–1, if the diffusion-limited rate constant of regeneration with I3– is 10–10 M–1 s–1.43 Therefore, the fast dye regeneration predicted is contradictory to the conventional dye regeneration mechanism in p-DSCs, in which reduced dye molecules transfer electrons to the I3 species in electrolyte.
To understand dye regeneration in p-DSCs, fs-TAS was applied to study the electron transfer process in NiO-PB6 and NiO-A-PB6 films after excitation with a 560 nm laser pulse (300 nJ/pulse). Reduced PB6 (PB6–) with an absorption peak around 650 nm was observed in NiO-PB6 film. In a NiO-A-PB6 sample, a similar absorption peak at 650 nm was observed after electron injection from excited PB6 (PB6*) molecules into the VB of NiO (see Figure S19). Thus, the kinetic decay of PB6– can be followed at 650 nm. There are two distinguishing charge-transfer processes causing the decay of PB6–. One is the charge recombination between PB6– and injected hole from PB6* (labeled as Path I in Figure 6 (right)); the other is charge injection from PB6– into NiOIBS-R (labeled as Path II in Figure 6 (right). In Figure 6 (left), the halftime of PB6– in NiO-PB6 film is ca. 6 ps, similar to previously reported results, whereas PB6– with t1/2 = 29 ps was confirmed in NiO-A-PB6 sample. With consideration of mono-/sub-monolayer of Al2O3, a negligible influence on electron tunneling was caused by the insulating layer of Al2O3. The enhanced halftime of PB6– in NiO-A-PB6 sample is attributed to the suppression on path II. Although removal of the intra-bandgap states indeed increased the halftime of PB6–, the long-lived PB6– clearly cannot efficiently transfer the electrons to I3– (dye regeneration), since a much lower Jsc was observed in Figure 4 (red line). Thus, in NiO-A-PB6-based p-DSCs, dye regeneration should be the main limiting factor on the unsatisfactory Jsc.
Figure 6.

Left: Kinetics traces of the fs-TAS signal of PB6– at 650 nm in NiO-PB6 and NiO-A-PB6 films. The halftime inserted was obtained directly from the raw data in Figure 6. The blue lines are from a triple-exponential fitting (fitting parameters are given in SI, p S13). Right: Simplified scheme of electrons transfer from PB6– to holes in VB labeled as Path I, to NiOIBS-R labeled as Path II.
In the NiO-PB6 film, it is more efficient for PB6– to inject electrons into NiOIBS-R by Path II (80%), leading to a shorter halftime of PB6–. The short-lived PB6–, however, can efficiently reduce I3–, leading to 2 orders of magnitude enhanced Jsc compared to that of NiO-A-PB6-based p-DSCs (Figure 4). The reason is that Path II is an intermediate step to reduce I3–. In other words, reduction of I3– requires two steps in NiO-based p-DSCs: electron injection into NiOIBS-R by Path II, resulting in NiOIBS-R–, and electron transfer from NiOIBS-R– into I3– to fulfill the reduction of I3–. Herein, Path II in NiO-PB6-based p-DSCs should be reasonably treated as a dye regeneration process, which is completely different with respect to that in NiO-A-PB6-based p-DSCs. In addition, intra-bandgap states in NiO-based DSC should not be simply ascribed to the charge recombination centers, leading to unsatisfactory solar-to-electricity conversion efficiency.
I3– species probably are in association with the NiO surface, similar to the reduction of I3– at the platinum electrode.44 Indirect evidence is from electrochemical reduction of I3– on NiO that is significantly affected by a mono-/sub-monolayer of Al2O3, implicating an inner-sphere reaction in which reactants, intermediates, or product often adsorb on the surface of electrodes.45 In previous findings, in dye-free NiO, the filled intra-bandgap states ((NiOIBS-R)−) recombine with holes on a 100-ns time scale, indicating that the lifetime of trapped electron is much long-lived compared to the lifetime (e.g., t1/2 = 6 ps) of PB6– on NiO.23 I3– species seem to have reasonable time to diffuse near NiOIBS-R– in order to complete electron transfer. In any case, reduced dye molecules will first transfer electrons to NiOIBS-R, and then (NiOIBS-R)− will transfer the electron to the associated I3– or solvated I3–. Here, we proposed two dye regeneration mechanisms in NiO-based p-DSCs, as shown in Schemes 1and 2.
Scheme 1.

Scheme 2.

Note that, in Scheme 1, the reduced dye (Dye–) transfers an electron into a NiOIBS-R site associated with I3– (a). The reduced surface state proceeds to reduce I3–, with the product being stabilized by the surface state (b). (NiOIBS-R)···I is reduced by a neighboring NiOIBS-R site trapped with an electron (c). In Scheme 2, the reduced dye injects an electron into a NiOIBS-R site (a). NiOIBS-R– is diffusionally quenched by I3– (b). I2–• is the diiodide radical from one-electron reduction.38,46 I3– in Scheme 1 is not necessary adsorbed on the NiOIBS-R, but should be near the NiOIBS-R. [Strictly, NiOIBS-R– should be written as (h+)NiOIBS-R–, since the injected hole from the excited dye should still exist inside NiO after electron injection from the reduced dye in Path II. When the electron transfers from NiOIBS-R– to I3–, the hole will diffuse to back contact to produce photocurrent in NiO-based p-DSCs.]
