

Abstract

The development of a fluorescent probe for a specific metal has required exquisite design, synthesis, and optimization of fluorogenic molecules endowed with chelating moieties with heteroatoms. These probes are generally chelation- or reactivity-based. Catalysis-based fluorescent probes have the potential to be more sensitive; however, catalytic methods with a biocompatible fluorescence turn-on switch are rare. Here, we have exploited ligand-accelerated metal catalysis to repurpose known fluorescent probes for different metals, a new approach in probe development. We used the cleavage of allylic and propargylic ethers as platforms that were previously designed for palladium. After a single experiment that combinatorially examined >800 reactions with two variables (metal and ligand) for each ether, we discovered a platinum- or copper-selective method with the ligand effect of specific phosphines. Both metal–ligand systems were previously unknown and afforded strong signals owing to catalytic turnover. The fluorometric technologies were applied to geological, pharmaceutical, serum, and live cell samples and were used to discover that platinum accumulates in lysosomes in cisplatin-resistant cells in a manner that appears to be independent of copper distribution. The use of ligand-accelerated catalysis may present a new blueprint for engineering metal selectivity in probe development.
Short abstract
Fluorescent imaging technologies were developed for platinum and copper based on ligand-accelerated catalysis and revealed unexpected distribution of these metals in cisplatin-resistant cancer cells.
Introduction
Increasing demands for detecting and quantifying heavy metals in science, engineering, and medicine require faster and less expensive methods. Quantifications of heavy metal continue to rely on atomic absorption spectroscopy and inductively coupled plasma mass spectroscopy (ICP-MS), which are expensive, time-consuming, and incompatible with on-site analyses.1,2 The use of fluorescence offers an inexpensive, fast, and on-site approach, with catalysis-based probe rivalling instrumental techniques for sensitivity.
Reactivity-based fluorescent probes3 recently garnered enormous interest.4−9 Generally, a specific functional group is installed in a fluorescent molecule to mask the fluorescence, and a chemical reaction with an analyte induces a structural change to emit fluorescence (Figure 1a). Substantial efforts are required to synthesize fluorescent probe candidates. As a departure from the well-established one-at-a-time approach, the Chang group employed a diversity-oriented synthesis, yielding multiple detection systems.10 In their approach, a library of probes was subjected to a library of potential analytes to discover a probe–analyte pair. However, the implementation of this approach in other laboratories remains to be seen, possibly due to the intensive synthetic work required.
Figure 1.
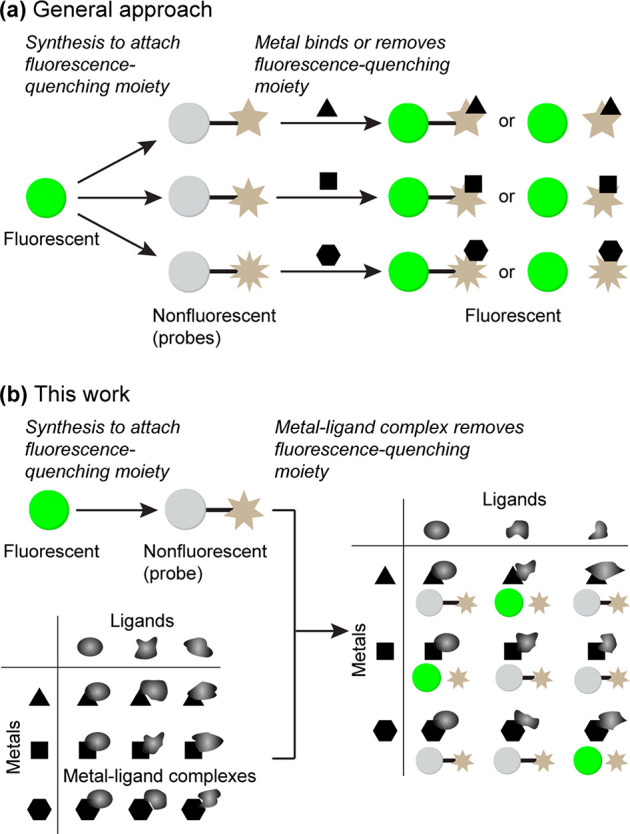
(a) General approach. A fluorescent molecule is derivatized into quenched molecules though chemical synthesis. Each metal-quenching moiety match generates fluorescence. (b) This work. One quenched molecule is screened against a combinatorial library of metal–ligand complexes. A specific metal generates fluorescence only in the presence of a specific ligand.
Meanwhile, the catalysis field witnessed advances by using high-throughput screenings. For example, the Burgess group screened 96 combinations of metals and ligands in less than a week.11 Additional examples of similar high-throughput screenings have been reported.12−15 Inspired by the successes in the catalysis field for ligand-accelerated reactions, we hypothesized that it would be plausible to develop novel metal-selective detection methods through combinatorial screening of metals and ligands (Figure 1b). We envisioned that a repurposing approach would bypass lengthy chemical synthesis to enable more widespread implementation. In this work, two probes for palladium, allyl Pittsburgh Green ether16 (APE, Figure 2a) and propargyl Pittsburgh Green ether17 (PPE, Figure 2b), are repurposed to selectively detect platinum or copper.
Figure 2.

(a) Fluorogenic conversion of APE to Pittsburgh Green. (b) Fluorogenic conversion of PPE to Pittsburgh Green. (c) The heat map of the metal vs ligand screening for the conversion of APE to Pittsburgh Green. (d) The heat map of the metal vs ligand screening for the conversion of PPE to Pittsburgh Green. For both heat maps, the colors indicate relative fluorescence intensities. Conditions: 1 μM metal, 20 μM APE or PPE, 200 μM ligand, 1:9 (v/v) DMSO/1.2 M phosphate buffer (pH 7.0), 25 °C, 2 h, n = 1. Row 37 contains no ligand. See Tables S1 and S2 for quantitative data. (e) Ligands used for the screenings.
Combinatorial screening of metals and ligands capitalizes upon the growing list of commercially available ligands to repurpose previously developed probes for new metals. Although intramolecular ligand effects were exploited in some instances,18,19 external-ligand-accelerated catalysis has not been used to achieve metal selectivity.
We focused on platinum and copper for two reasons. First, intracellular and solid tumor/organ accumulation of both metals plays an important role in human disease processes.20,21 Second, the two metals are linked in tumor cells; cisplatin dynamics and resistance have been linked to differential expression and activity of copper transporters.22 Therefore, there is a critical need to develop imaging techniques for cisplatin and copper in live cells, with a spatial resolution compatible with subcellular organelles.
Results and Discussion
Initial Screening
To test our hypothesis that novel metal selectivity can be discovered by a ligand–metal combinatorial approach, commercially available ligands 1–36 (Figure 2e) were added to mixtures of APE or PPE and metal ions in DMSO/pH 7 buffer (1:9) at 25 °C. Not surprisingly, many reaction conditions with palladium facilitated both the deallylation (Figure 2c) and depropargylation (Figure 2d) reaction. Hits were validated or invalidated as described in Figures S1 and S2. We discovered that the fluorescence signal from the reaction of platinum and APE in the presence of tris(4-fluorophenyl)phosphine 13 (TFPP) was the most prominent, in which the signal was 40-fold higher than the background signal and 10-fold higher that the corresponding signal from palladium. Because palladium is generally the most effective metal for allylic C–O bond cleavage,23 it is striking that TFPP rendered platinum more reactive than palladium for such cleavage. Phosphines 12, 18, and 34 also facilitated the deallylation, with the signal-to-background (S/B) ratios of 4.7, 4.5, and 11, respectively (Table S1). Five para-substituted phosphines, TFPP (13), 15, 18, 19, and 34 (F, OCH3, CH3, CF3, and H, respectively), generated the S/B ratios of 40, 1.0, 4.5, 1.1, and 11 with platinum. The Pt-TFPP catalysis was 4 times faster than the previously reported Pt-Ph3P catalysis.24 Bidentate phosphines, such as phosphine 11 (see Figure S3 for the proof for binding mode), did not facilitate the deallylation of APE.
With PPE, although palladium is generally the primary metal for propargylic C–O bond cleavage,17 we discovered that copper ions greatly increased the fluorescence signals in the presence of 4-(dimethylamino)phenyldiphenylphosphine (DMAPPP, 9). This phosphine did not render other metals, including palladium, reactive toward PPE. Copper-catalyzed propargylic C–O bond cleavage has been known in synthetic chemistry, but the ligands were mostly amines or amine–phosphine hybrids and were designed for stereoselectivity.25−29 Additionally, fluorescent probes for copper ions are primarily based on sulfides and amines as the binding motifs.30−34 The reaction of copper with PPE in the presence of phosphines 2, 15, 18, and 34 resulted in a fluorescence increase of 2-fold or more (Table S2). It appears that electron-rich phosphines were more effective. Importantly, these data support the aforementioned hypothesis, laying the foundation for a new platform for probe development. While the screening data present opportunities for other metal detection and catalysis, in this work, we decided to focus on platinum and copper with APE and PPE, respectively, for the biological reasons described below.
Method Optimization
Having discovered the Pt-TFPP and Cu-DMAPPP systems, we sought to optimize them. For the platinum method, we studied the effect of the salts, pH, temperature, and TFPP concentration. At pH 7, most salts shown in Figure 3a increased the reaction rate compared to no added salt, although none were better than the potassium phosphate buffer. There was little anion interference, except in the case of Br– and I–. As for the effect of pH, pH 7.8 is optimal (Figure 3b); the observed differences in the fluorescence intensity are not an artifact of the fluorescence of the released fluorophore at different pH values since the fluorescence of Pittsburgh Green is pH independent in the range tested.35 An increase in temperature also enhanced the reaction rate (Figure 3c). Figure 3d reveals that the reaction rates increased linearly, plateaued, and dropped as the TFPP concentrations increased. The rate decrease may be attributed to the formation of coordinatively saturated, catalytically inactive platinum species. This was further corroborated by the observation that Pt(PPh3)4 was ineffective at catalyzing the deallylation (Figure S4), reminiscent of the literature.35
Figure 3.

Optimization of reaction conditions for selectivity and sensitivity of APE toward Pt. n = 3. (a) Relative fluorescence intensity with various salts. Conditions: 510 nM Pt, 20 μM APE, 200 μM TFPP, 1:9 (v/v) DMSO/125 mM salt, pH 7.0, 25 °C, 10 min. (b) Relative fluorescence intensity at different pH values. Conditions: 510 nM Pt, 20 μM APE, 200 μM TFPP, 1:9 (v/v) DMSO/1.2 M phosphate, pH 7.0–9.0, 25 °C, 45 min. (c) Relative fluorescence intensity at different temperatures. Conditions: 250 nM Pt, 20 μM APE, 400 μM TFPP, 1:9 (v/v) DMSO/1.2 M phosphate, pH 7.8, 30 min. (d) Correlation between the concentrations of TFPP and the deallylation rate of APE. Conditions: 510 nM Pt, 20 μM APE, 1:9 (v/v) DMSO/1.2 M phosphate, pH 7.8, 25 °C, 1 h. n = 3. (e) Preincubation of various Pt species with TFPP. Conditions: 1 μM Pt, 20 μM APE, 200 μM TFPP, 1:9 (v/v) DMSO/1.2 M phosphate, pH 7.8, 25 °C, n = 3. (f) Reactions of cis-37 with TFPP or P(2-furyl)3 in CDCl3.
