

Abstract
Purpose:
Pro-inflammatory cytokines within the tumor microenvironment, such as IL-6, contribute to the maintenance of stem cells and promote their survival following treatment. The IL-6/STAT3 pathway is a key regulator of genes involved in cancer progression. Activation of STAT3 promotes expansion of cancer stem cells in triple negative breast cancer. Radiation has also been shown to expand cancer stem cell populations and can induce stemness in non-stem cells. However, the role of IL-6/STAT3 in radiation induced changes in cellular plasticity is unclear.
Materials and Methods:
Expression and secretion of IL-6 from triple negative breast cancer cell lines SUM159PT and MDA-MB-231 were determined after radiation treatment by real-time PCR and ELISA. Activation of STAT3 after radiation was determined by western blotting. Changes in cellular plasticity induced by radiation were determined by examining ALDEFLUOR activity, gene expression analysis of aldehyde dehydrogenase isoforms and mammosphere forming assays with and without the addition of STAT3 inhibitors. To determine the effect of radiation on non-stem cell populations, experiments were also carried out in ALDEFLUOR sorted cells.
Results:
Radiation induced an inflammatory response in both cell lines that resulted in activation of STAT3. Additionally, radiation induced a stem-like state as evidenced by an increased activity and expression of the ALDH isoforms ALDH1A1 and ALDH1A3, and increased self-renewal capabilities. Radiation increased ALDH activity and self-renewal in non-stem cell (ALDH−) populations, suggesting radiation induced cellular reprogramming. However, inhibition of STAT3 blocked the radiation-induced stem-like state in both ALDEFLUOR positive and negative populations, and enhanced radiosensitivity.
Conclusions:
Radiation-induced changes in cellular plasticity are STAT3 dependent and may be a potential target to reduce radioresistance in TNBC and improve treatment outcome.
Introduction
Triple negative breast cancer (TNBC) accounts for approximately 15% of all breast cancers and is defined by the absence of positive staining for estrogen and progesterone receptors, and lack amplification of HER2. TNBCs are considered aggressive tumors with a high degree of genomic instability and are associated with poor prognosis and early visceral metastasis, with survival rates for women who relapse within 5 years of treatment being significantly lower than those with hormone receptor positive breast cancer (Foulkes et al., 2010; Bianchini et al., 2016).
Resistance to treatment in breast cancer may be due, in part, to alterations in cellular plasticity. Changes in cellular state in response to stress may lead to the persistence of a subpopulation of tumor cells with stem-like features known as cancer stem cells (Risom et al., 2018). These radioresistant cells have the capability to self-renew and differentiate, allowing for re-population of a heterogenous tumor and are thought to be responsible for tumor growth, recurrence, and metastasis in breast cancer patients following treatment (Reya et al., 2001; Phillips et al., 2006; Rycaj & Tang, 2014; Arnold et al., 2015). Understanding mechanisms regulating changes in cellular plasticity in response to therapeutics may lead to better treatments for breast cancer.
Breast cancer stem cells are characterized by activity of aldehyde dehydrogenase (ALDH) (Ginestier et al., 2007). Previous studies have shown that breast cancer cells with high ALDH activity have enhanced tumorigenicity and a metastatic phenotype both in vitro and in vivo as evidenced by increased cellular proliferation, colony formation ability, tumor growth, and invasiveness of the cells (Charafe-Jauffret et al., 2009; Croker et al., 2009). In breast cancer patients, high expression of ALDH1, and thus a higher breast cancer stem cell population, was correlated with poorer prognosis, therapy resistance, early recurrence, and poor clinical outcome (Ginestier et al., 2007; Zhong et al., 2013; Kida et al., 2016),. Knockdown of ALDH or inhibiting ALDH activity in human breast cancer cell lines increased cellular sensitivity to radiation treatment, indicating breast cancer cells expressing high amounts of ALDH are more resistant to radiation (Croker & Allan, 2012; Croker et al., 2017). In addition to breast cancer stem cells surviving radiation treatment, previous in vitro studies have shown that radiation treatment can induce differentiated breast cancer cells to acquire a stem-like phenotype (Ghisolfi et al., 2012; Lagadec et al., 2012). Thus, radiotherapy contributes to the enrichment of the breast cancer stem cell population, which can alter patient response to treatment.
It has become apparent that the tumor microenvironment plays a large role in the maintenance and proliferation of breast cancer stem cells and contributes to treatment resistance in breast cancer patients (Arnold et al., 2015). Increased levels of cytokines within the tumor microenvironment in breast cancer patients are associated with poor clinical outcome (Benoy et al., 2004; Cho et al., 2013). Proinflammatory cytokines and growth factors released from tumor-associated macrophages, dendritic cells, and lymphocytes, which are recruited by the tumor following radiation treatment, promote survival and proliferation of breast cancer stem cells (Ginestier et al., 2010; Dethlefsen et al., 2013). In particular, the cytokine interleukin-6 (IL-6) has been found to promote survival of breast cancer stem cells. Radiation treatment has been found to induce secretion of IL-6 from cells within the breast tumor microenvironment (Dethlefsen et al., 2013; Di Maggio et al., 2015); however, effects of radiation induced inflammatory responses from tumor cells themselves and the effect on cancer stem cell populations is unclear.
One major downstream effector of IL-6 signaling, the signal transducer and activator of transcription 3 (STAT3), is a transcription factor which relays extracellular signals initiated by cytokines and growth factors to the nucleus and is a key regulator of genes involved in cancer progression (Dauer et al., 2005). STAT3 is known to be constitutively active in many solid tumors and has been shown to mediate tumor-promoting inflammation (Schetter et al., 2009) and expansion of the breast cancer stem cell population (Marotta et al., 2011; Chung et al., 2014), whereas inhibition of STAT3 activation decreases tumorigenicity of breast cancer cell lines (Lin et al., 2014). Activation of the IL-6/STAT3 pathway has also been shown to induce a stem-like phenotype in non-stem cells (Iliopoulos et al., 2011). In addition, previous studies have shown that inhibition of STAT3 resulted in enhanced radiosensitivity in human glioma cells and squamous cell carcinoma (Bonner et al., 2009; Gao et al., 2010), suggesting that the IL-6/STAT3 pathway could be an attractive therapeutic target to combat resistance to radiation treatment in breast cancer. Therefore, we wanted to investigate the involvement of the IL-6/STAT3 pathway in the radiation-induced expansion of the breast cancer stem cell population in TNBC and determine if inhibition of this pathway could alter the response to treatment.
In order to examine the involvement of IL-6/STAT3 pathway in radiation-induced changes of TNBC cells, we utilized small molecule inhibitors to the IL-6/STAT3 pathway, which target STAT3 directly. We demonstrate that radiation induces a stem-like phenotype in non-stem breast cancer cells as evidenced by an increase in ALDH activity and expression, colony formation capability, and mammosphere forming efficiency. This radiation-induced stem-like phenotype is blocked by direct inhibition of STAT3. These data suggest that direct targeting of STAT3 could potentially be used to prevent alterations in cellular phenotype induced by radiation, thus reducing populations of resistant stem-like cells, and improve treatment outcomes in breast cancer patients.