In n-DSCs, the I3– + 2e– → 3I– reaction (Re1) also happens as a charge recombination process between the injected electron in TiO2 and I3– in electrolyte,47 and the regeneration of I– by I3– reduction at the counter electrode.44 The charge recombination (electrons in TiO2 and I3–) has been proved significantly low (ms time scale48) in TiO2 matrix, but the reasons have not been well-understood. As mentioned by Meyer and co-workers: “why the injected electrons do not reduce I3– efficiently is unknown”,49 especially diffusing through tens of micrometers of TiO2. Willig stated that the concentration of the oxidized redox species involved in recombination should be small in the electrolyte. The bulk electrolyte I3– should not be the oxidized species, but a species after the I–I bond cleavage from I3–.50 Durrant experimentally proved that the recombination with I2 is more efficient than that with I3–.46 Meyer provided a reasonable speculation that the oxidative specie should be I2–• from dye regeneration in n-DSCs. However, accumulation of I2–• is impossible because of the rapid disproportionation of I2–•.51 These conclusions suggest that I3– cannot be the oxidative species that recombine with the electron in TiO2. But reducing I3– to regenerate I– is significantly efficient on the counter electrode with suitable catalyst, like Pt.44,52 It is interpreted as that activation barrier of iodine dissociation on platinized counter electrode should be lower than that of in TiO2.46 One electron reduction of I3– to I2–• seems to be the kinetic limiting process, requiring assistance of a suitable catalyst.
Surface modification of mesoporous NiO films changes their electrical conductivity dramatically. It may also change the charge transport behavior in DSC devices. Here, the charge transport behavior was characterized by deriving the charge transport time from transient photocurrents measurements. Otherwise identical NiO-PB6- and NiO-A-PB6-based p-DSCs were prepared to study charge transport in intact solar cells. NiO-PB6-A-based p-DSCs were also fabricated to further prove the influence of surface states on charge transport, for which an Al2O3 monolayer was deposited after PB6 sensitization. Since a typical high concentration of electrolyte (0.05 M I2 and 0.1 M LiI in ACN) was used for all the p-DSCs, the charge transport time in the devices can be reasonably approximated as the hole transport time based on an ambipolar diffusion model.53
The typical charge transport behavior in NiO-based p-DSCs is that hole transport time does not depend on light intensity.32,42,54−66 This is commonly explained by hole hopping on the NiO/electrolyte interface, first proposed by Boschloo and co-workers.32 Here, we further develop this hypothesis and try to draw a comprehensive physical picture in order to understand hole transport in NiO-based p-DSCs.
opping is an important charge transport mechanism in disordered semiconductors and is most often described by the Miller–Abrahams model.67,68 In this model, the carrier jump rate is exponentially dependent on the distance and energy difference between two localized sites. The most probable hopping events should occur around the quasi-Fermi level from the occupied sites to the empty ones (more empty sites around the occupied one). The quasi-Fermi level is light intensity dependent, since, with increasing the light intensity, more localized states can be filled by photoinduced charges from the deep traps to the shallow ones. This means the hopping rate could change with light intensity.
The hopping transport can also be simplified through an assumed concept called effective transport energy, which is the energy level responsible for charge transport. Charges localized in shallow traps need less time release to this effective transport level.67 The trapping/releasing events are affected by the quasi-Fermi level and light intensity. In NiO, the energy distribution of intra-bandgap states seems more approaching a Gaussian DOS. The distribution could change the charges trapping behavior, but it should not change the fact of light intensity dependence of the charge transport. In addition, Bisquert simulated the hopping transport in the Gaussian DOS, in which the diffusion coefficient still depends on light intensity.69
In NiO-PB6-based solar cells, the independence of transport time on light intensity is observed. Hole hopping should be the transport mechanism, since surface states dominate the conductivity. We therefore propose that the carriers trapped in surface states of NiO are not fully localized, but more delocalized because of the percolation effect.69 The densities of the surface states are estimated: 4 surface states per nm2 in NiO, and 1 surface state per nm2 in NiO-A (see SI). The concentration of surface states in NiO could surpass the percolation threshold.70 The percolation effect causes charges to be essentially delocalized on surface states. The activation energy of hopping between two surface states becomes much smaller. Therefore, the charge transport time is independent of the light intensity in the p-DSCs, as illustrated in Scheme 3.
Scheme 3. Proposed Charge Transport Mechanism in NiO- and NiO-A-Based p-DSCs: Left, Percolation Hoping Mechanism and Right, Multi-Trapping/Detrapping Transport.