To investigate the oxidation state of the reactive platinum species, Pt(0), Pt(II), and Pt(IV) were preincubated with TFPP, to which APE was added. We used Pt(CH2=CH2)(PPh3)2 as the Pt(0) species because Pt(PPh3)4 was unreactive. The fluorescence signals from the three platinum oxidation states overlapped (Figure 3e), suggesting that the catalytically active species is likely Pt(0); various Pt(IV) and Pt(II) species might be reduced by TFPP36,37 to the same TFPP-ligated Pt(0) species.
Next, we studied the dynamics of platinum complexes by means of 31P NMR spectroscopy and chose CDCl3 to dissolve the hydrophobic platinum species (Figures 3f, S5, and S6). The platinum complex cis-37 was treated with TFPP or P(2-furyl)3 (5, used as a control) in either a 1:2 or 1:4 mol ratio, affording a mixture of cis-Cl2Pt(PPh3)(TFPP) and cis-Cl2Pt(TFPP)2 (cis-38; see Figure S7 for the crystal structure) or a mixture of cis-Cl2Pt(PPh3)(P(2-furyl)3) and cis-Cl2Pt(P(2-furyl)3)2 (cis-39), respectively. TFPP was more efficient than P(2-furyl)3 at displacing PPh3 from cis-37 but was a weaker ligand than PPh3 for Pt(II). The weaker binding would promote dissociation of the ligand, generating coordinatively unsaturated, reactive platinum species in situ. TFPP not only enhances the reactivity of platinum toward APE but also suppresses that of palladium, which warrants further mechanistic studies in the future. The unique ability of TFPP to suppress the oxidative addition of Pd(0) has also been observed by others.38
To optimize the copper detection method for different applications from those for platinum, we studied the effect of cosolvent, pH, and buffer salt. To dissolve DMAPPP and active pharmaceutical ingredients (APIs; see below for applications), we tested N-methylpyrrolidone (NMP), DMSO, ethanol, and acetonitrile as potential cosolvents and found that ethanol interfered in the reaction the least (Figure 4a). Following this, commercially available phosphate buffers (pH 5–8) were diluted to a concentration of 50 mM. In the presence of PPE and DMAPPP at pH 7 and 8, both Cu(I) and Cu(II) showed nearly identical reactivity that correlated with pH (Figure 4b). This indicated that, in the presence of DMAPPP, both the Cu(II) and Cu(I) species form the same phosphine-ligated copper species. Subsequent experiments were performed using only Cu(II) due to better stability.
Figure 4.

(a) Effect of cosolvent on copper-catalyzed depropargylation. Conditions: 1 μM CuSO4, 20 μM PPE, 200 μM DMAPPP, 15:85 (v/v) NMP, DMSO, EtOH, or MeCN/1.2 M phosphate buffer, pH 7, 23 °C, 0.5 h, n = 3. (b) Effect of pH on reaction. 0 or 10 μM CuCl or CuCl2, 20 μM PPE, 200 μM DMAPPP, 1:9 (v/v) DMSO/50 mM, pH 5–8 phosphate buffer, 25 °C, 1 h, n = 3. (c) Effect of buffer salt on the reaction. 0 or 2 μM CuSO4, 20 μM PPE, 200 μM DMAPPP, 15:85 (v/v) EtOH/50 mM, pH 8.00 phosphate, 1 M, pH 8.5 sodium bicarbonate, 0.5 M borate, pH 8.5, or pH 10 carbonate buffers, 23 °C, 1 h, n = 3. (d) Effect of buffer salt after controlling for concentrations. 0 or 2 μM CuSO4, 20 μM PPE, 200 μM DMAPPP, 15:85 (v/v) EtOH/50 mM buffer, pH 8.00 phosphate, pH 8.5 sodium bicarbonate, or pH 8.5 borate, 24 °C, 1 h, n = 3. (e) Effect of cosolvent concentrations. 0 or 500 nM CuSO4, 20 μM PPE, 160 μM DMAPPP, 15–70% EtOH, 15 mM, pH 8.5 borate buffer, 25 °C, 0.5 h, n = 3. (f) Effect of DMAPPP concentrations. 0 or 500 nM CuSO4, 20 μM PPE, 160 μM DMAPPP, 15:85 (v/v) EtOH/500 mM borate buffer, pH 8.5, 25 °C, 0.5 h, n = 3.
We also tested the effect of the buffer salt on the reaction. To make the copper detection method readily available, we used only commercially available buffers; furthermore, on the basis of the results in Figure 4b, we tested buffers at or above pH 8. Of the buffer salts listed in Figure 4c, the borate buffer yielded the best result. The adjustment of the buffer concentrations to 50 mM revealed that the effect observed was due to the identity of the buffer salt and not the concentration of the buffer salts (Figure 4d); the carbonate buffer was not tested due to a lack of information from the manufacturer regarding the concentration of the salts. There was no difference in the fluorescence intensities when the borate buffer and phosphate buffer were used at different concentrations, but a dilution of the bicarbonate buffer significantly increased the fluorescence intensity (Figure 4d).
To further optimize the reaction rate, we studied the effect of both ethanol and DMAPPP concentration. As the concentration of ethanol was lowered, the rate of the reaction increased (Figure 4e). As the concentration of DMAPPP increased, the reaction rate increased linearly and then plateaued at higher concentrations (Figure 4f). We chose the 100–160 μM range for DMAPPP in further studies to minimize data fluctuation derived from the actual DMAPPP concentrations varying due to oxidation in air.
Metal Selectivity
Following our optimization experiments, we revisited the selectivity of each method against other metal ions. With APE, the optimized method was 12 times ((54.71 – 1.00)/(5.57 – 1.00)) more selective for platinum over palladium (Figure 5a). To our knowledge, this is the first example in which platinum is the most reactive metal with a fluorescent probe. Additionally, only Cu(I) and Cu(II) could convert PPE to Pittsburgh Green (Figure 5b).
Figure 5.

Relative reactivities of metals to (a) APE with TFPP or (b) PPE with DMAPPP. Conditions: 10 μM metals, 20 μM APE, 200 μM TFPP, 1:9 (v/v) DMSO/1.2 M phosphate buffer, pH 7.8, 25 °C, 10 min, n = 3 or 1 μM metals, 20 μM PPE, 160 μM DMAPPP, 15:85 (v/v) EtOH/500 mM borate buffer, pH 8.5, 26 °C, 20 min, n = 3.
Mechanism
The Pt-TFPP catalysis with APE presumably proceeds through the Tsuji-Trost-type mechanism.39 Our working hypothesis for the Cu-DMAPPP-PPE system is shown in Figure 6. Cu(II) is reduced to Cu(I) by DMAPPP and forms the phosphine–copper complex Cu-1, which coordinates with the alkyne to form copper acetylide Cu-2. With the electron-rich phosphine, the elimination of ArO– (Pittsburgh Green) would be facile. Subsequently, the cumulene-type intermediate Cu-3 reacts with an unidentified nucleophile to form the copper acetylide Cu-4. A nucleophile then attacks the copper to liberate the propargylated nucleophile and copper catalyst Cu-1. The greater efficiency of the fluorescence method with higher pH may be explained by the faster deprotonation with a stronger base or nucleophilic attack on the terminal carbon or on the copper atom. Figure 1b indicates that electron-rich phosphines are superior to electron-deficient phosphines possibly because electron-rich phosphine can facilitate the cleavage of the propargylic C–O bond when Cu-2 is converted to Cu-3. Unlike other copper-catalyzed propargylation methods performed in aprotic solvents,25−29 polydentate ligands did not accelerate the reaction in water in this study. Therefore, the copper-catalyzed propargylic C–O bond cleavage in water requires different ligand design principles.
Figure 6.
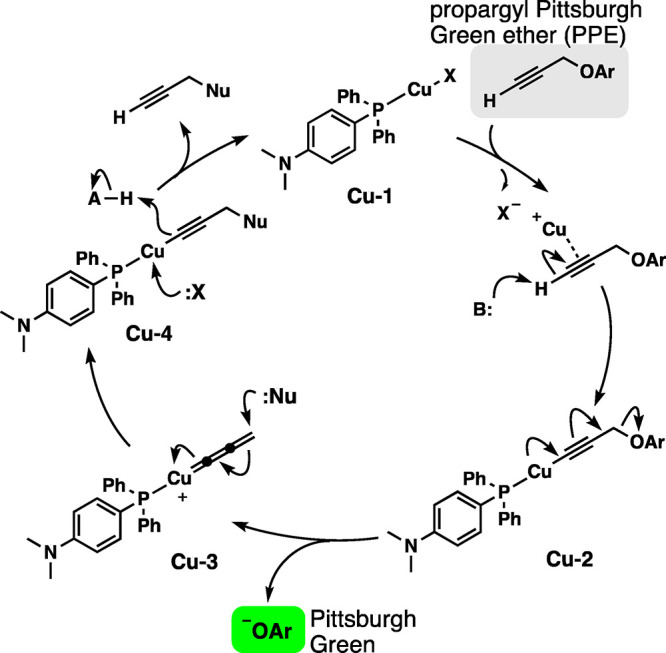
Proposed mechanism for the copper-catalyzed cleavage of the propargylic C–O bond.
Applications of the Platinum Method
As discussed in the Introduction, metal quantification currently requires instrument-intensive techniques. We asked whether the new fluorometric method could measure platinum concentrations. Figure 7a shows that the APE-TFPP-Pt method is quantitative. The limit of quantification (LOQ) and limit of detection (LOD) of the method with a 1 h incubation, on the basis of the standard error of the regressions, were 18.3 ± 0.1 and 5.50 ± 0.04 nM, respectively. With a standard curve (Figure S8), the turnover frequency was calculated to be 8.5 ± 3.4 h–1. Because the method is catalysis-based, allowing the reaction to proceed for longer periods of time might lower the LOQ and LOD. We assessed whether the method could accurately determine the concentrations of unknown samples. In a double-blind format, we treated platinum samples in the same manner as the samples for Figure 7a. The concentrations of platinum were known to those who prepared the samples but not known to the authors. We then generated a linear regression and determined the percent recovery of the unknowns. Percent recoveries (100% × experimental value/theoretical value) ranged from ∼90% to 115% in 4 of 6 samples, indicating that this method may be sufficiently accurate (Table S3).
Figure 7.

(a) Standard curve for Pt. Conditions: 20 μM APE, 400 μM TFPP, 1:9 (v/v) DMSO/1.2 M phosphate, pH 7.8, 25 °C, 1 h, n = 3. (b) Estimating the relative quantities of Pt in rock samples. Conditions: 100 mg of ore, 20 μM APE, 400 μM TFPP, 1:9 DMSO/1.2 M phosphate buffer, pH 7.8, 25–45 °C, n = 1. (c) Standard curve of cisplatin in serum. Conditions: 400 μM TFPP, 20 μM APE, 1:9 (v/v) DMSO/1.2 M phosphate buffer, pH 7.8, 45 °C, 1 h, n = 6.