Methods
Cell Culture and Inhibitors
The triple negative breast cancer cell line SUM159PT was obtained from Asterand and cultured in Hams F12 media supplemented with 10% FBS, 2 μg/ml insulin, 0.1 μg/ml hydrocortisone, and 1x antibiotic-antimycotic (Invitrogen). MDA-MB-231 cell line was purchased from ATCC (and cultured in RPMI media supplemented with 10% FBS and 1X antibiotic-antimycotic (Life Technologies). Cells were maintained at 37°C in 5% CO2. Stattic, an inhibitor of STAT3 activation and dimerization(Schust et al., 2006), was purchased from Tocris (#2798) and resuspended in DMSO at a stock concentration of 50 mM. C188-9, an inhibitor of STAT3(Lewis et al., 2015), was purchased from EMD Millipore (#573128) and resuspended in DMSO at a stock concentration of 100 mg/ml. STA-21, an inhibitor of STAT3 activation and dimerization(Bhasin et al., 2008), was purchased from Cayman Chemical (#14996) and resuspended in DMSO at a stock concentration of 50mM.
Separation of the ALDH+ and ALDH− Subpopulations of Breast Cancer Cells
The ALDEFLUOR kit (StemCell Technologies) was used to isolate cells into high or low ALDH enzymatic activity subpopulations. Briefly, cells were seeded at a density of 1 x 106 cells/100 mm dish. The following day, cells were trypsinized and resuspended in ALDEFLUOR assay buffer containing ALDH substrate (BAAA, 1 μmol/L per 1 x 106 cells) and incubated for 40 minutes at 37°C. As a control, cells in buffer containing substrate were incubated with 15 mmol/L diethylaminobenzaldehyde (DEAB), which inhibits ALDH activity. Cells were washed with ALDEFLUOR assay buffer and passed through a BD Falcon cell strainer cap tube for sorting. Percentage of ALDH positive cells were determined using a BD FACSAria II flow cytometer. Gates were set based on DEAB control having the percent of ALDH positive cells equal to 0.2%. Cells were sorted into ALDH negative (ALDH−) and ALDH positive (ALDH+) subpopulations and cultured for 48 hours for recovery. ALDH+ subpopulation was considered the top 10% of cells with the highest ALDH activity and ALDH− subpopulation was considered to be the bottom 10% of cells with the lowest ALDH activity. Cells were treated with radiation at doses of 2Gy, 4Gy and 8Gy an XRAD225 microirradiator (Precision X-ray). For fractionated experiments cells received a dose of 2Gy for 3 consecutive days. Cells were co-treated with 1 μM Stattic, 5 μM C188-9, 1 μM STA-21 at the time of radiation treatment or at the time of the last fraction for fractionated treatments. Cells were cultured for 5 days in the presence of STAT3 inhibitors. Two days prior to re-analysis of ALDH activity, cells were trypsinized and re-seeded at a density of 5 x 105 cells/100 mm dish and re-treated with appropriate inhibitor. Percentage of ALDH positive cells were determined 5 days post treatment. Gates were set for each plate based on DEAB control having 0.2% ALDH positive cells. Statistical Analysis using an unpaired T-test was performed using Graphpad Prism 6.0 software.
qPCR
SUM159PT cells were treated with 8 Gy of radiation and collected at indicated time points for mRNA expression analysis. RNA was extracted from cells using Trizol (Invitrogen) as described in manufacturer’s protocol. Briefly, cells were scraped in Trizol reagent and incubated at room temperature for 5 minutes to dissociate the nucleoprotein complex. Upon addition of chloroform, tubes were shaken vigorously to mix. Samples were centrifuged at 12,000xg for 15 minutes at 4°C. The aqueous phase was removed and combined with isopropanol, mixed, incubated at room temperature for 10 minutes and then centrifuged for 10 minutes at 12,000xg at 4°C. RNA pellets were washed with 75% ethanol and re-spun at 7500xg for 5 minutes at 4°C. Ethanol was removed and pellets were air-dried for 10 minutes. RNA pellets were dissolved in 30 μl of molecular grade water and treated with Ambion DNase I for 30 minutes at 37°C (ThermoFisher). cDNA was constructed from 3 μg of RNA using the SuperScript First-Strand Synthesis reverse transcriptase kit (Invitrogen). Primer sequences and source information were used as previously described(Opdenaker et al., 2015). Real-time PCR was performed with 50 ng of cDNA/well using Sybr green in an Applied Biosystems 7500 Fast Real-Time PCR System. Experiments were performed at least three times in triplicate. Statistical Analysis using a unpaired T-test was performed using Graph Pad prism 6.0.
ELISA
Supernatant from SUM159PT cells either untreated or treated with increasing doses of radiation were assayed with an IL-6 ELISA kit (R&D Systems) as described in the manufacturer’s protocol. Supernatants were collected 24, 48 or 72 hours post-irradiation and centrifuged to remove cells and debris. In addition, cells were trypsinized and counted by trypan blue exclusion using a Countess automated cell counter (Invitrogen) to normalize secreted IL-6 to total cell number. IL-6 ELISA was performed according to the manufacturer’s instructions. Briefly, 100 μl of supernatant or supplied standard was added to each well containing 100 μl of assay diluent and incubated at room temperature for 2 hours. After four washes, 200 μl of conjugate was added to each well and incubated at room temperature for an additional 2 hours. Following four additional washes, 200 μl of substrate was added to each well and incubated in the dark for 20 minutes. Finally, 50 μl of stop solution was added to each well and the absorbance was read at 450 nm. The standards were graphed and used to determine the concentration of IL-6 in each of the supernatant samples.
Western Blot Analysis
SUM159PT cells were treated and incubated as indicated and harvested in RIPA buffer with phosphatase and protease inhibitors. BCA assays were performed to determine protein concentration using Pierce BCA protein assay kit (ThermoFisher). Protein was diluted with 2x Lamelli buffer and loaded into 10% SDS-PAGE gels and ran at 100 volts for 2 hours. Gels were transferred to 0.2 μm nitrocellulose membrane at 100 volts for 1 hour in 1x Tris-glycine/methanol buffer. Following transfer, membranes were stained with Ponceau to ensure adequate transfer of proteins. Membranes were blocked in 5% non-fat dry milk in 1x TBS, 0.1% Tween for 1 hour at room temperature. Antibodies to STAT3 [79D7] (#4904, Cell Signaling), STAT3 (phospho Y705) [EP2147Y] (ab76315, Abcam), phosphoJAK2 (C80C3, Cell Signlaing) or β-actin (#sc-81178, Santa Cruz) were added overnight at 4°C with shaking. Blots were washed with 1x TBS, 0.1% Tween and incubated with secondary antibodies HRP AffiniPure donkey α-rabbit (#715-035-152, Jackson Immunoresearch Lab) or HRP AffiniPure donkey α-mouse (#715-035-150, Jackson Immunoresearch Lab) for 1 hour at room temperature. Membranes were washed, incubated with Pierce SuperSignal West Dura Extended Duration Substrate (ThermoFisher) and imaged on a G:BOX ChemiXP4 Imaging System (Syngene). Experiments were performed at least three times.
Targeted knock-down of STAT3
SUM159 cells were transiently transfected with Silencer pre-designed siRNA targeting the human STAT3 gene, or Stealth RNAi negative control siRNA (Invitrogen) using lipofectamine reagent (Invitrogen) according to the manufacturer’s insturctions. Cells were grown for 24-72 hours. Targeted knock-down of STAT3 was observed by western blotting at 24 hours and remained knocked-down for up to 72 hours. To determine the effect STAT3 knock-down on ALDEFLUOR positivity, the ALDEFLUOR assay was performed 48 hours post-transfection.