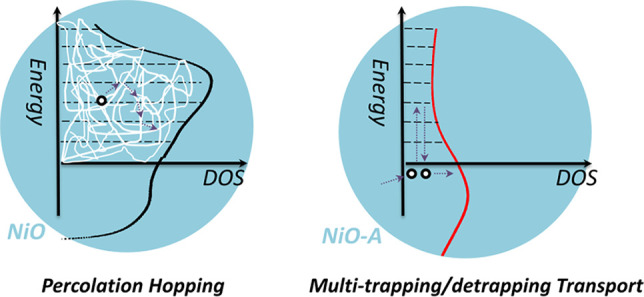
Cation adsorption is able to alter the transport time, but the process remains independent of light intensity, as reported in the literature.32 A similar behavior is the conductivity of NiO-PB6 films, decreasing by 4 times after dye adsorption. The non-dependence behavior on light intensity kept as well in NiO-PB6-based solar cells. These phenomena suggest that the percolation effect still holds since the surface states loss is not significant. Although the transport routes are likely altered, the concentration of surface states appears to still be above the percolation threshold. Thus, even though longer transport times are observed, they are still independent of light intensity.32,63
When the surface states are reduced significantly, the hole transport mechanism is highly different. In NiO-A films, the charge transport time is exponentially dependent on the light intensity, similar as in TiO2-based n-DSCs. As proved, 72% of the surface states were removed in NiO-A films, resulting in its DC dark conductivity decreases by 70 times. This could suggest that the concentration of surface states is lower than the percolation threshold. The DOS within the bandgap likely follow an exponential distribution in NiO-A film (see Figure 2). A multi-trapping/detrapping model,71 commonly applied in TiO2-based n-DSCs, can be applied to account for the hole transport in the NiO-A-based solar cells, as shown in Scheme 3.
In addition, the DC photoconductivity of NiO-PB6 and NiO-A-PB6 was investigated under one sun condition. No conductivity change is observed for the NiO-PB6 films under light or dark (Table S1). However, the photoconductivity of NiO-A-PB6 is increased by a factor of 2 over its dark conductivity, which is likely from an increased hole concentration in the VB of NiO under illumination.
As for NiO-PB6-A, PB6 dye adsorption can occupy part of the surface states, but the influence on surface states is less than that of the ALD Al2O3, as confirmed by the different conductivity responses. Thus, the pre-adsorbed PB6 dyes are analogous as a protection layer of surface states before ALD of Al2O3. The hole transport time also shows an exponential dependence on light intensity. Here, an interesting difference is that hole transport time in NiO-PB6-A device is less dependent on the light intensity than that in NiO-A-PB6. This is reflected by the slope of NiO-PB6-A being smaller than that in NiO-A-PB6, but higher than that in NiO-PB6 in Figure 7. Note that the transport time on both NiO-A-PB6 and NiO-PB6-A is 1–2 orders of magnitude longer than the transport time on NiO-PB6 at a low light intensity. At high light intensity, the transport times in different samples get closer. This could indicate that the hole mobility in the VB of NiO-A-PB6 is comparable with that in surface states of NiO. Jsc ≈ 200 μA·cm–2 is detected at the high intensity (186 mW·cm–2) in NiO-A-PB6-based devices, and Jsc exhibits the tendency to approach a higher value with greater light intensity, considering the slope of NiO-A-PB6 (Figure 7).
Figure 7.

Transport time as a function of current density (which is linearly dependent on light intensity) in the differently prepared p-DSCs.
On the basis of the aforementioned discussions, we will make some comments on two applications of mesoporous NiO films-based devices, (1) p-DSCs and (2) p-type dye-sensitized photoelectrochemical cells (p-DSPECs) for H2 evolution. The low performance of p-DSCs is widely known, although it has been studied for 20 years.1 In this work, we found that surface states affect the dye regeneration and the reduction of I3– species. In addition, the photoinjected holes in the VB of NiO are able to oxidize I3– or I–, as confirmed by the two oxidation peaks of the CV (Figure 5). This can cause charge recombination during hole diffusion through the NiO matrix. Hole transport involves hopping among the surface states on NiO, which could even enhance the charge recombination between holes in NiO and electrolytes. The surface states of NiO seem able to influence many electronic properties of mesoporous NiO films. For instance, high surface states will improve hole transport rate, facilitate dye regeneration, and increase Jsc, but they lead to more charge recombination and a lower Voc as well. Balancing all the parameters simultaneously is contradictory. This could be the reason why it is so challenging to enhance the performance of p-DSCs.
Similar to p-DSCs, the performance of p-DSPECs is also unsatisfactory. It is difficult for the photocatalytic current density for H2 evolution to surpass 100 μA·cm–2 under one sun condition.29 H2 evolution from a dye-sensitized NiO photocathode system could be limited by catalysts or dye loading, but is also likely affected by the NiO itself. One possibility is that the surface states of NiO could be involved in the catalytic reaction, no matter if there is an extra catalyst on the surface. Simonov and co-workers have shown that dye-sensitized mesoporous NiO films have the ability to catalyze the H2 evolution reaction without an additional catalyst. They concluded that the interfaces are the photoactive sites for H2 evolution. More specifically, the metal Ni is suggested as the catalyst formed during the H2 evolution reaction.14 In this work, photocurrent was observed from NiO-PB6 and NiO-A-PB6 photocathodes in a p-DSPECs setup (Figure S24). Photocurrent on NiO-PB6 is enhanced with decreasing pH. In the NiO-A-PB6 photocathode, photocurrent is much lower and independent of pH. This indicates that surface states could be involved in the catalytic cycle of the H2 evolution reaction.