Having demonstrated the quantitative nature of the technology with a simple matrix, we tested whether we could quantify platinum in the presence of several metals in rock extracts. After confirming the accuracy with a standard procedure for metal extraction with aqua regia (Figure S10), we attempted to directly detect platinum without the acid digestion. Approximately 100 mg of several balled-milled rock samples was mixed with a solution of APE and TFPP for 2 h; platinum concentrations quantified by the fire assay were mildly correlated with fluorescence intensities (Figure 7b). This indicates that the fluorescence method has a potential to be used on site for the direct (i.e., no aqua regia) estimation of platinum in ores.
We then turned to platinum in medicine. Cisplatin is the most frequently used anticancer drug in the clinic and is used to treat up to 50% of cancer patients.40 Despite this, clinicians do not quantify cisplatin concentration in treated patients. A method that could quantify cisplatin in serum samples on site would allow clinicians to personalize dosage for patients. In effect, the APE-TFPP method could detect cisplatin (Figure S11). To determine whether the method can quantify clinically relevant, protein-free platinum,41−43 we precipitated proteins from serum and spiked the serum with cisplatin; spiking protein-free serum in this manner replicates clinically relevant free, soluble cisplatin. Figure 7c shows a linear correlation between cisplatin concentration and fluorescence intensity. The LOQ and LOD of the method based on the standard error of the regression were 410 ± 8 and 120 ± 2 nM, respectively, which is comparable to atomic emission spectroscopy (LOD ∼ 260 nM platinum in sera1).
Applications of the Copper Method
We monitored the copper-catalyzed reaction with PPE over time (Figure 8a). Using 200 nM Cu(II) and 20 μM PPE, we determined that, after 1 h, the reaction product (Pittsburgh Green) was produced at approximately 480 nM concentration and continued to be produced. Therefore, the turnover frequency was 2.6 ± 0.3 h–1, indicating that the method was catalytic.
Figure 8.

(a) Kinetic study to determine turnover frequency. Conditions: 200 nM CuSO4, 20 μM PPE, 160 μM DMAPPP, 15:85 (v/v) EtOH/500 mM borate buffer, pH 8.5, 30 °C, n = 3. (b) Copper concentration dependence to determine the limit of detection and limit of quantification. Conditions: 20 μM PPE, 100 μM DMAPPP, 15:85 (v/v) EtOH/50 mM borate buffer, pH 8.5, 2 h, 25 °C, n = 8 for plate reader, n = 4 for fluorometer. (c) Visualization of the relative concentrations of copper in drinking water samples. (d) Structures of compounds used in (e) and (f). (e, f) Quantification of copper in drug-like samples. Conditions: 30 ppm (e) or 300 ppm (f) Cu(II) in the solid phase, 20 μM PPE, 160 μM DMAPPP, 14.2:3.3:85 (v/v/v) EtOH/DMSO/500 mM borate buffer, pH 8.5, 24 °C, 1 h, n = 3. (g) Detection of copper in SOD. Conditions: 0 or 0.5 mg/mL SOD, 0.5 μM PPE, 10 μM DMAPPP, 0.5:0.5:99 EtOH/DMSO/500 mM borate buffer, pH 8.5, 25 °C, n = 2. (h) Distribution of copper between water and CHCl3 in the presence of DMAPPP. Partition conditions: CHCl3 and aqueous, pH 7 buffer, 0 or 5 μM CuSO4 in H2O. Analysis by fluorescence: 20 μM PPE, 160 μM DMAPPP, 15:85 (v/v) EtOH/500 mM borate buffer, pH 8.5, 1 h, 25 °C, n = 3.
Figure 8b shows the linear relationship between copper concentration and fluorescence intensity. A linear regression analysis using the standard error of the regression revealed the LOD and LOQ to be 0.25 ± 0.04 and 0.83 ± 0.12 nM, respectively, with a fluorometer.
We then applied the method to the detection of copper in drinking water. The limit defined by the United States Environmental Protection Agency for the concentration of copper in drinking water is 20 μM. We obtained three samples: (1) drinking water filtered through a 14-week old water filter, (2) drinking water filtered through a new, unused water filter, and (3) unfiltered drinking water from a water fountain in our building. We compared these with solutions containing 0 and 5 μM CuSO4 in distilled ultrapure water as negative and positive controls, respectively, which showed a marked difference (Figure 8c). We could visualize that all of the drinking water samples contained copper. The unfiltered water was the most fluorescent of the water samples, followed by the water filtered through the 14-week old filter; the new, unused filter had the lowest fluorescence intensity of the three samples, although it was still more fluorescent than the negative control sample. Importantly, the method could visually detect micromolar concentrations of copper below the government’s mandated limit.
The development of a high-throughput method to quantify trace copper in synthetic samples would be a step toward Principle 11 of the 12 principles of green chemistry.44 The federal guideline in the United States USP ⟨232⟩ indicates that the copper concentration in the solid state of an API must be below 300 ppms (ppm). To test whether the PPE-DMAPPP method can quantify such low concentrations of copper in drug-like material, we chose 12 commercially available functionalized compounds (Figure 8d) treated with copper at concentrations of 30 and 300 ppm in the solid phase (see the Supporting Information for sample preparation). With the final copper concentrations of 7.1 or 71 ng mL–1 in the assay solution, the fluorescence intensity correlated with the positive control (“no drug” in Figure 8e,f) despite the presence of the drug-like compounds, including N-acetylcysteine; the only exception to this was with 2,2′-bipyridyl, which interfered with the method. Excluding the 2,2′-bipyridyl, the average percent recoveries for the samples with 30 and 300 ppm copper were 112 ± 19% and 92 ± 10%, respectively. These results were consistent across repeated experiments. This result indicates that the method may be applied in the pharmaceutical process chemistry. We compared our method with ICP-OES for imidazole and 2,2′-bipyridyl; although ICP-OES could determine copper concentrations in the 300 ppm samples, it failed to quantify the metal in the corresponding 30 ppm samples. Thus, at least for these samples, the fluorometric method is more sensitive than ICP-OES.
Next, we asked whether the PPE-DMAPPP method could detect copper in three commercially available copper-containing enzymes: superoxide dismutase (SOD), ascorbate oxidase (AO), and bilirubin oxidase (BO). We reproducibly observed that the fluorescence increased only in the presence of SOD (Figures 8g, S12, and S13). Thus, compatibility with copper-containing enzymes is enzyme specific. This may be due to the accessibility of the enzyme-bound copper ions; SOD has a substrate-binding site with copper that apparently remains accessible,45 while BO only transiently opens to allow the substrate to bind.46 Since AO and BO belong to the same family of enzymes, a similar dynamic may apply to AO.47,48
We became concerned that copper ions might be excreted from cells when bound to DMAPPP. As a model system, CuSO4 was suspended in a biphasic system (aqueous pH 7 buffer and CHCl3) in the presence or absence of DMAPPP. Despite our concern, the copper concentrations did not increase in the organic phase when DMAPPP was present (Figure 8h), indicating that the putative copper–DMAPPP complex may not be membrane permeable or may not be sufficiently stable.
Rescreening Ligands for Biological Compatibility
Due to the low solubility of TFPP and DMAPPP in water that proved to be incompatible with live cell imaging in our preliminary studies, we rescreened metal ligands for reactivity with APE and platinum or PPE and copper in Hanks’s Balanced Salt Solution with 1% DMSO for 1 h. This screening revealed that Danphos (12) and 20 accelerated platinum-catalyzed deallylation, with Danphos showing the greatest kinetic competence (Figure 9a). Both phosphines were substantially more water-soluble than TFPP, leading to a more biocompatible imaging method. Among several ligands that accelerated the copper-catalyzed depropargylation of PPE, phosphines 34 and 49 showed greater reactivity than DMAPPP, with 49 (Ellman ligand) being the most efficient (Figure 9b).
Figure 9.

Rescreening ligands under biologically compatible conditions. (a) Heat map with APE and platinum. (b) Heat map with PPE and copper. (c) Ligands used for (a) and (b). Conditions: 1 μM Pt2+ or Cu2+, 100 μM ligand, 20 μM APE or PPE, 1:99 DMSO/HBSS, 25 °C, n = 2.
Cellular Imaging Experiments
Copper and cisplatin are intimately linked in the context of cancer biology insofar as cisplatin trafficking is at least partially dependent on endogenous copper transporters.49−51 Cisplatin binds both copper transporters and copper chaperones, and differential expression and/or activity of some transporters (e.g., ATP7B) have been linked to the development of cisplatin resistance in head and neck squamous cell carcinoma (HNSCC).52 However, understanding of cisplatin resistance mechanisms continues to elude researchers,53,50 in part due to an inability to nondestructively study cisplatin under biological conditions. Tagging platinum complexes with fluorescent molecules is not fluorogenic.54−56 Fluorogenic detection systems compatible with viable cells would greatly advance preclinical studies of cisplatin response and resistance and facilitate the development of personalized therapeutic approaches that match cisplatin dosing with relative tumor sensitivity.57,53
We thus sought to determine whether our probes would illuminate the fates of intracellular cisplatin and copper in living tumor cells to understand cisplatin resistance. We used widefield microscopy with Danphos 12 and APE to image platinum distribution in two HNSCC cell lines, HN30 (cisplatin sensitive) and HN30 R8 (cisplatin resistant), developed in the Sandulache62 group (see the Experimental Section). Following overnight cisplatin exposure, intracellular fluorescence significantly increased (Figure 10a). The analysis of 5 images with 50–70 cells per image showed that HN30 cells exhibited stronger fluorescence signals than HN30 R8 (219 ± 0.8 for HN30, 156 ± 0.96 for HN30 R8, mean ± standard deviation; t test, p < 0.0005). This is consistent with reports that cisplatin-resistant cells can export or sequester cisplatin.58−62
Figure 10.

(a) Widefield fluorescence images of cisplatin-treated HN30 and HN30 R8 cells. For statistical analysis, n = 5 (5 stage positions, with 50–70 cells per image). (b) Confocal fluorescence images of platinum in HN30 and HN30 R8 cells and mean pixel intensity of HN30 and HN30 R8 cells treated with 0 or 8 μM cisplatin. (c) Confocal fluorescence images of copper in HN30 and HN30 R8 cells and mean pixel intensity of HN30 and HN30 R8 cells. For (b) and (c), cells were stained with Hoechst 33342 and 10 kDa Texas Red dextran and loaded with APE and Danphos for platinum imaging or PPE and Ellman ligand for copper imaging. Conditions: 0 or 8 μM cisplatin, 20 μM APE or PPE, 800 μM Danphos or 50 μM phosphine Ellman ligand, 0.1% DMSO in DMEM with penicillin/streptomycin, 30 min, 37 °C. Scale bars = 25 μm.