Clonogenic Assays
Cells were seeded into 6 well plates at a density of 100-5000 cells/well. Cells were treated with 2, 4,or 8 Gy and/or 1 μM Stattic or 5 μM C188-9 and cultured for 7 days. Colonies were fixed with formalin, stained with 0.5% crystal violet, rinsed and were counted. Colony-forming efficiency (colonies counted/cells plated) was determined. Experiments were performed three times in triplicate. Statistical analysis was performed using one-way ANOVA followed by a post-hoc Tukey’s T test using GraphPad Prism software 6.0.
Sphere Forming Assays
SUM159PT and MDA-MB-231 cells were sorted as described above and seeded into 96 well low attachment plates at densities of 1000-5000 cells/well in mammocult media (MEM supplemented with 1x B27, 20 ng/ml epidermal growth factor (EGF), and 20 ng/ml basic fibroblast growth factor (bFGF)) to promote the formation of spheres. Following seeding, cells were treated with 8 Gy radiation and/or 1 μM Stattic or 5 μM C188-9. Cells were continually treated with either inhibitor throughout the assay. Spheres were allowed to form for 10 days, trypsinized, counted and re-plated in sphere media with or without inhibitors to STAT3 to obtain secondary spheres. Secondary spheres were counted at least 10 days after plating. Sphere forming efficiency was determined as the number of spheres counted/ the number of cells plated. Experiments were performed three times in triplicate. Statistical analysis was performed using one-way ANOVA with a post-hoc Tukey’s T test with GraphPad Prism software 6.0.
Apoptosis assays
For cells grown in monolayers, cells were seeded in 96 well plates at a density of 1000 cells per well. Cells were grown overnight and treated with 8Gy radiation or 1micromolar Stattic and grown for 48 hours. For cells grown in mammospheres, cells were plated in mammosphere forming conditions as described above. Spheres were allowed to grow for 10 days and then treated with radiation or 1 micromolar Stattic. Capsase activation was determined using the Caspase-Go 3/7 assay (Promega) according to the manufacturer’s instructions. To account for differences in cell number, protein concentration was determined using a BCA assay, and capsase activity was normalized to total protein concentration.
Results
Radiation increases ALDH activity and isoform expression in breast cancer cells
In order to confirm that radiation can alter the cancer stem cell population of TNBC, the enzymatic activity of the cancer stem cell marker, aldehyde dehydrogenase (ALDH) was determined in SUM159PT cells. These cells were derived from a primary breast tumor and exhibit claudin-low features (Prat et al., 2013). ALDH activity was measured by ALDEFLUOR assay and cells were sorted into ALDH positive (ALDH+) and ALDH negative (ALDH−) subpopulations using flow cytometry. Prior to sorting, 19-30% of cells were ALDH positive (Supplemental figure 1A). In both the ALDH+ and ALDH− subpopulations, treatment with 8 Gy of radiation significantly increased the percentage of ALDH positive cells. ALDEFLUOR positive cells increased from a mean of 29.88% (±SEM 2.187%) to 46.68% (±SEM 6.413%, p=.04) in the ALDH+ subpopulation (Figure 1A and B). Interestingly, an increase in ALDH activity was also observed in the ALDH− population rising from a mean of 0.86% (± SEM 0.14%) to 4.7% (±SEM 0.6597%, p=.0004) (Figure 1C and D), indicating that radiation treatment induced a rise in ALDH activity in previously negative breast cancer cells. Increases in ALDH activity were also observed in ALDH− cells at a dose of 4Gy (Figure 1E, supplemental figure 1B), but to a lesser extent than those observed at 8Gy. Although no increases were observed with a dose of 2Gy, substantial increases in ALDH activity were observed with repeated fractions of 2Gy, indicating that increases in ALDH activity may be related to the total radiation dose ( Figure 1E, supplemental figure 1C).
Figure 1.

Radiation increases ALDH activity and isoform expression in breast cancer cells. Flow cytometry analysis of ALDH and activity following radiation treatment in ALDH-sorted populations. SUM159 cells were sorted into (A&B) ALDH-positive and (C&D) ALDH-negative subpopulations using FACS and irradiated with 8 Gy. Cells were re-analyzed by flow cytometry 5 days post-irradiation. Gates were set for each plate based on DEAB control having 0.2% ALDH positive cells. In both subpopulations, radiation increased the percentage of ALDH positive cells. (E). Graph showing increases in ALDH activity in ALDH− cells treated with the indicated doses of radiation. Cells were treated and analyzed as described in the previous experiments. Graphs represent average of three separate experiments. Error bars indicate standard deviation. *p<.01, **p<.001.
ALDH has multiple isoforms that are expressed to varying degrees depending on the tissue and tumor type. Two isoforms, ALDH1A1 and ALDH1A3, have been found to be responsible for ALDH activity measured in the ALDEFLUOR assay, with ALDH1A3 being the dominant isoform in primary breast cancer cell lines while ALDH1A1 is associated with metastasis (Croker et al., 2017), (Marcato et al., 2011). Since radiation caused an increase in measured ALDH activity, we sought to determine if radiation increased the expression of ALDH1A1 and ALDH1A3 in cell lines derived from primary and metastatic tumors. Unsorted SUM159PT and MDA-MB-231 cells were treated with 8 Gy radiation for increasing time periods and transcript levels of each isoform were determined using qPCR. In SUM159PT cells, ALDH1A1 and ALDH1A3 expression began to increase at 48 hours post-radiation treatment and continued to be elevated at five days post-radiation treatment, with ALDH1A1 expression increasing 3-fold and ALDH1A3 expression increasing 4-fold compared to untreated levels (Figure 2A and B). Similar results were observed in MDA-MD-231 cells where significant radiation-induced fold-changes in ALDH1A1 were observed at both 48 hours and 5 days (Figure 2C). However, these cells demonstrated less upregulation of ALDH1A3 with only a mean 1.4 fold-increase observed at 5 days (Figure 2D). These results indicate that radiation may increase both ALDH isoform expression and activity in both primary and metastatic claudin-low tumors, although the expression of ALDH isoforms may differ depending on the tumor origin.
Figure 2.
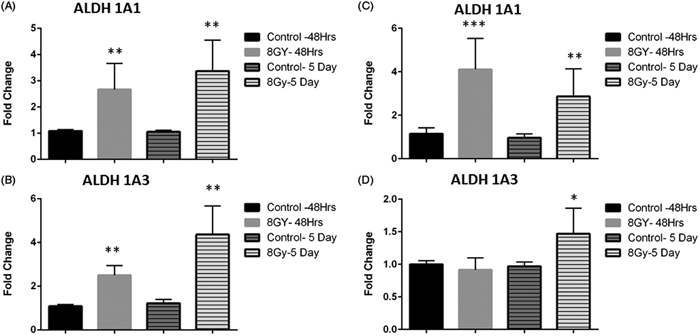
Radiation induces expression of ALDH isoforms. SUM159 (A–B) and MDA-MB-231 (C–D) cells were treated with 8 Gy for indicated time points, RNA was extracted and expression of the ALDH isoforms, ALDH1A1 (A&C) and ALDH1A3 (B&D) were assessed with real-time PCR. Graphs represent averages of three separate experiments. Error bars represent standard deviation. *p<.01, **p<.001, ***p<.001.
