

Abstract

The only recently discovered gem-hydrogenation of internal alkynes is a fundamentally new transformation, in which both H atoms of dihydrogen are transferred to the same C atom of a triple bond while the other position transforms into a discrete metal carbene complex. [Cp*RuCl]4 is presently the catalyst of choice: the resulting piano-stool ruthenium carbenes can engage a tethered alkene into either cyclopropanation or metathesis, and a prototypical example of such a reactive intermediate with an olefin ligated to the ruthenium center has been isolated and characterized by X-ray diffraction. It is the substitution pattern of the olefin that determines whether metathesis or cyclopropanation takes place: a systematic survey using alkenes of largely different character in combination with a computational study of the mechanism at the local coupled cluster level of theory allowed the preparative results to be sorted and an intuitive model with predictive power to be proposed. This model links the course of the reaction to the polarization of the double bond as well as to the stability of the secondary carbene complex formed, if metathesis were to take place. The first application of “hydrogenative metathesis” to the total synthesis of sinularones E and F concurred with this interpretation and allowed the proposed structure of these marine natural products to be confirmed. During this synthesis, it was found that gem-hydrogenation also provides opportunities for C–H functionalization. Moreover, silylated alkynes are shown to participate well in hydrogenative metathesis, which opens a new entry into valuable allylsilane building blocks. Crystallographic evidence suggests that the polarized [Ru–Cl] bond of the catalyst interacts with the neighboring R3Si group. Since attractive interligand Cl/R3Si contacts had already previously been invoked to explain the outcome of various ruthenium-catalyzed reactions, including trans-hydrosilylation, the experimental confirmation provided herein has implications beyond the present case.
Introduction
The catalytic gem-hydrogenation of internal alkynes is a fundamentally new reactivity mode: it allows both H atoms of dihydrogen to be transferred to the same C atom of a triple bond, whereas the second C atom is concomitantly transformed into a discrete metal carbene.1 This unprecedented outcome was originally discovered during mechanistic studies into the equally perplexing trans-hydrogenation of alkynes with the aid of [Cp*Ru]-based catalysts (Scheme 1).2−4
Scheme 1. Link between trans-Hydrogenation and gem-Hydrogenation.
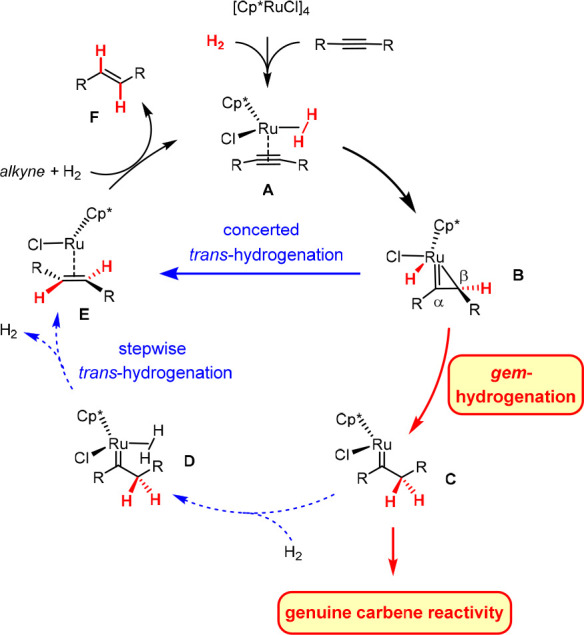
The steps of the trans-hydrogenation pathway downstream of C that need to be blocked or outperformed in order to harness genuine carbene chemistry are shown as dotted arrows.
The reaction commences with the upload of the substrate and H2 onto the ruthenium catalyst as shown in A, followed by a first hydrogen transfer to the π system activated by the carbophilic metal fragment.5 If the second H atom is delivered to the Cα position, the resulting ruthenacyclopropene B(6) evolves in a concerted manner into complex E, from which the E-olefin F is released. Competing transfer to Cβ, however, constitutes the actual “gem-hydrogenation” event: the resulting piano-stool ruthenium carbene C flanked by the newly formed methylene group can also transform into alkene F, provided that a second molecule of H2 binds to the metal center to lower the barriers. For an unbiased substrate such as 2-butyne, the two interwoven pathways were computed to have basically the same probability;2,3 polarization of the triple bond and/or incorporation of a propargylic substituent able to ligate the Ru center tips the scales such that gem-hydrogenation becomes the dominant or even exclusive course.2,3 Moreover, a distinct correlation between the electronic properties of the ancillary CpX ligand and the ease of gem-hydrogenation was established.7
Details apart, the generation of discrete metal carbene complexes via gem-hydrogenation of an alkyne provides an attractive outlook:1 if one is able to either block or outcompete the steps downstream of C that lead to the E-alkene F, this process might be diverted and genuine carbene chemistry might be harnessed (Scheme 1). To this end, we pursued different lines of research and were able to achieve a proof of concept in more than one format: novel hydrogenative skeletal rearrangements,3 hydrogenative heterocycle syntheses,3,8 counterintuitive “hydrogenative cyclopropanation” reactions,3,9,10 and even a “hydrogenative metathesis” manifold have been discovered.9,11 The present report is focused on this last transformation, which converts an enyne into a cycloalkene rather than into a 1,3-diene as conventional enyne metathesis would.12,13 Therefore, it embodies a new paradigm in metathesis. A combined experimental and computational approach provided insights into the factors controlling the reaction outcome and led to a refined mechanistic picture of this unorthodox transformation.
Results and Discussion
Reaction Development
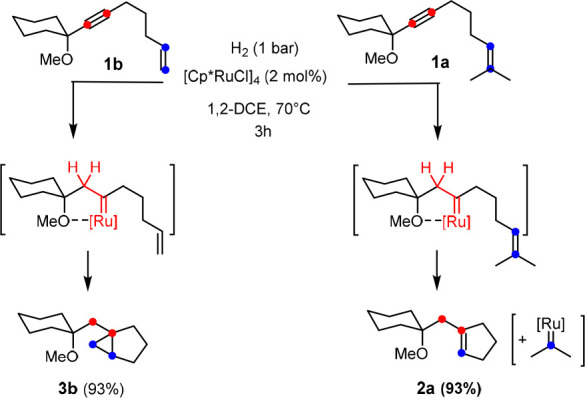
Cognizant of the need to steer gem-hydrogenation with the aid of a propargylic substituent,1−3 we initially chose enynes of type 1 as model substrates. As previously described, the outcome of the reaction is largely determined by the substitution of the olefinic site (Scheme 2):9 while hydrogenation of 1b comprising a terminal alkene with [Cp*RuCl]4 as a precatalyst in 1,2-dichloroethane at an elevated temperature furnished cyclopropane 3b, its sibling 1a containing two methyl substituents on the alkene gave the metathesis product 2a under otherwise identical conditions.14,15 This strikingly divergent behavior proved to be general;9 it came as a surprise in view of earlier literature reports which had found that putative carbene complexes of the type [Cp*Ru(Cl)(=CR2)] generated in situ from diazo precursors are highly competent in cyclopropanation but essentially failed to effect olefin metathesis.16−21
Scheme 2. Representative Example Showing the Effect of the Degree of Substitution on the Reaction Outcome.
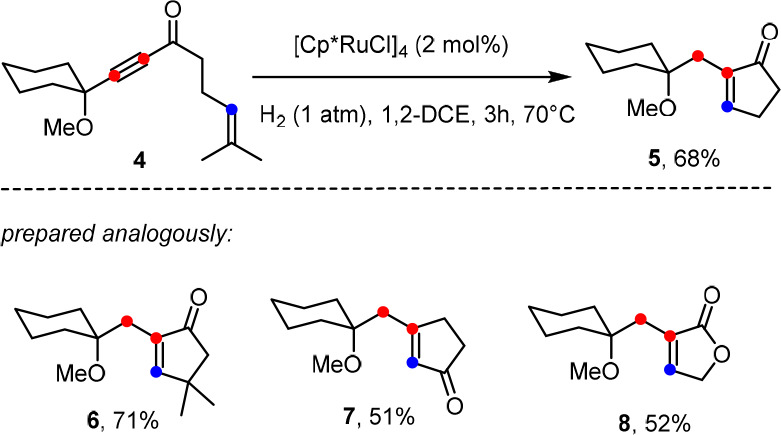
These hydrogenative transformations work well as long as the propargylic substituent is a tertiary ether, silyl ether, or acetal, as illustrated by a range of diverse products (see ref (9) and the Supporting Information). For the time being, the reactions are limited to the formation of five- and six-membered rings; all attempts at making larger cycles have so far met with failure likely because of the kinetic handicap in closing medium-sized or macrocyclic rings, which allows the competing trans-hydrogenation of the enyne via C and D (with/without over-reduction) to prevail. Moreover, the use of substrates containing (electron-rich) arenes, 1,3-dienes, or related motifs that bind more tightly to the [Cp*Ru] fragment than to the triple bond to be gem-hydrogenated was usually met with failure; likewise, competing cycloisomerization was observed for certain enyne substrates (for representative examples, see the Supporting Information).22 On the other hand, it is noteworthy that all products formed by “hydrogenative metathesis” are trisubstituted olefins, which are not necessarily easy to make even with the aid of classical (first-generation) Grubbs-type catalysts; this is particularly true for trisubstituted butenolides or cyclic ketones such as 5–8, which posed significant challenges in the past but are well within the reach of this new methodology (Scheme 3).12,23,24
Scheme 3. Trisubstituted Cyclic Ketones and Butenolides Formed by “Hydrogenative Metathesis”.
Total Synthesis of Sinularones E and F by Hydrogenative Metathesis
To assess this valuable aspect more closely, two unusual cyclopentenone derivatives isolated from a Sinularia octocoral collected off Hainan Island were chosen for a first application of the novel hydrogenative metathesis in the realm of natural product chemistry.25,26 The assignment of the relative stereochemistry of sinularones E and F, which differ only in the configuration of C7, is solely based on computed 13C NMR shifts and computed specific rotations and hence mandates experimental confirmation. From a synthetic viewpoint, these targets provide a rigorous testing ground for the new methodology because the gem-dimethyl group flanking the trisubstituted alkene to be metathesized entails notable steric hindrance (Scheme 4). Since hydrogenative metathesis requires a noncoordinating solvent, the presence of a cis-configured tetrahydrofuran moiety in the target compound was another point of concern, because it might entail the formation of an unfavorable six-membered chelate complex with the transient Lewis acidic carbene center.27
Scheme 4. Structures and Retrosynthetic Analysis of Sinularones E and F.