Reports using indirect methods suggested that cisplatin can be sequestered in lysosomes via ATP7B as a mode of drug resistance.63,64 We performed colocalization studies using 10 kDa dextran conjugated with Texas Red as a fluid phase marker for endocytosis using high speed confocal microscopy (Figure 10b); 10 kDa dextran is a polysaccharide that is sequestered in the lysosomes of cells, and the conjugate has been used to track lysosomes in living cells.65,66 Using this approach, we observed a greater increase in fluorescence in the cisplatin-resistant cells treated with cisplatin: with bright, punctate fluorescence observed within the cytoplasm in both the red and green channels. There was good correlation between the fluorescence produced by the reaction of APE and the fluorescence from Texas Red dextran, such that the Pearson’s correlation coefficient between these was 0.7–0.8 in the cisplatin-resistant cells treated with 8 μM cisplatin. More detailed studies of colocalization using area overlap analysis confirmed that 100% of the red lysosomal structures was positive for the green signal from the APE. In contrast, the fluorescence produced by the reaction of APE was more diffuse and did not appear to form bright puncta consistent with lysosomal accumulation in untreated HN30 R8 cells, suggesting that lysosomal perturbation contributes to cisplatin resistance, consistent with previous studies.58−60
Our combined data using widefield and confocal microscopy to image platinum suggest that cisplatin-sensitive cells accumulate more platinum throughout the entire cell, eventually resulting in cell death. Meanwhile, cisplatin-resistant cells sequester platinum within lysosomes, allowing these cells to evade cisplatin toxicity. The lysosomal sequestration of platinum may be caused by overexpression or activity of the copper transporter ATP7B. Danphos likely enters the lysosomes via passive diffusion; if Danphos were to be internalized via an endocytotic process, we would not see green fluorescence within the lysosomes because Danphos would be encapsulated in newly formed lysosomes that do not contain platinum because platinum was removed prior to the addition of the phosphine. Additionally, in that case, these newly formed vesicles would not be stained with Texas Red dextran and would not account for the colocalization observed.
We were unable to image cisplatin nuclear uptake in the current study. There are several explanations for this. First, the nuclear cisplatin concentration may fall below the detection threshold for our technique.62 Second, either the probe or phosphine may not reach the nucleus. Third, APE and Danphos may penetrate the nuclear envelope, but DNA-bound platinum may be catalytically incompetent.
Next, we sought to determine whether the differential cisplatin localization correlated with copper localization. Toward this objective, the first step was to determine whether the PPE-Ellman ligand technology can detect intracellular copper ions at the basal level. Despite many detection methods for copper,67 it remains highly challenging to image the metal at the basal level.68,69 In effect, as the cell images and the graph in Figure S14 show, only the combination of PPE and Ellman ligand caused increases in fluorescence emissions, supporting the notion that the fluorescence stems from endogenous copper at the basal level (t test, p value 0.0004). The PPE-Ellman ligand method was also effective with HepG2 cells (Figure S15). This imaging protocol revealed that the basal copper level was slightly higher for HN30 R8 cells (Figure 10c). There were no differences in the localizations of platinum and copper, nor were there differences in the localization between the resistant and sensitive cell lines. Pittsburgh Green formed from the reaction of PPE and copper was found in 100% of the lysosomes in cisplatin-resistant and -sensitive cells. The percentage of fluorescence emissions (overlap area) associated with the lysosomes averaged 8.9 ± 0.9% for Pittsburgh Green from APE and 6.0 ± 1.1% for Pittsburgh Green from PPE.
Currently, cisplatin resistance is believed to be acquired, at least in part, by decreased expression of copper importers (e.g., Ctr1 and Ctr2)64 or increased expression of copper exporters (e.g., ATP7A and ATP7B).70 In this work, we imaged copper and cisplatin cellular uptake and cellular localization. Together, our results indicate that there is a difference in total and localized platinum accumulation in cisplatin-sensitive and -resistant cells, while copper levels remain nearly the same. Furthermore, since the distribution of copper ions across cellular compartments was similar between the cisplatin-sensitive and the cisplatin-resistant HNSCC cells, we conclude that acquisition of cisplatin resistance does not necessarily perturb basal copper levels or intracellular distribution despite the role of copper transporters. These new insights warrant further investigations with patient-derived samples for generality. By imaging intracellular uptake and distribution of these two metals, we have eliminated some confounders of previous studies based on large probes to determine localization and the mainly inferential approaches that relied on gene expression and protein levels as an indirect measure of metal trafficking and transport.71−73
Conclusions
The combinatorial screening of ligands and metals led to the discovery and development of sensitive and selective fluorescence methods for platinum and copper ions. Specific phosphines selectively accelerated platinum and copper catalysis for high sensitivity and suppressed the reactivity of other metals including palladium for the allylic and propargylic ethers, respectively. The methods were sufficiently quantitative for the total platinum or copper in complex samples relevant to pharmaceutical production, medicine, and geological exploration. Whether the method can be applied to copper-bound enzymes depends on the enzyme structure. The probes demonstrated utility for biological applications, by providing imaging of intracellular platinum and copper ion distribution in human cancer cells. Using this technique, we demonstrated that the intracellular distributions of platinum and copper ions were independent of each other in cisplatin-resistant cancer cells. Importantly, copper ions were not enriched in the lysosomes of HN30 R8 cells compared to those of HN30 cells. This novel discovery and the gained ability to dissect the underlying mechanisms using the current probe-based approach represent an important step toward understanding and overcoming cisplatin resistance in solid tumors. It is not yet clear whether our method detects labile or protein-bound copper ions inside the cell, and further investigation is needed to address this question. Given the fact that nearly 30 fluorometric methods developed by others are based on palladium-catalyzed allylic C–O bond cleavage, we envision that their methods can also be repurposed for platinum. The tunability of metal selectivity may be applied to other fluorescent probes, which can rapidly broaden applicability for various chemical and biological applications.
Experimental Section
Fluorescence Measurements
All fluorescence measurements, unless otherwise stated, were performed on a Modulus II Microplate Multimode Reader. Fluorescence was measured after exciting samples with 490 nm light, and emission was measured between 510 and 570 nm. All fluorescence measurements were obtained at room temperature.
Screening Ligands versus Metals (Figure 2c,d)
Ligands used in the screen included tri-o-tolylphosphine, 3-(diphenylphosphino)bezenesulfonic acid sodium salt, 1,3,5-triaza-7-phosphaadamantane, 1,1′-bis(diphenylphosphino)ferrocene, tri(2-furyl)phosphine, 1,2-bis(diphenylphosphino)ethane, bis(4-carboxyphenyl)(4-trifluoromethylphenyl)phosphine, tris(2,4-di-tert-butylphenyl)phosphite, 4-(dimethylamino)phenyl-diphenylphosphine, 1,4-bis(diphenylphosphino)butane, 1,3-bis(diphenylphosphino)propane, bis(3-sulfonatophenyl)(3,5-ditrifluoromethylphenyl)phosphine disodium salt, tris(4-fluorophenyl)phosphine, (±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl, tris(4-methoxyphenyl)phosphine, 2-(di-tert-butylphosphine)biphenyl, (1R,2R)-(+)-1,2-diaminocyclohexane-N,N′-bis(2-diphenylphosphino-1-naphthoyl), tri-p-tolylphosphine, tris(4-trifluoromethylphenyl)phosphine, triphenylphosphine-3,3′,3′’-trisulfonic acid trisodium salt, tri-tert-butylphosphonium tetrafluoroborate, bis(diphenylphosphino)methane, di-tert-butylmethylphosphonium tetrafluoroborate, bis(2-diphenylphosphinophenyl)ether, tris(2-cyanoethyl)phosphine, thymine, 2-amino-1,3-propanediol, cytosine, imidazole, 1,3-diphenylthiourea, thiophene, thiazole, pyridine, triphenylphosphine, indole-3-carboxylic acid, and 2,2′-bipyridyl.
In separate vials, 20–30 mg of each ligand was dissolved in DMSO containing 250 ppm butylated hydroxyl toluene (BHT) to a final concentration of 20 mM ligand. Solutions containing the ligands were prepared by mixing 1.23 M phosphate buffer (pH 7, 177.2 mL), DMSO (19.424 mL), and 8.0 mM APE or PPE in DMSO (556 μL). Aliquots of these mixtures (4.944 mL) and 20 mM ligands and 250 ppm BHT in DMSO (55.5 μL) were combined into 15 mL conical tubes to prepare the working solutions. Aliquots of these working solutions (180 μL) were transferred to black, round-bottom 96-well plates.
Metals used in the experiments included AgNO3, AuCl3, CdCl2·2.5H2O, CeCl3·3H2O, CoCl2·6H2O, CrCl2, Na2Cr2O7, CuCl, CuCl2·2H2O, FeSO4, FeCl3, HgCl2, MgCl2·6H2O, K2MnO4, NiCl2, Pb(OAc)2, RhCl(PPh3)3, Rh(OAc)2 dimer, RhCl3·xH2O (45.5% Rh), RuCl3, Sr(NO3)2, and ZnCl2.
In separate vials, 10 mM metal solutions were prepared by mixing 5–20 mg of the listed metals with dilute acid; all metals were dissolved in 3% HCl (TraceMetal grade), with the exception of AgNO3, Sr(NO3)3, and RhCl(PPh3)3. AgNO3 and Sr(NO3)3 were dissolved in 5% HNO3 (TraceMetal grade); RhCl(PPh3)3 was dissolved in DMSO, and the working solutions were adjusted to account for the solvent. The metal solutions were then diluted to 100 μM in 3% HCl or 5% HNO3 (TraceMetal grade) or DMSO.
100 μM Pd2+ and Pt2+ solutions were prepared by diluting Pd and Pt standard solution (1000 ppm = 9.4 or 5.1 mM for Pd and Pt, respectively). For Pd, the 9.4 mM standard (21 μL) was diluted with 5% HNO3 (1.98 mL, TraceMetal grade). For Pt, the 5.1 mM standard (39 μL) was diluted with 3% HCl (1.96 mL, TraceMetal grade).
To perform the assay, 10 μM metal solutions (20 μL) were transferred to the prepared working solutions (180 μL) in the wells. The fluorescence was measured immediately and again after allowing the reaction to proceed for 1 and 2 h at 25 °C.
Effect of Salt (Figure 3a)
125 mM solutions of K2CO3, Na2SO3, K3PO4, NaF, NaCl, NaBr, NaI, NaOAc, NH4OAc, NaNO2, and NaNO3 were prepared. The pH of all solutions was adjusted to 7.0 with NaOH and HCl solutions. Each solution (8.86 mL) was mixed with 800 μM APE in DMSO (278 μL), 20 mM TFPP and 250 ppm BHT in DMSO (111 μL), and DMSO (721 μL) to generate new working solutions. Each working solution (180 μL) was transferred to black, round-bottom 96-well plates. A solution of 5.1 μM Pt in 0.3% HCl (20 μL) was then transferred to the wells. The reaction mixture was incubated at 25 °C for 1 h before the fluorescence was measured. Average and standard deviation values are shown in Figure 3a, and the data set with K3PO4 is normalized to 1.
Effect of pH (Figure 3b)
Aliquots of 1.2 M, pH 7 potassium phosphate buffer (∼50 mL) were transferred to 50 mL conical tubes, and the pH of these solutions was adjusted to 7–9 using NaOH and HCl. The resulting solutions (160 μL) were transferred to a black, round-bottom 96-well plate. A mixture of 800 μM APE in DMSO (480 μL), 20 mM TFPP and 250 ppm BHT in DMSO (192 μL), DMSO (1248 μL), and 5.1 μM Pt standard in 0.3% HCl (1920 μL) was prepared in a 5 mL centrifuge tube. This mixture (40 μL) was transferred to the 96-well plate. The reaction mixture was incubated at 25 °C for 30 min before the fluorescence was measured. Average and standard deviation values are shown in Figure 3b, and the data set at pH 7.0 is normalized to 1.
