

Abstract
Genomic instability (GI) is a hallmark of cancer and plays a central role in breast cancer initiation and development1,2. The success of Poly-ADP ribose polymerase inhibitors in the treatment of homologous recombination (HR)-deficient breast cancers exemplifies the utility of synthetic lethal genetic interactions in the treatment of GI-driven breast cancer3. Given that HR-defects are present in only a subset of breast cancers, there is a need to identify additional GI-driver mechanisms, and targeted strategies to exploit these defects in cancer treatment. Here, we identify that centrosome-depletion induces synthetic lethality in cancer cells harbouring the 17q23 amplicon, a recurrent copy number aberration (CNA) that defines ~9% of all breast tumours and is associated with high GI4-6. Specifically, small-molecule inhibition of Polo-like kinase 4 (PLK4) leads to centrosome depletion that triggers mitotic catastrophe in cells harbouring amplicon-directed overexpression of TRIM37. To explain this effect, we identify TRIM37 as a negative regulator of centrosomal pericentriolar material (PCM). In 17q23-amplified cells, elevated TRIM37 blocks the formation of non-centrosomal PCM foci, structures with microtubule nucleating capacity that are required for successful cell division in the absence of centrosomes. Lastly, we find TRIM37 overexpression causes GI by delaying centrosome maturation and separation at mitotic entry and thereby increasing the frequency of mitotic errors. Collectively, these findings highlight TRIM37-dependent GI as a putative driver event in 17q23-amplified breast cancer and provide a rationale for centrosome-targeting therapeutics in their treatment.
Main Text
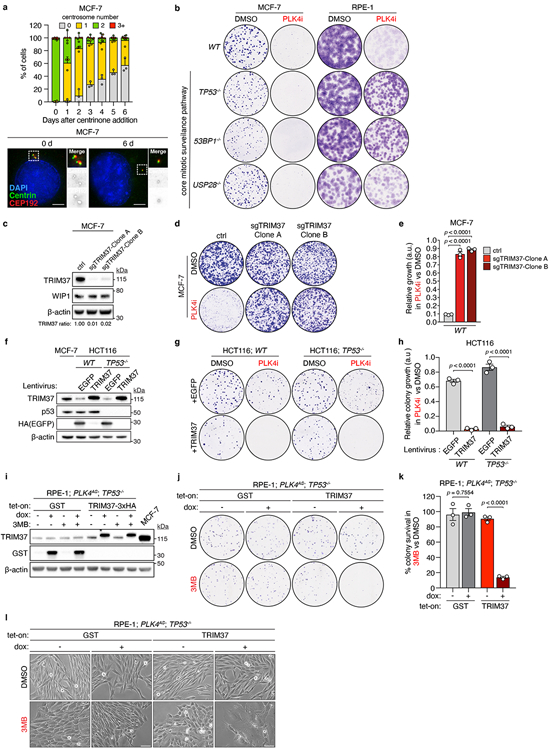
Many cancer cells can proliferate without centrosomes7,8. However, while evaluating the response of cell lines to centrosome loss, we discovered that MCF-7 human breast adenocarcinoma cells were hypersensitive to centrosome loss induced by treatment with the PLK4 inhibitor (PLK4i) centrinone7. Progressive centrosome loss induced upon centrinone treatment in MCF-7 cells (Extended Data Fig. 1a) blocked the proliferation of MCF-7 cells within 3 days (Fig. 1a), and greatly reduced clonogenic survival (Fig. 1c). In non-transformed cells, centrosome depletion with centrinone leads to mitotic surveillance pathway activation, which triggers p53-dependent growth arrest through USP28 and 53BP17,9-13. However, the sensitivity of MCF-7 to centrinone treatment was found to be independent of this pathway (Fig. 1a, Extended Data Fig. 1b).
Figure 1. PLK4 inhibition is synthetic lethal with TRIM37 amplification.
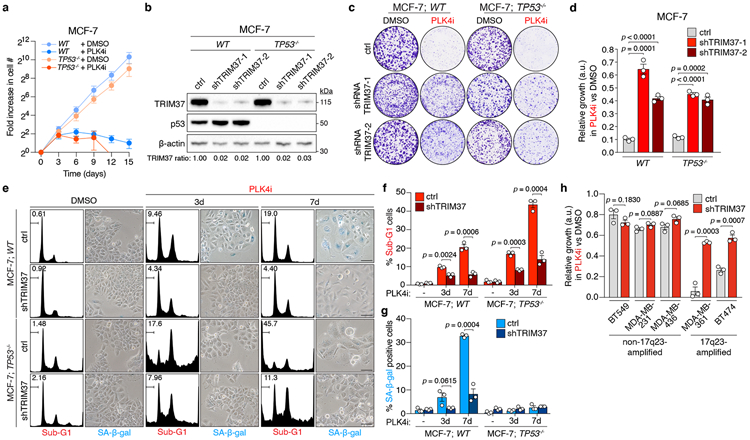
(A) Fold increase in cell number after centrinone (125 nM) addition. n = 3, biological replicates. Mean ± s.e.m.
(B) Immunoblot showing TRIM37 protein levels in WT and TP53−/− MCF-7 cells stably expressing a control or one of two independent TRIM37-targeting shRNAs. β-Actin, loading control. Representative data; n = 3, biological replicates. For gel source data, see Supplementary Figure 1.
(C) Representative data of a 10 day clonogenic survival of indicated MCF-7 cell lines treated with DMSO (control) or centrinone (PLK4i, 125 nM).
(D) Quantification of (C), n = 3, biological replicates. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(E) MCF-7 cells treated with DMSO (control) or centrinone (PLK4i, 125 nM) were analysed for DNA content, and stained for senescence-associated β-galactosidase expression. Representative data of n = 3, biological replicates. Scale bars, 100 μm.
(F) Percentage of sub-G1 cells from (E). n = 3, biological replicates. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(G) Quantification of the percentage of SA-β-gal positive cells from (E). n = 3, biological replicates, each comprising ≥ 200 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
biological replicates. P values, paired two-tailed t-test. Mean ± s.e.m. AS, analogue sensitive.
(H) Quantification of clonogenic survival data for 17q23-amplified and non-17q23-amplified breast cancer cells transduced with a TRIM37-shRNA or vector control and treated with DMSO (control) or centrinone (PLK4i, 125 nM). n = 3 biological replicates. P values, unpaired two-tailed t-test. Mean ± s.e.m.
We considered whether the genetic background of MCF-7 underlies their hypersensitivity to PLK4i-induced centrosome depletion. MCF-7 harbour the 17q23 breast cancer amplicon, a 3-4 MB recurrent CNA found in ~9% of all primary breast cancer tumours4,14. 17q23 amplification also represents the defining feature of IntClust1 tumours, a subset of primarily ER-positive, luminal B type breast cancers, detected following the genomic and transcriptomic profiling of >2,000 primary breast tumours in the METABRIC project4,5,15. Of the ~40 protein-coding genes located within the 17q23 amplicon16-18, we noted TRIM37, a gene implicated in centrosome function19,20. TRIM37 knockout leads to PCM accumulation and accelerated spindle assembly in acentrosomal cells19. We therefore hypothesized that conversely, high TRIM37 levels could reduce PCM-mediated microtubule nucleation, thereby sensitizing cells to centrosome loss. To test this, we transduced WT and TP53−/− MCF-7 cell lines with lentiviruses encoding control or TRIM37-targeting shRNAs, and monitored growth in the presence or absence of centrinone. In both cell lines, efficient TRIM37 depletion using two different shRNAs restored cell-growth in centrinone when compared to controls (Fig. 1b-d). Similarly, disruption of TRIM37 with CRISPR/Cas9 conferred resistance to centrinone in two MCF-7 clones (Extended Data Fig. 1c-e). Centrinone treatment induced senescence in MCF-7 cells, evidenced by a time-dependent increase in cell-flattening and senescence-associated β-galactosidase expression, or cell death, as marked by an accumulation of cells with a sub-G1 DNA content (Fig. 1e-g). Centrinone treatment also inhibited the proliferation of TP53−/− MCF-7, primarily inducing cell death (Fig. 1e, f). Centrinone-induced senescence or cell death in MCF-7 cell lines was suppressed by the depletion of TRIM37 (Fig. 1e-g), suggesting elevated TRIM37 expression to be synthetic lethal with PLK4 inhibition.
To test whether TRIM37 overexpression sensitizes cells to PLK4 inhibition, control (EGFP) or TRIM37 transgenes were introduced into HCT116, a human colorectal carcinoma cell-line insensitive to centrosome loss7. Overexpression of TRIM37 at levels comparable to MCF-7 (Extended Data Fig. 1f) inhibited clonogenic survival in centrinone-treated HCT116, while only modestly affecting the growth of DMSO-treated controls (Extended Data Fig. 1g, h). To ascertain whether the synthetic lethal effect of centrinone was specific to PLK4 inhibition, we overexpressed TRIM37 in PLK4AS TP53−/− RPE-1 cells, which exclusively express analogue-sensitive (AS) PLK413. In these cells, neither doxycycline-induced TRIM37-overexpression alone, nor the inhibition of PLK4AS with the bulky ATP analogue 3MB-PP1, affected cell proliferation (Extended Data 1i-l). By contrast, 3MB-PP1 treatment resulted in cell flattening and dramatically reduced colony survival in TRIM37-overexpressing PLK4AS TP53−/− RPE-1, but not cells overexpressing a control GST transgene (Extended Data Fig. 1i-l). This confirms that the specific inhibition of PLK4 kills cells with elevated TRIM37 expression.