The required diastereomeric enyne substrates were readily attained starting from the cheap furan 11 (Scheme 5). On hydrogenation over Rh/Al2O3, saturation of the aromatic ring precedes reduction of the ketone and hence allows the cis-configured tetrahydrofuran 12 to be obtained on a gram scale, provided the reaction is properly monitored (see the Supporting Information).28,29 The subsequent addition of acetylide needed careful optimization because 12 is readily enolized on treatment with commercial HC≡CMgBr at low temperature, leading to a complex mixture comprising the self-aldolization product 13 (mixture of isomers) as major constituent. The problem was remedied by the addition of LaCl3·2LiCl to temper the basicity of the organometallic reagent.30,31 Under these conditions, the propargyl alcohols 14 were obtained in 74% combined yield (66% on a 7.4 mmol scale). As one might expect from a highly oxophilic Lewis acid, the major diastereomer 14a formed in the presence of La(3+) corresponds to the Cram-chelate adduct.32 The epimers 14a and 14a′ are readily separable, and both yielded single crystals suitable for X-ray diffraction (see the Supporting Information);33 as the corresponding carbinol center in sinularones E and F is the one that needs experimental proof, a sound basis for the completion of the total synthesis was reached.
Scheme 5.

Reagents and conditions: (a) H2 (1 atm), Rh/Al2O3 (1 mol %), Et2O, 59% (16 mmol scale); (b) HC≡CMgBr, THF, −78 °C → RT, 44% (dr = 1.4:1); (c) LaCl3·2LiCl, THF, then HC≡CMgBr, 0 °C, 66–74% (dr = 4:1) (gram scale); (d) SEMCl, iPrNEt2, CH2Cl2, 94%; (e) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C → RT, 97%; (f) TMSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 92%; (g) nBuLi, THF, 0 °C, then 18, −78 °C (−50 °C) → RT, 52% (15b), 61% (15c), 65% (15d); (h) [Cp*RuCl]4 (2 mol %), H2 (1 atm), 1,2-dichloroethane, 70 °C, 68% (NMR, dr = 6:1); (i) 19 (10 mol %), nBu4NCl (12 mol %), H2 (1 atm), 1,2-dichloroethane, 70 °C, 76% (R = TMS); (j) TBAF, THF, 0 °C, 88%; (k) TMSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 89%; (l) nBuLi, THF, 0 °C, then 18, −78 °C → 0 °C, 78%; (m) 19 (10 mol %), nBu4NCl (12 mol %), H2 (1 atm), 1,2-dichloroethane, 70 °C, 67%; (n) TBAF, THF, 0 °C, 97%.
In order to steer the projected carbene formation to the distal site of the triple bond of enyne 15 and hence ensure that the envisaged hydrogenative metathesis reaction proceeds with the desired regioselectivity, the tert-alcohol group must be protected.34 A priori, different silyl ethers or a SEM-acetal are adequate for this purpose and should be cleavable after the event under conditions that are sufficiently mild not to destroy the elimination-prone tert-alcohol in the resulting products. Therefore, compound 14a was transformed into 14b-d and these building blocks then coupled with Weinreb amide 18, which in turn is available from methyl vinyl ketone by following a literature route.35
With the three different enynes 15b–d in hand, it was possible to test the hydrogenative metathesis as the key step of the synthesis. We were surprised to find that hydrogenation of the SEM derivative 15b under standard conditions using [Cp*RuCl]4 as the catalyst furnished only trace amounts of the desired cyclopentenone; rather, compound 16 formed by C–H insertion of the transient carbene into the methylene subunit of the SEM group was the major product. While no such reactivity had been noticed before for any acetal-protected model compound,9 this finding prompted us to revisit catalytic hydrogenative C–H functionalization (see below). The silyl ether analogues 15c,d were both much better behaved, especially when complex 19 bearing a less electron rich CpX ligand was used as the catalyst in combination with nBu4NCl.36 Under these conditions, competing over-reduction was almost completely suppressed (<5%) and the desired metathesis products were obtained in 78% and 76% yields, respectively, despite the crowded situation. The cyclization of the diastereomeric enyne 17 was similarly productive. Deprotection with TBAF furnished sinularones F (10) and E (9), the NMR spectra of which matched the reported data (for details, see the Supporting Information).25 Therefore, we conclude that the assignment of these diastereomeric marine secondary metabolites, which had been largely based on a comparison of simulated and recorded spectra,25 is indeed correct.
Hydrogenative C–H Insertion
As previously reported, both hydrogenative metathesis and hydrogenative cyclopropanation reactions of several acetal-containing substrates are high yielding;9 side products derived from competing C–H functionalization have not been noticed,37 despite the fact that related piano-stool ruthenium carbenes formed by other means have previously been used exactly for this purpose.21,38,39 To explain why C–H insertion interfered with productive metathesis upon hydrogenation of enyne 15b, we reasoned that the flanking carbonyl group might play a pivotal role, in that it renders the transient carbene more highly electrophilic.
Several substrates were made to test this hypothesis (Scheme 6). Hydrogenations of ynones 20a,b and 23a,b bearing different protecting groups on the propargylic alcohol substituent under standard conditions all led to C–H insertion with formation of the corresponding tetrahydrofuran derivatives. The reactions proceed with high diastereoselectivity in favor of the cis isomers, as deduced from the NMR data; for 24b, the assignment was confirmed by X-ray crystallography (see the Supporting Information). In contrast, the analogous ester (25) or amide (26) derivatives failed to afford the corresponding C–H-insertion products but merely succumbed to (over)reduction. Computations for substrate 20a suggest that the insertion reactions proceed via the concerted transition state H (for details, see the Supporting Information); a direct interaction of the carbonyl oxygen atom with the site of C–H bond cleavage, as previously proposed for related transformations involving ruthenium carbenes generated by diazo decomposition,40 does not seem to play a role in this case.
Scheme 6.

Reagents and conditions: (a) [Cp*RuCl]4 (2 mol %), H2 (1 atm), 1,2-dichloroethane, 3 h, 70 °C.
The product equilibrates in CDCl3 to reach this dr after 5 h.
Hydrogenative Metathesis of Enynes without Propargylic Substituents
Silylated alkynes are another class of substrates amenable to gem-hydrogenation.41 As one might expect, the reaction is highly regioselective in that the ruthenium carbene I is formed at the internal position. This regiochemical course likely reflects the hyperconjugation of the silyl group with the emerging electrophilic carbene center and predisposes the resulting α-silylated carbene to subsequent 1,2-silyl migration with the selective formation of alkenylsilanes.41
It was shown that the migratory aptitude of the silyl group is strongly dependent on the nature of the substituents at silicon in that one aryl substituent was found necessary to render the rearrangement facile.41 In consideration thereof, we reasoned that it should be possible to outperform the 1,2-silyl shift and engage an α-silylated carbene of type I primarily formed in productive metathesis or cyclopropanation, depending on the substitution of the olefin. The examples compiled in Scheme 7 show that this is indeed the case. Hydrogenative metathesis hence opens an entry into variously functionalized allylsilanes. The hydrogenation of enyne 35 proves that the underlying concept is not limited to silylated substrates. As expected, the reaction worked well on a gram scale, as illustrated by the formation of product 38 (the structure of this compound in the solid state is contained in the Supporting Information). In this particular reaction, complex 39 was isolated as a byproduct, which is presumably formed by oxidative cyclization of two ynoates followed by reductive elimination of the resulting ruthenacycle to give the cyclobutadiene ligand.42,43 While this bias is perhaps unsurprising for an electron-deficient substrate, it constitutes a major catalyst deactivation pathway because it traps no less than ∼50% of the initial ruthenium loading. Moreover, the fact that 39 is obtained as a single regioisomer is deemed highly significant.
Scheme 7.

Reagents and conditions: [Cp*RuCl]4 (2 mol %), H2 (1 atm), 1,2-dichloroethane, 3h, 70 °C. Reagents and conditions in the case of 28b: 19 (20 mol %), nBu4NCl (25 mol %), H2 (1 atm), 1,2-dichloroethane, 8 h, 70 °C.
The structure of 39 in the solid state (Figure 1) features surprisingly close contacts between the chloride ligand on ruthenium and the neighboring silyl groups.44,45 Specifically, the distances between the −Cl substiuent and the H atoms directed toward it are all notably short (2.8–3.1 Å; sum of van der Waals radii: 3.6 Å)46a and likely mediate, at least in part, the interaction. The Cl1···Si1 (3.43 Å) and Cl1···Si2 (3.56 Å) distances are also well below the sum of the van der Waals radii (4.22,46a 3.85 Å46b). Interestingly, they fall into the range previously computed for an attractive through-space [Ru–Cl]···Si contact of this type (3.4 Å);48 however, the C–Si–C angles (average 108.5°) hardly deviate from the ideal tetrahedral geometry. The 29Si NMR shift might also mirror the interaction, although this data point cannot be considered a firm proof (for details, see the Supporting Information).45,48
Figure 1.
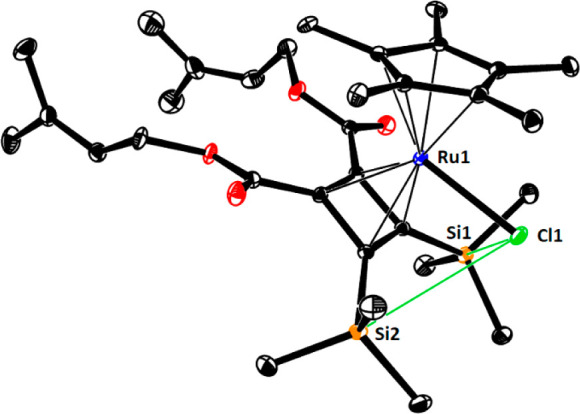
Structure of complex 39 in the solid state. Only one of the two independent molecules in the unit cell is shown, and hydrogen atoms are omitted for clarity. The green lines indicate attractive interactions between the polarized [Ru–Cl] bond and the −Si(CH3)3 groups, partially mediated via the hydrogen atoms (cf. the text). For the full structure, see the Supporting Information.
Under the proviso that attractive interactions of this type are operative during the upload of the substrate onto the catalyst and the ensuing transition state leading to the metallacycle, the head-to-head alignment of the silyl groups in 39 is readily explained. This observation has implications beyond this specific case: such interligand interactions had previously been invoked to explain the regiochemical course of trans-hydrosilylation reactions of unsymmetrical alkynes (Scheme 8, top) and related reactions catalyzed by [Cp*RuCl]-based catalysts.47−49 Moreover, they allow an otherwise unexplained observation reported in the literature to be rationalized: why the coupling of allyl alcohol with TMS-acetylene on the one hand and tert-butylacetylene on the other hand proceed at the opposite end of the triple bond despite the comparable steric demand (Scheme 8, bottom).50,51 Complex 39 hence corroborates the notion that interligand interactions involving a polarized [Ru–Cl] unit are a powerful yet perhaps still underappreciated control element for different ruthenium-catalyzed transformations.21a,43,47−49
Scheme 8. Examples from the Literature in which the Reaction Outcome Is Likely Determined by Cl···Si Interactions.