Effect of Temperature (Figure 3c)
The working solution was prepared by mixing 1.2 M, pH 7 phosphate buffer (8.86 mL), 800 μM APE in DMSO (278 μL), 40 mM TFPP and 250 ppm BHT in DMSO (111 μL), and DMSO (721 μL) in a 15 mL conical tube. The working solution (180 μL) was transferred to black, round-bottom 96-well plates. A solution of 2.5 μM Pt in 0.2% HCl (20 μL) was then transferred to the wells. The samples were sealed to prevent evaporation and incubated at 4, 25, or 45 °C. After 30 min, the fluorescence was measured. Average and standard deviation values are shown in Figure 3c, and the data set at 25 °C is normalized to 1.
Effect of TFPP Concentration (Figure 3d)
A stock working solution was prepared by mixing 1.2 M, pH 7 phosphate buffer (1.28 mL), 800 μM APE in DMSO (40 μL), 20 mM tris(4-fluorophenyl) phosphine (TFPP) and 250 ppm BHT in DMSO (64 μL), and DMSO (56 μL) in a 5 mL centrifuge tube. 2-Fold serial dilutions were performed using 22.2 μM APE in 11.1% DMSO in 1.2 M, pH 7 phosphate buffer as the diluent. The working solutions containing different concentrations of TFPP (180 μL) were transferred to a black, round-bottom 96-well plate. Additionally, a 20 mM stock solution of TFPP was prepared by dissolving TFPP (12.1 mg; 38 μmol) in DMSO containing 250 ppm BHT (1.91 mL). 1.11-Fold serial dilutions were performed using DMSO as the diluent; in total, 32 solutions were prepared with concentrations of TFPP ranging from 0 to 20 mM. To these TFPP solutions (10 μL), DMSO (65 μL), 800 μM APE in DMSO (25 μL), and 1.2 M, pH 7 phosphate buffer (800 μL) were added in this sequence. The newly prepared working solutions containing different concentrations of TFPP (180 μL) were transferred to a black, round-bottom 96-well plate. A solution of 5.1 μM Pt in 0.3% HCl (20 μL) was added to each well. The reaction mixture was incubated at 25 °C for 1 h before the fluorescence was measured. Average and standard deviations are shown in Figure 7b, and the data set with 200 μM TFPP is normalized to 1.
Kinetics of Various Pt Species (Figure 3e)
Platinum solutions were made by dissolving ethylene bis(triphenylphosphine)Pt(0) (3.2 mg; 4.3 μmol), K2PtCl4 (7.0 mg, 17 μmol), or K2PtCl6 (8.8 mg, 18 μmol) in either DMSO or water to a final concentration of 20 μM. Ethylene bis(triphenylphosphine)Pt(0) was dissolved in DMSO; K2PtCl4 and K2PtCl6 were dissolved in water. The ethylene bis(triphenylphosphine)Pt(0) sample was mixed with an equal volume of water, while the K2PtCl4 and K2PtCl6 samples were mixed with equal volumes of DMSO. The resulting 10 μM Pt solutions in 1:1 water/DMSO (20 μL) were transferred to a black, round-bottom 96-well plate. A solution of 20 mM TFPP (2 μL) was transferred to the Pt samples, which was then incubated at 27 °C for 1 h. Following this, a solution containing 22.5 μM APE in 10.1% DMSO in 1.2 M, pH 7 phosphate buffer (178 μL) was transferred to the wells. The fluorescence was measured immediately and again every 10 min for 100 min. The difference between the first fluorescence measurement and each 10 min thereafter was recorded.
Reactions of cis-37 with TFPP or 5 (Figure 3f)
They are described in the Supporting Information.
Effect of Cosolvent on Copper-Catalyzed Depropargylation (Figure 4a)
A 1 mM ligand solution of 4-(dimethylamino)phenyl-diphenylphosphine was prepared in DMSO (2.14 mL), and a 15.7 mM solution of CuSO4 was prepared by dissolving 27.8 mg of CuSO4 in Millipore water (11.12 mL). This was then diluted to 10 μM CuSO4 (10 mL) using Millipore H2O as the diluent. A solution of 8 mM propargyl Pittsburgh Green ether (10 μL) was dissolved in each of the following cosolvents: EtOH, DMSO, NMP, and MeCN (90 μL) in separate 2 mL microcentrifuge tubes. A working solution was prepared for each cosolvent containing 1.2 M, pH 7 phosphate buffer (1.5 mL), the cosolvent (230 μL), 1 mM 4-(dimethylamino)phenyl-diphenylphosphine (20 μL), and 800 μM PPE (50 μL). Each working solution (180 μL) was added to six wells in a black 96-well plate. To the first three wells, Millipore water (20 μL) was added, and 10 μM CuSO4 (20 μL) was added to other three wells. The fluorescence was measured immediately and in 30 min.
Effect of pH on Reaction (Figure 4b)
Commercial pH 5, 6, 7, and 8 phosphate buffers (Fisher Scientific) were diluted to 50 mM. Four different working solutions were prepared by mixing the buffers (1.92 mL), DMSO (60 μL), 800 μM PPE in DMSO (60 μL), and 4 mM DMAPPP in DMSO (120 μL). The working solutions (180 μL) were then added to a black 96-well plate. A solution of 10 mM CuCl or CuCl2 in 3% HCl was diluted to 100 μM using water as the diluent. These solutions (20 μL) were then added to the wells, and the fluorescence was measured.
Effect of Buffer Salt on Reaction (Figure 4c)
Different buffers were purchased from Fisher Scientific. These were 50 mM phosphate, pH 8.5 buffer (Fisher Scientific catalog #SB112-500), 1 M, pH 8.5 sodium bicarbonate buffer (Fisher Scientific catalog #AAJ60408AK), 0.5 M, pH 8.5 borate buffer (Fisher Scientific catalog #AAJ60803AK), and pH 10 carbonate buffer (Fisher Scientific catalog #AC258605000).
Four different working solutions were prepared by mixing the buffers (1.20 mL), EtOH (184 μL), 800 μM PPE in EtOH (40 μL), and 20 mM DMAPPP in DMSO (16 μL). The working solutions (180 μL) were then added to a black 96-well plate. A solution of 0 or 20 μM CuSO4 in distilled ultrapure water (20 μL) was then added to the wells, and the fluorescence was measured.
Effect of Buffer Salt after Controlling for Concentrations (Figure 4d)
The buffers purchased for the experiment testing the effect of different buffer salts were diluted to 50 mM. Three different working solutions were prepared by mixing the buffers (1.20 mL), EtOH (184 μL), 800 μM PPE in EtOH (40 μL), and 20 mM DMAPPP in DMSO (16 μL). The working solutions (180 μL) were then added to a black 96-well plate. A solution of 0 or 20 μM CuSO4 in distilled ultrapure water (20 μL) was then added to the wells, and the fluorescence was measured.
Effect of Cosolvent Concentrations (Figure 4e)
Seven different working solutions were prepared, varying only the concentration of EtOH. Solutions containing distilled ultrapure water (1197, 1097, 897, 697, 497, 297, or 97 μL) and EtOH (250, 350, 550, 750, 950, 1150, or 1350 μL) were prepared, to which 500 mM, pH 8.5 borate buffer (53 μL), 800 μM PPE in EtOH (50 μL), and 10 mM DMAPPP in DMSO (40 μL) were also added. The working solutions (180 μL) were added to a black 96-well plate. A solution of 0 or 5 μM CuSO4 in distilled ultrapure water (20 μL) was then added to the wells, and the fluorescence was measured.
Effect of DMAPPP Concentrations (Figure 4f)
The working solution was prepared by mixing 500 mM, pH 8.5 borate buffer (2.368 mL), EtOH (400 μL), 800 μM PPE in EtOH (80 μL), and 10 mM DMAPPP in DMSO (32 μL). 2-Fold serial dilutions of the working solution were performed using a solution of 800 μM PPE in EtOH (300 μL), DMSO (120 μL), EtOH (1.500 mL), and 500 mM, pH 8.5 borate buffer (8.880 mL) as a diluent. Each dilution of the working solution (180 μL) was transferred to a black, round-bottom 96-well plate. The final concentration of the DMAPPP ranged from 0 to 200 μM.
A solution of 0 or 5 μM CuSO4 (20 μL) was then transferred to the wells containing the working solution. The fluorescence was measured immediately after the addition of the copper solution and again after 30 min. The fluorescence intensities after 30 min were reported. Average and standard deviation values are shown in Figure 4f.
Metal Selectivity for the APE-TFPP System (Figure 5a)
The working solution was prepared by mixing 1.2 M, pH 7.8 phosphate buffer (12.00 mL), 800 μM APE in DMSO (375 μL), 40 mM TFPP and 250 ppm BHT in DMSO (150 μL), and DMSO (975 μL). The resulting working solution (180 μL) was transferred to black, round-bottom 96-well plates. 100 μM metal solutions (20 μL) were then transferred to the wells, and the fluorescence was measured immediately and again 10 min later at 25 °C. Average and standard deviation values are shown in Figure 5a, and the data set with no metal is normalized to 1.
Metal Selectivity of the PPE-DMAPPP System (Figure 5b)
The working solution was prepared by mixing 500 mM, pH 8.5 borate buffer (13.5 mL), EtOH (2.51 mL), 8.0 mM PPE in EtOH (45 μL), and 20 mM DMAPPP in DMSO (144 μL) in a conical tube. The working solution (180 μL) was then added to a black 96-well plate. The metal solutions used for the combinatorial screen were then diluted to 10 μM using water as the diluent. The metal solutions (20 μL) were then added to the wells, and the fluorescence was measured.
Establishing a Pt Standard Curve (Figure 7a)
The working solution was prepared by mixing 1.2 M, pH 7.8 phosphate buffer (17.72 mL), 800 μM APE in DMSO (556 μL), 40 mM TFPP and 250 ppm BHT in DMSO (222 μL), and DMSO (1.44 mL). The resulting working solution (180 μL) was transferred to black, round-bottom 96-well plates.
1.5-Fold serial dilutions of the Pt standard were performed from 5.1 μM in 0.3% HCl using ultrapure water as the diluent. The Pt solutions (20 μL) were then transferred to the wells containing the working solution. The fluorescence was measured immediately after the addition of the Pt solutions and again after incubation at 25 °C for 1 h. The difference in the fluorescence intensities was reported. Average, standard deviation values, and linear regression are shown in Figure 7a.
Limit of Detection and Limit of Quantification Calculations (Figure 7a)
A linear regression was performed using GraphPad Prism 8. The LOD was determined by multiplying the standard error of the regression by 3 and dividing by the slope of the regression line; the LOQ was determined by multiplying the standard error of the regression by 10 and dividing by the slope of the regression line.
Quantification of Pt in Rock Samples (Figure 7b)
Approximately 100 mg of ore samples A–M from Stillwater74 were transferred into glass vials. The working solution was prepared by the addition of 1.2 M, pH 7.8 phosphate buffer (6.40 mL), DMSO (700 μL), 8.0 mM APE in DMSO (20.0 μL), and 40 mM TFPP in DMSO (80.0 μL). The working solution was added to the vials to a mixture of 100 mg of ore in 500 μL of working solution. The vials were then placed in a sonicator for 2 h. The temperature of the water in the sonicator increased from 25 to 45 °C over the course of the experiment. The samples were then centrifuged. The supernatant (100 μL) was placed in the wells of a black 96-well plate, and the fluorescence was measured.