CFI-400945 is a PLK4 inhibitor that also targets Aurora B and is in clinical trials for patients with breast cancer21-23. We therefore tested the effect of CFI-400945 on the proliferation of MCF-7. Treatment with centrinone, CFI-400945, or the Aurora B inhibitor ZM44749 all potently inhibited clonogenic survival in MCF-7 (Extended Data Fig. 2a, b). However, depletion of TRIM37 only restored the proliferation of cells treated with centrinone, but not CFI-400945 or ZM447439. In cells treated with CFI-400945 or ZM447439, DNA content analysis revealed an accumulation of polyploid cells, a readout of Aurora B kinase inhibition (Extended Data Fig. 2c). In contrast, centrinone treatment did not increase the fraction of polyploid cells, consistent with the known selectivity of this compound for inhibiting PLK4 and not Aurora B24. This shows that inhibitor selectivity towards PLK4, and not other kinases, is crucial for the synthetic lethal killing of cells overexpressing TRIM37.
To determine whether PLK4i-induced cell-killing was common to 17q23-amplified breast cancer cell lines, we tested the effect of centrinone on the viability of BT474 and MDA-MB-361, two additional 17q23-amplified breast cancer cell lines that overexpress TRIM3725,26, and compared responses with a control panel of non-17q23-amplified breast cancer cell lines (BT549, MDA-MB-231 and MDA-MB-436) with normal TRIM37 expression (Extended Data Fig. 3a). As expected, PLK4 inhibition only minimally affected clonogenic survival across the control cell line panel, with TRIM37-depletion conferring no added resistance (Fig. 1h, Extended Data Fig. 3b). In contrast, both 17q23-amplified cell lines were hypersensitive to PLK4i, with centrinone treatment inducing growth arrest, morphological aberrations, and cell death (Fig. 1h, Extended Data Fig. 3b-d). Again, these effects were suppressed by stable TRIM37-knockdown, confirming that the synthetic lethal effect of PLK4i treatment across multiple 17q23-amplified cell lines depended on TRIM37 overexpression.
To test whether TRIM37-overexpression could be predictive for PLK4-inhibitor-induced cell-killing in patient-derived organoid (PDO) models of breast cancer, we examined the centrinone-sensitivity of human breast cancer PDO 3D cultures with high or low-level TRIM37 mRNA expression (Extended Data Fig. 3e). TRIM37 mRNA expression was only partly predictive of TRIM37 protein levels (Extended Data Fig. 3g). Nevertheless, of four established cultures, two PDOs with high-TRIM37 protein levels and one with intermediate-TRIM37 protein levels were sensitive to nanomolar doses of centrinone. In contrast, one PDO with low-TRIM37 protein levels remained insensitive to centrinone at sub-1μM concentrations (Extended Data Fig. 3f, g). These PDO centrinone sensitivity profiles resembled the 3D-culture responses of 17q23 amplified (MCF-7, BT-474) and non-amplified (MDA-MB-231, BT-549) breast tumour cell lines to centrinone (Extended Data Fig. 3h). Taken together, our cell-line and PDO experiments underscore the utility of PLK4-specific inhibitors in the killing of TRIM37-amplified breast cancer cells.
To interrogate how PLK4 inhibition triggers growth defects in MCF-7 cells, we performed time-lapse microscopy to track the fates of control and centrinone-treated TP53−/− MCF-7 (Fig. 2a, Supplementary Videos 1-4). While control cells progressed through mitosis normally, 47% of centrinone-treated cells formed short bipolar spindles that collapsed and remained arrested in mitosis or slipped out of mitosis without undergoing anaphase (Fig. 2b-d). Importantly, TRIM37-depletion rescued robust bipolar spindle formation in centrinone-treated MCF-7 cells, and almost completely reversed the effects of centrinone in prolonging mitosis and inducing cell division errors in MCF-7 (Fig. 2b-d). Thus, elevated TRIM37 expression antagonizes spindle assembly in the absence of centrosomes, resulting in mitotic catastrophe.
Figure 2. PLK4 inhibition triggers mitotic catastrophe in TRIM37 amplified cancer cells.
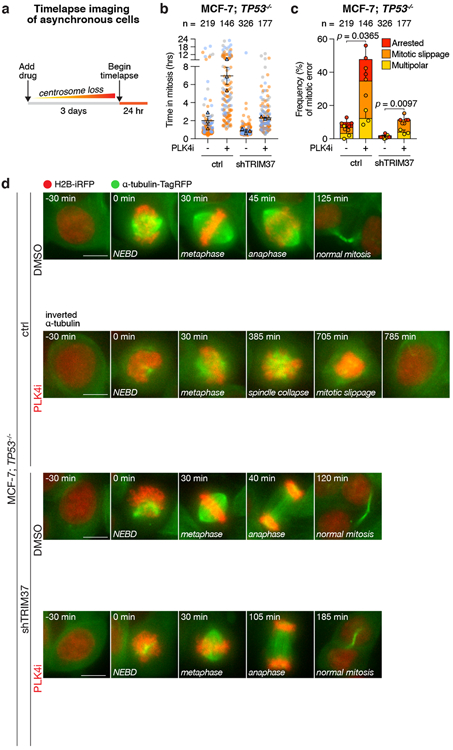
(A) Experimental schematic.
(B) Quantification of mitotic duration in control TP53−/− MCF-7 cells compared to those expressing a TRIM37-shRNA. Cells were treated with DMSO (control) or centrinone (PLK4i, 125 nM) for 3 days prior to imaging. Triangles represent the mean for each biological replicate; coloured circles show individual data points from each of the replicates. Data acquired across n = 3, biological replicates, each with >40 cells. Mean ± s.e.m.
(C) Quantification of mitotic phenotypes from (B) in control TP53−/− MCF-7 cells compared to those expressing a TRIM37-shRNA. Data acquired across n = 3, biological replicates, each with >40 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(D) Representative time-lapse images of mitotic progression in DMSO or centrinone (125 nM)-treated TP53−/− MCF-7 cells expressing vector control or TRIM37-shRNA. Representative data; n = 3, biological replicates. Cells are labelled with H2B-iRFP and TagRFP-α-tubulin. Scale bars, 5 μm.
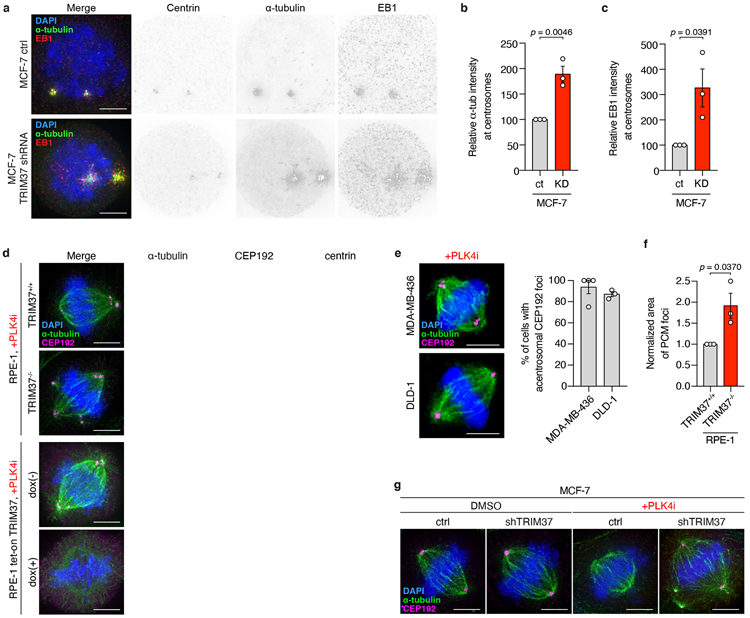
To understand how TRIM37 inhibits spindle assembly, we identified proximity interaction partners by expressing mTurbo-tagged TRIM37 and performing proximity-dependent biotin labelling in RPE-127,28. After background subtraction, we identified 184 TRIM37 proximity interaction partners, including 7 known interactors29 (Extended Data Fig. 4a, c & Extended Data Table 1). Gene ontology analysis showed a striking enrichment of centrosome proteins within these interactors (Extended Data Fig. 4b). This was corroborated by the localisation of a pool of endogenous TRIM37 in close proximity of the centrosome and the enrichment of biotinylated proteins at the centrosomes of mTurbo-TRIM37-expressing RPE-1 (Extended Data Fig. 4d, e). Among the most enriched proximity interactors of TRIM37 was CEP192, a core PCM component that accumulates in non-centrosomal foci in TRIM37 knockout cells19. The interaction between TRIM37 and CEP192 was confirmed by co-immunoprecipitation (Extended Data Fig. 4f). Neither Histone H2A, nor the peroxisome protein PEX5, two previously reported TRIM37 substrates30,31, were among the labelled interactors, confirming centrosome proteins to be primary TRIM37 proximity-interaction partners.
To test if TRIM37 regulates the abundance of PCM proteins, we monitored the effect of altered TRIM37 expression on the cell-wide levels of three PCM scaffolding proteins: CEP192, PCNT, and CDK5RAP2. Acute overexpression of TRIM37 in RPE-1 cells dramatically reduced the abundance of all three PCM proteins (Fig. 3a). Proteasome blockade with MG132 prevented the reduction in PCM protein abundance, suggesting that TRIM37 directs the degradation of these proteins via the ubiquitin-proteasome pathway. In agreement, the E3 ligase activity of TRIM37 was critical for PCM protein degradation, as both catalytically inactive (C18R)30,31 and predicted ubiquitin-binding and transfer defective (R67A)32 RING domain mutants of TRIM37, failed to reduce PCM protein levels (Fig. 3a). Having established that TRIM37 directs PCM protein proteolysis, we next investigated whether 17q23-amplification status correlated with reduced PCM levels. 17q23-amplified cell lines with TRIM37 overexpression were found to have lower cell-wide levels of CEP192, PCNT, and CDK5RAP2 compared to non-17q23-amplified counterparts (Extended Data Fig. 4g). The levels of CEP192, PCNT, and CDK5RAP2 were also reduced at mitotic centrosomes in control MCF-7, but restored following TRIM37-depletion to levels comparable to that seen in RPE-1 (Fig. 3b, c). Microtubule regrowth assays showed that mitotic centrosomes in TRIM37-depleted MCF-7 nucleated nearly twice the amount of α-tubulin compared to control cells (Extended Data Fig. 5a, b). Similarly, the levels of EB1, a plus-end tracking marker of growing microtubules, were increased by >3-fold at the centrosomes of TRIM37-depleted cells (Extended Data Fig. 5a, c).