Radiation induces IL-6-JAK2-STAT3 signaling which is required for ALDH activity
It has been demonstrated that differentiated cancer cells can convert to stem-like cancer cells and that expression and secretion of pro-inflammatory cytokines, such as interleukin-6 (IL-6), play a role in the inducible formation of breast cancer stem cells (Kim et al., 2013). In order to determine if the IL-6 signaling pathway plays a role in the radiation-induced increase of the breast cancer stem cell population that we observed, we analyzed the expression of IL-6, as well as other pro-inflammatory cytokines that are downstream of IL-6 signaling, interleukin-8 (IL-8) and interleukin-1 beta (IL-1β), following irradiation with 8 Gy. Radiation induced gene expression of all three cytokines at 48 hours post-treatment in both SUM159PT and MDA-MB-231 cells. Expression of all cytokines was further elevated five days post-radiation treatment in both cell lines demonstrating a sustained inflammatory state within the irradiated cells (Figure 3 A-D).
Figure 3.
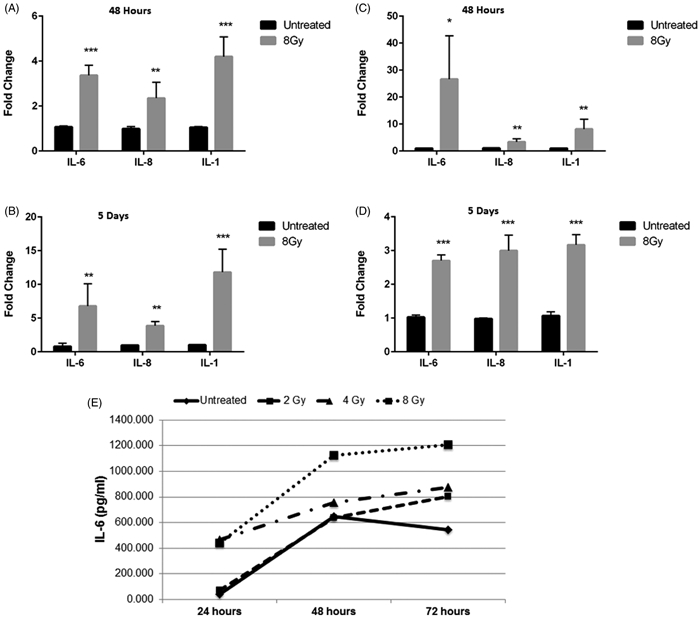
Radiation induces release of inflammatory cytokines. (A&B) SUM159 and (C&D) MDA-MB-231 cells were analyzed at 48 hrs (A&C) or 5 days (B&D) after radiation with 8 Gy. RNA was extracted and expression of the interleukins, IL-6, IL-8, and IL-1b was assessed with real-time PCR. Graphs represent averages of three separate experiments. Error bars represent standard deviation. *p<.01, **p<.001, ***p<.001. (E) Secretion of IL-6 from SUM159 cells treated with increasing doses of radiation at 24, 48, or 72 hours was assessed by ELISA. Secretion of IL-6 was radiation dose- and time-dependent in SUM159 cells. This experiment was repeated in duplicate.
In order to confirm that the enhanced gene expression of interleukins observed after radiation treatment correlated with cytokine secretion, release of IL-6 from SUM159PT cells treated with increasing doses of radiation was determine via ELISA. A radiation dose- and time-dependent increase in IL-6 secretion was observed from SUM159PT cells (Figure 3E), indicating that radiation induces both expression and secretion of IL-6.
Binding of IL-6 to its receptor results in the activation of the Janus family Kinases (JAKs) which then phosphorylate and activate a family of transcription factors known as the signal transducer and activator of transcriptions (STATs). In particular, STAT3 has been shown to be constitutively active in tumors and is important in the survival, proliferation, and maintenance of cancer stem cells (Banerjee & Resat, 2016). In claudin-low cell lines and in vivo models, STAT3 is preferentially activated within stem cell compartments and has been shown to support self-renewal and tumor initiating capabilities (Wei et al., 2014). To determine if activity of ALDH is dependent on STAT3 activation, we tested the effect of targeted knock-down of STAT3 on ALDEFLUOR activity. Our results show that transfection of SUM159 cells with siRNA targeting STAT3 leads to a significant decrease ALDH activity, (Figure 4A, Supplemental Figure 2A) indicating that STAT3 expression is required for ALDH activity. Additionally, the role of STAT3 activation on ALDH activity was determined using small molecule inhibitors of STAT3. Stattic and C-188-9 are selective small molecules that bind to the SH2 domain of STAT3 and block both activation and dimerization (Schust et al., 2006),(Redell et al., 2011). Treatment of cells with either inhibitor resulted in a significant decrease in STAT3 phosphorylation without altering total STAT3 protein (Supplemental figure 2B and C). Treatment of both SUM159PT and MDA-MB-231 cells with Stattic significantly decreased measured ALDH activity, (Figure 4B&C). These results were confirmed with another inhibitor of STAT3 dimerization, C188-9 (Figure 4B&C), indicating that STAT3 expression and activation is required for ALDH activity.
Figure 4.

STAT3 is required for ALDH activity and is activated upon treatment with radiation. (A) Sum159 cells were treated with siRNA to STAT3 or Control siRNA and ALDH activity was determined 48 hours following knockdown. (B&C) Sum159 (B) and MDA-MB-231 (C) cells were treated with inhibitors to STAT3 and analyzed 48 hours after treatment for ALDH activity. (D&E) Activation of JAK2 and STAT3 following 8 Gy radiation STAT3 was assessed via Western blot for increases in phosphorylation. (D). SUM159 cells or MD-MB-231 (E) were treated with 8 Gy of radiation for 24 hours prior to lysing. Blots were probed for phosphorylated STAT3, or phosphorylated JAK2. Beta-actin was used as a loading control. Relative protein levels of phosphorylated STAT3 were normalized to betaactin.
As STAT3 is required for ALHD activity, we next sought to determine if radiation increased activation of the IL-6-JAK-STAT3 axis. We observed increases in phosphorylation of JAK2 48 hours after radiation. Likewise, we observed robust increases in STAT3 phosphorylation in both SUM159 and MDA-MB-231 cells, indicating that STAT3 was activated following radiation treatment (Figure 4D and E).
Inhibition of STAT3 blocks radiation-induced increases in ALDH activity and reduces radiation induced expression of stemness genes
In order to determine if the radiation-induced increase in ALDEFLUOR activity is dependent on STAT3 activation, cells were sorted as previously described, treated with radiation with or without the presence of STAT3 inhibitors. Consistent with previous results, an increase in ALDEFLUOR activity was observed in ALDH− cells treated with 8Gy radiation compared to untreated cells. Treatment with Stattic or C-188-9 significantly blocked this increase returning ALDH activity levels to near that of untreated cells (Figure 5A and B). Similar results were observed in ALDH+ cells Figure 5C. Although a slight decrease in viability was noted with the combination of STAT3 inhibitors and radiation, a more dramatic decrease in viability was observed with the combination of the combination of STAT3 knockdown and radiation resulted in few attached cells 24-48 hours after treatment. Further analysis of ALDH activity in STAT3 siRNA transfected cells was not possible. These results indicate that radiation-induced changes in ALDH activity require STAT3 activation, and that STAT3 expression may be required for radiation resistance.
Figure 5.