Standard Curve of Cisplatin in Serum (Figure 7c)
Protein-free serum was prepared by mixing 1:2 serum to EtOH. The mixture was centrifuged at 14 000 rpm in an Eppendorf Centrifuge 5417R for 1 h at 4 °C. Cisplatin (5.0 mg, 17 μmol) was dissolved in DMSO (1.67 mL) to prepare a 10 mM solution, which was diluted to a concentration of 1.0 mM. The cisplatin solution was added to protein-free serum in a 1:999 v/v ratio, resulting in a final concentration of 1.0 μM cisplatin in protein-free serum. 1.11-Fold serial dilutions were performed using this sample as the highest concentration and protein-free serum as the diluent. The working solution was prepared in the same way as in the standard curve experiment. The working solution (180 μL) was transferred to the cisplatin-serum samples (20 μL) in black, round-bottom 96-well plates, and the fluorescence was measured immediately. Following the initial measurement, the plates were covered to prevent evaporation and incubated at 45 °C for 1 h before measuring the fluorescence again. The difference in fluorescence intensities was reported.
Kinetic Study (Figure 8a)
A standard curve was prepared for dichlorofluorescein (DCF) by dissolving the solid in EtOH to a concentration of 20 mM. The DCF was then diluted to 500 μM in EtOH. A stock solution was prepared by adding 500 mM, pH 8.5 borate buffer (600 μL), EtOH (81.6 μL), 500 μM DCF in EtOH (32 μL), and 20 mM DMAPPP in DMSO (6.4 μL). This stock solution (200 μL) was then transferred to a black 96-well plate.
The diluent was prepared by mixing 500 mM, pH 8.5 borate buffer (9.00 mL), EtOH (1.70 mL), and 20 mM DMAPPP in DMSO (96 μL). The diluent (100 μL) was transferred to the black 96-well plate. 2-Fold serial dilutions were performed by removing 100 μL of the stock solution from the wells and adding it to the wells containing the diluent. Following this, an additional 100 μL of the diluent was added to the wells for a total volume of 200 μL. The fluorescence of these solutions was measured, and a standard curve was generated.
The working solution was prepared by mixing 500 mM, pH 8.5 borate buffer (1.20 mL), EtOH (187 μL), 800 μM PPE in EtOH (40 μL), and 20 mM DMAPPP in DMSO (12.8 μL) in a conical tube. The working solution (180 μL) was then added to a black 96-well plate. A solution of 0 or 2.0 μM Cu2+ (20 μL) was then added to the wells, and the fluorescence was measured every 5 min for 2 h.
The fluorescence value was converted to a concentration of Pittsburgh Green using the linear regression generated from the standard curve of DCF. The fluorescence vs time was then plotted.
Copper Concentration Dependence to Determine the Limit of Detection and Limit of Quantification (Figure 8b)
The working solution was prepared by mixing 500 mM, pH 8.5 borate buffer (16.280 mL), EtOH (2.750 mL), 800 μM PPE in EtOH (550 μL), 20 mM DMAPPP in DMSO (120 μL), and DMSO (110 μL). The resulting working solution (180 μL) was transferred to a black, round-bottom 96-well plate.
1.11-Fold serial dilutions of CuSO4 in water were performed from 250 nM. The fluorescence was measured immediately after the addition of copper and every 5 min for 2 h. The fluorescence intensities at 2 h were reported. Average, standard deviation values, and linear regression are shown in Figure 8b. For lower concentrations of copper, a HoribaMax Fluorometer (excitation 490 nm, emission 526–528 nm) was used.
Visualization of the Relative Concentrations of Copper in Drinking Water Samples (Figure 8c)
Three fountain water samples were prepared; the fountain water was kept unfiltered, filtered through a 14-week old drinking water filter, or filtered through a new, unused drinking water filter. The working solution was prepared by the addition of 0.5 M, pH 8.5 borate buffer (2.10 mL), EtOH (814.5 μL), 800 μM PPE in EtOH (7.5 μL), and 20 mM DMAPPP in DMSO (24 μL). Dowex M4195 resin was added to the working solution. The working solution (500 μL) was transferred to vials. 0 or 5 μM CuSO4 in distilled Milli-Q water or the fountain water samples (500 μL) were added, and the fluorescence was observed over time.
Quantification of Copper in Drug-Like Compounds (Figure 8e,f)
Pyridine, yohimbine, 2,2′-bipyridyl, indole, morpholine, 2-butyn-1-ol, N-acetylcysteine, quinine, imidazole, pyrazole, propynol, and bromobenzene were dissolved in DMSO to a concentration of 10 mg/mL. Ten mg/mL of the drug-like samples (1.00 mL) were mixed with 0.006 or 0.06 mg/mL CuSO4 in water (50 μL).
Distilled Milli-Q water (15.0 μL) was added to the wells of a 96-well plate. Ten mg/mL of 12 different drug-like samples spiked with 0.006 mg/mL Cu2+ (5.00 μL) was then added to the wells. The working solution was prepared by mixing 0.5 M, pH 8.5 borate buffer (7.50 mL), EtOH (1.17 mL), 800 μM PPE in EtOH (250 μL), and 20 mM DMAPPP in DMSO (80 μL). The working solution (180 μL) was transferred to the wells, and the fluorescence was measured.
Detection of Copper in Copper-Containing Enzymes (Figure 8g)
The working solution was prepared by mixing 0.5 M, pH 8.5 borate buffer (1068 μL), 100 μM PPE in EtOH (6.00 μL), and 2.0 mM DMAPPP in DMSO (6.00 μL). Dowex M4195 resin was added to the working solution. The working solution (270 μL) was transferred to cuvettes. A 100 mM phosphate buffer or 5 mg/mL superoxide dismutase in 100 mM, pH 7.8 phosphate buffer, bilirubin oxidase in 100 mM, pH 8.4 phosphate buffer, or ascorbate oxidase in 100 mM, pH 7.0 phosphate buffer (30 μL) was added, and the fluorescence was observed over time. The fluorescence was measured using a Horiba FluoroMax-3 fluorometer with 2 nm slit widths (excitation 490 nm, emission 515 nm).
Distribution of Copper between Water and CHCl3 in the Presence of DMAPPP (Figure 8h)
Three suspensions were prepared from these: 10 mM DMAPPP in CHCl3 (1.0 mL) and 5 mM CuSO4 in 1 mM phosphate buffer (pH 7.0, 1.0 mL), CHCl3 (1.0 mL) and 5 mM CuSO4 in 1 mM phosphate buffer (pH 7.0, 1.0 mL), and 10 mM DMAPPP in CHCl3 (1.0 mL) and 1 mM phosphate buffer (pH 7.0, 1.0 mL). The suspensions were vortexed to ensure proper partitioning of the CuSO4 and DMAPPP. Aliquots of the aqueous and organic layers of each suspension (100 μL each) were then taken and evaporated. The residue following evaporation was resuspended in HBSS (100 μL).
The working solution was prepared by mixing 50 mM, pH 8.5 borate buffer (2.96 mL), EtOH (500 μL), 800 μM PPE in EtOH (100 μL), and 10 mM DMAPPP in DMSO (40 μL). The working solution (180 μL) was transferred to the wells of a black 96-well plate. The resuspended samples (20 μL) were then added to the wells, and the fluorescence was measured.
Screening Ligands for Biologically Compatible Conditions (Figure 9)
Solutions (20 nM) of ligands were prepared in DMSO and stored overnight at −20 °C. Working solutions were prepared by mixing HBSS (1.068 mL), 3.6 mM APE or PPE in DMSO (6 μL), and 0 or 20 mM ligands in DMSO (6 μL). The working solutions (180 μL) were transferred to the wells of a black 96-well plate. Pt or Cu (10 μM) was added to the wells, and the fluorescence was measured immediately and again 30 and 60 min later at 25 °C.
HN30 R8 Cell Line Description
The parental HN30 cell line (head and neck squamous cell carcinoma, HNSCC) was obtained from an established cell line bank in the laboratory of Dr. Jeffrey N. Myers under approved institutional protocols and authenticated using short tandem repeat analysis every 3 months. Cisplatin-resistant HN30 R8 cells were generated from HN30 by gradually increasing the cisplatin concentration in culture media from 0.3 to 8 μM, respectively, at a rate of approximately 1 μM every 2 weeks. HN30 R8 cells were maintained in growth media containing 8 μM cisplatin for the remainder of the experimental period, except when cisplatin was withdrawn for specific individual experiments. HN30 R8 cells were generated at ∼21 weeks following initial cisplatin treatment.
Widefield and Swept Field Confocal Fluorescence Imaging of Cisplatin and Copper in HN30 and HN30 8R Cells (Figure 10)
HN30 or HN30 R8 cells were seeded in MatTek dishes and incubated for 2 days in a water-saturated, 5% CO2 atmosphere. The media was then replaced with media containing 5 μg/mL Texas Red dextran for 3 h. The media was again replaced with media containing 0 or 8 μM cisplatin in 0.1% DMSO in DMEM containing 10% FBS and penicillin/streptomycin for Figure 10b or DMEM containing 10% FBS and penicillin/streptomycin for Figure 10c and allowed to incubate overnight. Following the overnight incubation, the media containing metal was removed and fresh media was added. Cells were then loaded with 5 μg/mL Hoechst 33342 and 800 μM Danphos or 50 μM Ellman ligand in DMEM with penicillin/streptomycin for at least 30 min. The media was then removed, and DMEM with penicillin/streptomycin (2.00 mL) was transferred to each dish prior to imaging. APE or PPE (8.0 mM) in DMSO (2.5 μL) was transferred to the dish during imaging. Time-lapse data were acquired on a Nikon Ti stand equipped with a 60× (1.4 NA) objective and scientific CMOS camera (Hamamatsu ORCA-Flash 4.0). Experiments were performed twice with 3–5 stage positions collected per dish. Data were acquired and analyzed using NIS Elements (Nikon Inc., Melville NY). For the intensity measurements shown in Figure 10b,c, the images were segmented on the basis of intensity (thresholded) and mean pixel intensity within the binary mask was expressed in arbitrary units. Co-localization of the Pittsburgh Green product (green) with the lysosomal marker (red) was assessed using area overlap analysis.75 Each channel (green and red) was first segmented on the basis of emission intensity to generate a binary mask. A binary “having” statement was used to quantify the pixels that were positive for both channels (overlapping red and green).
Safety Statement
Aqua regia must be handled with caution.
Acknowledgments
This work was supported by research grants from the US National Science Foundation (Grants CHE-0911092 and CHE-1506942), Merck Research Laboratories, and Aging Institute of the University of Pittsburgh to K. Koide. We thank Melissa L. Campbell and Miho Naruse (University of Pittsburgh) for preliminary results and producing the data shown in Figure 8b, respectively. V.C.S. is supported by National Institute of Dental and Craniofacial Research (NIDCR) R03DE028858.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00676.
Validation studies performed with APE and PPE, platinum binding of bidentate ligands, reaction of cis-37 with TFPP or 5 in CDCl3, X-ray structure of cis-dichloro-bis(tris(4-fluorophenyl)phosphine)platinum, standard curve used for the calculation of turnover frequency, effect of NaBH4, verification of the platinum method in rock samples, detection of copper in enzymes, widefield imaging of copper in HeLa cells, confocal imaging of copper in HepG2 cells, and tables of raw fluorescence data (PDF)
CIF data (CIF) for cis-38 (cis-PtCl2(TFP)2) (CIF)
The authors declare no competing financial interest.