Figure 3. PCM sequestration by TRIM37 drives mitotic catastrophe in acentrosomal cells.
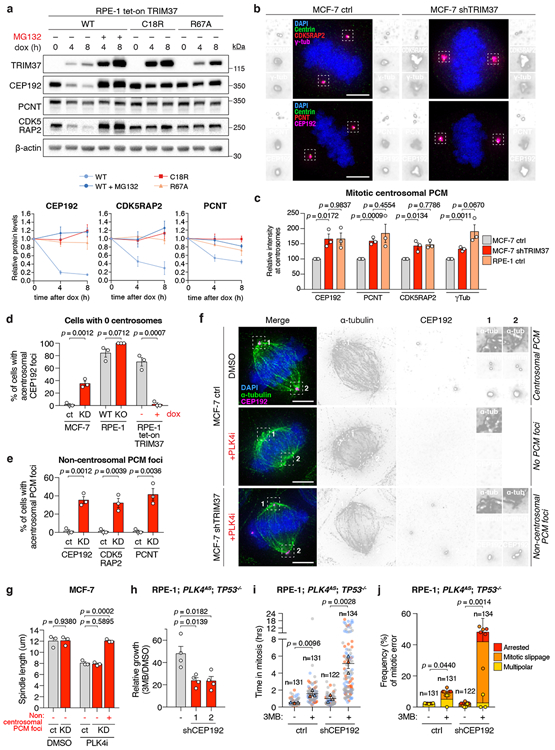
(A) Top, immunoblot showing PCM levels following overexpression of wild-type TRIM37(WT), RING domain mutant TRIM37(C18R), or ubiquitin-transfer defective mutant TRIM37(R67A). For gel source data, see Supplementary Figure 1. Bottom, normalised PCM levels relative to 0 h, representative of n = 3, biological replicates. Mean ± s.e.m.
(B) Centrosomal PCM levels in mitotic control or TRIM37-depleted cells. Representative images, n = 3, biological replicates. Scale bars, 5 μm.
(C) Quantification of centrosomal PCM signal in mitotic cells. n = 3, biological replicates. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(D) Quantification of mitotic CEP192 foci in centrinone-treated cells lacking centrosomes. n = 3, biological replicates, each comprising >30 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(E) Quantification of mitotic PCM foci in centrinone-treated cells lacking centrosomes. n = 3, biological replicates, each comprising >30 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(F) Representative images for (E). Scale bars, 5 μm.
(G) Quantification of mitotic spindle length in control and TRIM37-shRNA expressing cells. n = 3, biological replicates, each comprising >10 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(H) Relative growth of 3MB-PP1-treated control or CEP192-shRNA expressing cells. n = 4, biological replicates. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(I) Mitotic duration in of the cells described in (H) expressing H2B-EGFP and TagRFP-tubulin. Cells were grown in DMSO or 3MB-PP1 for 3 days prior to imaging. Triangles, mean for each biological replicate; coloured circles, individual data points from each replicate. n = 3, biological replicates, each comprising >30 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(J) Frequency of mitotic errors quantified in the same samples as described in (I). n = 3, biological replicates, each comprising >30 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
Fixed cell analysis revealed that acentrosomal RPE-1, DLD-1 and MDA-MB-436 cells that express low levels of TRIM37 formed PCM aggregates in >80% of mitotic cells. However, these PCM foci were absent from MCF-7 and TRIM37-overexpressing RPE-1 (Fig. 3d-f, Extended Data Fig. 5d, e). Hence, we asked if the role of TRIM37 in controlling PCM abundance could modulate the assembly of non-centrosomal PCM foci. Depleting TRIM37 enabled the formation of PCM foci in acentrosomal MCF-7 (Fig. 3d-f) and increased the penetrance and size of these structures in acentrosomal RPE-1 (Fig. 3d, Extended Data Fig. 5d, f). Centrosome loss also reduced the length of the mitotic spindle in MCF-7 (Fig. 3g, Extended Data Fig. 5g). However, TRIM37 depletion enabled acentrosomal MCF-7 with PCM foci to generate spindle lengths that matched those of untreated MCF-7. Thus, TRIM37-overexpression suppresses the formation of non-centrosomal PCM foci, leading to spindle assembly defects.
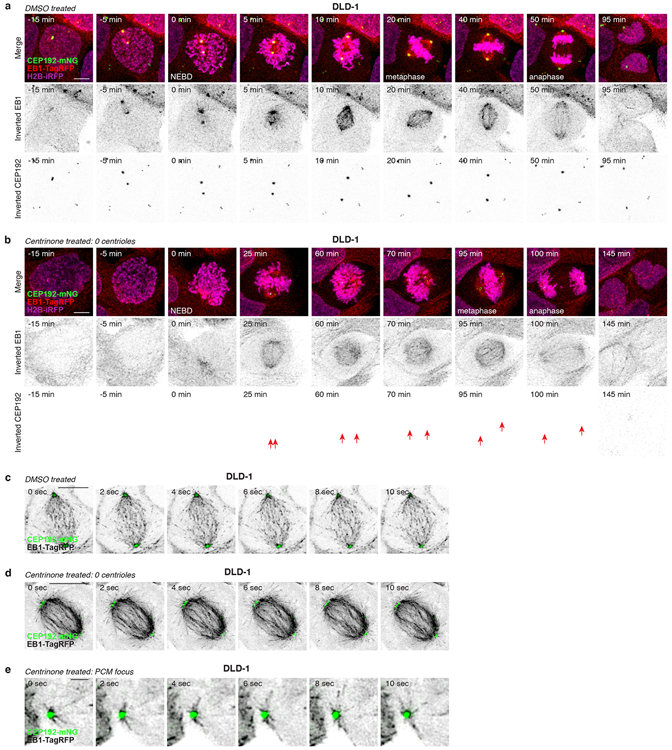
To define the spatial and temporal assembly properties of non-centrosomal PCM foci, we generated DLD-1 cells expressing endogenously tagged CEP192-mNeonGreen to mark PCM material. In control DLD-1, CEP192-mNeonGreen localised to the centrosomes in interphase and increased in intensity by ~3-fold during mitosis (Extended Data Fig. 6a, Supplementary Video 5). By contrast, CEP192-mNeonGreen was diffusely localised in acentrosomal DLD-1 throughout interphase but assembled into multiple non-centrosomal PCM foci during early prometaphase (Extended Data Fig. 6b, Supplementary Video 6). These PCM foci coalesced into spindle poles at metaphase and subsequently disassemble upon mitotic exit.
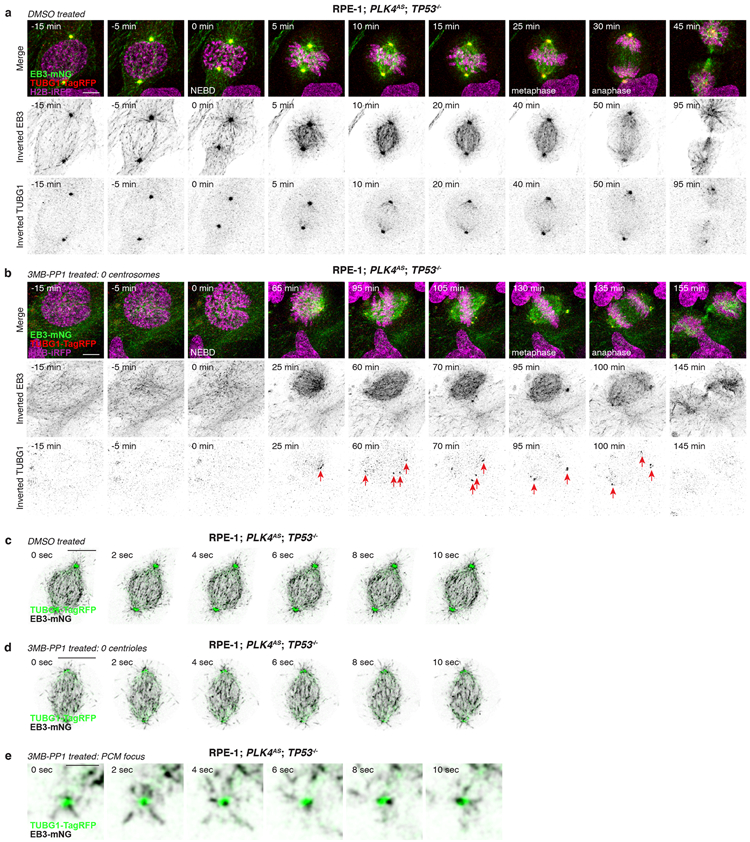
Non-centrosomal PCM foci often resided at the centre of microtubule asters (Extended Data Fig. 5d, e, g). To examine whether these structures could promote microtubule nucleation, we performed live-cell confocal imaging on acentrosomal DLD-1 expressing endogenously tagged CEP192-mNeonGreen and EB1-TagRFP. In untreated controls, EB1-TagRFP tracked the growing ends of microtubules nucleated by the centrosomes (Extended Data Fig. 6c, Supplementary Video 7). In acentrosomal DLD-1, non-centrosomal PCM foci nucleated microtubules and were incorporated into the mitotic spindle (Extended Data Fig 6d, Supplementary Video 8). Importantly, microtubule nucleation by non-centrosomal PCM foci preceded their incorporation into the spindle (Extended Data Fig 6e, Supplementary Video 9). Similar results were obtained with acentrosomal RPE-1 co-expressing EB1-mNeonGreen to track the plus-end tip of microtubules and γTubulin-TagRFP to mark acentrosomal PCM foci (Extended Data Fig 7, Supplementary Videos 10-14). We conclude that in cells that lack centrosomes, non-centrosomal PCM foci form specifically in mitosis and are required for efficient microtubule nucleation and robust bipolar spindle assembly.