Conclusion
In this paper, we applied a modified ALD procedure to achieve a better control on the surface passivation of mesoporous NiO films by a monolayer of Al2O3. This results in a removal of 72% of the surface states of NiO. The resultant NiO-A films showed ca. 70 times lower DC dark conductivity compared to NiO films. This implies that surface states, and not the bulk, are responsible for the conductivity of mesoporous NiO films. A much lower photocurrent is observed in NiO-A-based dye-sensitized solar cells with I–/I3– redox electrolyte. This is ascribed to the fact that dye regeneration requires the assistance of intra-bandgap states on NiO. Specifically, dye regeneration is finished by injection of an electron into NiOIBS-R on NiO, which then transfers to I3–. Additionally, a more complete charge transport mechanism is suggested to explain the independence of hole transport time on light intensity in p-DSCs. Hole transport on NiO is proposed as a percolation hopping process which can be significantly affected by the concentration of surface states. After 72% of surface states are removed in NiO-A films (1 surface state per nm2), the dependence of hole transport time on light intensity transformed into an exponential dependence, commonly observed in TiO2-based n-DSCs. We suggest that the hole transport mechanism is altered from the percolation hopping into a multi-trapping/detrapping mechanism.
Acknowledgments
The work is financially supported by Göran Gustafssons Stiftelse and Uppsala University. L.T. thanks the China Scholarship Council (CSC) for the scholarship support. We thank Qitao Hu (UU) for repeating photoconductivity measurements, Dr. Tomas Kubart (UU) for help with sputtering NiO, Dr. Palas Baran Pati for synthesis of the PB6 dye, Prof. Anders Hagfeldt (EPFL) for helpful discussions, Dr. Shihuai Wang (UU) for kind support with dry ACN and glovebox, and Dr. Carl Hägglund (UU) for helpful discussions on ALD.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c08886.
XRD of NiO films; XPS spectra of different samples; the function of extracted charges on Voc and IPCE from both NiO- and NiO-A-based p-DSCs; schematic drawing of resistance measurements and conductivity data of different samples; and cyclic voltammetry and transient spectroscopy data, including Figures S1–S24, Scheme S1, and Tables S1–S3 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- He J.; Lindström H.; Hagfeldt A.; Lindquist S.-E. Dye-sensitized nanostructured p-type nickel oxide film as a photocathode for a solar cell. J. Phys. Chem. B 1999, 103, 8940–8943. 10.1021/jp991681r. [DOI] [Google Scholar]
- Li L.; Duan L.; Wen F.; Li C.; Wang M.; Hagfeldt A.; Sun L. Visible light driven hydrogen production from a photo-active cathode based on a molecular catalyst and organic dye-sensitized p-type nanostructured NiO. Chem. Commun. 2012, 48, 988–990. 10.1039/C2CC16101J. [DOI] [PubMed] [Google Scholar]
- Poizot P.; Laruelle S.; Grugeon S.; Dupont L.; Tarascon J. Nano-sized transition-metal oxides as negative-electrode materials for lithium-ion batteries. Nature 2000, 407, 496. 10.1038/35035045. [DOI] [PubMed] [Google Scholar]
- Morin F. Electrical properties of NiO. Phys. Rev. 1954, 93, 1199. 10.1103/PhysRev.93.1199. [DOI] [Google Scholar]
- Irwin M. D.; Buchholz D. B.; Hains A. W.; Chang R. P.; Marks T. J. p-Type semiconducting nickel oxide as an efficiency-enhancing anode interfacial layer in polymer bulk-heterojunction solar cells. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2783–2787. 10.1073/pnas.0711990105. [DOI] [Google Scholar]
- Rubinstein M.; Kodama R.; Makhlouf S. A. Electron spin resonance study of NiO antiferromagnetic nanoparticles. J. Magn. Magn. Mater. 2001, 234, 289–293. 10.1016/S0304-8853(01)00313-4. [DOI] [Google Scholar]
- Ichiyanagi Y.; Wakabayashi N.; Yamazaki J.; Yamada S.; Kimishima Y.; Komatsu E.; Tajima H. Magnetic properties of NiO nanoparticles. Phys. B 2003, 329, 862–863. 10.1016/S0921-4526(02)02578-4. [DOI] [Google Scholar]