Inhibition of STAT3 blocks radiation induced increases in ALDH activity in sorted cells. (A) Representative flow cytometry analysis of ALDH activity in ALDH-negative subpopulation following STAT3 inhibition and radiation treatment. SUM159 cells were sorted into ALDH negative subpopulation using FACS and cultured for two days to allow recovery from sorting. ALDH activity was measured by ALDEFLUOR assay and percentage of ALDH-positive cells was determined by flow cytometry. (B&C) SUM159 cells were sorted into ALDH− (B) or ALDHþ (C) populations and were treated with 8 Gy of radiation and/or STAT3 inhibitors Stattic, or C188-9 and cultured for five days with inhibitors before reassessing ALDH activity. (D). Sum159 cells were treated with 8 Gy with and without Stattic. Cells were grown for 5 days in the presence of Stattic and RNA was obtained. Quantitative PCR was performed using primers to Nanog, Oct-4 and Sox-2. Graphs represents average of three separate experiments. Error bars represent standard deviation. *p<.01, **p<.001, ***p<.001.
STAT3 has previously been shown to directly regulate transcription of genes such as nanog, oct-4 and Sox-2 which are important for maintenance of embryonic and adult stem cells as well as cancer stem cells (Do et al., 2013). Therefore, we sought to determine the effect of radiation on these genes. As shown in figure 5D, treatment of cells with 8Gy leads to a significant upregulation of Nanog and Oct-4 gene expression 48hrs post-radiation treatment compared to untreated cells. This increase was inhibited when cells were co-treated with Stattic (Figure 5D).
Inhibition of STAT3 reduces mammosphere forming capabilities and and sensitizes breast cancer stem-like cells to radiation
Since cancer stem cells have the long-term ability to self-renew and proliferate, we sought to determine the effect of STAT3 inhibition on colony formation capability and mammosphere forming efficiency with and without radiation. As shown in Figure 6A, treatment with either Stattic or C188-9 alone significantly reduced colony formation compared to untreated cells in both SUM159 and MDA-MD-231 cells. However, STAT3 inhibition sensitized the colony forming cells to radiation in a dose dependent manner, suggesting STAT3 signaling is associated with survival and/or expansion of colony forming cells after radiation (Figure 6A). To further support the involvement of STAT3 in the radiation-induced self-renewal of breast cancer cells, we examined secondary mammosphere forming efficiency of SUM159PT and MDA-MB-231 cells treated with Stattic and/or radiation. Treatment with 8 Gy radiation increased sphere formation, whereas Stattic alone decreased sphere forming capability. The combination of radiation and STAT3 inhibition significantly decreased the number of mammospheres, indicating STAT3 is necessary for the increase in self-renewal capacity of breast cancer cells following radiation treatment (Figure 6B and C). To further examine changes in cellular plasticity upon radiation treatment, mammosphere assays were also carried out in ALDH+ and ALDH− populations. As expected, a significantly higher sphere forming efficiency was observed in ALDH+ sorted cells compared to ALDH− or mock-sorted cells, indicating that this cellular population has a higher degree of intrinsic self-renewal capabilities (Supplemental Figure 3A). A slight increase in mammosphere forming ability was observed in ALDH+ cells treated with radiation (Supplemental Figure 3B). ALDH− populations from both cell lines formed few spheres. However, mammosphere formation was significantly increased after radiation in both cell lines. This increase was blocked in cells cultured with Stattic,indicating a STAT3 dependent increase in mammosphere forming capabilities following radiation (Figure 6B,C and D).
Figure 6.

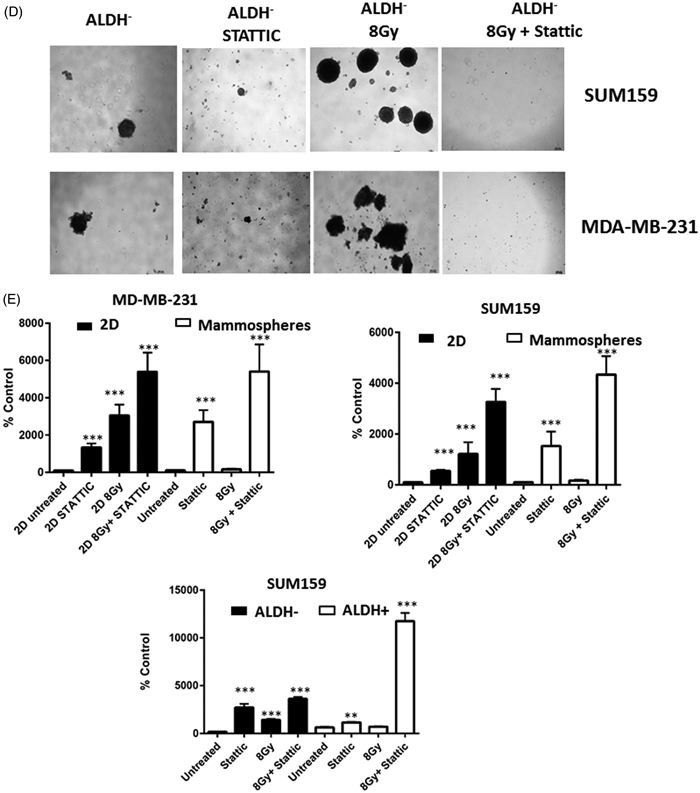
Inhibition of STAT3 sensitizes cells to radiation and decreases mammopshere forming ability. (A) Clonogenic analysis of SUM159 or MDA-MB-231 cells following treatment with 2,4 or 8 Gy of radiation, 1 lM Stattic, 5 lM C188-9, or a combination of radiation and inhibitor. Colonies (>50 cells) were counted and data is reported as colony-forming efficiency (colonies counted/cells plated). Error bars represent standard deviation. Inhibition of STAT3 reduced colony-forming ability and sensitized cells to radiation. ***p<.0001. The effect of STAT3 inhibition on secondary mammosphere forming ability was assessed in the total or ALDH− populations of (B) SUM159 cells and (C) MDA-MB-231 cells following treatment with 8 Gy of radiation and/or 1 lM Stattic or 5 lM C188-9. Cells were continuously treated with inhibitor for 10 days. Data are reported as mammosphere forming efficiency, which is the number of spheres counted/number of cells plated and normalized to untreated cells. Graphs represent average of three separate experiments. Error bars represent standard deviation. *p<.01, **p<.001, ***p<.001. (D). Images of secondary mammospheres in ALDH− populations. (E). Caspase3/7 activation assays in total populations or mammospheres. Cells or mammospheres were treated with radiation or static and caspase3/7 activation was assessed using a luminescence assay 48 hours post-treatment. Data are expressed as % luminescence of untreated cells. Data were normalized to protein concentration in mammosphere assays. Graphs represent average of technical replicates from two separate experiments. Error bars represent standard deviation. *p<.01, **p<.001, ***p<.001.
As STAT3 activation has been shown to occur preferentially in stem-like cells (Wei, 2014), we sought to determine if STAT3 is required for survival of stem cell populations upon treatment with radiation and thus may be affecting survival as well as self-renewal capabilties. To this end, we analyzed apoptosis via activation of Caspae3/7 in mammospheres and ALDH sorted cells 48 hours after treatment with radiation with or without STATTIC. As shown in Figure 6E, mammospheres derived from both SUM-159 and MDA-MD-231 cell lines showed significantly less apoptosis upon treatment with radiation than monolayer (2D) counterparts. However, treatment with STATTIC significantly increased radiation-induced apoptosis in both 2D and mammosphere cultures. However, this effect was most pronounced in cells cultured in stem-cell enriching mammosphere cultures. Similarly, ALDH+ cells were less likely to undergo radiation induced apoptosis than ALDH− cells. However, caspase activation was significantly increased in this population with a combination of radiation and STATTIC treatment compared to the ALDH− population (Figure 6E). These findings indicate that activation of STAT3 may be necessary for increased self-renewal capabilities of non-stem cells, but may also be required for radiation-resistance of breast cancer stem-like cells.