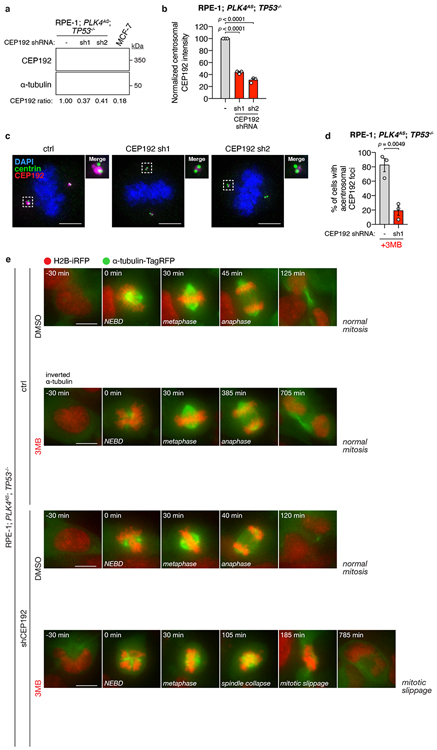
To test if PCM depletion could explain the synthetic lethal effect of centrosome loss in cells with TRIM37 overexpression, we depleted the TRIM37 target CEP192 in TP53−/− PLK4AS RPE-1 (Extended Data Fig. 8a-c). As predicted, depletion of CEP192 sensitized these cells to centrosome loss (Fig. 3h). 3MB-PP1 treatment in CEP192-depleted RPE-1 cells also recapitulated the mitotic phenotypes observed in acentrosomal MCF-7, including a reduced frequency of non-centrosomal CEP192 foci, prolonged mitotic duration, and a dramatic increase in mitotic errors (Fig. 3i-j, Extended Data Fig. 8d-e, Supplementary Videos 15-19). We conclude that TRIM37-dependent PCM depletion in mitosis leads to delayed and inefficient microtubule nucleation, defects that can be exploited to induce mitotic catastrophe upon pharmacological depletion of centrosomes.
17q23-amplicon positive breast cancer comprises highly proliferative ER-positive/luminal B tumours characterized by high levels of GI4,5. However, the mechanisms and driver genes responsible for GI in 17q23-amplified tumours remain undefined. The high genomic instability burden in MCF-7 drives their rapid genetic diversification in culture33, leading us to consider the contribution of TRIM37 overexpression to this process. Previous work showed delayed centrosome separation increases the rates of kinetochore mis-attachments and mitotic errors in cancer cells34-36. We therefore investigated whether increased TRIM37 expression during mitosis modulated the timing of centrosome maturation and separation. In contrast to RPE-1, where TRIM37 was transcriptionally downregulated in G2 phase and mitosis (Extended Data Fig. 9a-e), TRIM37 protein expression persisted throughout the cell cycle in MCF-7 (Extended Data Fig. 9d). TRIM37 depletion in MCF-7 accelerated centrosome maturation by 20 min in late G2 phase (Extended Data Fig. 10a, d; Supplementary Videos 19-20), and increased centrosome separation at mitotic entry (Extended Data Fig. 10b). Conversely, TRIM37 overexpression in RPE-1 delayed centrosome maturation in late G2 phase by 17 min (Extended Data Fig. 10c, e; Supplementary Videos 21-22). Collectively, these data show that TRIM37-driven suppression of PCM assembly delays microtubule nucleation and centrosome separation in late G2.
To test if TRIM37 overexpression-dependent delays in centrosome separation at mitotic entry could cause mitotic errors, we compared the effect of TRIM37 expression levels on the timing of centrosome separation, mitotic duration and frequency of mitotic errors in 17q23-amplified (MCF-7 and MDA-MB-361) versus non-amplified (MDA-MB-231 and MDA-MB-436) breast cancer cell lines. TRIM37-depletion increased the distance between the centrosomes at early prophase and reduced mitotic duration in the 17q23-amplified cancer cell lines (Fig. 4a, b). By contrast, TRIM37 knockdown did not alter centrosome separation timing or mitotic duration in non-17q23-amplified breast cancer cells (Fig. 4a, b). Importantly, TRIM37 depletion also reduced the frequency of mitotic errors in 17q23-amplified breast cancer cells, but trended towards increasing the rate of cell division errors in non-17q23-amplified cancer cells (Fig. 4c). These data show that overexpression of TRIM37 delays the timing of centrosome separation at mitotic entry in 17q23-amplified breast cancer cells and suggest that this delay in centrosome separation promotes genetic instability by increasing the frequency of mitotic errors.
Figure 4. TRIM37 overexpression delays centrosome separation in late G2 phase and promotes mitotic errors.
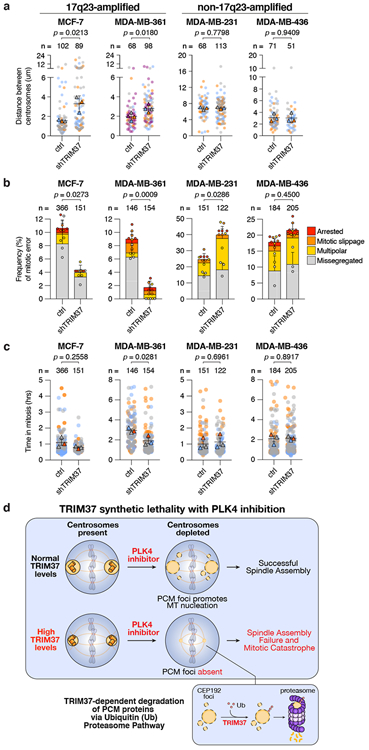
(A) Quantification of the distance between the two centrosomes at NEBD in control MCF-7, MDA-MB-361, MDA-MB-231, and MDA-MB-436 cells compared to those expressing a TRIM37-shRNA. Data acquired across n = ≥3, biological replicates, each with 8 - 45 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(B) Quantification of mitotic phenotypes in control MCF-7, MDA-MB-361, MDA-MB-231, and MDA-MB-436 cells compared to those expressing a TRIM37-shRNA. Data acquired across n = 3, biological replicates, each with >40 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(C) Quantification of mitotic duration in control MCF-7, MDA-MB-361, MDA-MB-231, and MDA-MB-436 cells compared to those expressing a TRIM37-shRNA. Triangles represent the mean for each biological replicate; coloured circles show individual data points from each of the replicates. Data acquired across n = 3, biological replicates, each with >40 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(D) A model illustrating the synthetic lethal effect of PLK4 inhibition with TRIM37 overexpression in 17q23-amplified breast cancer cells.
We propose that TRIM37 normally acts to inhibit the assembly of non-centrosome associated PCM material into structures that would otherwise compromise mitotic fidelity. Consequently, the centrosomal defects that accompany TRIM37-amplification may fuel the stochastic mitotic errors that contribute to the high genomic instability burden in 17q23-amplified breast tumours and could drive tumour evolution. We also propose centrosome depletion as a therapeutic strategy to kill cancers that overexpress TRIM37: high TRIM37 levels reduces PCM protein availability, thereby impeding the formation of non-centrosomal PCM foci; assemblies we propose to be required for mitosis in the absence of centrosomes (Fig. 4d). Our work therefore indicates that the inhibition of PLK4, or other regulators of centrosome duplication or assembly, represents a promising strategy to selectively target breast cancers, or other tumours37, driven by 17q23-amplification.
Methods
Cell lines and culture conditions
MCF-7, MDA-MB-231, MDA-MB-436, DLD-1 and HCT-116 cells were grown in DMEM medium (Corning Cellgro) containing 10% fetal bovine serum (Sigma), 100 U/ml penicillin, 100 U/ml streptomycin and 2 mM L-glutamine. hTERT RPE-1 cells were grown in DMEM:F12 medium (Corning Cellgro) containing 10% fetal bovine serum (Sigma), 0.348% sodium bicarbonate, 100 U/ml penicillin, 100 U/ml streptomycin and 2 mM L-glutamine. MDA-MB-361 cells were grown in DMEM medium (ThermoFisher Scientific) containing 20% fetal bovine serum (Sigma), 100 U/ml penicillin, 100 U/ml streptomycin and 2 mM L-glutamine. BT474 and BT549 cells were grown in RPMI 1640 medium (ThermoFisher Scientific) containing 10% fetal bovine serum (Sigma), 100 U/ml penicillin, 100 U/ml streptomycin and 2 mM L-glutamine and 10 ug/ml bovine insulin (Sigma). All cell lines were maintained at 37°C in a 5% CO2 atmosphere with 21% oxygen and routinely checked for mycoplasma contamination.
Centrinone sensitivity in 3D tumour cell line and Patient-Derived Organoid (PDO) cultures
Human breast tumour samples were obtained from adult female patients after informed consent as part of a non-interventional clinical trial (BTBC study REC no.: 13/LO/1248, IRAS ID 131133; Principal Investigator: Prof. Andrew Tutt; Study Title: “Analysis of functional immune cell stroma and malignant cell interactions in breast cancer in order to discover and develop diagnostics and therapies in breast cancer subtypes”). This study had local research ethics committee approval and was conducted adhering to the principles of the Declaration of Helsinki. Specimens were collected from surgery and transported immediately. A clinician histopathologist or pathology-trained technician identified and collected tumour material into basal culture media. Tumour samples were coarsely minced with scalpels and then dissociated using a Gentle MACS dissociator (Miltenyi). The resulting cell suspension was mechanically disrupted, filtered and centrifuged. Resulting cell pellets were then plated into 3D cultures at approximately 1 × 103 to 2 × 103 cells per ul in Ocello PDX media (OcellO B.V) and hydrogel as described previously38-40. All cultures were maintained in humidified incubators at 37°C, 5% CO2. For centrinone sensitivity analysis, PDOs (between passage 10 and 25) and tumour cell lines (STR typed every five passages to confirm identity) were dissociated to single cell populations using TrypLE (Life Technologies). Cell suspensions were then dispensed into 384 well plates in Ocello PDX media and hydrogel. 24 hrs after seeding, cultures were treated with centrinone (diluted in 0.1 % v/v DMSO) and then continuously cultured for a total of 13 days, with drug-containing media being replenished every 4 days. After this point, cell viability was estimated using Cell Titer Glo 3D (CTG, Promega) as per the manufacturer’s guidelines. CTG luminescence was measured using a Victor X5 plate reader (Perkin Elmer). Data are presented as % survival normalised to cells exposed to 0.1 % v/v DMSO alone.