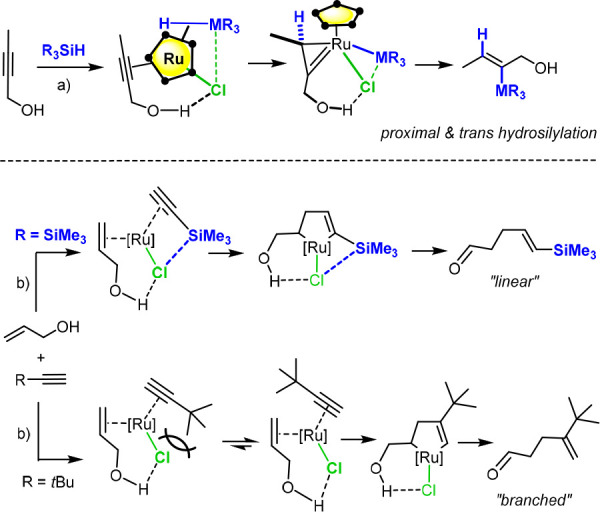
Reagents and conditions: (a) [Cp*RuCl]4 cat., CH2Cl2 (or pentane), see ref (47); (b) [Cp*Ru(cod)Cl] cat., neat, see ref (50). • denotes a CMe edge of the Cp* ring in the Newman projection of the loaded catalyst and the ensuing ruthenacyclopropene.
A Loaded Carbene Complex
As a prelude for the mechanistic studies, we sought rigorous confirmation that hydrogenative metathesis as described herein proceeds via discrete ruthenium carbenes formed by gem-hydrogeantion as the key reactive intermediates. To this end, enyne 40—which is just one methylene group shorter than the model substrate 1a—was hydrogenated with stoichiometric [Cp*RuCl]4 (0.25 equiv) as the precatalyst with the hope of forming a metastable carbene complex amenable to full characterization (Scheme 9):52 the shortened tether should retard or even prevent intramolecular metathesis from occurring (a strained cyclobutene ring would be formed), whereas the bulk in the first coordination sphere about the metal disfavors competing intermolecular reactions.
Scheme 9. Preparation of a Loaded Carbene Complex by gem-Hydrogenation.

This expectation proved correct in that gem-hydrogenation of enyne 40 led to the clean formation of an intermediate 41 (≥95%, NMR), which proved stable enough in solution for full characterization by NMR at low temperature and could even be isolated in the form of single crystals suitable for X-ray diffraction.53 The unit cell contains no less than six independent molecules, and structure elucidation faced an additional complication of a slight modulation of the structure along the c unit cell axis in that the individual molecules are partially replaced by their enantiomers (note that the complex is chiral-at-metal; for a discussion, see the Supporting Information); despite these complications, the structural attributes of the complex in the solid state appear to be unambiguous (Figure 2). As expected, gem-hydrogenation steered by the −OMe substituent has led to the regioselective formation of a piano-stool ruthenium carbene at the distal alkyne C atom. In the resulting 18-electron complex 41, the tethered olefin, though trisubstituted, has displaced the ether ligand in complex J, which is thought to be initially formed by virtue of the steering −OMe substituent. The C4–C5 double bond is almost orthogonal to the C1–Ru1 carbene unit and experiences significant back-donation of electron density from the metal into the empty π* ligand orbital, as manifested in the elongated C4–C5 bond (1.39(4) Å) and in the fact that the two methyl substituents (C6, C7) appear slightly out of plane; both features indicate notable rehybridization of the olefin. The structure in solution (CD2Cl2) is almost certainly very similar: only one of the two methyl groups on the olefin shows a NOE contact with the methyl substituents of the Cp* ligand, suggesting that the orientation perpendicular to the C1–Ru1 vector observed in the solid state is retained (Scheme 9). The signal at δC 368.4 ppm confirms the (Fischer) carbene nature of 41;54 it is actually more deshielded than related half-sandwich ruthenium carbenes of type J in which the lateral −OMe substituent remains ligated to the metal center.3,7 The massive high-field shift of the alkene signals to δC 79.4/79.7 ppm proves that the π system is tightly bound to the metal, even at ambient temperature, whereas the −OMe is off.55 This detailed structural portrayal of complex 41 forms a calibration point for the mechanistic discussion summarized below.
Figure 2.
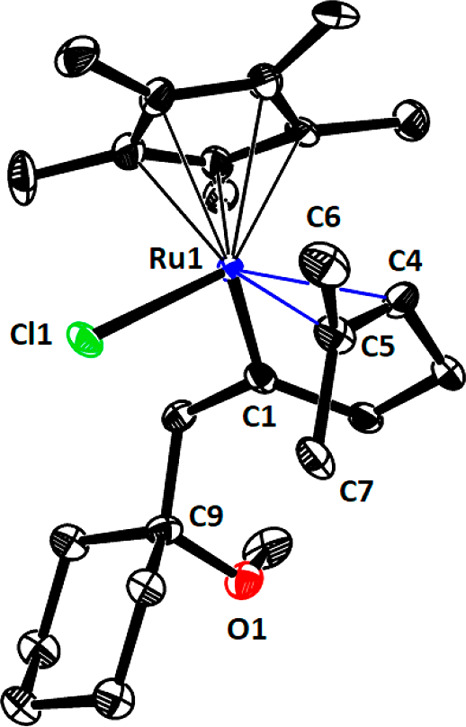
Structure of one of the six independent molecules of complex 41 in the solid state. Hydrogen atoms and slight disorder of Ru1 and Cl1 over two positions are not shown for clarity; for the entire structure and a brief discussion, see the Supporting Information.
Fate of the Secondary Carbene
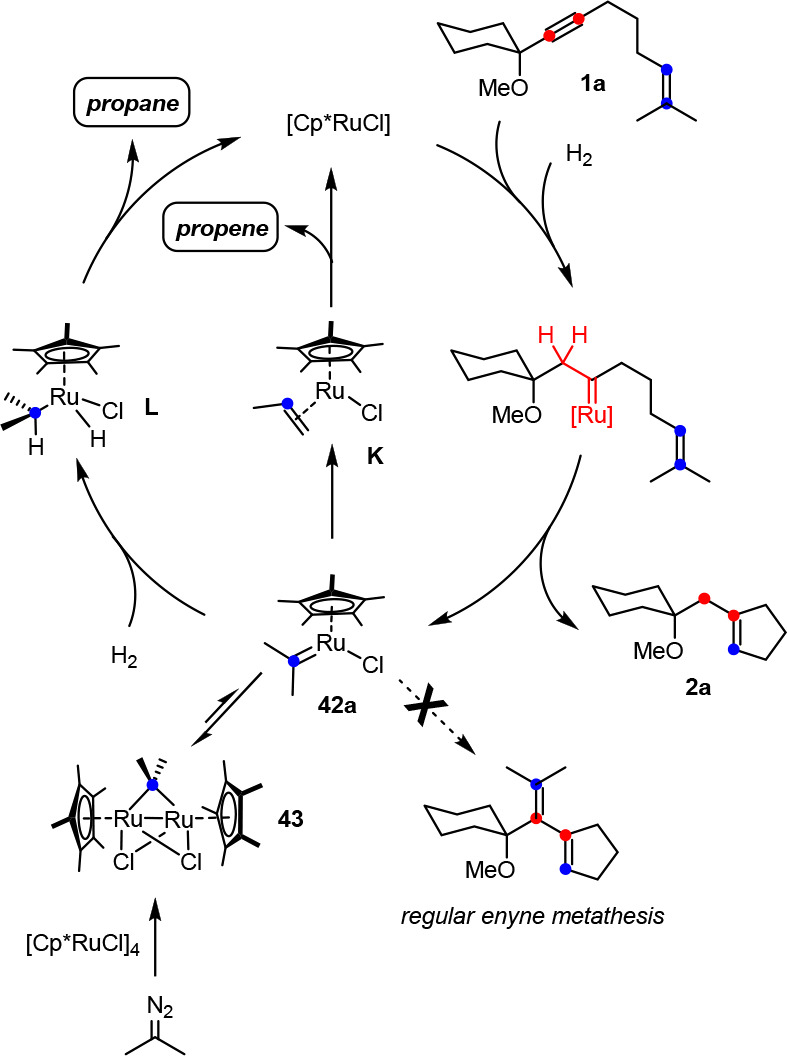
The isolation of 41 is consistent with the hypothesis that [Cp*RuCl] is the catalytically active species that entails gem-hydrogenation and is accountable for the conversion of an enyne such as 1a into a cycloalkene 2a as described herein. The actual metathesis step, however, generates a secondary carbene of type 42, which must be reconverted into this active catalyst at a rate that is faster than its addition to the triple bond of unreacted substrate; otherwise, “hydrogenative metathesis” transmutes into an ordinary enyne metathesis with formation of a 1,3-diene product.12,13
Privileged substrates such as 1a with two methyl groups on the alkene release 42a as the secondary carbene (Scheme 10). We have previously shown that 42a traps free [Cp*RuCl] to give the binuclear complex 43, held together by two bridging chloride ligands and the now equally bridging carbene unit (see the Supporting Information).9,5643, however, is most likely a dormant rather than active species, and the turnover-limiting step likely comes after its formation.9 At elevated temperatures, which are usually necessary for hydrogenative metathesis to proceed at a reasonable rate, 43 is in equilibrium with monomeric 42a: it is this latter species which reacts under a hydrogen atmosphere via K and L to give propene and propane, respectively, which represent by far the major components of the volatile fraction, as confirmed by headspace GC analysis (see the Supporting Information).9
Scheme 10. Fate of the Secondary Carbene in the Case of an Enyne Substrate with a Dimethylated Olefin.
Alkene Substitution as the Key Determinant
The representative example depicted in Scheme 2 had shown that the reaction outcome is critically dependent on the degree of substitution of the alkene unit of a given substrate. For this perplexing result, it was also deemed necessary to investigate the influence of the nature of the substituents. In this context it is important to note that changes in the substitution pattern do not only alter the sterics about and electronics of the double bond to be metathesized or cyclopropanated; in the case of metathesis, the substituents also profoundly affect the constitution and stability of the secondary carbene 42, which must be recycled into the actual catalyst sufficiently quickly.
The examples compiled in Schemes 11 and 12 are relevant in mechanistic terms. First and foremost, the outcome of the hydrogenation with [Cp*RuCl] is obviously not a matter of whether the double bond of the enyne is electron-rich or electron-deficient. The comparison of substrates 1e with an enol ether and 1f containing a gem-difluoro group at the terminus illustrates this aspect: though very different in electronic terms, both substrates undergo metathesis to afford product 2a. The resulting secondary carbenes 42e,f both carry heteroatom substituents as stabilizing π-donor ligands;54,57,58 this thermodynamic aspect notwithstanding, the good leaving-group properties of the heteroelements open low-lying decomposition pathways which outcompete reconversion into the propagating species.59 Therefore, a stoichiometric amount of [Cp*RuCl]4 was necessary to reach full conversion in both cases; as one might expect, the same is true for the alkenyl chloride derivative 1g.60 All other reactions shown in Scheme 11 are catalytic processes that proceed well under the standard conditions outlined above.
Scheme 11.