Discussion
Our findings indicate that radiation induces an inflammatory microenvironment resulting in increased STAT3 phosphorylation which promotes changes stem cell plasticity and radiation resistance in claudin-low TNBC cell lines. Additionally, we show that radiation leads to increased expression of at least two isoforms of ALDH, ALD1A1 and ALDH1A3, which have been shown to be responsible for the majority of the ALDH activity observed in breast cancer (Marcato et al., 2011). Although induction of ALDEFLUOR activity was most pronounced upon treatment with high doses of radiation, such as those used in hypofractionated protocols, our findings also show that increases in ALDEFLUOR activity may occur at lower doses, and may be related to cumulative radiation dose.
High ALDH activity has been shown to protect cells from oxidative stress and may provide protection from radiation-induced reactive oxygen species (ROS) (Zhang et al., 2010; Croker & Allan, 2012). Our findings indicate that STAT3 signaling is required to maintain ALDH activity in breast cancer, as treatment with inhibitors to STAT3 activation significantly reduced both constitutive and radiation-induced ALDH activity. STAT3 activity has also been shown to directly promote transcription of ALDH1A3 (Canino et al., 2015). Thus, activation of STAT3 by radiation may promote increased expression and activity of ALDH1A1 and ALDH1A3, leading to a reduction of ROS and increased radiation resistance. Likewise, as study by Wang et al showed that targeted knockdown of STAT3 resulted in reduction of genes involved in various metabolic pathways, including ALDH2, another isoform of aldehyde dehydrogenase that has been implicated in ALDEFLUOR activity and breast cancer self renewal (Marcato et al., 2011; Thomas et al., 2016) In this study, STAT3 activation was shown regulate lipid metabolism which is required for maintenance of breast cancer stem cells and chemoresistance (Wang et al., 2018).
STAT3 has been shown to regulate stemness, particularly in claudin-low models of breast cancer and is required for self-renewal and tumor initiating capability (Wei et al., 2014). In this study, Wei et al., demonstrated enhanced expression of phosphorylated STAT3 in breast cancer stem-like and tumor initiating cells (Wei et al., 2014). We observed STAT3-dependent increases in both ALDH activity and secondary mammosphere forming efficiency in claudin-low breast cancer cells after radiation treatment, providing support for the hypothesis that radiation enhances the stem cell compartment. Moreover, although increases in STAT3 dependent self-renewal and ALDH activity were observed stem-like ALDH+ population, increases were more striking in the non-stem-like ALDH− subpopulations, indicating that STAT3 is involved in cellular reprogramming of ALDH− cells upon radiation-induced cellular damage. Additionally, our findings show that STAT3 activation enhances radiation-induced apoptosis in breast mammospheres and ALDH+ cells. These findings show that STAT3 activation may not only be involved in cellular reprogramming, but may confer a protective advantage to breast stem-like cells.
Lagadec et al.(Lagadec et al., 2012) reported that radiation-induced conversion of non-stem cells to stem cells is dependent on activation of the Notch signaling pathway. Although the exact interactions between IL-6/STAT3 and Notch signaling in TNBC is unclear, previous studies have indicated that STAT3 activation may occur downstream of Notch signaling in breast cancer and lead to promotion of a cancer stem cell phenotype through induction of IL-1β and NF-kβ (Mazzone et al., 2010; Zhang et al., 2014). On the other hand, constitutive activation of NF-kβ and STAT3 in glioblastoma cancer stem cells has been found to regulate expression of a set of genes that drive activation of Notch signaling and thus maintenance of the stem cell phenotype (Garner et al., 2013). In breast cancer, IL-6 promotes cancer stem cell survival through a number of pathways, including Notch(Guo et al., 2011). Moreover, IL-6 can induce mammosphere formation in breast cancer cells (Sansone et al., 2007) and, through Notch-1, promote breast cancer bone metastasis (Sethi et al., 2011). In addition, STAT3-dependent activation of Notch signaling has been observed after fractionated radiation treatment of breast cancer cells (Kim et al., 2016). Kim et al., demonstrated that radiation-induced IL-6/JAK/STAT3 signaling was required for Notch-driven induction of epithelial-mesenchymal transition (EMT) in breast cancer cells after radiation treatment and an IL-6 neutralizing antibody was shown to decrease both STAT3 phosphorylation and radiation induced Notch-2, Jagged1 and DLL4 expression. Our findings show that radiation-induced cellular plasticity is dependent on STAT3. Although further work needs to be done to assess the interactions between the two pathways, the cellular reprogramming observed in our study may be due, in part, to STAT3 induction of Notch signaling.
Growing evidence suggests that treatment-resistant breast cancer stem cells are responsible for tumor growth, recurrence, and metastasis, however, how these cells evade treatment is unclear. The IL-6 STAT3 pathway has been shown to promote growth of cancer stem cells in many solid tumors (Lee et al., 2011),. Additionally, STAT3 activation within the tumor has been shown to promote an immunosuppressive microenvironment. Targeted deletion of STAT3 in the PyVmT mammary tumor model lead to increased immune infiltration and regression of early breast cancer lesions (Jones et al., 2016),. Furthermore, sustained STAT3 activation in tumor cells contributes to release of immunosuppressive factors such as IL-10 and IL-6 which promote expansion regulatory T cells and myeloid derived suppressor cells (Lee et al., 2011; Su et al., 2018) Although radiation has been shown to promote priming of T cell responses in some cases (Arnold et al., 2018), the role of radiation-Induced IL-6 Stat3 signaling on the tumor microenvironment is unclear. However, targeting IL-6/STAT3 may lead to anti-tumor effects on both tumor and infiltrating immune cells, leading to reduced populations of resistant stem-like cells, and enhanced radiation induced immune responses.
Although preclinical investigations have shown the utility of agents targeting the IL-6-JAK-STAT3, clinical data in solid tumors is still forthcoming. Early phase clinical trials of Tocilizumab, a monoclonal antibody that disrupts IL-6 classic and trans-signaling is in early phase trials for treatment of tumors such as pancreatic and ovarian cancer (Goumas et al., 2015; Yanaihara et al., 2016). Clinical investigation of JAK inhibitors has been limited due to significant toxicity including dose-limiting myelosuppression, as well as neurological adverse events (Harrison & Vannucchi; Loh et al.). However, data from a phase II study in metastatic cancer patients has shown that a combination of the JAK2 inhibitor ruxolitinib and chemotherapy to be well tolerated (Hurwitz et al.). Additionally, several STAT3 SH2 domain antagonists including OPB-31121 and C188-9 are also in early trials (Bharadwaj et al.; Oh et al.). Although possible anti-tumor activity has been demonstrated in patients with solid tumors, significant dose-limiting side effects such as peripheral neuropathy have been observed(Okusaka et al.) Additionally, a phase I dose-escalation trial with the small molecule OPB-111077 demonstrated limited toxicity with disease stabilization when delivered as a monotherapy in a range of advanced solid tumors (Tolcher et al.).. Although clinical studies have so far been limited in breast cancer, our findings indicate that STAT3 inhibitors may lead to improved tumor outcomes, when used in combination with radiation treatment in triple negative breast cancer.
Supplementary Material
Acknowledgements
This project was supported by a grant from the National Institute of General Medical Sciences – NIGMS (P20 GM103446) and U54-GM104941 from the National Institutes of Health and the state of Delaware.