Gene targeting and stable cell lines
To generate CRISPR/Cas9-mediated knockout lines, gene-specific sgRNAs (TP53Δ, 5′-gtgcagctgtgggttgattc-3′; TP53BP1Δ, 5′-gaacgaggagacggtaatagt-3′; USP28Δ, 5′-tgccattgctttgagtctac-3′) were cloned into a modified pX330 vector (#42230; Addgene) containing a puromycin resistance cassette. Cells were transiently transfected (Fugene HD, Promega) with the pX330 plasmids and positive selection of transfected cells was performed 2 days after transfection with 2.0 ug/ml puromycin. Monoclonal cell lines were isolated by limiting dilution. The presence of gene-disrupting indels in edited cell lines was confirmed by Sanger sequencing, and the ablation of protein production was assessed by immunoblotting.
To generate EB1-TagRFP, H2B-iRFP and TagRFP-tubulin or EGFP-tubulin labelled cell lines, ORFs were cloned into FUGW lentiviral vectors. Lentivirus was produced as described below. MCF-7 cells were transduced with H2B-iRFP and TagRFP-tubulin, RPE-1 cells were transduced with H2B-iRFP and EGFP-tubulin, and DLD-1; CEP192-mNeonGreen cells were transduced with H2B-iRFP and EB1-TagRFP, MDA-MB-361, MDA-MB-231 and MDA-MD-436 cells were transduced with H2B-iRFP. Polyclonal populations of cells expressing both markers were used directly or isolated using FACS.
To generate EB3-mNeonGreen and γ-tubulin-TagRFP labelled cell lines, the two ORFs separated by a T2A sequence were cloned into a CMV-puro lentiviral vector. Lentivirus was produced as described below. RPE-1 cells were transduced with EB3-mNeonGreen and γ-tubulin-TagRFP dual expressing lentivirus, and polyclonal populations of cells expressing both markers were selected using puromycin.
To generate TRIM37-overexpressing cell lines, the TRIM37 ORF was cloned into a constitutive or tet-inducible lentiviral vector. The C18R and R67A mutations were introduced using PCR-directed mutagenesis and verified by Sanger sequencing. Lentivirus was produced as described below. Cells were transduced and stable polyclonal populations of cells selected and maintained in the presence of 1.0 μg/ml puromycin.
To create the DLD-1; CEP192-mNeonGreen cell line, an sgRNA targeting the CEP192 translational stop codon (5′- cgactaattggtgaagctct-3′) was cloned into a pX459 vector (#62988; Addgene). To generate the CEP192 repair vector, we cloned a 2x mNeonGreen tag followed by a T2A-neomycin and a translational stop codon into a modified pUC vector. 475 bp 5′ and 462 bp 3′ homology arms were PCR amplified from genomic DLD-1 DNA and cloned on either side of the central 2x mNeonGreen-T2A-Neomycin cassette. DLD-1 cells were transiently transfected (X-tremeGENE HP, Roche) with the pX459 plasmid and repair vector. Selection of transfected cells was performed 5 days after transfection with 400 ug/ml G418.
RNA interference
shRNAs targeting TRIM37 (TRIM37-1, 5’-tcgagaatatgatgctgtg-3’; TRIM37-2, 5’-aggactttgctggaggtta-3’) were cloned into the pGIPz (Thermofisher Scientific) vector. shRNAs targeting CEP192 (CEP192-1, 5’-cctgttacataaaccagagat-3’; CEP192-2, 5’-gaggcatcagttaatactgat-3’) were cloned into pLKO.1. Stable shRNA-mediated knockdown (KD) cell lines were generated by lentivirus-mediated transduction. Polyclonal populations of cells were subsequently selected and maintained in the presence of puromycin (1.0 μg/ml). Knock down efficiency was assessed by immunoblotting.
Lentiviral production and transduction
Lentiviral expression vectors were co-transfected into 293FT cells with the lentiviral packaging plasmids psPAX2 and pMD2.G (Addgene #12260 and #12259). Briefly, 3 × 106 293FT cells were seeded into a Poly-L-Lysine coated 10 cm culture dish the day before transfection. For each 10 cm dish the following DNA were diluted in 0.6 mL of OptiMEM (Thermo Fisher Scientific): 4.5 μg of lentiviral vector, 6 μg of psPAX2 and 1.5 μg of pMD2.G. Separately, 72 μl of 1 μg/μl 25 kDa polyethylenimine (PEI; Sigma) was diluted into 1.2 mL of OptiMEM, briefly vortexed, and incubated at room temperature for 5 min. After incubation, the DNA and PEI mixtures were combined, briefly vortexed, and incubated at room temperature for 20 min. During this incubation, the culture media was replaced with 17 ml of pre-warmed DMEM + 1% FBS. The transfection mixture was then added drop-wise to the 10 cm dish. Viral particles were harvested 48 hr after the media change and filtered through a 0.45 μm PVDF syringe filter. The filtered supernatant was either concentrated in 100 kDa Amicon Ultra Centrifugal Filter Units (Millipore) or used directly to infect cells. Aliquots were snap-frozen and stored at −80°C. For transduction, lentiviral particles were diluted in complete growth media supplemented with 10 μg/ml polybrene (Sigma) and added to cells.
Chemical inhibitors
3MB-PP1 (Millipore) was dissolved in dimethyl sulfoxide (DMSO) and used at a final concentration of 10 μM, and centrinone (a gift from Karen Oegema, Ludwig Institute for Cancer Research, CA) was dissolved in DMSO and used at a final concentration of 125 nM unless otherwise indicated. CFI-400945 (Cayman Chemicals) was dissolved in DMSO and used at a final concentration of 50 or 500 nM. ZM447439 (Cayman Chemicals) was dissolved in DMSO and used at a final concentration of 2 μM. MG132 (Sigma) was dissolved in DMSO and used at a final concentration of 10 μM. CHX (VWR International) was dissolved in DMSO and used at a final concentration of 100 μg/mL. RO-3306 (Sigma) was dissolved in DMSO and used at a final concentration of 9 μM. Nutlin-3 (Cayman Chemicals) was dissolved in ethanol and used at a final concentration of 4 μM.
TRIM37 RNA abundance in PDOs
RNA from PDO cell pellets was extracted using the RNeasy kit (Qiagen) according to the manufacturer’s instructions. Quality and quantity of RNA were assessed using a Qubit and Bioanalyzer (Agilent). NEBNEXT Ultra II Directional RNA and polyA RNA selection kits (Illumina) were used to generate paired-end sequencing libraries which were sequenced on an Illumina NovaSeq 6000 S2 platform. Paired-end reads were aligned to the human reference genome GRCh38 using STAR v2.5.1b41 using “quantMode GeneCounts” and “twopassMode” basic alignment settings. Feature quantification was performed using GENCODE (v22) GTF file. Post alignment quality control was performed using RseQC (v2.6.3)42. Data was normalised using edgeR’s TMM method (trimmed mean of M-values)43 and TRIM37 mRNA expression values converted into Z scores adjusted to the median of all mRNA species in the sample.
BioID sample preparation, mass spectrometry (MS), and data analysis
To generate cell lines for BioID, puro-sensitive RPE-1 cells were transduced with lentivirus containing tet-inducible miniTurbo control, or miniTurbo-TRIM37 constructs. 48 hr after transduction, cells were selected in 2.5 μg/ml puromycin for 2 days. Cells were then expanded into 7 × 15 cm dishes. One day prior to biotin labelling, 1 μg/ml doxycycline was added to induce expression of miniTurbo constructs. 24 hrs after the start of induction, with cells at ~60% confluency, 10 uM dimethylenastron (Sigma) was added to cell culture medium to block cells in mitosis. After 2 hours of mitotic block, medium was supplemented with 250 uM D-biotin (P212121; prepared as 250 mM stock in DMSO) to initiate labelling of proximity interactors. After 4 hours of biotin labelling, mitotic samples were harvested by mitotic shake-off, while remaining interphase cells were harvested by scraping. All samples were transferred to 15 ml conicals and rinsed four times with PBS to remove excess biotin. Cell pellets were lysed in ~1.5 ml lysis buffer (all buffer recipes are found in44) by gentle pipetting followed by sonication. Lysates were clarified by centrifugation at 16,000 g for 10 min at 4°C. To enrich for biotinylated material, 60 μl of streptavidin agarose bead resin (Pierce) was washed with lysis buffer, then incubated with clarified lysates, rotating at 4°C, overnight. Samples are then washed for 10 min each with a series of four wash buffers that decrease in detergent concentration. Beads were then washed a final two times in 1x PBS, left in ~60 μl volume PBS, then frozen until ready for analysis by the mass spectrometry facility.