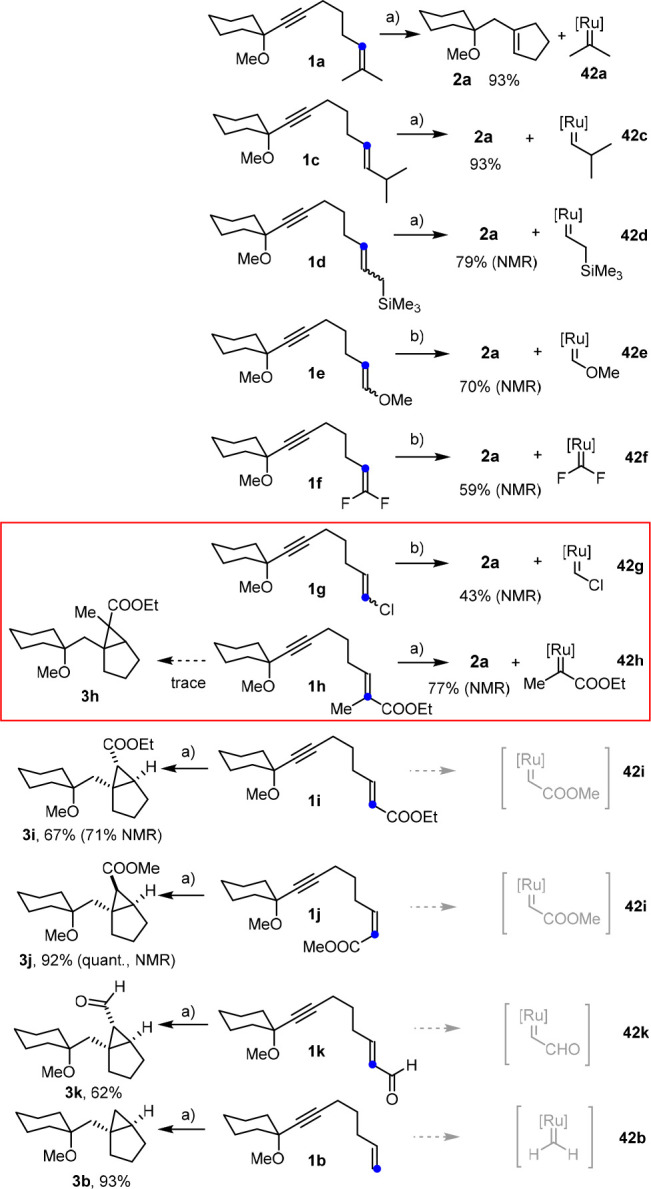
Reagents and conditions: (a) [Cp*RuCl]4 (2 mol %), H2 (1 atm), 1,2-dichloroethane, 3 h, 70 °C; (b) [Cp*RuCl]4 (0.25 equiv), H2 (1 atm), 1,2-dichloroethane, 3 h, RT. The color-coded C atom denotes the more nucleophilic position of the alkene. The secondary carbenes are the proposed (catalytic) intermediates, which are shown because their stability and fate are thought to determine the course of the overall transformation (metathesis versus cyclopropanation; see the text). [Ru] = Cp*RuCl.
Scheme 12.
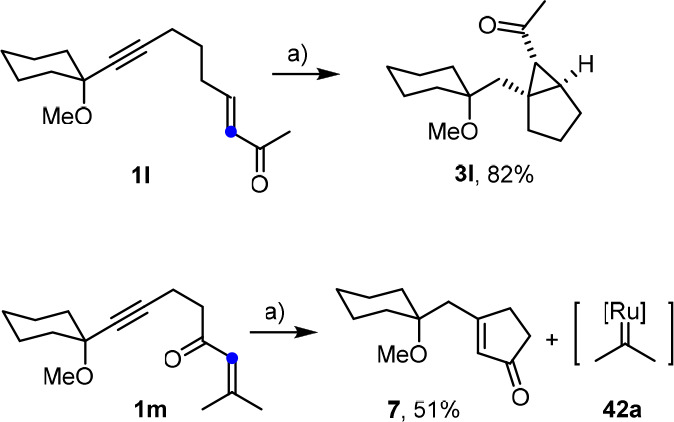
Reagents and conditions: (a) [Cp*RuCl]4 (2 mol %), H2 (1 atm), 1,2-dichloroethane, 3 h, 70 °C. The color-coded C atom denotes the more nucleophilic position of the alkene.
The divergent behavior of the difluoroalkene 1f and compound 1b containing an ordinary terminal olefin is also informative: because hydrogen and fluorine are not overly different in size but the outcome of the reactions is opposite, one must conclude that the product-determining step of the catalytic cycle is (largely) governed by electronic rather than steric factors. This notion is corroborated by a comparison of 1a, 1c, and 1d, which shows that disubstitution of the olefin is also not mandatory. Equally remarkable is the striking switch observed for enoates 1h and 1i, which differ only in the presence or absence of a single methyl group: whereas the former undergoes hydrogenative metathesis to give 2a and just a trace of what seems to be the corresponding cyclopropane, the nor-methyl derivative 1i succumbs to clean hydrogenative cyclopropanation with formation of 3i. The reaction is stereospecific in that the Z isomer 1j provides the diastereomeric cyclopropane 3j, which speaks for a highly ordered selectivity-determining transition state (for details, see Computational Studies and Mechanistic Discussion). The compatibility of an aldehyde group as shown by the hydrogenative conversion of 1k into 3k is yet another noteworthy aspect.
Additional information can be deduced from the reactions shown in Scheme 12: although both substrates are enones, hydrogenation under standard conditions takes an entirely different course in that 1l furnishes cyclopropane 3l as a single diastereomer, whereas 1m undergoes metathesis with formation of the cyclopentenone derivative 7.
At first sight, these examples speak for a correlation between the polarization of the double bond and the reaction outcome. If one takes the chemical shift as a proxy,61 the trisubstituted alkene in 1a is more nucleophilic at the internal position, whereas its cousin 1b is actually more nucleophilic at the terminus. If then—in accordance with “Markovnikov’s rule”—the prime site of attack of the olefin onto an electrophilic Fischer-type carbene changes,62 one might assume that 1b engages in a 6-endo-trig transition state that leads to the cyclopropane by an outer-sphere process. In contrast, 1a seems poised for an ordinary Chauvin-type mechanism via a [2 + 2] cycloaddition/cycloreversion mechanism that ultimately results in metathesis.12 Under this proviso, one would expect that all enynes of type 1, in which the position of the alkene proximal to the ruthenium carbene is the more nucleophilic site, will undergo metathesis, whereas the opposite polarization results in cyclopropanation. This model concurs with the fact that the hydrogenations of substrates 1e and 1f comprising olefins as different as an enol ether and a difluoroalkene take the same course and both result in metathesis; likewise, it predicts that the ordinary terminal alkene 1b and the much more electron deficient enal 1k both afford the corresponding cyclopropanes.
However, Scheme 11 shows two cases, which cast doubt on the view that this interpretation assuming a competing outer-sphere/inner-sphere mechanism captures the full picture. Specifically, alkenyl chloride 1g and the trisubstituted enoate 1h both show the “distal pattern” yet undergo metathesis (at least as the major reaction channel).63 In this context, it is also necessary to re-evaluate the small number of related examples documented in the literature in which stoichiometric reactions of preformed Fischer carbene complexes with enynes resulted in either cyclopropanation or metathesis depending on the substitution pattern (and/or the solvent).64−66 Fully convincing explanations have not been published, but it has been speculated that the stability of the secondary carbene generated in the case of metathesis might play a role in determining the reaction outcome. Although this looks—a priori—like a thermodynamic argument, it cannot be discounted in the first place but deserves further consideration.
Computational Studies and Mechanistic Discussion
To complement the experimental data and draw a more accurate picture of the underlying mechanism(s), detailed computational studies were carried out using DLPNO-CCSD(T)/def2-TZVPP single-point energies on top of B3LYP-D3/def2-TZVP(-f) geometry optimization.67−70 The perturbative triples correction was calculated using the so-called semicanonical approximation.71 Solvation effects were included at the DFT level using the implicit solvation model C-PCM (CH2Cl2).72,73 An initial exploration of the chemical space74 was carried out using the semiempirical tight-binding based quantum chemistry method GFN2-xTB.75
Substrates 1a, 1b, 1e, 1f, and 1i were chosen for this computational survey. First, extensive conformation sampling showed that in all cases the tethered olefin is η2-ligated to the Ru center in the most stable carbene complex A formed during the gem-hydrogenation step, independent of the electronic character of the double bond. Complexes of type A, as the starting point for the computations, nicely correspond to compound 41 characterized by X-ray diffraction (see Figure 2). The further evolution into the cyclopropane by an outer-sphere mechanism, as considered in our preliminary communication with the explicit caveat that more detailed scrutiny is necessary,9,62 has a prohibitively high activation barrier (>35 kcal mol–1) and can hence be safely disregarded.
Rather, A first converts into a “kite-shaped” metallacycle A′before the pathway bifurcates, in which all three C atoms entertain bonding interactions to the metal (Figure 3). Depending on the specific substitution pattern, A′ is either a regular minimum on the potential energy surface or just an “inflection point”, as in the case of enyne 1b with the terminal alkene shown in Figure 3.76 This distorted metallacycle A′ can cyclorevert via TSAB to afford the (invariant) metathesis product C and the corresponding secondary carbene; alternatively, it can evolve via TSAD into the “regular” metallacycle D, which undergoes reductive elimination and releases cyclopropane F (this step is facilitated by agostic interactions in the transition state TSDE and the resulting adduct complex E).77 For enyne 1b with the terminal alkene, TSAD leading to the cyclopropane is 1.9 kcal mol–1 lower in Gibbs free energy than TSAB en route to the metathesis product; this computational result is in excellent agreement with the experimentally observed outcome.78 For the highly ordered character of the transition state leading to D and of the subsequent reductive elimination step, it is readily understood why cyclopropanation reactions of substituted alkenes proceed stereospecifically (compare 1i and 1j in Scheme 11; additional examples are contained in ref (9)).
Figure 3.
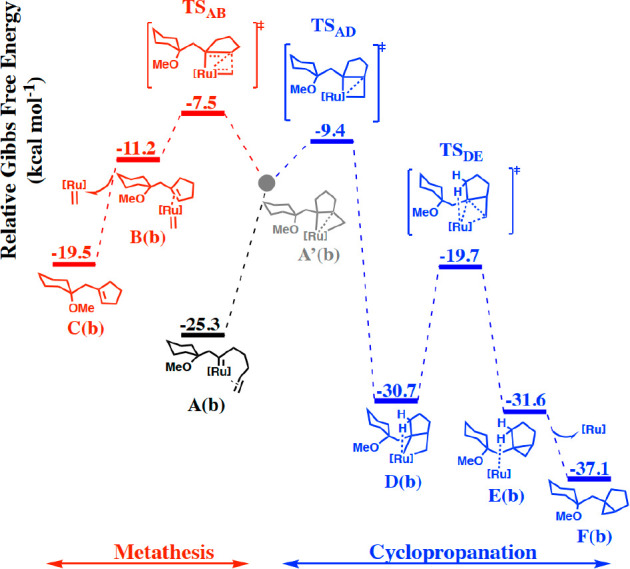
Metathesis versus cyclopropanation pathways for a carbene complex derived from enyne 1b with a terminal alkene; as a reference energy we used the substrate and the [Cp*RuCl] catalyst. For the sake of simplicity, only the key intermediates and transition states along the minimum energy pathways are shown.