- Corrigan D. A. The catalysis of the oxygen evolution reaction by iron impurities in thin film nickel oxide electrodes. J. Electrochem. Soc. 1987, 134, 377–384. 10.1149/1.2100463. [DOI] [Google Scholar]
- Li Y.-F.; Selloni A. Mechanism and activity of water oxidation on selected surfaces of pure and Fe-doped NiO x. ACS Catal. 2014, 4, 1148–1153. 10.1021/cs401245q. [DOI] [Google Scholar]
- Friebel D.; Louie M. W.; Bajdich M.; Sanwald K. E.; Cai Y.; Wise A. M.; Cheng M.-J.; Sokaras D.; Weng T.-C.; Alonso-Mori R.; et al. Identification of highly active Fe sites in (Ni, Fe)OOH for electrocatalytic water splitting. J. Am. Chem. Soc. 2015, 137, 1305–1313. 10.1021/ja511559d. [DOI] [PubMed] [Google Scholar]
- Sumikura S.; Mori S.; Shimizu S.; Usami H.; Suzuki E. Syntheses of NiO nanoporous films using nonionic triblock co-polymer templates and their application to photo-cathodes of p-type dye-sensitized solar cells. J. Photochem. Photobiol., A 2008, 199, 1–7. 10.1016/j.jphotochem.2008.04.007. [DOI] [Google Scholar]
- Li Z.; Niu W.; Zhou L.; Yang Y. Phosphorus and aluminum codoped porous NiO nanosheets as highly efficient electrocatalysts for overall water splitting. ACS Energy Lett. 2018, 3, 892–898. 10.1021/acsenergylett.8b00174. [DOI] [Google Scholar]
- Yoshinobu J.; Ballinger T.; Xu Z.; Jänsch H.; Zaki M.; Xu J.; Yates J. Jr Ultraviolet photodesorption of CO from NiO as measured by infrared spectroscopy. Surf. Sci. 1991, 255, 295–302. 10.1016/0039-6028(91)90686-M. [DOI] [Google Scholar]
- Hoogeveen D. A.; Fournier M.; Bonke S. A.; Nattestad A.; Mishra A.; Bäuerle P.; Spiccia L.; Mozer A. J.; Simonov A. N. Origin of photoelectrochemical generation of dihydrogen by a dye-sensitized photocathode without an intentionally introduced catalyst. J. Phys. Chem. C 2017, 121, 25836–25846. 10.1021/acs.jpcc.7b08067. [DOI] [Google Scholar]
- Kytin V.; Dittrich T.; Bisquert J.; Lebedev E.; Koch F. Limitation of the mobility of charge carriers in a nanoscaled heterogeneous system by dynamical Coulomb screening. Phys. Rev. B: Condens. Matter Mater. Phys. 2003, 68, 195308. 10.1103/PhysRevB.68.195308. [DOI] [Google Scholar]
- Ching W.; Lin C. C.; Guttman L. Structural disorder and electronic properties of amorphous silicon. Phys. Rev. B 1977, 16, 5488. 10.1103/PhysRevB.16.5488. [DOI] [Google Scholar]
- Tench D. M.; Yeager E. Capacitance measurements on lithiated nickel oxide electrodes. J. Electrochem. Soc. 1973, 120, 164–171. 10.1149/1.2403414. [DOI] [Google Scholar]
- Niklasson G. A.; Granqvist C. G. Electrochromics for smart windows: thin films of tungsten oxide and nickel oxide, and devices based on these. J. Mater. Chem. 2007, 17, 127–156. 10.1039/B612174H. [DOI] [Google Scholar]
- Boschloo G.; Hagfeldt A. Spectroelectrochemistry of nanostructured NiO. J. Phys. Chem. B 2001, 105, 3039–3044. 10.1021/jp003499s. [DOI] [Google Scholar]
- Marrani A. G.; Novelli V.; Sheehan S.; Dowling D. P.; Dini D. Probing the redox states at the surface of electroactive nanoporous NiO thin films. ACS Appl. Mater. Interfaces 2014, 6, 143–152. 10.1021/am403671h. [DOI] [PubMed] [Google Scholar]
- D’Amario L.; Antila L. J.; Pettersson Rimgard B.; Boschloo G.; Hammarström L. Kinetic evidence of two pathways for charge recombination in NiO-based dye-sensitized solar cells. J. Phys. Chem. Lett. 2015, 6, 779–783. 10.1021/acs.jpclett.5b00048. [DOI] [PubMed] [Google Scholar]
- Smeigh A. L.; Le Pleux L.; Fortage J.; Pellegrin Y.; Blart E.; Odobel F.; Hammarström L. Ultrafast recombination for NiO sensitized with a series of perylene imide sensitizers exhibiting Marcus normal behaviour. Chem. Commun. 2012, 48, 678–680. 10.1039/C1CC16144J. [DOI] [PubMed] [Google Scholar]
- D’Amario L.; Föhlinger J.; Boschloo G.; Hammarström L. Unveiling hole trapping and surface dynamics of NiO nanoparticles. Chem. Sci. 2018, 9, 223–230. 10.1039/C7SC03442C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise C. F.; Mayer J. M. Electrochemically Determined O–H Bond Dissociation Free Energies of NiO Electrodes Predict Proton-Coupled Electron Transfer Reactivity. J. Am. Chem. Soc. 2019, 141, 14971–14975. 10.1021/jacs.9b07923. [DOI] [PubMed] [Google Scholar]