In preparation for mass spectrometry, proteins were reduced with 1.75 μl 15 mg/mL DTT in 10 mM TEAB, shaking at 56°C for 50 min. Samples were then cooled to RT, the pH adjusted to 8 with 500 mM TEAB buffer, and alkylated with 1.8 μl 36 mg/mL iodoacetomide in 100 mM TEAB for 20 min at RT, in the dark. Next, 20 ng/μl trypsin (Promega) was added to proteolyze the samples at 37°C, overnight. Supernatant was collected, and the beads were washed with 0.1x TFA three times, with washes added to supernatant. The pH was adjusted to acidic range, and peptides desalted on u-HLB Oasis plates, eluted with 60% acetonitrile/0.1% TFA, and dried. 10% desalted peptides were analysed on Nano LC-MS/MS on Q Exactive Plus (Thermo) in FTFT mode. MS/MS data was searched with Mascot via PD2.2 against RefSeq2017_83 human species database and a small enzyme and standard (BSA) containing database using the FilesRC option, with mass tolerance 3 ppm on precursors and 0.01 Da on fragments, and annotating variable modifications such as oxidation on M, carbamidomethyl C, deamidation NQ, with and without Biotin K. Mascot .dat files were 1) compiled in Scaffold and 2) processed in PD2.2 to identify peptides and proteins using Percolator as a PSM validator.
Protein hits only identified in miniTurbo-TRIM37 BioID, and hits whose spectral counts in miniTurbo-TRIM37 BioID were 2-fold greater than those of mTurbo alone were considered as candidates for TRIM37 interaction. The filtered list of BioID hits was annotated with Gene Ontology (GO) terms via the Panther classification system45 and analysed using the statistical overrepresentation test (binomial) to derive P values46.
Antibody techniques
For immunoblot analyses, protein samples were separated by SDS-PAGE, transferred onto nitrocellulose membranes with a Trans-Blot Turbo Transfer System (BioRad) and then probed with the following primary antibodies: YL1/2 (rat anti-α-tubulin, ThermoFisher Scientific, MA1-80017, 1:3000), TRIM37 (rabbit, Bethyl, A301-174A, 1:1000), p53 (mouse, Dako, M7001, 1:1000), β-actin (mouse, Sigma, A1978, 1:1000), HA-11 (mouse, BioLegend, 901501, 1:1000), GST (mouse, Sigma, G1160, 1:1000), CEP192 (rabbit, home-made, 1:1000), CDK5RAP2 (rabbit, Millipore, 06-1398, 1:2500), Pericentrin (rabbit, Abcam, ab4448, 1:2500), cyclin A (mouse, SantaCruz Biotechnology, sc-53228, 1:1000), phosphorylated Histone H3 (rabbit, Millipore, 06-570, 1:2000). Proteins were then detected using HRP-conjugated anti-mouse (goat, ThermoFisher Scientific, 31432, 1:1000) or anti-rabbit (goat, ThermoFisher Scientific, 31462, 1:10000) secondary antibodies and enhanced chemiluminescence (Clarity, Bio-Rad). Signals were visualised and acquired using the Gel Doc™XRSystem (Bio-Rad).
For immunofluorescence, cells were grown on 18-mm glass coverslips and fixed for 10 min in either 4% formaldehyde at room temperature, or 100% ice-cold methanol at −20°C for 10 minutes. Cells were blocked in 2.5% FBS, 200 mM glycine, and 0.1% Triton X-100 in PBS for 1 hour. Antibody incubations were conducted in the blocking solution for 1 hour. DNA was stained with DAPI and cells were mounted in ProLong Gold Antifade (Invitrogen). Staining was performed with the following primary antibodies: Centrin (mouse, Millipore, 04-1624, 1:1000), CDK5RAP2 (rabbit, Millipore, 06-1398, 1:2500), γ-tubulin-Cy5 (directly-labelled goat, raised against the following peptide: CDEYHAATRPDYISWGTQEQ, this study, 1:1000), Pericentrin (rabbit, Abcam, ab4448, 1:2500), CEP192-Cy5 (directly-labelled goat, raised against CEP192 a.a. 1-211, this study, 1:1000), YL1/2 (rat anti-α-tubulin, ThermoFisher Scientific, MA1-80017, 1:3000), EB1 (mouse, Santa Cruz, sc-47704, 1:200).
Immunofluorescence images were collected using a Deltavision Elite system (GE Healthcare) controlling a Scientific CMOS camera (pco.edge 5.5). Acquisition parameters were controlled by SoftWoRx suite (GE Healthcare). Images were collected at room temperature (25°C) using an Olympus 40× 1.35 NA, 60× 1.42 NA or Olympus 100× 1.4 NA oil objective at 0.2 μm z-sections. Images were acquired using Applied Precision immersion oil (N=1.516). For quantitation of signal intensity at the centrosome, deconvolved 2D maximum intensity projections were saved as 16-bit TIFF images. Signal intensity was determined using ImageJ by drawing a circular region of interest (ROI) around the centriole (ROI S). A larger concentric circle (ROI L) was drawn around ROI S. ROI S and L were transferred to the channel of interest and the signal in ROI S was calculated using the formula IS − [(IL − IS/AL − AS) × AS], where A is area and I is integrated pixel intensity.
To measure the distance between two centrosomes in prophase, cells were fixed in 4% formaldehyde at room temperature for 10 minutes. Coverslips were blocked and stained as above with the following primary antibodies: CENP-F (sheep, a gift from Stephen Taylor at the University of Manchester, 1:2000), Phospho-Histone H3 (mouse, Cell Signaling, 9701, 1:2000), and CEP192 (rabbit, a gift from Karen Oegema at the University of California at San Diego, this study, 1:2000). Phospho-Histone H3 positive cells with a nuclear envelope localised CENP-F signal were selected for analysis. The distance between two centrosomes was measured from 3D-image stacks using Imaris (Bitplane) software.
Live cell microscopy
Fluorescent cell lines were seeded into either 4-chamber, 35 mm glass bottom culture dishes (Greiner) or 4-well chamber slides (Ibidi) and maintained at 37°C in an environmental control station. Long-term time-lapse imaging was performed using a Deltavision Elite system (GE Healthcare) controlling a Scientific CMOS camera (pco.edge 5.5.). Images were acquired with an Olympus 40× 1.4 NA oil objective. Every 5 minutes, 7 × 3 μm z-sections were acquired in respective fluorescent channels and by DIC. Time-lapse imaging of PCM foci and EB1 or EB3 comets was performed using a Lecia SP-8 confocal microscope, equipped with a resonance scanner, and 405 nm, 488 nm, 552 nm and 638 nm laser lines. Images were acquired with a Leica 40× 1.3 NA or 63× 1.4 NA oil objectives. For time-lapse imaging of PCM foci, images were captured every 5 minutes in 20 × 1 μm z-sections. For time-lapse imaging of EB1 or EB3 comets, images were collected every 2 seconds in a single z-plane. Movies were deconvolved using the LIGHTNING adaptive approach and assembled and analysed in FIJI. Mitotic duration was calculated as the time taken from nuclear envelope breakdown to the onset of anaphase.
Microtubule regrowth assay
Cells were treated with 3.3 μM nocodazole for 1 hr to disrupt the microtubule network, then quickly rinsed 3x with warmed media (37°C) to remove drug. Cells were then incubated in warmed media for 90 seconds to allow microtubule regrowth, fixed in 100% ice-cold methanol for 10 minutes, and processed as described above for immunofluorescence. For fluorescence intensity quantification, images were analysed in ImageJ using a circular area of 5 μm to quantify α-tubulin and EB1 signals around centrosomes. Background fluorescence using a circle of corresponding size was subtracted from each measurement.
PLK4i survival assays
For short-term survival assays, cells seeded in triplicate at 1.25 × 104 cells/well in 6-well plates were treated with either DMSO control or PLK4i (10 μM 3MB-PP1, or 125 nM centrinone) 16 h later. After the indicated number of days, cells were fixed and stained using 0.5% (w/v) crystal violet in 20% (v/v) methanol for 5 minutes. Excess of reagent was extensively washed with distilled water and plates dried overnight. For quantification, bound crystal violet was dissolved in 10% (v/v) acetic acid in dH2O and absorbance of 1:50 dilutions were measured at 595 nm in a WPA S800 Spectrawave spectrophotometer (Biochrom Ltd, Cambridge, UK). OD595 was measured as a quantitative metric of relative growth.
For long-term clonogenic survival assays, 500 cells were seeded in a 10 cm2 culture dish in triplicate and left to adhere overnight. Cells were treated the next day and left to grow for ∼14 days or until colonies were visible by eye. Plates were then stained with crystal violet dye (Sigma) and colonies counted. The relative colony growth was assessed relative to DMSO control plates.
RNA extraction and RT-qPCR
Total RNA was extracted using the RNeasy Plus Mini kit (Qiagen) and reverse transcription was performed using the iScript™ cDNA Synthesis Kit (Bio-Rad) following manufacturer’s protocol. TRIM37 transcripts were measured by qPCR in triplicate on a CFX96 Real-Time Analyzer (Bio-Rad) using Quantifast SYBR Green reagent (QIAGEN), normalised to reference gene HBMS and quantified using the ΔΔCt method to obtain relative expression. Thermocycling conditions were set as follows: 1 cycle (95 °C for 5mins), 40 cycles (95 °C for 15s, 58 °C for 60s). Primer sequences: TRIM37 forward (5’-TCAGCTGTATTAGGCGCTGG-3’), TRIM37_reverse (5’-ACTTCTTCTGCCCAACGACA-3’), HBMS_forward (5’- GCCCTGGAGAAGAATGAA-3’), HBMS_reverse (5’-GGTGAAAGACAACAGCATC-3’).