This basic scenario remains the same for all substrates investigated (1a, 1b, 1e, 1f, and 1i), but the substituents at the alkene terminus and the electronic character of the alkene massively affect the barrier heights. The case of the “isosteric” difluoroalkene derivative 1f is representative (Figure 4; for the other cases, see the Supporting Information): the distorted metallacycle A′(f) formed before the pathways bifurcate is a true intermediate rather than an inflection point. Once it is reached, metathesis is almost barrierless and outcompetes cyclopropanation; this computed outcome is again in accord with experiment.
Figure 4.
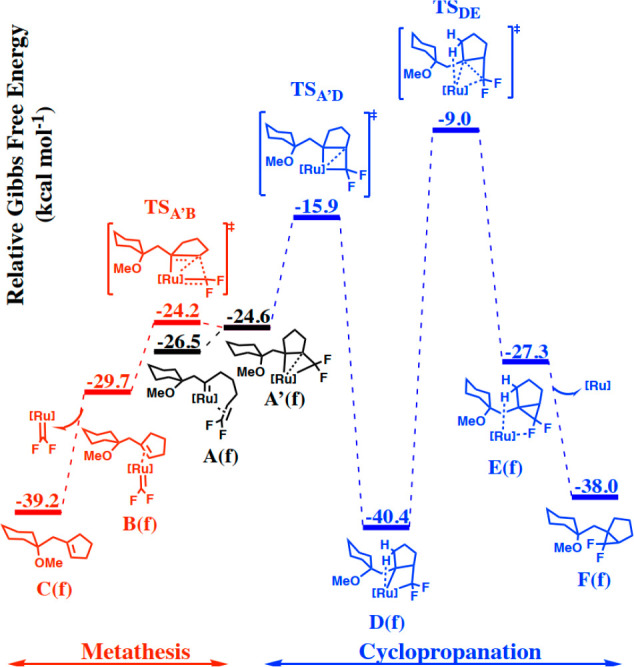
Metathesis versus cyclopropanation pathways for a carbene complex derived from enyne 1f with a difluoroalkene moiety; as a reference energy, we used the substrate and the [Cp*RuCl] catalyst.
It is significant that the barriers for cyclopropanation are fairly “insensitive” even for enynes comprising olefins as different as a terminal and a difluorinated alkene (15.9 and 10.6 kcal mol–1, respectively).79 In striking contrast, the barriers for metathesis are massively affected in that ΔG⧧ decreases from 17.8 kcal mol–1 for 1b to only 2.3 kcal mol–1 for 1f. The trend that the barrier for cycloreversion of the distorted metallacycle and hence metathesis is particularly responsive to changes of the substitution pattern and/or polarization of the olefin (as manifested in the NPA charges) pertains to all substrates investigated (Table 1): in essence, it is this effect that determines the outcome.80
Table 1. Computed Barriers That Determine the Evolution of the Ruthenium Carbene Complex A Primarily Formeda.
|
Q |
outcome |
|||||||
|---|---|---|---|---|---|---|---|---|
| substrate | terminus | at C1 | at C2 | ΔG⧧(TSAB)b (kcal mol–1) | ΔG⧧(TSAD)c (kcal mol–1) | ΔG(A→C)d (kcal mol–1) | predicted | exptl |
| 1b | –CH=CH2 | –0.39 | –0.17 | 17.8 | 15.9 | +5.8 | cycloprop | cycloprop |
| 1i | –CH=CHCOOEt | –0.32 | –0.08 | 21.4 | 11.2 | +6.6 | cycloprop | cycloprop |
| 1a | –CH=CMe2 | +0.02 | –0.20 | 10.8 | 21.6 | –4.1 | metathesis | metathesis |
| 1f | –CH=CF2 | +0.72 | –0.36 | 2.3 | 10.6 | –12.7 | metathesis | metathesis |
| 1e | –CH=CH(OMe) | +0.12 | –0.28 | 3.7 | 15.5 | –7.7 | metathesis | metathesis |
Q denotes the NPA (natural population analysis) charge at the “terminal” (C1) and internal (C2) atom of the olefin.
TSAB refers to the cycloreversion step and hence the reaction channel resulting in metathesis.
TSAD is the first transition state toward cyclopropanation, which may or may not be the highest barrier of this pathway; see the Supporting Information.
Thermochemistry associated with the transformation of the primary carbene into the metathesis product 2a together with the corresponding secondary carbene (C).
This computational result warrants further consideration, as does that fact that the path leading to the cyclopropane passes through two distinctly different metallacycles, whereas metathesis involves only one. It is well established in the literature that metallacyclobutanes in general fall into two different categories (Figure 5).81−83 One type features a significant agostic interaction between the metal and the Cβ atom, which in turn results in a short M···Cβ contact, weakened Cα–Cβ bonds, and notable alkylidene character at Cα/Cα′; metallacycles of this sort are prone to cycloreversion. The other type of metallacyclobutanes, in contrast, lacks the M···Cβ agostic interaction and the other characteristics that it entails; most notably, their Cα atoms do not have any significant alkylidene character. Intermediates of this latter type easily succumb to β-H-elimination reactions or reductive elimination.
Figure 5.
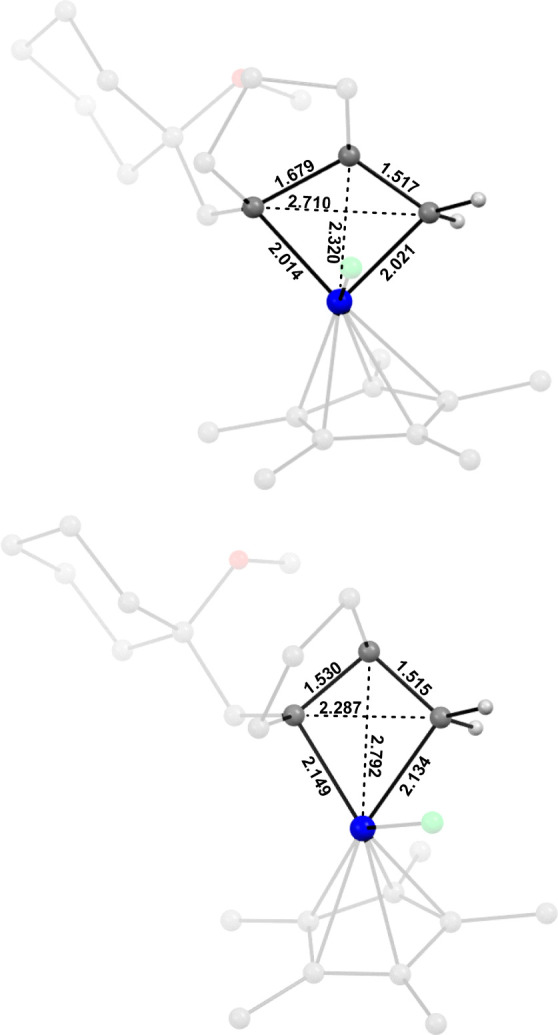
Computed structures of the ruthenacyclobutane entites of complexes A′(b) (top) and D(b) (bottom) derived from enyne 1b with the terminal alkene. Distances are given in Å.
Figure 5 depicts the computed ruthenacyclobutane substructure of complexes A′(b) and D(b) derived from enyne 1b. From the overall geometries and metric data it is obvious that they correspond very well to the two extremes: for the “kite-shaped” metallacycle A′(b) to transform into D(b), it must undergo a change in bonding (loss of the β-agostic interaction) accompanied by a significant change in geometry, which actually brings the Cα/Cα′ atoms closer together (for further details, see the Supporting Information). Only after D(b) is reached can reductive elimination with formation of the cyclopropane occur. It is intuitive that any substitution pattern on the ring that increases the alkylidene character of the Ru–Cα bond of A′(b) will disfavor this process relative to [2 + 2] cycloreversion: a π-donor substituent and/or an appropriately polarized double bond fall into this category.54−57
It is equally important to recognize that a (heteroelement) substituent at Cα that increases the carbene (alkylidene) character of the metallacycle A′ and therefore favors metathesis also translates into the thermodynamically more stable secondary carbene 42. The significant lowering of the barrier heights of TSAB on the metathesis pathway can hence be seen as an illustration of the Bell–Evans–Polanyi principle, which links the energy of the transition state to the energy of the subsequent intermediate (Figure 6).84 Since all hydrogenations shown in Scheme 11 form the same metathesis product 2a, it is the stability of the secondary carbene 42a–k which makes the key difference because it manifests itself already in the transition state: in cases in which the secondary carbene is destabilized (shaded in gray), TSAB is expected to be high in energy and metathesis is hence prevented.
Figure 6.

Illustration of the Bell–Evans–Polanyi principle. The barrier height for cycloreversion (ΔG⧧(TSAB)) correlates with the stability of the secondary carbene complex released together with the invariant metathesis product ((ΔG(A → C)): a more exergonic cycloreversion process exhibits a lower activation barrier (the energies relative to the initial carbene complex A).
In a view through this lens, one may conclude that the original proposals made in the literature that the polarization of the double bond of the substrate or the stability of the secondary carbene determines the course of the reaction are both correct in a way, even though it took the computations to understand why that is the case. The insight that the Bell–Evans–Polanyi principle intimately connects the substitution pattern with the critically responsive barrier height of the metathesis pathway results in a rather intuitive model based on an assessment of the substrate’s ground state, which allows the outcome of the reactions to be predicted with confidence; it forms a sound basis that should encourage the practitioners to implement gem-hydrogenations into increasingly complex settings. In any case, ongoing work in this laboratory intends to widen the scope of this unusual novel entry into the realm of transition-metal carbenes and to refine our understanding of the underlying principles.
Conclusions
The gem-hydrogenation of internal alkynes is a fundamentally new transformation that gives access to discrete metal carbene complexes for use in synthesis. For substrates that contain a tethered alkene, the piano-stool carbene intermediates derived from [Cp*RuCl] as the privileged catalyst can undergo cyclopropanation or metathesis reactions. The outcome is largely determined by the substitution pattern of the olefin, which is a perplexing result at first sight. A systematic investigation in combination with computational study allowed the seemingly bewildering results to be sorted and an intuitive model to be deduced, which allows the course of the reaction to be predicted on the basis of the substitution pattern of the substrate. The computational data find excellent correspondence in a half-sandwich carbene complex, in which the olefinic partner is coordinated to the ruthenium center, which represents the “loaded” catalyst and which was fully characterized by spectroscopic and crystallographic means. A first application to the total synthesis of two diastereomeric marine natural products illustrates the virtues of this methodology, as does a new hydrogenative entry into allylsilanes, which are valuable building blocks in organic synthesis.
Acknowledgments
Generous financial support by the MPG is gratefully acknowledged. We thank Dr. A. Guthertz and Mr. T. Biberger for early experimental contributions and discussions and all analytical departments of our Institute for valuable support, especially Mr. J. Rust and Prof. C. W. Lehmann, for excellent X-ray service.
Supporting Information Available
(PDF) Copies of (PDF) Crystallographic data (CIF) The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c07808.