- Flynn C. J.; McCullough S. M.; Oh E.; Li L.; Mercado C. C.; Farnum B. H.; Li W.; Donley C. L.; You W.; Nozik A. J.; et al. Site-selective passivation of defects in NiO solar photocathodes by targeted atomic deposition. ACS Appl. Mater. Interfaces 2016, 8, 4754–4761. 10.1021/acsami.6b01090. [DOI] [PubMed] [Google Scholar]
- Flynn C. J.; McCullough S. M.; Li L.; Donley C. L.; Kanai Y.; Cahoon J. F. Passivation of nickel vacancy defects in nickel oxide solar cells by targeted atomic deposition of boron. J. Phys. Chem. C 2016, 120, 16568–16576. 10.1021/acs.jpcc.6b06593. [DOI] [Google Scholar]
- Biswas S.; Husek J.; Londo S.; Baker L. R. Ultrafast Electron Trapping and Defect-Mediated Recombination in NiO Probed by Femtosecond Extreme Ultraviolet Reflection–Absorption Spectroscopy. J. Phys. Chem. Lett. 2018, 9, 5047–5054. 10.1021/acs.jpclett.8b01865. [DOI] [PubMed] [Google Scholar]
- Petersson J.; Eklund M.; Davidsson J.; Hammarström L. Ultrafast Electron Transfer Dynamics of a Zn (II) porphyrin– Viologen Complex Revisited: S2 vs S1 Reactions and Survival of Excess Excitation Energy. J. Phys. Chem. B 2010, 114, 14329–14338. 10.1021/jp911686z. [DOI] [PubMed] [Google Scholar]
- Xu B.; Tian L.; Etman A. S.; Sun J.; Tian H. Solution-processed nanoporous NiO-dye-ZnO photocathodes: Toward efficient and stable solid-state p-type dye-sensitized solar cells and dye-sensitized photoelectrosynthesis cells. Nano Energy 2019, 55, 59–64. 10.1016/j.nanoen.2018.10.054. [DOI] [Google Scholar]
- D’Amario L.; Jiang R.; Cappel U. B.; Gibson E. A.; Boschloo G.; Rensmo H.; Sun L.; Hammarström L.; Tian H. Chemical and physical reduction of high valence Ni states in mesoporous NiO film for solar cell application. ACS Appl. Mater. Interfaces 2017, 9, 33470–33477. 10.1021/acsami.7b01532. [DOI] [PubMed] [Google Scholar]
- Fabregat-Santiago F.; Mora-Seró I.; Garcia-Belmonte G.; Bisquert J. Cyclic voltammetry studies of nanoporous semiconductors. Capacitive and reactive properties of nanocrystalline TiO2 electrodes in aqueous electrolyte. J. Phys. Chem. B 2003, 107, 758–768. 10.1021/jp0265182. [DOI] [Google Scholar]
- Zhu H.; Hagfeldt A.; Boschloo G. Photoelectrochemistry of mesoporous NiO electrodes in iodide/triiodide electrolytes. J. Phys. Chem. C 2007, 111, 17455–17458. 10.1021/jp077134k. [DOI] [Google Scholar]
- Prasittichai C.; Hupp J. T. Surface modification of SnO2 photoelectrodes in dye-sensitized solar cells: significant improvements in photovoltage via Al2O3 atomic layer deposition. J. Phys. Chem. Lett. 2010, 1, 1611–1615. 10.1021/jz100361f. [DOI] [Google Scholar]
- Tian L.; Föhlinger J.; Pati P. B.; Zhang Z.; Lin J.; Yang W.; Johansson M.; Kubart T.; Sun J.; Boschloo G.; et al. Ultrafast dye regeneration in a core–shell NiO–dye–TiO2 mesoporous film. Phys. Chem. Chem. Phys. 2018, 20, 36–40. 10.1039/C7CP07088H. [DOI] [PubMed] [Google Scholar]
- D’Amario L.; Boschloo G.; Hagfeldt A.; Hammarstrom L. Tuning of conductivity and density of states of NiO mesoporous films used in p-type DSSCs. J. Phys. Chem. C 2014, 118, 19556–19564. 10.1021/jp504551v. [DOI] [Google Scholar]
- Wu M.; Lin X.; Wang Y.; Wang L.; Guo W.; Qi D.; Peng X.; Hagfeldt A.; Grätzel M.; Ma T. Economical Pt-free catalysts for counter electrodes of dye-sensitized solar cells. J. Am. Chem. Soc. 2012, 134, 3419–3428. 10.1021/ja209657v. [DOI] [PubMed] [Google Scholar]
- Duan Y.; Tang Q.; Liu J.; He B.; Yu L. Transparent metal selenide alloy counter electrodes for high-efficiency bifacial dye-sensitized solar cells. Angew. Chem., Int. Ed. 2014, 53, 14569–14574. 10.1002/anie.201409422. [DOI] [PubMed] [Google Scholar]
- Boschloo G.; Hagfeldt A. Characteristics of the iodide/triiodide redox mediator in dye-sensitized solar cells. Acc. Chem. Res. 2009, 42, 1819–1826. 10.1021/ar900138m. [DOI] [PubMed] [Google Scholar]
- Quintana M.; Edvinsson T.; Hagfeldt A.; Boschloo G. Comparison of dye-sensitized ZnO and TiO2 solar cells: studies of charge transport and carrier lifetime. J. Phys. Chem. C 2007, 111, 1035–1041. 10.1021/jp065948f. [DOI] [Google Scholar]