Flow cytometry
G0/G1, S and G2/M cell cycle profiles were assessed using 5’-Bromouridine (BrdU) incorporation and propidium iodide (PI) staining. Cells were pulsed with 10 μM BrdU (Sigma-Aldrich) for 30 min, trypsinised, and washed with 1% BSA in PBS (1500 rpm, 5 min) before being fixed in 70% ethanol. DNA denaturation was performed using a solution of 0.2 mg/ml of pepsin (Sigma-Aldrich) in 2 M HCl for 20 min at RT. Cells were washed twice with PBS and re-suspended in a solution containing anti-BrdU-FITC conjugated antibody (rat, Bio-Rad, MCA2060FT, 1:100) in 0.5% (v/v) Tween-20, 0.5% (v/v) BSA in PBS and incubated for 1 h in the dark. For mitotic phase cell population (M) determination, cells fixed in 70% ethanol were permeabilised with 0.2% Tween-20 in 2 M HCl for 10 min. Cells were then stained with anti-phosphorylated Histone H3 (Ser10) antibody (mouse, Cell Signaling Technology, 9706, 1:50) in 1% (v/v) BSA in PBS for 3 h. Cells were then washed twice with PBS, re-suspended in a solution containing anti-mouse Alexa Fluor 488 conjugated antibody (goat, ThermoFisher Scientific, A-11029, 1:250) in 1% (v/v) BSA in PBS and incubated for 1 h in the dark. For total DNA staining, including those used for sub-G1 population determination and ploidy analyses, a 20 min incubation at 37°C in a solution of PI/RNaseA (10 μg/ml and 0.1 mg/ml respectively) in PBS was performed. Samples were analysed using an Attune NxT flow cytometer (Life Technologies) and data processing was done using FlowJo software.
Senescence-associated β-galactosidase (SA-β-Gal) staining
SA-β-gal activity of DMSO or centrinone-treated MCF-7 cells was assessed using a staining kit (Cell Signaling, #9860), as per manufacturer’s protocol. Stained cells were imaged with a Nikon wide-field TE2000U Microscope at 200x magnification. For quantification, up to 200 cells per condition were counted across multiple fields to determine the percentage of SA-β-gal positive cells.
Extended Data
Extended Data Figure 1 (related to Figure 1). TRIM37 overexpression in HCT116 and RPE-1 cells recapitulates synthetic lethality with centrosome loss.
(A) Top, centrosome number distribution in interphase MCF-7 cells at various times after addition of 125 nM centrinone. Mean ± s.e.m. Bottom, representative images of centrosome staining (centrioles labelled by Centrin, and PCM labelled by CEP192). Data, n = 3, biological replicates, each comprising >100 cells.
(B) Representative data of a 14 day clonogenic survival assay of MCF-7 and RPE-1 cells with the indicated genotypes treated with DMSO (control) or 125 nM centrinone (PLK4i). n = 3, biological replicates.
(C) Immunoblot showing TRIM37 protein levels in two WT MCF-7 clones stably expressing control or a TRIM37-targeting sgRNA. β-Actin, loading control. Representative data; n = 3, biological replicates. For gel source data, see Supplementary Figure 1.
(D) Representative data of a 10 day clonogenic survival of indicated MCF-7 cell lines treated with DMSO (control) or 125 nM centrinone (PLK4i).
(E) Quantification of n = 3, biological replicates in (D). P values, unpaired two-tailed t-test. Mean ± s.e.m.
(F) Immunoblot of lysates prepared from WT and TP53−/− HCT116 cells expressing a control (EGFP) or TRIM37 transgene. MCF-7 cells were used as a reference for TRIM37 protein overexpression in a 17q23-amplified cell line. β-Actin, loading control. Representative data; n = 3, biological replicates. For gel source data, see Supplementary Figure 1.
(G) Representative data of a 14 day clonogenic survival assay of HCT116 cells treated with DMSO (control) or 125 nM centrinone (PLK4i).
(H) Quantification of n = 3, biological replicates in (G). P values, unpaired two-tailed t-test. Mean ± s.e.m.
(I) Immunoblot showing doxycycline-induced GST or TRIM37 expression in RPE-1; PLK4AS; TP53−/− cells. β-Actin, loading control. Representative data; n = 3, biological replicates. For gel source data, see Supplementary Figure 1.
(J) Representative data of a 14 day colony survival assay of RPE-1; PLK4AS; TP53−/− cells expressing doxycycline-inducible GST (control) or TRIM37 transgenes, treated with DMSO (control) or 3MB-PP1. AS, analogue sensitive.
(K) Quantification of n = 3, biological replicates in (J). P values, unpaired two-tailed t-test. Mean ± s.e.m.
(L) Representative images of RPE-1; PLK4AS; TP53−/− cells in (J). Scale bars, 100 μm.
Extended Data Figure 2. Inhibitor selectivity for PLK4, and not other kinases, is required for the synthetic lethal killing of cells overexpressing TRIM37.
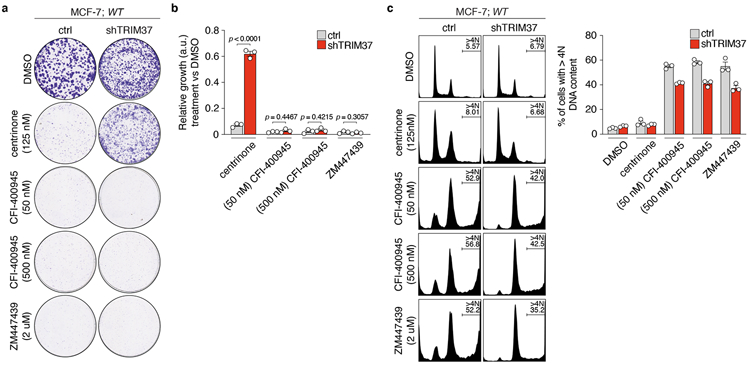
(A) Representative data of a 10 day clonogenic survival of indicated MCF-7 cell lines treated with DMSO (control), centrinone, CFI-400945 or ZM447439.
(B) Quantification of (A), n = 3, biological replicates. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(C) Left, representative flow cytometric analysis of DNA content in MCF-7 cells treated with DMSO (control), centrinone, CFI-400945 or ZM447439 for 3 days. Right, quantification of the percentage of cells with >4N DNA content (polyploidy). n = 3, biological replicates. Mean ± s.e.m.
Extended Data Figure 3 (related to Figure 1). Additional characterization of TRIM37 expression and synthetic lethality in breast cancer cell lines and patient-derived organoids (PDOs).
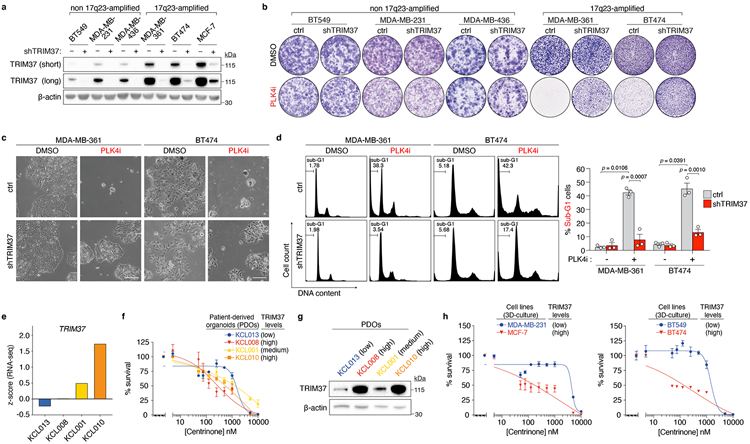
(A) Immunoblot showing TRIM37 protein levels in the indicated 17q23-amplified cell lines (MDA-MB-361, BT474, MCF-7) and non-17q23-amplified cell lines (BT549, MDA-MB-231, MDA-MB-436) expressing control, or TRIM37-targeting, shRNA. β-Actin, loading control. Representative data; n = 3, biological replicates. For gel source data, see Supplementary Figure 1.
(B) Clonogenic survival of 17q23-amplified and non-17q23-amplified cell lines treated with DMSO (control) or 125 nM centrinone (PLK4i). Representative data; n = 3, biological replicates.
(C) Images of DMSO or PLK4i-treated MDA-MB-361 and BT474 cells expressing control, or TRIM37-targeting, shRNA. Scale bars, 200 μm. Representative data; n = 3, biological replicates.
(D) Left, Representative flow cytometric DNA content analysis in DMSO or PLK4i-treated MDA-MB-361 and BT474 cells. Percentages of sub-G1 events are indicated. Right, percentage of sub-G1 cells across n = 3, biological replicates. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(E) TRIM37 gene expression in PDOs. Gene expression is reported as a z-score derived from RNA-Seq datasets across n = 22, independent biological samples.
(F) Viability of patient-derived breast tumour organoids following a 14 day exposure to the indicated concentrations of centrinone. Data from n = 2 biological replicates are shown. Mean ± s.e.m.
(G) Immunoblot showing TRIM37 protein levels in PDOs. β-Actin, loading control. Data from n = 1, biological replicate. For gel source data, see Supplementary Figure 1.
(H) Viability of 3D-cultures of the indicated cell lines following a 14 day exposure to the indicated concentrations of centrinone. Left panel, n = 2, biological replicates, Mean ± s.e.m. Right panel, n = 4, technical replicates, Mean ± s.e.m.
Extended Data Figure 4 (related to Figure 3). TRIM37 localises to centrosomes, where it interacts with, and regulates the abundance of PCM proteins.
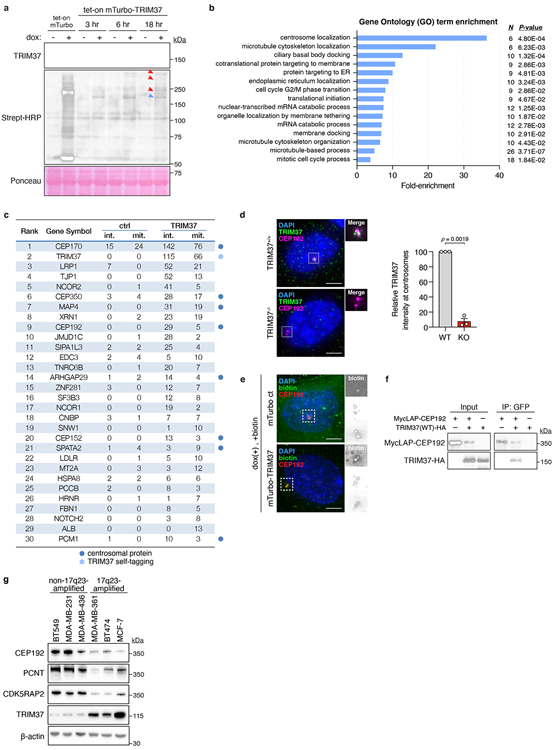
(A) Immunoblot showing TRIM37 and biotinylated proximity interactors. Ponceau-stained blot indicates loading. Data is from a single experiment performed in duplicate. For gel source data, see Supplementary Figure 1.