Experimental section containing supporting X-ray structures and crystallographic information, characterization data, and computational details (PDF)
NMR spectra of new compounds (PDF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fürstner A. trans-Hydrogenation, gem-Hydrogenation, and trans-Hydrometalation of Alkynes: An Interim Report on an Unorthodox Reactivity Paradigm. J. Am. Chem. Soc. 2019, 141, 11–24. 10.1021/jacs.8b09782. [DOI] [PubMed] [Google Scholar]
- Leutzsch M.; Wolf L. M.; Gupta P.; Fuchs M.; Thiel W.; Farès C.; Fürstner A. Formation of Ruthenium Carbenes by gem-Hydrogen Transfer to Internal Alkynes: Implications for Alkyne trans-Hydrogenation. Angew. Chem., Int. Ed. 2015, 54, 12431–12436. 10.1002/anie.201506075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthertz A.; Leutzsch M.; Wolf L. M.; Gupta P.; Rummelt S. M.; Goddard R.; Farès C.; Thiel W.; Fürstner A. Half-Sandwich Ruthenium Carbene Complexes Link trans-Hydrogenation and gem-Hydrogenation of Internal Alkynes. J. Am. Chem. Soc. 2018, 140, 3156–3159. 10.1021/jacs.8b00665. [DOI] [PubMed] [Google Scholar]
- Radkowski K.; Sundararaju B.; Fürstner A. A Functional-Group Tolerant Catalytic trans-Hydrogenation of Alkynes. Angew. Chem. 2013, 125, 373–378. 10.1002/ange.201205946. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Davies P. W. Catalytic Carbophilic Activation: Catalysis by Platinum and Gold π-Acids. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]
- A ruthenacyclopropane had previously been proposed as the key intermediate of related trans-hydrosilylation reactions. See:Chung L. W.; Wu Y.-D.; Trost B. M.; Ball Z. T. A Theoretical Study on the Mechanism, Regiochemistry, and Stereochemistry of Hydrosilylation Catalyzed by Cationic Ruthenium complexes. J. Am. Chem. Soc. 2003, 125, 11578–11582. 10.1021/ja034833b. [DOI] [PubMed] [Google Scholar]
- Biberger T.; Gordon C.; Leutzsch M.; Peil S.; Guthertz A.; Copéret C.; Fürstner A. Alkyne gem-Hydrogenation: Formation of Pianostool Ruthenium Carbene Complexes and Analysis of Their Chemical Character. Angew. Chem., Int. Ed. 2019, 58, 8845–8850. 10.1002/anie.201904255. [DOI] [PubMed] [Google Scholar]
- Peil S.; Fürstner A. Mechanistic Divergence in the Hydrogenative Synthesis of Furans and Butenolides: Ruthenium Carbenes Formed by gem-Hydrogenation or via Carbophilic Activation of Alkynes. Angew. Chem., Int. Ed. 2019, 58, 18476–18481. 10.1002/anie.201912161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peil S.; Guthertz A.; Biberger T.; Fürstner A. Hydrogenative Cyclopropanation and Hydrogenative Metathesis. Angew. Chem., Int. Ed. 2019, 58, 8851–8856. 10.1002/anie.201904256. [DOI] [PubMed] [Google Scholar]
- These reactions are counterintuitive in that cyclopropanes are usually cleaved rather than formed by catalytic hydrogenation.
- For a second, light-driven system, see:Biberger T.; Zachmann R. J.; Fürstner A. Angew. Chem., Int. Ed. 2020, 59, 18423–18429. 10.1002/anie.202007030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handbook of Metathesis, 2nd ed.; Grubbs R. H., O’Leary D. J., Eds.; Wiley-VCH: Weinheim, 2015. [Google Scholar]
- a Diver S. T.; Giessert A. J. Enyne Metathesis (Enyne Bond Reorganization). Chem. Rev. 2004, 104, 1317–1382. 10.1021/cr020009e. [DOI] [PubMed] [Google Scholar]; b Hansen E. C.; Lee D. Search for Solutions to the Reactivity and Selectivity Problems in Enyne Metathesis. Acc. Chem. Res. 2006, 39, 509–519. 10.1021/ar050024g. [DOI] [PubMed] [Google Scholar]
- Analogous substrates with a single Me– (Et−) group on the alkene furnished product mixtures; see ref (9).
- Prenyl groups and related 1,1-dimethylated alkenes are good substrates in Grubbs catalysis as well. Compare:; a Bahou K. A.; Braddock D. C.; Meyer A. G.; Savage G. P. Kinetic Benchmarking Reveals the Competence of Prenyl Groups in Ring-Closing Metathesis. Org. Lett. 2017, 19, 5332–5353. 10.1021/acs.orglett.7b02492. [DOI] [PubMed] [Google Scholar]; b Fürstner A.; Dierkes T.; Thiel O. R.; Blanda G. Total Synthesis of (−)-Salicylihalamide. Chem. - Eur. J. 2001, 7, 5286–5298. . [DOI] [PubMed] [Google Scholar]
- a Monnier F.; Vovard-Le Bray C.; Castillo D.; Aubert V.; Derien S.; Dixneuf P. H.; Toupet L.; Ienco A.; Mealli C. Selective Ruthenium-Catalyzed Transformations of Enynes with Diazoalkanes into Alkenylbicyclo][3.1.0]hexanes. J. Am. Chem. Soc. 2007, 129, 6037–6049. 10.1021/ja0700146. [DOI] [PubMed] [Google Scholar]; b Vovard-Le Bray C.; Derien S.; Dixneuf P.; Murakami M. A Direct Synthesis of Alkenyl Alkylidene Bicyclo[3.1.0]hexane Derivatives via Ruthenium(II)-Catalysed Bicyclisation of Allenynes. Synlett 2008, 2008, 193–196. 10.1055/s-2008-1032015. [DOI] [Google Scholar]
- Reference (16a) contains a single example which gave a product mixture formed by cyclopropanation and competing metathesis; interestingly, the substrate contained a 1,1-dimethylated alkene unit. See also:Basato M.; Tubaro C.; Biffis A.; Bonato M.; Buscemi G.; Lighezzolo F.; Lundardi P.; Vianini C.; Benetollo F.; Del Zotto A. Reactions of Diazo Compounds with Alkenes Catalysed by [RuCl(cod)(Cp)]: Effect of the Substituents in the Formation of Cyclopropanation or Metathesis Products. Chem. - Eur. J. 2009, 15, 1516–1526. 10.1002/chem.200801211. [DOI] [PubMed] [Google Scholar]
- Tanaka D.; Sato Y.; Mori M. Unpredicted Cyclization of an Enyne Having a Keto-Carbonyl Group on an Alkyne Using a Ruthenium Catalyst under Ethylene Gas. Organometallics 2006, 25, 799–801. 10.1021/om058053i. [DOI] [Google Scholar]
- Trost B. M.; Breder A.; O’Keefe B. M.; Rao M.; Franz A. W. Propargyl Alcohols as β-Oxocarbenoid Precursors for the Ruthenium-Catalyzed Cyclopropanation of Unactivated Olefins by Redox Isomerization. J. Am. Chem. Soc. 2011, 133, 4766–4769. 10.1021/ja200971v. [DOI] [PubMed] [Google Scholar]
- For [Cp*Ru]-derived ruthenacyclopentadienes as dicarbene equivalents in cyclopropanation, see:; a Yamamoto Y.; Arakawa T.; Ogawa R.; Itoh K. Ruthenium(II)-Catalyzed Selective Intramolecular [2 + 2+2] Alkyne Cyclotrimerizations. J. Am. Chem. Soc. 2003, 125, 12143–12160. 10.1021/ja0358697. [DOI] [PubMed] [Google Scholar]; b Schmid R.; Kirchner K. Ruthenium-Mediated C–C Coupling Reactions of Alkynes – The Key Role of Ruthenacyclopentatriene Complexes. Eur. J. Inorg. Chem. 2004, 2004, 2609–2626. 10.1002/ejic.200400219. [DOI] [Google Scholar]
- For reviews of yet other reactivity of such species, see:; a Padin D.; Varela J. A.; Saá C. Cp*RuCl-Vinyl Carbenes: Two Faces and the Bifunctional Role in Catalytic Processes. Chem. - Eur. J. 2020, 26, 7470–7478. 10.1002/chem.202000391. [DOI] [PubMed] [Google Scholar]; b Padin D.; Varela J. A.; Saá C. Recent Advances in Ruthenium-Catalyzed Carbene/Alkyne Metathesis (CAM) Transformations. Synlett 2020, 31, 1147–1157. 10.1055/s-0039-1690861. [DOI] [Google Scholar]; c Dey S.; De Sarkar S. Synthetic Applications of Vinyl Ruthenium Carbenes Derived from Diazoalkanes and Alkynes. Adv. Synth. Catal. 2017, 359, 2709–2722. 10.1002/adsc.201700622. [DOI] [Google Scholar]; d Pei C.; Zhang C.; Qian Y.; Xu X. Catalytic Carbene/Alkyne Metathesis (CAM): A Versatile Strategy from Alkyne Bifunctionalization. Org. Biomol. Chem. 2018, 16, 8677–8685. 10.1039/C8OB02420K. [DOI] [PubMed] [Google Scholar]; e Torres O.; Pia-Quintana A. The Rich Reactivity of Transition Metal Carbenes with Alkynes. Tetrahedron Lett. 2016, 57, 3881–3891. 10.1016/j.tetlet.2016.07.029. [DOI] [Google Scholar]
- For representative examples showing the tight binding of arenes or dienes to [CpXRu] fragments, see:; a Gill T. P.; Mann K. R. Photochemical Properties of the Cyclopentadienyl(η6-benzene)ruthenium(II) cation. The Synthesis and Reactions of a Synthetically Useful Intermediate: the Cyclopentadienyltris(acetonitrile)ruthenium(II) cation. Organometallics 1982, 1, 485–488. 10.1021/om00063a014. [DOI] [Google Scholar]; b Fagan P. J.; Ward M. D.; Calabrese J. C. Molecular Engineering of Solid-State Materials: Organometallic Building Blocks. J. Am. Chem. Soc. 1989, 111, 1698–1719. 10.1021/ja00187a024. [DOI] [Google Scholar]; c Steines S.; Englert U.; Drießen-Hölscher B. Stereoselective Catalytic Hydrogenation of Sorbic Acid and Sorbic Alcohol with New Cp*Ru Complexes. Chem. Commun. 2000, 217–218. 10.1039/a909355i. [DOI] [Google Scholar]
- Fürstner A. Olefin Metathesis and Beyond. Angew. Chem., Int. Ed. 2000, 39, 3012–3043. . [DOI] [PubMed] [Google Scholar]
- See also:Fürstner A.; Thiel O. R.; Ackermann L.; Schanz H.-J.; Nolan S. P. Ruthenium Carbene Complexes with N,N′-Bis(mesityl)imidazol-2-ylidene Ligands: RCM Catalysts of Extended Scope. J. Org. Chem. 2000, 65, 2204–2207. 10.1021/jo9918504. [DOI] [PubMed] [Google Scholar]
- Shi H.; Yu s.; Liu D.; van Ofwegen L.; Proksch P.; Lin W. Sinularones A-I, New Cyclpentenone and Butenolide Derivatives from a Marine Soft Coral Sinularia sp. and Their Antifouling Activity. Mar. Drugs 2012, 10, 1331–1344. 10.3390/md10061331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent study on another secondary metabolite derived from a Sinularia sp, see:Meng Z.; Fürstner A. Total Synthesis of (−)-Sinulariadiolide. A Transannular Approach. J. Am. Chem. Soc. 2019, 141, 805–809. 10.1021/jacs.8b12185. [DOI] [PubMed] [Google Scholar]
- The formation of kinetically stable chelate complexes is a major issue for first-generation Grubbs catalysts. See:; a Fürstner A.; Langemann K. Total Synthesis of (+)-Ricinelaidic Acid Lactone and of (−)-Gloeosporone Based on Transition Metal Catalyzed C–C-Bond Formations. J. Am. Chem. Soc. 1997, 119, 9130–9136. 10.1021/ja9719945. [DOI] [Google Scholar]; b Fürstner A.; Thiel O. R.; Lehmann C. W. Study Concerning the Effects of Chelation on the Structure and Catalytic Activity of Ruthenium Carbene Complexes. Organometallics 2002, 21, 331–335. 10.1021/om0108503. [DOI] [Google Scholar]
- Compare:Arco M. J.; Trammell M. H.; White J. D. Synthesis of (±)-Nonactic Acid. J. Org. Chem. 1976, 41, 2075–2083. 10.1021/jo00874a001. [DOI] [PubMed] [Google Scholar]
- Zaera F. The Surface Chemistry of Metal-Based Hydrogenation Catalysis. ACS Catal. 2017, 7, 4947–4967. 10.1021/acscatal.7b01368. [DOI] [Google Scholar]
- Krasovskiy A.; Kopp F.; Knochel P. Soluble Lanthanide Salts (LnCl3·2LiCl) for the Improved Addition of Organomagnesium Reagents to Carbonyl Compounds. Angew. Chem., Int. Ed. 2006, 45, 497–500. 10.1002/anie.200502485. [DOI] [PubMed] [Google Scholar]
- See also:Jung M. E.; Pontillo J. Synthetic Approach to Analogues of the Original Structure of Sclerophytin A. J. Org. Chem. 2002, 67, 6848–6851. 10.1021/jo016246j. [DOI] [PubMed] [Google Scholar]
- a Mengel A.; Reiser O. Around and Beyond Cram’s Rule. Chem. Rev. 1999, 99, 1191–1223. 10.1021/cr980379w. [DOI] [PubMed] [Google Scholar]; b Reetz M. T. Chelation or Non-Chelation Control in Addition Reactions of Chiral α- and β- Alkoxy Carbonyl Compounds. Angew. Chem., Int. Ed. Engl. 1984, 23, 556–569. 10.1002/anie.198405561. [DOI] [Google Scholar]
- Interestingly, 14a′ forms a cyclic dimeric aggregate in the solid state via intermolecular hydrogen bonding, whereas 14a leads to a hydrogen-bonded polymeric array; see the Supporting Information.