- Abayev I.; Zaban A.; Kytin V. G.; Danilin A. A.; Garcia-Belmonte G.; Bisquert J. Properties of the electronic density of states in TiO2 nanoparticles surrounded with aqueous electrolyte. J. Solid State Electrochem. 2007, 11, 647–653. 10.1007/s10008-006-0220-1. [DOI] [Google Scholar]
- Nelson J.; Eppler A. M.; Ballard I. M. Photoconductivity and charge trapping in porous nanocrystalline titanium dioxide. J. Photochem. Photobiol., A 2002, 148, 25–31. 10.1016/S1010-6030(02)00035-7. [DOI] [Google Scholar]
- Tian L.; Törndahl T.; Lin J.; Pati P. B.; Zhang Z.; Kubart T.; Hao Y.; Sun J.; Boschloo G.; Tian H. Mechanistic Insights into Solid-State p-Type Dye-Sensitized Solar Cells. J. Phys. Chem. C 2019, 123, 26151–26160. 10.1021/acs.jpcc.9b08251. [DOI] [Google Scholar]
- Zhou G. Q.; Zhong W. Z. Diffusion-Controlled Reactions of Enzymes: A Comparison between Chou’s Model and Alberty-Hammes-Eigen’s Model. Eur. J. Biochem. 1982, 128, 383–387. 10.1111/j.1432-1033.1982.tb06976.x. [DOI] [PubMed] [Google Scholar]
- Hauch A.; Georg A. Diffusion in the electrolyte and charge-transfer reaction at the platinum electrode in dye-sensitized solar cells. Electrochim. Acta 2001, 46, 3457–3466. 10.1016/S0013-4686(01)00540-0. [DOI] [Google Scholar]
- Bard A. J. Inner-sphere heterogeneous electrode reactions. Electrocatalysis and photocatalysis: the challenge. J. Am. Chem. Soc. 2010, 132, 7559–7567. 10.1021/ja101578m. [DOI] [PubMed] [Google Scholar]
- Green A. N.; Chandler R. E.; Haque S. A.; Nelson J.; Durrant J. R. Transient absorption studies and numerical modeling of iodine photoreduction by nanocrystalline TiO2 films. J. Phys. Chem. B 2005, 109, 142–150. 10.1021/jp047414p. [DOI] [PubMed] [Google Scholar]
- Nakade S.; Kanzaki T.; Kubo W.; Kitamura T.; Wada Y.; Yanagida S. Role of electrolytes on charge recombination in dye-sensitized TiO2 solar cell (1): the case of solar cells using the I–/I3– redox couple. J. Phys. Chem. B 2005, 109, 3480–3487. 10.1021/jp0460036. [DOI] [PubMed] [Google Scholar]
- Van de Lagemaat J.; Park N.-G.; Frank A. Influence of electrical potential distribution, charge transport, and recombination on the photopotential and photocurrent conversion efficiency of dye-sensitized nanocrystalline TiO2 solar cells: a study by electrical impedance and optical modulation techniques. J. Phys. Chem. B 2000, 104, 2044–2052. 10.1021/jp993172v. [DOI] [Google Scholar]
- Farnum B. H.; Gardner J. M.; Meyer G. J. Flash-quench technique employed to study the one-electron reduction of triiodide in acetonitrile: evidence for a diiodide reaction product. Inorg. Chem. 2010, 49, 10223–10225. 10.1021/ic1015466. [DOI] [PubMed] [Google Scholar]
- Schwarzburg K.; Willig F. Origin of photovoltage and photocurrent in the nanoporous dye-sensitized electrochemical solar cell. J. Phys. Chem. B 1999, 103, 5743–5746. 10.1021/jp990312s. [DOI] [Google Scholar]
- Rowley J. G.; Farnum B. H.; Ardo S.; Meyer G. J. Iodide chemistry in dye-sensitized solar cells: making and breaking I– I bonds for solar energy conversion. J. Phys. Chem. Lett. 2010, 1, 3132–3140. 10.1021/jz101311d. [DOI] [Google Scholar]
- Pichot F.; Gregg B. A. The photovoltage-determining mechanism in dye-sensitized solar cells. J. Phys. Chem. B 2000, 104, 6–10. 10.1021/jp993035y. [DOI] [Google Scholar]
- Kopidakis N.; Schiff E. A.; Park N.-G.; Van de Lagemaat J.; Frank A. Ambipolar diffusion of photocarriers in electrolyte-filled, nanoporous TiO2. J. Phys. Chem. B 2000, 104, 3930–3936. 10.1021/jp9936603. [DOI] [Google Scholar]
- Huang Z.; Natu G.; Ji Z.; He M.; Yu M.; Wu Y. Probing the low fill factor of NiO p-type dye-sensitized solar cells. J. Phys. Chem. C 2012, 116, 26239–26246. 10.1021/jp310053f. [DOI] [Google Scholar]
- Ji Z.; Natu G.; Wu Y. Cyclometalated ruthenium sensitizers bearing a triphenylamino group for p-type NiO dye-sensitized solar cells. ACS Appl. Mater. Interfaces 2013, 5, 8641–8648. 10.1021/am402263q. [DOI] [PubMed] [Google Scholar]