Footnotes
Disclosure of interest
The authors report no conflicts of interest
References
- Arnold KM, Flynn NJ, Raben A, Romak L, Yu Y, Dicker AP, Mourtada F & Sims-Mourtada J. (2018). The Impact of Radiation on the Tumor Microenvironment: Effect of Dose and Fractionation Schedules. Cancer Growth Metastasis 11, 1179064418761639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold KM, Opdenaker LM, Flynn D & Sims-Mourtada J. (2015). Wound healing and cancer stem cells: inflammation as a driver of treatment resistance in breast cancer. Cancer Growth Metastasis 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee K & Resat H. (2016). Constitutive activation of STAT3 in breast cancer cells: A review. Int J Cancer 138, 2570–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoy IH, Salgado R, Van Dam P, Geboers K, Van Marck E, Scharpe S, Vermeulen PB & Dirix LY. (2004). Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin Cancer Res 10, 7157–7162. [DOI] [PubMed] [Google Scholar]
- Bharadwaj U, Eckols TK, Xu X, Kasembeli MM, Chen Y, Adachi M, Song Y, Mo Q, Lai SY & Tweardy DJ. Small-molecule inhibition of STAT3 in radioresistant head and neck squamous cell carcinoma. Oncotarget 7, 26307–26330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhasin D, Cisek K, Pandharkar T, Regan N, Li C, Pandit B, Lin J & Li PK. (2008). Design, synthesis, and studies of small molecule STAT3 inhibitors. Bioorg Med Chem Lett 18, 391–395. [DOI] [PubMed] [Google Scholar]
- Bianchini G, Balko JM, Mayer IA, Sanders ME & Gianni L. (2016). Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol 13, 674–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner JA, Trummell HQ, Willey CD, Plants BA & Raisch KP. (2009). Inhibition of STAT-3 results in radiosensitization of human squamous cell carcinoma. Radiother Oncol 92, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canino C, Luo Y, Marcato P, Blandino G, Pass HI & Cioce M. (2015). A STAT3-NFkB/DDIT3/CEBPbeta axis modulates ALDH1A3 expression in chemoresistant cell subpopulations. Oncotarget 6, 12637–12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D & Wicha MS. (2009). Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res 69, 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YA, Sung MK, Yeon JY, Ro J & Kim J. (2013). Prognostic role of interleukin-6, interleukin-8, and leptin levels according to breast cancer subtype. Cancer Res Treat 45, 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SS, Giehl N, Wu Y & Vadgama JV. (2014). STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. Int J Oncol 44, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker AK & Allan AL. (2012). Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44(+) human breast cancer cells. Breast Cancer Res Treat 133, 75–87. [DOI] [PubMed] [Google Scholar]
- Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA & Allan AL. (2009). High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med 13, 2236–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker AK, Rodriguez-Torres M, Xia Y, Pardhan S, Leong HS, Lewis JD & Allan AL. (2017). Differential Functional Roles of ALDH1A1 and ALDH1A3 in Mediating Metastatic Behavior and Therapy Resistance of Human Breast Cancer Cells. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer DJ, Ferraro B, Song L, Yu B, Mora L, Buettner R, Enkemann S, Jove R & Haura EB. (2005). Stat3 regulates genes common to both wound healing and cancer. Oncogene 24, 3397–3408. [DOI] [PubMed] [Google Scholar]
- Dethlefsen C, Hojfeldt G & Hojman P. (2013). The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res Treat 138, 657–664. [DOI] [PubMed] [Google Scholar]
- Di Maggio FM, Minafra L, Forte GI, Cammarata FP, Lio D, Messa C, Gilardi MC & Bravata V. (2015). Portrait of inflammatory response to ionizing radiation treatment. J Inflamm (Lond) 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do DV, Ueda J, Messerschmidt DM, Lorthongpanich C, Zhou Y, Feng B, Guo G, Lin PJ, Hossain MZ, Zhang W, Moh A, Wu Q, Robson P, Ng HH, Poellinger L, Knowles BB, Solter D & Fu XY. (2013). A genetic and developmental pathway from STAT3 to the OCT4-NANOG circuit is essential for maintenance of ICM lineages in vivo. Genes Dev 27, 1378–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes WD, Smith IE & Reis-Filho JS. (2010). Triple-negative breast cancer. N Engl J Med 363, 1938–1948. [DOI] [PubMed] [Google Scholar]
- Gao L, Li F, Dong B, Zhang J, Rao Y, Cong Y, Mao B & Chen X. (2010). Inhibition of STAT3 and ErbB2 suppresses tumor growth, enhances radiosensitivity, and induces mitochondria-dependent apoptosis in glioma cells. Int J Radiat Oncol Biol Phys 77, 1223–1231. [DOI] [PubMed] [Google Scholar]
- Garner JM, Fan M, Yang CH, Du Z, Sims M, Davidoff AM & Pfeffer LM. (2013). Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappaB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J Biol Chem 288, 26167–26176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisolfi L, Keates AC, Hu X, Lee DK & Li CJ. (2012). Ionizing radiation induces stemness in cancer cells. PLoS One 7, e43628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS & Dontu G. (2007). ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D, Guan JL, Dontu G & Wicha MS. (2010). CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest 120, 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumas FA, Holmer R, Egberts JH, Gontarewicz A, Heneweer C, Geisen U, Hauser C, Mende MM, Legler K, Rocken C, Becker T, Waetzig GH, Rose-John S & Kalthoff H. (2015). Inhibition of IL-6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. Int J Cancer 137, 1035–1046. [DOI] [PubMed] [Google Scholar]
- Guo S, Liu M & Gonzalez-Perez RR. (2011). Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta 1815, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C & Vannucchi AM. Ruxolitinib: a potent and selective Janus kinase 1 and 2 inhibitor in patients with myelofibrosis. An update for clinicians. Ther Adv Hematol 3, 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, Wade SM 3rd, Nemunaitis JJ, Stella PJ, Pipas JM, Wainberg ZA, Manges R, Garrett WM, Hunter DS, Clark J, Leopold L, Sandor V & Levy RS. Randomized, Double-Blind, Phase II Study of Ruxolitinib or Placebo in Combination With Capecitabine in Patients With Metastatic Pancreatic Cancer for Whom Therapy With Gemcitabine Has Failed. J Clin Oncol 33, 4039–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos D, Hirsch HA, Wang G & Struhl K. (2011). Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A 108, 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LM, Broz ML, Ranger JJ, Ozcelik J, Ahn R, Zuo D, Ursini-Siegel J, Hallett MT, Krummel M & Muller WJ. (2016). STAT3 Establishes an Immunosuppressive Microenvironment during the Early Stages of Breast Carcinogenesis to Promote Tumor Growth and Metastasis. Cancer Res 76, 1416–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida K, Ishikawa T, Yamada A, Shimada K, Narui K, Sugae S, Shimizu D, Tanabe M, Sasaki T, Ichikawa Y & Endo I. (2016). Effect of ALDH1 on prognosis and chemoresistance by breast cancer subtype. Breast Cancer Res Treat 156, 261–269. [DOI] [PubMed] [Google Scholar]