During the review process, a biocompatible platinum-catalyzed bond cleavage76 and ligand effects on a palladium-based fluorescent probe for carbon monoxide have been published.77
Supplementary Material
References
- Ingle J. D. Jr.; Crouch S. R.. Spectrochemical Analysis; Prentice Hall: Upper Saddle River, New Jersey, USA, 1988. [Google Scholar]
- Vandecasteele C.; Vanhoe H.; Dams R. Inductively coupled plasma mass spectrometry of biological samples. Invited lecture. J. Anal. At. Spectrom. 1993, 8, 781–786. 10.1039/ja9930800781. [DOI] [Google Scholar]
- Chae M. Y.; Czarnik A. W. Fluorometric chemodosimetry. Mercury(II) and silver(I) indication in water via enhanced fluorescence signaling. J. Am. Chem. Soc. 1992, 114, 9704–9705. 10.1021/ja00050a085. [DOI] [Google Scholar]
- Li X.; Gao X.; Shi W.; Ma H. Design strategies for water-soluble small molecular chromogenic and fluorogenic probes. Chem. Rev. 2014, 114, 590–659. 10.1021/cr300508p. [DOI] [PubMed] [Google Scholar]
- Pershagen E.; Borbas K. E. Designing reactivity-based responsive lanthanide probes for multicolor detection in biological systems. Coord. Chem. Rev. 2014, 273, 30–46. 10.1016/j.ccr.2013.10.012. [DOI] [Google Scholar]
- Aron A. T.; Ramos-Torres K. M.; Cotruvo J. A.; Chang C. J. Recognition- and reactivity-based fluorescent probes for studying transition metal signaling in living systems. Acc. Chem. Res. 2015, 48, 2434–2442. 10.1021/acs.accounts.5b00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y. H.; Lee D. Y.; Wang J. L.; Li G. H.; Yu J. H.; Lin W. Y.; Yoon J. Y. Development of fluorescent probes based on protection-deprotection of the key functional groups for biological imaging. Chem. Soc. Rev. 2015, 44, 5003–5015. 10.1039/C5CS00103J. [DOI] [PubMed] [Google Scholar]
- Singha S.; Jun Y. W.; Sarkar S.; Ahn K. H. An endeavor in the reaction-based approach to fluorescent probes for biorelevant analytes: Challenges and achievements. Acc. Chem. Res. 2019, 52, 2571–2581. 10.1021/acs.accounts.9b00314. [DOI] [PubMed] [Google Scholar]
- Chang C. J.; James T. D.; New E. J.; Tang B. Z. Activity-based sensing: Achieving chemical selectivity through chemical reactivity. Acc. Chem. Res. 2020, 53, 1. 10.1021/acs.accounts.9b00542. [DOI] [PubMed] [Google Scholar]
- Yun S.-W.; Kang N.-Y.; Park S.-J.; Ha H.-H.; Kim Y. K.; Lee J.-S.; Chang Y.-T. Diversity oriented fluorescence library approach (DOFLA) for live cell imaging probe development. Acc. Chem. Res. 2014, 47, 1277–1286. 10.1021/ar400285f. [DOI] [PubMed] [Google Scholar]
- Burgess K.; Lim H. J.; Porte A. M.; Sulikowski G. A. New catalysts and conditions for a C-H insertion reaction identified by high throughput catalyst screening. Angew. Chem., Int. Ed. Engl. 1996, 35, 220–222. 10.1002/anie.199602201. [DOI] [Google Scholar]
- Robbins D. W.; Hartwig J. F. A simple, multidimensional approach to high-throughput discovery of catalytic reactions. Science 2011, 333, 1423–1427. 10.1126/science.1207922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitrago Santanilla A.; Regalado E. L.; Pereira T.; Shevlin M.; Bateman K.; Campeau L. C.; Schneeweis J.; Berritt S.; Shi Z. C.; Nantermet P.; et al. Nanomole-scale high-throughput chemistry for the synthesis of complex molecules. Science 2015, 347, 49–53. 10.1126/science.1259203. [DOI] [PubMed] [Google Scholar]
- Renom-Carrasco M.; Lefort L. Ligand libraries for high throughput screening of homogeneous catalysts. Chem. Soc. Rev. 2018, 47, 5038–5060. 10.1039/C7CS00844A. [DOI] [PubMed] [Google Scholar]
- Isbrandt E. S.; Sullivan R. J.; Newman S. G. High throughput strategies for the discovery and optimization of catalytic reactions. Angew. Chem., Int. Ed. 2019, 58, 7180–7191. 10.1002/anie.201812534. [DOI] [PubMed] [Google Scholar]
- Song F. L.; Garner A. L.; Koide K. A highly sensitive fluorescent sensor for palladium based on the allylic oxidative insertion mechanism. J. Am. Chem. Soc. 2007, 129, 12354–12355. 10.1021/ja073910q. [DOI] [PubMed] [Google Scholar]
- Santra M.; Ko S. K.; Shin I.; Ahn K. H. Fluorescent detection of palladium species with an O-propargylated fluorescein. Chem. Commun. 2010, 46, 3964–3966. 10.1039/c001922d. [DOI] [PubMed] [Google Scholar]
- Heffern M. C.; Park H. M.; Au-Yeung H. Y.; Van de Bittner G. C.; Ackerman C. M.; Stahl A.; Chang C. J. In vivo bioluminescence imaging reveals copper deficiency in a murine model of nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 14219–14224. 10.1073/pnas.1613628113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. Y.-S.; Posimo J. M.; Lee S.; Tsang T.; Davis J. M.; Brady D. C.; Chang C. J. Activity-based ratiometric FRET probe reveals oncogene-driven changes in labile copper pools induced by altered glutathione metabolism. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 18285–18294. 10.1073/pnas.1904610116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capone K.; Azzam R. K. Wilson’s Disease: A review for the general pediatrician. Pediatr. Annals 2018, 47, E440–E444. 10.3928/19382359-20181026-01. [DOI] [PubMed] [Google Scholar]
- Gillison M. L.; Trotti A. M.; Harris J.; Eisbruch A.; Harari P. M.; Adelstein D. J.; Jordan R. C. K.; Zhao W.; Sturgis E. M.; Burtness B.; et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): a randomised, multicentre, non-inferiority trial. Lancet 2019, 393, 40–50. 10.1016/S0140-6736(18)32779-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makovec T. Cisplatin and beyond: molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. 10.2478/raon-2019-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; VanVranken D. L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 1996, 96, 395–422. 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- Garner A. L.; Koide K. Fluorescent method for platinum detection in buffers and serums for cancer medicine and occupational hazards. Chem. Commun. 2009, 83–85. 10.1039/B817220J. [DOI] [PubMed] [Google Scholar]
- Huang G. X.; Cheng C.; Ge L.; Guo B. B.; Zhao L.; Wu X. Y. Trialkyl methanetricarboxylate as dialkyl malonate surrogate in copper-catalyzed enantioselective propargylic substitution. Org. Lett. 2015, 17, 4894–4897. 10.1021/acs.orglett.5b02463. [DOI] [PubMed] [Google Scholar]
- Cheng C.; Ge L.; Lu X. H.; Huang J. P.; Huang H. C.; Chen J.; Cao W. G.; Wu X. Y. Cu-Pybox catalyzed synthesis of 2,3-disubstituted imidazo[1,2-a]pyridines from 2-aminopyridines and propargyl alcohol derivatives. Tetrahedron 2016, 72, 6866–6874. 10.1016/j.tet.2016.09.013. [DOI] [Google Scholar]
- Shao L.; Wang Y. H.; Zhang D. Y.; Xu J.; Hu X. P. Desilylation-activated propargylic transformation: Enantioselective copper-catalyzed [3 + 2] cycloaddition of propargylic esters with β-naphthol or phenol derivatives. Angew. Chem., Int. Ed. 2016, 55, 5014–5018. 10.1002/anie.201510793. [DOI] [PubMed] [Google Scholar]
- Shao L.; Zhang D.-Y.; Wang Y.-H.; Hu X.-P. Enantioselective copper-catalyzed propargylic etherification of propargylic esters with phenols promoted by inorganic base additives. Adv. Synth. Catal. 2016, 358, 2558–2563. 10.1002/adsc.201600284. [DOI] [Google Scholar]
- Tsuchida K.; Senda Y.; Nakajima K.; Nishibayashi Y. Construction of chiral tri- and tetra-arylmethanes bearing quaternary carbon centers: Copper-catalyzed enantioselective propargylation of indoles with propargylic esters. Angew. Chem., Int. Ed. 2016, 55, 9728–9732. 10.1002/anie.201604182. [DOI] [PubMed] [Google Scholar]
- Yang L. C.; McRae R.; Henary M. M.; Patel R.; Lai B.; Vogt S.; Fahrni C. J. Imaging of the intracellular topography of copper with a fluorescent sensor and by synchrotron x-ray fluorescence microscopy. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11179–11184. 10.1073/pnas.0406547102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.; Miller E. W.; Pralle A.; Isacoff E. Y.; Chang C. J. A selective turn-on fluorescent sensor for imaging copper in living cells. J. Am. Chem. Soc. 2006, 128, 10–11. 10.1021/ja055064u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrni C. J. Synthetic fluorescent probes for monovalent copper. Curr. Opin. Chem. Biol. 2013, 17, 656–662. 10.1016/j.cbpa.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotruvo J. A. Jr.; Aron A. T.; Ramos-Torres K. M.; Chang C. J. Synthetic fluorescent probes for studying copper in biological systems. Chem. Soc. Rev. 2015, 44, 4400–4414. 10.1039/C4CS00346B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M. T.; McCallum A. M.; Fahrni C. J. Rational design of a water-soluble, lipid-compatible fluorescent probe for Cu(I) with sub-part-per-trillion sensitivity. Chem. Sci. 2016, 7, 1468–1473. 10.1039/C5SC03643G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T.; Miyamoto Y.; Ipposhi J.; Nakahara Y.; Utsunomiya M.; Mashima K. Platinum-catalyzed direct amination of allylic alcohols under mild conditions: Ligand and microwave effects, substrate scope, and mechanistic study. J. Am. Chem. Soc. 2009, 131, 14317–14328. 10.1021/ja9046075. [DOI] [PubMed] [Google Scholar]
- Malatesta L.; Cariello C. Platinum(0) compounds with triarylphosphines and analogous ligands. J. Chem. Soc. 1958, 2323–2328. 10.1039/jr9580002323. [DOI] [Google Scholar]
- Sen A.; Halpern J. Role of transition metal-dioxygen complexes in catalytic-oxidation - catalysis of oxidation of phosphines by dioxygen adducts of platinum. J. Am. Chem. Soc. 1977, 99, 8337–8339. 10.1021/ja00467a046. [DOI] [Google Scholar]
- Niemeyer Z. L.; Milo A.; Hickey D. P.; Sigman M. S. Parameterization of phosphine ligands reveals mechanistic pathways and predicts reaction outcomes. Nat. Chem. 2016, 8, 610–617. 10.1038/nchem.2501. [DOI] [PubMed] [Google Scholar]
- Pohorilets I.; Tracey M. P.; LeClaire M. J.; Moore E. M.; Lu G.; Liu P.; Koide K. Kinetics and inverse temperature dependence of a Tsuji-Trost reaction in aqueous buffer. ACS Catal. 2019, 9, 11720–11733. 10.1021/acscatal.9b03011. [DOI] [Google Scholar]