- Kang S. H.; Neale N. R.; Zhu K.; Halverson A. F.; Yan Y.; Frank A. J. The effect of a metallic Ni core on charge dynamics in CdS-sensitized p-type NiO nanowire mesh photocathodes. RSC Adv. 2013, 3, 13342–13347. 10.1039/c3ra41216d. [DOI] [Google Scholar]
- Kang S. H.; Zhu K.; Neale N. R.; Frank A. J. Hole transport in sensitized CdS–NiO nanoparticle photocathodes. Chem. Commun. 2011, 47, 10419–10421. 10.1039/c1cc13932k. [DOI] [PubMed] [Google Scholar]
- Klein Y.; Marinakis N.; Constable E.; Housecroft C. A phosphonic acid anchoring analogue of the sensitizer P1 for p-type dye-sensitized solar cells. Crystals 2018, 8, 389. 10.3390/cryst8100389. [DOI] [Google Scholar]
- Le Pleux L.; Smeigh A. L.; Gibson E.; Pellegrin Y.; Blart E.; Boschloo G.; Hagfeldt A.; Hammarström L.; Odobel F. Synthesis, photophysical and photovoltaic investigations of acceptor-functionalized perylene monoimide dyes for nickel oxide p-type dye-sensitized solar cells. Energy Environ. Sci. 2011, 4, 2075–2084. 10.1039/c1ee01148k. [DOI] [Google Scholar]
- Li L.; Gibson E. A.; Qin P.; Boschloo G.; Gorlov M.; Hagfeldt A.; Sun L. Double-layered NiO photocathodes for p-type DSSCs with record IPCE. Adv. Mater. 2010, 22, 1759–1762. 10.1002/adma.200903151. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Wei L.; Yuan S.; Ren X.; Zhao Y.; Wang Z.; Zhang M.; Shi L.; Li D. The effect of Ni (CH3COO)2 post-treatment on the charge dynamics in p-type NiO dye-sensitized solar cells. J. Mater. Sci. 2015, 50, 6668–6676. 10.1007/s10853-015-9221-8. [DOI] [Google Scholar]
- Mori S.; Fukuda S.; Sumikura S.; Takeda Y.; Tamaki Y.; Suzuki E.; Abe T. Charge-transfer processes in dye-sensitized NiO solar cells. J. Phys. Chem. C 2008, 112, 16134–16139. 10.1021/jp803919b. [DOI] [Google Scholar]
- Natu G.; Hasin P.; Huang Z.; Ji Z.; He M.; Wu Y. Valence band-edge engineering of nickel oxide nanoparticles via cobalt doping for application in p-type dye-sensitized solar cells. ACS Appl. Mater. Interfaces 2012, 4, 5922–5929. 10.1021/am301565j. [DOI] [PubMed] [Google Scholar]
- Wang H.; Zeng X.; Huang Z.; Zhang W.; Qiao X.; Hu B.; Zou X.; Wang M.; Cheng Y.-B.; Chen W. Boosting the photocurrent density of p-type solar cells based on organometal halide perovskite-sensitized mesoporous NiO photocathodes. ACS Appl. Mater. Interfaces 2014, 6, 12609–12617. 10.1021/am5025963. [DOI] [PubMed] [Google Scholar]
- Wei L.; Jiang L.; Yuan S.; Ren X.; Zhao Y.; Wang Z.; Zhang M.; Shi L.; Li D. Valence band edge shifts and charge-transfer dynamics in Li-doped NiO based p-type DSSCs. Electrochim. Acta 2016, 188, 309–316. 10.1016/j.electacta.2015.12.026. [DOI] [Google Scholar]
- Tian L.; Föhlinger J.; Zhang Z.; Pati P. B.; Lin J.; Kubart T.; Hua Y.; Sun J.; Kloo L.; Boschloo G. Solid state p-type dye sensitized NiO–dye–TiO2 core–shell solar cells. Chem. Commun. 2018, 54, 3739–3742. 10.1039/C8CC00505B. [DOI] [PubMed] [Google Scholar]
- Arkhipov V.; Emelianova E.; Heremans P.; Adriaenssens G. Equilibrium hopping conductivity in disordered materials. J. Optoelectron. Adv. Mater. 2002, 4, 425–436. [Google Scholar]
- Bisquert J. Hopping transport of electrons in dye-sensitized solar cells. J. Phys. Chem. C 2007, 111, 17163–17168. 10.1021/jp077419x. [DOI] [Google Scholar]
- Bisquert J. Interpretation of electron diffusion coefficient in organic and inorganic semiconductors with broad distributions of states. Phys. Chem. Chem. Phys. 2008, 10, 3175–3194. 10.1039/b719943k. [DOI] [PubMed] [Google Scholar]
- Benkstein K. D.; Kopidakis N.; Van de Lagemaat J.; Frank A. J. Influence of the percolation network geometry on electron transport in dye-sensitized titanium dioxide solar cells. J. Phys. Chem. B 2003, 107, 7759–7767. 10.1021/jp022681l. [DOI] [Google Scholar]
- Bisquert J.; Fabregat-Santiago F.; Mora-Sero I.; Garcia-Belmonte G.; Gimenez S. Electron lifetime in dye-sensitized solar cells: theory and interpretation of measurements. J. Phys. Chem. C 2009, 113, 17278–17290. 10.1021/jp9037649. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.