- Unprotected propargyl alcohols favor carbene formation at the proximal C-atom or provide mixtures; cf. ref (3).
- The Weinreb amide was obtained from the ethyl ester (MeNH(OMe)·HCl, iPrMgCl, THF, – 20°C 95%); see the Supporting Information. For the preparation of the ester, see:Kelkar S. V.; Arbale A. A.; Joshi G. S.; Kulkarni G. H. Claisen Orthoester Rearrangement Reaction with Seondary & Tertiary Allylic Alcohols: Synthesis of 1,2-Seco-Crysanthemates and Their Structural Analogues. Synth. Commun. 1990, 20, 839–847. 10.1080/00397919008052329. [DOI] [Google Scholar]
- Spectroscopic and electrochemical data showed that less electron rich CpX ligands favor gem-hydrogenation and render the resulting carbenes more stable; see ref (7). In line with this notion, the combination of 19 and nBu4NCl often leads to superior results; see ref (9).
- A singular attempt at triggering C–H insertion by alkyne gem-hydrogenation failed; see ref (3).
- a Cambeiro F.; López S.; Varela J. A.; Saá C. Cyclization by Cationic Ruthenium Carbene Insertion into Csp3–H Bonds. Angew. Chem., Int. Ed. 2012, 51, 723–727. 10.1002/anie.201107344. [DOI] [PubMed] [Google Scholar]; b Cambeiro F.; Martínez-Núñez E.; Varela J. A.; Saá C. DFT and Kinetic Monte Carlo Study of TMS-Substituted Ruthenium Vinyl Carbenes: Key Intermediates for Stereoselective Cyclizations. ACS Catal. 2015, 5, 6255–6262. 10.1021/acscatal.5b01333. [DOI] [Google Scholar]
- For a general review, see:Doyle M. P.; Duffy R.; Ratnikov M.; Zhou L. Catalytic Carbene Insertion into C–H Bonds. Chem. Rev. 2010, 110, 704–724. 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- Tortoreto C.; Achard T.; Zeghida W.; Austeri M.; Guénée L.; Lacour J. Enol Acetal Synthesis through Carbenoid C–H Insertion into Tetrahydrofuran Catalyzed by CpRu Complexes. Angew. Chem., Int. Ed. 2012, 51, 5847–5851. 10.1002/anie.201201541. [DOI] [PubMed] [Google Scholar]
- Song L.; Feng Q.; Wang Y.; Ding S.; Wu Y.-D.; Zhang X.; Chung L. W.; Sun J. Ru-Catalyzed Migratory Geminal Semihydrogenation of Internal Alkynes to Terminal Olefins. J. Am. Chem. Soc. 2019, 141, 17441–17451. 10.1021/jacs.9b09658. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Frederiksen M. U.; Rudd M. T. Ruthenium-Catalyzed Reactions – A Treasure Trove of Atom-Economic Transformations. Angew. Chem., Int. Ed. 2005, 44, 6630–6666. 10.1002/anie.200500136. [DOI] [PubMed] [Google Scholar]
- Rummelt S. M.; Cheng G.-J.; Gupta P.; Thiel W.; Fürstner A. Hydroxy-Directed Ruthenium-Catalyzed Alkene/Alkyne Coupling: Increased Scope, Stereochemical Implications, and Mechanistic Rationale. Angew. Chem., Int. Ed. 2017, 56, 3599–3604. 10.1002/anie.201700342. [DOI] [PubMed] [Google Scholar]; corrigendum:Angew. Chem., Int. Ed. 2017, 56, 5652. 10.1002/anie.201703197
- An analogous head-to-head complex derived from TMS-acetylene was characterized by X-ray diffraction, but the −Cl····Si(CH3)3 contact and its possible implications have not been discussed. See:Gemel C.; LaPensée A.; Mauthner K.; Mereiter K.; Schmid R.; Kirchner K. The Substitution Chemistry of RuCp* (temeda)Cl. Monatsh. Chem. 1997, 128, 1189–1199. 10.1007/BF00807250. [DOI] [Google Scholar]
- Direct contacts between −Cl and the Si atom were observed in σ-silane complexes such as [Cp*RuCl(R3P)(σ-H–SiCl3)]. See:Gutsulyak D. V.; Churakov A. V.; Kuzmina L. G.; Howard J. A. K.; Nikonov G. I. Steric and Electronic Effects in Half-Sandwich Ruthenium Silane σ-Complexes with Si–H and Si–Cl Interligand Interactions. Organometallics 2009, 28, 2655–2657. 10.1021/om801192b. [DOI] [Google Scholar]
- a Rahm M.; Hoffmann R.; Ashcroft N. W. Atomic and Ionic Radii of Elements 1–96. Chem. - Eur. J. 2016, 22, 14625–14632. 10.1002/chem.201602949. [DOI] [PubMed] [Google Scholar]; b Bondi A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. 10.1021/j100785a001. [DOI] [Google Scholar]
- Rummelt S. M.; Radkowski K.; Rosca D.-A.; Fürstner A. Interligand Interactions Dictate the Regioselectivity of trans-Hydrometalations and Related Reactions Catalyzed by [Cp*RuCl]. Hydrogen Bonding to a Chloride Ligand as a Steering Principle in Catalysis. J. Am. Chem. Soc. 2015, 137, 5506–5519. 10.1021/jacs.5b01475. [DOI] [PubMed] [Google Scholar]
- Rosca D.-A.; Radkowski K.; Wolf L. M.; Wagh M.; Goddard R.; Thiel W.; Fürstner A. Ruthenium-Catalyzed Alkyne trans-Hydrometalation: Mechanistic Insights and Preparative Implications. J. Am. Chem. Soc. 2017, 139, 2443–2455. 10.1021/jacs.6b12517. [DOI] [PubMed] [Google Scholar]
- Le Paih J.; Vovard-Le Bray C.; Dérien S.; Dixneuf P. H. Ruthenium-Catalyzed Synthesis of Functional Conjugated Dienes via Addition of Two Carbene Units to Alkynes. J. Am. Chem. Soc. 2010, 132, 7391–7397. 10.1021/ja101064b. [DOI] [PubMed] [Google Scholar]
- Dérien S.; Jan D.; Dixneuf P. H. Ruthenium-Catalyzed Coupling of Allyl Alcohol with Alkynes: A New Route to γ,ô-Unsaturated Acetals and Aldehydes. Tetrahedron 1996, 52, 5511–5524. 10.1016/0040-4020(96)00185-8. [DOI] [Google Scholar]
- For the interpretation of this result in terms of interligand interactions, see ref (47).
- For related tactics to study intermediates derived from Grubbs catalysts, see:; a Tallarico J. A.; Bonitatebus P. J.; Snapper M. L. Ring-Opening Metathesis. A Ruthenium Catalyst Caught in the Act. J. Am. Chem. Soc. 1997, 119, 7157–7158. 10.1021/ja971285r. [DOI] [Google Scholar]; b Anderson D. R.; Hickstein D. D.; O’Leary D. J.; Grubbs R. H. Model Compounds of Ruthenium-Alkene Intermediates in Olefin Metathesis Reactions. J. Am. Chem. Soc. 2006, 128, 8386–8387. 10.1021/ja0618090. [DOI] [PubMed] [Google Scholar]; For an unconstrained example, see:; c van der Eide E. F.; Romero P. E.; Piers W. E. Generation and Spectroscopic Characterization of Ruthenacyclobutane and Ruthenium Olefin Carbene Intermediates Relevant to Ring Closing Metathesis Catalysis. J. Am. Chem. Soc. 2008, 130, 4485–4491. 10.1021/ja710364e. [DOI] [PubMed] [Google Scholar]
- Upon standing in solution, carbene 41 slowly converts into an isomeric alkene via a 1,2-H shift from the less hindered methylene group; cf. the Supporting Information.
- Crabtree R. H.The Organometallic Chemistry of the Transition Metals, 4th ed.; Wiley: Hoboken, NJ, 2005. [Google Scholar]
- A dynamic behavior at ambient temperature is manifested in a line broadening of one of the H atoms of the methylene group formed by gem-hydrogenation, which in turn implies that the side chain is dangling; even though this motion is frozen out at – 20 °C, the missing NOE signal between the −OMe group and the methyl substituents of the Cp* ring speak against any interaction between the ether and the metal center, as do the high-field resonances of the bound alkene that persist over the entire temperature range.