- Kim RK, Kaushik N, Suh Y, Yoo KC, Cui YH, Kim MJ, Lee HJ, Kim IG & Lee SJ. (2016). Radiation driven epithelial-mesenchymal transition is mediated by Notch signaling in breast cancer. Oncotarget 7, 53430–53442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT & Lee YJ. (2013). Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal 25, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadec C, Vlashi E, Della Donna L, Dekmezian C & Pajonk F. (2012). Radiation-induced reprogramming of breast cancer cells. Stem Cells 30, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Pal SK, Reckamp K, Figlin RA & Yu H. (2011). STAT3: a target to enhance antitumor immune response. Curr Top Microbiol Immunol 344, 41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KM, Bharadwaj U, Eckols TK, Kolosov M, Kasembeli MM, Fridley C, Siller R & Tweardy DJ. (2015). Small-molecule targeting of signal transducer and activator of transcription (STAT) 3 to treat non-small cell lung cancer. Lung Cancer 90, 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Hutzen B, Lee HF, Peng Z, Wang W, Zhao C, Lin HJ, Sun D, Li PK, Li C, Korkaya H, Wicha MS & Lin J. (2014). Evaluation of STAT3 signaling in ALDH+ and ALDH+/CD44+/CD24− subpopulations of breast cancer cells. PLoS One 8, e82821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh ML, Tasian SK, Rabin KR, Brown P, Magoon D, Reid JM, Chen X, Ahern CH, Weigel BJ & Blaney SM. A phase 1 dosing study of ruxolitinib in children with relapsed or refractory solid tumors, leukemias, or myeloproliferative neoplasms: A Children's Oncology Group phase 1 consortium study (ADVL1011). Pediatr Blood Cancer 62, 1717–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcato P, Dean CA, Pan D, Araslanova R, Gillis M, Joshi M, Helyer L, Pan L, Leidal A, Gujar S, Giacomantonio CA & Lee PW. (2011). Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells 29, 32–45. [DOI] [PubMed] [Google Scholar]
- Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, Wu Z, Gonen M, Mulvey LA, Bessarabova MO, Huh SJ, Silver SJ, Kim SY, Park SY, Lee HE, Anderson KS, Richardson AL, Nikolskaya T, Nikolsky Y, Liu XS, Root DE, Hahn WC, Frank DA & Polyak K. (2011). The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest 121, 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzone M, Selfors LM, Albeck J, Overholtzer M, Sale S, Carroll DL, Pandya D, Lu Y, Mills GB, Aster JC, Artavanis-Tsakonas S & Brugge JS. (2010). Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl Acad Sci U S A 107, 5012–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Lee SH, Han SW, Kim MJ, Kim TM, Kim TY, Heo DS, Yuasa M, Yanagihara Y & Bang YJ. Phase I Study of OPB-31121, an Oral STAT3 Inhibitor, in Patients with Advanced Solid Tumors. Cancer Res Treat 47, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okusaka T, Ueno H, Ikeda M, Mitsunaga S, Ozaka M, Ishii H, Yokosuka O, Ooka Y, Yoshimoto R, Yanagihara Y & Okita K. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol Res 45, 1283–1291. [DOI] [PubMed] [Google Scholar]
- Opdenaker LM, Modarai SR & Boman BM. (2015). The Proportion of ALDEFLUOR-Positive Cancer Stem Cells Changes with Cell Culture Density Due to the Expression of Different ALDH Isoforms. Cancer Stud Mol Med 2, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips TM, McBride WH & Pajonk F. (2006). The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst 98, 1777–1785. [DOI] [PubMed] [Google Scholar]
- Prat A, Karginova O, Parker JS, Fan C, He X, Bixby L, Harrell JC, Roman E, Adamo B, Troester M & Perou CM. (2013). Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res Treat 142, 237–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB & Tweardy DJ. (2011). Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood 117, 5701–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF & Weissman IL. (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. [DOI] [PubMed] [Google Scholar]
- Risom T, Langer EM, Chapman MP, Rantala J, Fields AJ, Boniface C, Alvarez MJ, Kendsersky ND, Pelz CR, Johnson-Camacho K, Dobrolecki LE, Chin K, Aswani AJ, Wang NJ, Califano A, Lewis MT, Tomlin CJ, Spellman PT, Adey A, Gray JW & Sears RC. (2018). Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat Commun 9, 3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycaj K & Tang DG. (2014). Cancer stem cells and radioresistance. Int J Radiat Biol 90, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, Ceccarelli C, Santini D, Paterini P, Marcu KB, Chieco P & Bonafe M. (2007). IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest 117, 3988–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schetter AJ, Nguyen GH, Bowman ED, Mathe EA, Yuen ST, Hawkes JE, Croce CM, Leung SY & Harris CC. (2009). Association of inflammation-related and microRNA gene expression with cancer-specific mortality of colon adenocarcinoma. Clin Cancer Res 15, 5878–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schust J, Sperl B, Hollis A, Mayer TU & Berg T. (2006). Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 13, 1235–1242. [DOI] [PubMed] [Google Scholar]
- Sethi N, Dai X, Winter CG & Kang Y. (2011). Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 19, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YL, Banerjee S, White SV & Kortylewski M. (2018). STAT3 in Tumor-Associated Myeloid Cells: Multitasking to Disrupt Immunity. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas ML, de Antueno R, Coyle KM, Sultan M, Cruickshank BM, Giacomantonio MA, Giacomantonio CA, Duncan R & Marcato P. (2016). Citral reduces breast tumor growth by inhibiting the cancer stem cell marker ALDH1A3. Mol Oncol 10, 1485–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolcher A, Flaherty K, Shapiro GI, Berlin J, Witzig T, Habermann T, Bullock A, Rock E, Elekes A, Lin C, Kostic D, Ohi N, Rasco D, Papadopoulos KP, Patnaik A, Smith L & Cote GM. A First-in-Human Phase I Study of OPB-111077, a Small-Molecule STAT3 and Oxidative Phosphorylation Inhibitor, in Patients with Advanced Cancers. Oncologist 23, 658–e672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y, Somlo G, Jandial R, Ann D, Hanash S, Jove R & Yu H. (2018). JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab 27, 136–150 e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Tweardy DJ, Zhang M, Zhang X, Landua J, Petrovic I, Bu W, Roarty K, Hilsenbeck SG, Rosen JM & Lewis MT. (2014). STAT3 signaling is activated preferentially in tumor-initiating cells in claudin-low models of human breast cancer. Stem Cells 32, 2571–2582. [DOI] [PubMed] [Google Scholar]
- Yanaihara N, Hirata Y, Yamaguchi N, Noguchi Y, Saito M, Nagata C, Takakura S, Yamada K & Okamoto A. (2016). Antitumor effects of interleukin-6 (IL-6)/interleukin-6 receptor (IL-6R) signaling pathway inhibition in clear cell carcinoma of the ovary. Mol Carcinog 55, 832–841. [DOI] [PubMed] [Google Scholar]
- Zhang M, Shoeb M, Goswamy J, Liu P, Xiao TL, Hogan D, Campbell GA & Ansari NH. (2010). Overexpression of aldehyde dehydrogenase 1A1 reduces oxidation-induced toxicity in SH-SY5Y neuroblastoma cells. J Neurosci Res 88, 686–694. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhao X, Shao S, Zuo X, Ning Q, Luo M & Gu S. (2014). Notch1 induces epithelial-mesenchymal transition and the cancer stem cell phenotype in breast cancer cells and STAT3 plays a key role. Int J Oncol 46, 1141–1148. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Lin Y, Shen S, Zhou Y, Mao F, Guan J & Sun Q. (2013). Expression of ALDH1 in breast invasive ductal carcinoma: an independent predictor of early tumor relapse. Cancer Cell Int 13, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.