- Ghosh S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. 10.1016/j.bioorg.2019.102925. [DOI] [PubMed] [Google Scholar]
- Peng B.; English M. W.; Boddy A. V.; Price L.; Wyllie R.; Pearson A. D. J.; Tilby M. J.; Newell D. R. Cisplatin pharmacokinetics in children with cancer. Eur. J. Cancer 1997, 33, 1823–1828. 10.1016/S0959-8049(97)00341-9. [DOI] [PubMed] [Google Scholar]
- Urien S.; Brain E.; Bugat R.; Pivot X.; Lochon I.; Van M. L.; Vauzelle F.; Lokiec F. Pharmacokinetics of platinum after oral or intravenous cisplatin: a phase 1 study in 32 adult patients. Cancer Chemother. Pharmacol. 2005, 55, 55–60. 10.1007/s00280-004-0852-8. [DOI] [PubMed] [Google Scholar]
- Sprowl J. A.; Gregorc V.; Lazzari C.; Mathijssen R. H.; Loos W. J.; Sparreboom A. Associations between ABCC2 polymorphisms and cisplatin disposition and efficacy. Clin. Pharmacol. Ther. 2012, 91, 1022–1026. 10.1038/clpt.2011.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erythropel H. C.; Zimmerman J. B.; de Winter T. M.; Petitjean L.; Melnikov F.; Lam C. H.; Lounsbury A. W.; Mellor K. E.; Janković N. Z.; Tu Q. S.; et al. The Green ChemisTREE: 20 years after taking root with the 12 principles. Green Chem. 2018, 20, 1929–1961. 10.1039/C8GC00482J. [DOI] [Google Scholar]
- Tainer J. A.; Getzoff E. D.; Richardson J. S.; Richardson D. C. Structure and Mechanism of Copper, Zinc Superoxide-Dismutase. Nature 1983, 306, 284–287. 10.1038/306284a0. [DOI] [PubMed] [Google Scholar]
- Cracknell J. A.; McNamara T. P.; Lowe E. D.; Blanford C. F. Bilirubin oxidase from Myrothecium verrucaria: X-ray determination of the complete crystal structure and a rational surface modification for enhanced electrocatalytic O-2 reduction. Dalton T. 2011, 40, 6668–6675. 10.1039/c0dt01403f. [DOI] [PubMed] [Google Scholar]
- Messerschmidt A.; Huber R. The Blue Oxidases, Ascorbate Oxidase, Laccase and Ceruloplasmin - Modeling and Structural Relationships. Eur. J. Biochem. 1990, 187, 341–352. 10.1111/j.1432-1033.1990.tb15311.x. [DOI] [PubMed] [Google Scholar]
- Sakurai T.; Kataoka K. Basic and applied features of multicopper oxidases, CueO, bilirubin oxidase, and laccase. Chem. Rec. 2007, 7, 220–229. 10.1002/tcr.20125. [DOI] [PubMed] [Google Scholar]
- Ishida S.; Lee J.; Thiele D. J.; Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 14298–14302. 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.; Ding Q.; Zhang Z.; Wang S.; Zhong B.; Huang X.; Shao Z. PTBP1 modulates osteosarcoma chemoresistance to cisplatin by regulating the expression of the copper transporter SLC31A1. J. Cell. Mol. Med. 2020, 24, 5274–5289. 10.1111/jcmm.15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Kang Y.; Chen L.; Wang H.; Liu J.; Zeng S.; Yu L. The drug-resistance mechanisms of five platinum-based antitumor agents. Front. Pharmacol. 2020, 11, 343. 10.3389/fphar.2020.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas A.; Duvvuri U.; Kiselyov K. Copper-dependent ATP7B up-regulation drives the resistance of TMEM16A-overexpressing head-and-neck cancer models to platinum toxicity. Biochem. J. 2019, 476, 3705–3719. 10.1042/BCJ20190591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. 10.1016/j.phrs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Tracey M. P.; Pham D.; Koide K. Fluorometric imaging methods for palladium and platinum and the use of palladium for imaging biomolecules. Chem. Soc. Rev. 2015, 44, 4769–4791. 10.1039/C4CS00323C. [DOI] [PubMed] [Google Scholar]
- White J. D.; Haley M. M.; DeRose V. J. Multifunctional Pt(II) reagents: Covalent modifications of Pt complexes enable diverse structural variation and in-cell detection. Acc. Chem. Res. 2016, 49, 56–66. 10.1021/acs.accounts.5b00322. [DOI] [PubMed] [Google Scholar]
- Farrer N. J.; Griffith D. M. Exploiting azide-alkyne click chemistry in the synthesis, tracking and targeting of platinum anticancer complexes. Curr. Opin. Chem. Biol. 2020, 55, 59–68. 10.1016/j.cbpa.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalami Y.; de Castro G.; Bernard-Marty C.; Awada A. Management of head and neck cancer in elderly patients. Drugs Aging 2009, 26, 571–583. 10.2165/11316340-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Safaei R.; Katano K.; Larson B. J.; Samimi G.; Holzer A. K.; Naerdemann W.; Tomioka M.; Goodman M.; Howell S. B. Intracellular localization and trafficking of fluorescein-labeled cisplatin in human ovarian carcinoma cells. Clin. Cancer Res. 2005, 11, 756–767. [PubMed] [Google Scholar]
- Safaei R.; Larson B. J.; Cheng T. C.; Gibson M. A.; Otani S.; Naerdemann W.; Howell S. B. Abnormal lysosomal trafficking and export of cisplatin in drug-resistant ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604. 10.1158/1535-7163.MCT-05-0102. [DOI] [PubMed] [Google Scholar]
- Nilsson C.; Roberg K.; Grafstrom R. C.; Ollinger K. Intrinsic differences in cisplatin sensitivity of head and neck cancer cell lines: Correlation to lysosomal pH. Head Neck-J. Sci. Spec. 2010, 32, 1185–1194. 10.1002/hed.21317. [DOI] [PubMed] [Google Scholar]
- Zhitomirsky B.; Assaraf Y. G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. 10.1016/j.drup.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Yu W.; Chen Y.; Putluri N.; Coarfa C.; Robertson M. J.; Putluri V.; Stossi F.; Dubrulle J.; Mancini M. A.; Pang J. C.; Nguyen T.; Baluya D.; Myers J. N.; Lai S. Y.; Sandulache V. C. Acquisition of Cisplatin Resistance Shifts Head and Neck Squamous Cell Carcinoma Metabolism toward Neutralization of Oxidative Stress. Cancers 2020, 12, 1670. 10.3390/cancers12061670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadini-Buoninsegni F.; Bartolommei G.; Moncelli M. R.; Inesi G.; Galliani A.; Sinisi M.; Losacco M.; Natile G.; Arnesano F. Translocation of polatinum anticancer drugs by human copper ATPases ATP7A and ATP7B. Angew. Chem., Int. Ed. 2014, 53, 1297–1301. 10.1002/anie.201307718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Shanbhag V.; Wang Y.; Lee J.; Petris M. A role for the ATP7A copper transporter in tumorigenesis and cisplatin resistance. J. Cancer 2017, 8, 1952–1958. 10.7150/jca.19029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miedel M. T.; Rbaibi Y.; Guerriero C. J.; Colletti G.; Weixel K. M.; Weisz O. A.; Kiselyov K. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J. Exp. Med. 2008, 205, 1477–1490. 10.1084/jem.20072194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba Q. L.; Raghavan G.; Kiselyov K.; Yang G. Whole-cell scale dynamic organization of lysosomes revealed by spatial statistical analysis. Cell Rep. 2018, 23, 3591–3606. 10.1016/j.celrep.2018.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman G.; Iniya M.; Anand T.; Kotla N. G.; Sunnapu O.; Singaravadivel S.; Gulyani A.; Chellappa D. Chemically diverse small molecule fluorescent chemosensors for copper ion. Coord. Chem. Rev. 2018, 357, 50–104. 10.1016/j.ccr.2017.11.020. [DOI] [Google Scholar]
- Krishnamoorthy L.; Cotruvo J. A.; Chan J.; Kaluarachchi H.; Muchenditsi A.; Pendyala V. S.; Jia S.; Aron A. T.; Ackerman C. M.; Vander Wal M. N.; et al. Copper regulates cyclic-AMP-dependent lipolysis. Nat. Chem. Biol. 2016, 12, 586–592. 10.1038/nchembio.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M. T.; Bourassa D.; Harankhedkar S.; McCallum A. M.; Zlatic S. A.; Calvo J. S.; Meloni G.; Faundez V.; Fahrni C. J. Ratiometric two-photon microscopy reveals attomolar copper buffering in normal and Menkes mutant cells. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 12167–12172. 10.1073/pnas.1900172116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samimi G.; Safaei R.; Katano K.; Holzer A. K.; Rochdi M.; Tomioka M.; Goodman M.; Howell S. B. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin. Cancer Res. 2004, 10, 4661–4669. 10.1158/1078-0432.CCR-04-0137. [DOI] [PubMed] [Google Scholar]
- Kishimoto S.; Yasuda M.; Fukushima S. Changes in the expression of various transporters as influencing factors of resistance to cisplatin. Anticancer Res. 2017, 37, 5477–5484. 10.21873/anticanres.11977. [DOI] [PubMed] [Google Scholar]
- Konishi M.; Imai A.; Fujii M.; Sugimoto K.; Katakami N.; Imai Y.; Kamoshida S. Correlation of Expression Levels of Copper Transporter 1 and Thymidylate Synthase with Treatment Outcomes in Patients with Advanced Non-small Cell Lung Cancer Treated with S-1/Carboplatin Doublet Chemotherapy. Asian Pac. J. Cancer Prev. 2018, 19, 435–441. 10.22034/APJCP.2018.19.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y.-H.; Kuo C.; Kuo M. T.; Chen H. H. W. Modulating chemosensitivity of tumors to platinum-based antitumor drugs by transcriptional regulation of copper homeostasis. Int. J. Mol. Sci. 2018, 19, 1486. 10.3390/ijms19051486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. M.; Koide K. A high-throughput method to detect palladium in ores. Ind. Eng. Chem. Res. 2013, 52, 8612–8615. 10.1021/ie400959z. [DOI] [Google Scholar]
- Dunn K. W.; Kamocka M. M.; McDonald J. H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol.: Cell Physiol. 2011, 300, C723–C742. 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira B. L.; Stenton B. J.; Unnikrishnan V. B.; de Almeida C. R.; Conde J.; Negrão M.; Schneider F. S. S.; Cordeiro C.; Ferreira M. G.; Caramori G. F.; et al. Platinum-triggered bond-cleavage of pentynoyl amide and N-propargyl handles for drug-activation. J. Am. Chem. Soc. 2020, 142, 10869–10880. 10.1021/jacs.0c01622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morstein J.; Hofler D.; Ueno K.; Jurss J. W.; Walvoord R. R.; Bruemmer K. J.; Rezgui S. P.; Brewer T. F.; Saitoe M.; Michel B. W.; Chang C. J.. Ligand-directed approach to activity-based sensing: Developing palladacycle fluorescent probes that enable endogenous carbon monoxide detection. J. Am. Chem. Soc. 2020, 10.1021/jacs.0c06405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.