- For complexes of this type characterized by X-ray crystallography, see ref (8) and the following:; a Gagné M. R.; Grubbs R. H.; Feldman J.; Ziller J. W. Catalytic Activity of a Well-Defined Binuclear Ruthenium Alkylidene Complex. Organometallics 1992, 11, 3933–3935. 10.1021/om00060a003. [DOI] [Google Scholar]; b Risse J.; Dutta B.; Solari E.; Scopelleti R.; Severin K. Contribnutions to the Chemistry of the Cyclopentadienyl Complex [Cp∧RuCl2]2 (“Cp-roof” = η5-1-Methoxy-2,4-tert-butyl-3-neopentyl-cyclopentadienyl). Z. Anorg. Allg. Chem. 2014, 640, 1322–1329. 10.1002/zaac.201400048. [DOI] [Google Scholar]
- For stability scales that show the effect of π-donor substituents on free carbenes, see:; a Mueller P. H.; Rondan N. G.; Houk K. N.; Harrison J. F.; Hooper D.; Willen B. H.; Liebmann J. F. Carbene Singlet-Triplet Gaps. Linear Correlations with Substituent π Donation. J. Am. Chem. Soc. 1981, 103, 5049–5052. 10.1021/ja00407a015. [DOI] [Google Scholar]; b Mieusset J.-L.; Brinker U. H. The Carbene Reactivirty Surface: A Classification. J. Org. Chem. 2008, 73, 1553–1558. 10.1021/jo7026118. [DOI] [PubMed] [Google Scholar]
- For a recent case study and a compilation of pertinent literature, see:Tskhovrebov A. G.; Lingnau J. B.; Fürstner A. Gold Difluorocarbenoid Complexes: Spectroscopic and Chemical Profiling. Angew. Chem., Int. Ed. 2019, 58, 8834–8838. 10.1002/anie.201903957. [DOI] [PubMed] [Google Scholar]
- In this context, it is relevant to note that Grubbs-type catalysts with a heteroatom at the carbene center are readily formed but have (multiple) low-energy decomposition pathways. See:Louie J.; Grubbs R. H. Metathesis of Electron Rich Olefins: Structure and Reactivity of Electron-rich Carbene Complexes. Organometallics 2002, 21, 2153–2164. 10.1021/om011037a. [DOI] [Google Scholar]
- Macnaughtan M. L.; Johnson M. J. A.; Kampf J. W. Olefin Metathesis Reactions with Vinyl Halides: Formation, Observation, Interception and Fate of the Ruthenium-Monohalomethylidene Moiety. J. Am. Chem. Soc. 2007, 129, 7708–7709. 10.1021/ja0715952. [DOI] [PubMed] [Google Scholar]
- For an instructive case study, see:Schnatter W. F. K.; Rogers D. W.; Zavitsas A. A. Electrophilic Addition to Alkenes: The Relation between Reactivity and Enthalpy of Hydrogenation: Regioselectivity is Determined by the Stability of the Two Conceivable Products. Chem. - Eur. J. 2015, 21, 10348–10361. 10.1002/chem.201500314. [DOI] [PubMed] [Google Scholar]
- Electrophilic carbenes can react with an alkene to generate reactive intermediates of largely carbenium ion character; see ref (57) and the following:Brookhart M.; Kegley S. E.; Husk G. R. Transfer of Ethylidene from η6-C5H5(CO)2Fe=CHCH3+ to Para-Substituted Styrenes. Loss of Stereochemistry about the Cα–Cβ Double Bond of cis-β-Deuterio-p-methoxystyrene. Organometallics 1984, 3, 650–652. 10.1021/om00082a033. [DOI] [Google Scholar]
- The mass balance of the stoichiometic reaction of alkenyl chloride 1g is poor, but no other discrete product could be isolated from the crude reaction mixture.
- a Korkowski P. F.; Hoye T. R.; Rydberg D. B. Fischer Carbene Mediated Conversion of Enynes to Bi- and Tricyclic Cyclopropane-Containing Carbon Skeletons. J. Am. Chem. Soc. 1988, 110, 2676–2678. 10.1021/ja00216a066. [DOI] [Google Scholar]; b Hoye T. R.; Suriano J. A. Reactions of Pentacarbonyl(1-methoxyethylidene)-molybdenum and – tungsten with α,ω-Enynes: Comparison with the Chromium Analogue and Resulting Mechanistic Ramifications. Organometallics 1992, 11, 2044–2050. 10.1021/om00042a019. [DOI] [Google Scholar]; c Mori M.; Watanuki S. New Syntheses of Pyrrolidine Derivatives via the Chromacyclobutanes Generated from Enynes and Fischer Carbene Complexes. J. Chem. Soc., Chem. Commun. 1992, 1082–1083. 10.1039/c39920001082. [DOI] [Google Scholar]; d Casey C. P.; Hornung N. L.; Kosar W. P. Intramolecular Cyclopropanation and Olefin Metathesis Reactions of (CO)5W=C(OCH2CH2CH=CHOCH3)C6H4-p-CH3. J. Am. Chem. Soc. 1987, 109, 4908–4916. 10.1021/ja00250a025. [DOI] [Google Scholar]; e Barluenga J.; Andina F.; Aznar F.; Valdés C. New Cascade Processes on Group 6 Fischer-Type Carbene Complexes: Cyclopropanation and Metathesis Reactions. Org. Lett. 2007, 9, 4143–4146. 10.1021/ol701604g. [DOI] [PubMed] [Google Scholar]
- Catalytic manifestations of such divergent reactivity are exceedingly rare; cf. ref (17). We know of only one example in which a classical Grubbs catalyst engaged an enyne into metathesis/cyclopropanation:; a Peppers B. P.; Diver S. T. Tandem Cyclopropanation/Ring-Closing Metathesis of Dienynes. J. Am. Chem. Soc. 2004, 126, 9524–9525. 10.1021/ja049079o. [DOI] [PubMed] [Google Scholar]; Furthermore, remotely related examples are known in which a Grubbs catalyst is used to decompose a diazo derivative to give a new carbene complex that then leads to cyclopropanation/metathesis cascades. See:; b Alcaide B.; Almendros P.; Luna A. Chem. Rev. 2009, 109, 3817–3858. 10.1021/cr9001512. [DOI] [PubMed] [Google Scholar]
- Noels A. F.; Demonceau A.; Carlier E.; Hubert A. J.; Márquez-Silva R.-L.; Sánchez-Delgado R. A. Competitive Cyclopropanation and Cross-metathesis Reactions of Alkenes Catalysed by Diruthenium Tetrakis Carboxylates. J. Chem. Soc., Chem. Commun. 1988, 783–784. 10.1039/C39880000783. [DOI] [Google Scholar]
- Neese F. The ORCA Program System. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Riplinger C.; Pinski P.; Becker U.; Valeev E. F.; Neese F. Sparse Maps – A Systematic Infrastructure for Reduced-scaling Electronic Structure Methods. II. Linear Scaling Domain Based Pair Natural Orbital Coupled Cluster Theory. J. Chem. Phys. 2016, 144, 024109. 10.1063/1.4939030. [DOI] [PubMed] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Riplinger C.; Sandhoefer B.; Hansen A.; Neese F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. 10.1063/1.4821834. [DOI] [PubMed] [Google Scholar]
- Barone V.; Cossi M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. 10.1021/jp9716997. [DOI] [Google Scholar]
- With regard to the solvation correction, SMD and C-PCM solvation schemes provided essentially the same results for all relevant stationary points on the potential energy surface; for details, see the Supporting Information.
- Yepes D.; Neese F.; List B.; Bistoni G. Unveiling the Delicate Balance of Steric and Dispersion Interactions in Organocatalysis Using High-Level Computational Methods. J. Am. Chem. Soc. 2020, 142, 3613–3625. 10.1021/jacs.9b13725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bannwarth C.; Ehlert S.; Grimme S. GFN2-xTB – An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Corrections. J. Chem. Theory Comput. 2019, 15, 1652–1671. 10.1021/acs.jctc.8b01176. [DOI] [PubMed] [Google Scholar]; b Grimme S. Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations. J. Chem. Theory Comput. 2019, 15, 2847–2862. 10.1021/acs.jctc.9b00143. [DOI] [PubMed] [Google Scholar]
- Even for metallacycles derived from classical Grubbs carbenes the surrounding energy barriers can vanish, especially in exergonic reactions. See:Adlhart C.; Chen P. Mechanism and Activity of Ruthenium Olefin Metathesis Catalysts: The Role of Ligands and Substrates from a Theoretical Perspective. J. Am. Chem. Soc. 2004, 126, 3496–3510. 10.1021/ja0305757. [DOI] [PubMed] [Google Scholar]
- Similar agostic interactions have previously been proposed in cyclopropanation reactions of piano-stool ruthenium carbenes generated with the aid of diazoalkanes; see ref (16a).
- For the sake of completeness we add that Figure 3 must not be misinterpreted in that cyclopropane formation is also favored on thermodynamic grounds. Note that the metathesis pathway is not yet closed at the stage where product C and the secondary carbene [Cp*Ru(Cl)(=CH2)] are formed; rather, the latter must be reconverted into the propagating species by subsequent hydrogenation, which is known to be a highly exothermic process (cf. refs (2 and 3)).
- The higher barrier for substrate 1a carrying two methyl substituents at the olefin is tentatively ascribed to steric hindrance in the reductive elimination step.
- The barrier TSDE for reductive elimination of the ordinary metallacycle D with formation of the cyclopropane is also very responsive to the changes and can even be higher than that of TSAD (see Figure 4); in no case investigated, however, does TSAD ultimately determine the reaction outcome.
- Gordon C. P.; Yamamoto K.; Liao W.-C.; Allouche F.; Andersen R. A.; Copéret C.; Raynaud C.; Eisenstein O. Metathesis Activity Encoded in the Metallacyclobutane Carbon-13 NMR Shift Tensors. ACS Cent. Sci. 2017, 3, 759–768. 10.1021/acscentsci.7b00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Remya P. R.; Suresh C. H. Hypercoordinate β-Carbon in Grubbs and Schrock Olefin Metathesis Metallacycles. Dalton Trans. 2015, 44, 17660–17672. 10.1039/C5DT02801A. [DOI] [PubMed] [Google Scholar]; b Suresh C. H. Nature of α,β-CCC Agostic Bonding in Metallacyclobutanes. J. Organomet. Chem. 2006, 691, 5366–5374. 10.1016/j.jorganchem.2006.07.030. [DOI] [Google Scholar]
- Etienne M.; Weller A. S. Intramolcular C–C Agostic Complexes: C–C Sigma Interactions by Another Name. Chem. Soc. Rev. 2014, 43, 242–259. 10.1039/C3CS60295H. [DOI] [PubMed] [Google Scholar]
- a Evans M. G.; Polanyi M. Inertia and Driving Force of Chemical Reactions. Trans. Faraday Soc. 1938, 34, 11–24. 10.1039/tf9383400011. [DOI] [Google Scholar]; b Hammond G. S. A Correlation of Reaction Rates. J. Am. Chem. Soc. 1955, 77, 334–338. 10.1021/ja01607a027. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.