(B) Gene ontology analysis of mass spectrometry data.
(C) Thresholded mass spectrometry results displaying the top 30 proximity interactors by spectral count. Interactors were filtered to isolate those with >2x more peptides in the mTurbo-TRIM37 sample compared to control.
(D) Left, immunofluorescence of TRIM37 in TRIM37+/+ and TRIM37−/− RPE-1 cells. Scale bars, 5 μm. Right, quantification of TRIM37 intensity at the centrosome in RPE-1 cells. n = 3, biological replicates, each comprising >40 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(E) Immunofluorescence of biotin-labelled proteins in mTurbo cell lines. Representative data; n = 3. Scale bars, 5 μm.
(F) Co-immunoprecipitation showing the interaction of TRIM37 with CEP192. Representative data; n = 3, biological replicates. For gel source data, see Supplementary Figure 1.
(G) Immunoblot showing the levels of TRIM37 and PCM components in non-17q23-amplified versus 17q23-amplified cell lines. β-Actin, loading control. Representative data; n = 3, biological replicates. For gel source data, see Supplementary Figure 1.
Extended Data Figure 5 (related to Figure 3). TRIM37 suppresses microtubule nucleation by the centrosome and supresses the formation of non-centrosomal PCM foci.
(A) Microtubule regrowth following nocodazole washout in control and TRIM37-shRNA expressing MCF-7 mitotic cells. Representative images from (B). n = 3, biological replicates. Scale bars, 5 μm.
(B) Quantification of microtubule regrowth following nocodazole washout in control and TRIM37-shRNA expressing MCF-7 mitotic cells. n = 3, biological replicates, each with >25 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(C) Quantification of centrosomal EB1 intensity following nocodazole washout in control and TRIM37-shRNA expressing MCF-7 mitotic cells. n = 3, biological replicates, each with >25 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(D) Representative images of mitotic PCM foci in acentrosomal RPE-1 cells described in Figure 3d. n = 3, biological replicates. Scale bars, 5 μm.
(E) Left, representative images of mitotic PCM foci in acentrosomal MDA-MB-436 and DLD-1 cells. Scale bars, 5 μm. Right, Quantification of mitotic PCM foci in centrinone-treated MDA-MB-436 and DLD-1 cells that lacked centrosomes. n = >3, biological replicates, each comprising ≥84 cells for DLD-1 cells and ≥ 6 cells for MDA-MB-436 cells. Mean ± s.e.m.
(F) Quantification of CEP192 foci area in RPE-1 TRIM37+/+ vs TRIM37−/− cells in (D). n = 3, biological replicates, each comprising >20 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(G) Representative images for spindle length analysis in indicated MCF-7 cells described in Figure 3g. n = 3, biological replicates. Scale bars, 5 μm.
Extended Data Figure 6 (related to Figure 3). Non-centrosomal PCM foci nucleate microtubules and contribute to spindle assembly in DLD-1 cells.
(A) Representative time-lapse images of mitosis in control DLD-1 cells. n = 3, biological replicates. Scale bar, 5 μm.
(B) Representative time-lapse images of PCM foci formation during mitosis in acentrosomal DLD-1 cells. Scale bar, 5 μm. n = 3, biological replicates. Arrows indicate PCM foci.
(C) Representative time-lapse images of microtubule nucleation from centrosomes in the mitotic spindle of control DLD-1 cells. n = 3, biological replicates. Scale bar, 5 μm.
(D) Representative time-lapse images of microtubule nucleation from PCM foci incorporated into the mitotic spindle of acentrosomal DLD-1 cells. n = 3, biological replicates. Scale bar, 5 μm.
(E) Representative time-lapse images of microtubule nucleation from a PCM focus prior to its incorporation into the mitotic spindle of the acentrosomal cell shown in (D). n = 3, biological replicates. Scale bar, 1 μm.
Extended Data Figure 7 (related to Figure 3). Non-centrosomal PCM foci nucleate microtubules and contribute to spindle assembly in RPE-1 cells.
(A) Representative time-lapse images of mitosis in control RPE-1; PLK4AS; TP53−/− cells. n = 3, biological replicates. Scale bar, 5 μm.
(B) Representative time-lapse images of PCM foci formation during mitosis in acentrosomal RPE-1; PLK4AS; TP53−/− cells. n = 3, biological replicates. Scale bar, 5 μm. Arrows indicate PCM foci.
(C) Representative time-lapse images of microtubule nucleation from centrosomes in the mitotic spindle of control RPE-1; PLK4AS; TP53−/− cells. n = 3, biological replicates. Scale bars, 5 μm.
(D) Representative time-lapse images of microtubule nucleation from PCM foci incorporated into the mitotic spindle of acentrosomal RPE-1; PLK4AS; TP53−/− cells. n = 3, biological replicates. Scale bar, 5 μm.
(E) Representative time-lapse images of microtubule nucleation from a PCM focus prior to its incorporation into the mitotic spindle in acentrosomal RPE-1; PLK4AS; TP53-/−. n = 3, biological replicates. Scale bar, 1 μm.
Extended Data Figure 8 (related to Figure 3). Depletion of CEP192 in RPE-1 cells recapitulates the synthetic lethal mitotic phenotypes observed in high-TRIM37 expressing cells.
(A) Immunoblot showing the CEP192 levels in indicated control and CEP192-depleted RPE-1; PLK4AS; TP53−/− cells. α-tubulin, loading control. For gel source data, see Supplementary Figure 1.
(B) Quantification of mitotic centrosomal CEP192 signal in the same cells as described in (A). n = 3, biological replicates, each comprising >30 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(C) Representative images of centrosomal CEP192 in the same cells as described in (A). Scale bars, 5 μm.
(D) Quantification of the percentage of 3MB-PP1 treated RPE-1; PLK4AS; TP53−/− cells with acentrosomal mitotic CEP192 PCM foci. n = 3, biological replicates, each comprising >30 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(E) Representative time-lapse images of mitotic progression in DMSO or 3MB-treated control and CEP192-depleted RPE-1; PLK4AS; TP53−/− cells. Cells are labelled with H2B-iRFP and tagRFP-tubulin. n = 3, biological replicates.
Extended Data Figure 9 (related to Figure 4). Cell cycle regulation of TRIM37 expression.
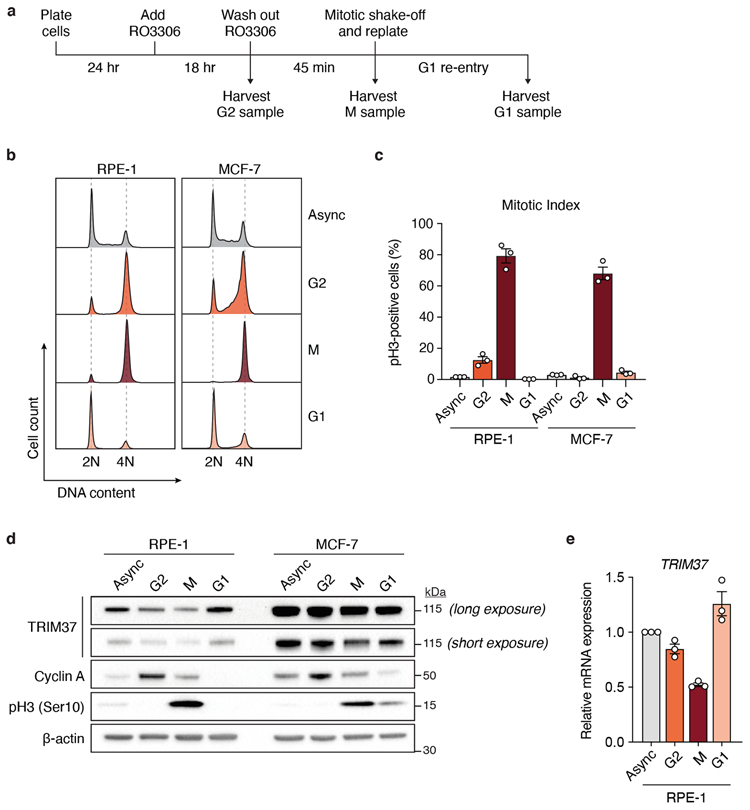
(A) Schematic of the experimental protocol used for cell cycle synchronisation. Samples were subjected to dual flow cytometry staining of phospho-Histone Ser10 (pH3) to mark mitotic cells and propidium iodide (PI) to determine synchronisation efficiency. M, mitotic phase.
(B) Flow cytometric DNA content analysis of samples harvested according to (A). Left, RPE-1; Right, MCF-7. Async, asynchronous.
(C) Mitotic index of cell cycle samples as determined by the percentage of pH3-positive cells with 4N DNA content. n = 3, biological replicates. Mean ± s.e.m.
(D) Immunoblot showing endogenous TRIM37, cyclin A and pH3 in samples analysed in (B). β-actin, loading control. For gel source data, see Supplementary Figure 1.
(E) RT-qPCR analysis indicating relative TRIM37 mRNA expression in RPE-1 cells analysed in (B). Data was normalised to TRIM37 mRNA expression in asynchronous cells. n = 3, biological replicates. Mean ± s.e.m.
Extended Data Figure 10 (related to Figure 4). TRIM37 overexpression delays centrosome maturation in G2/M phase.
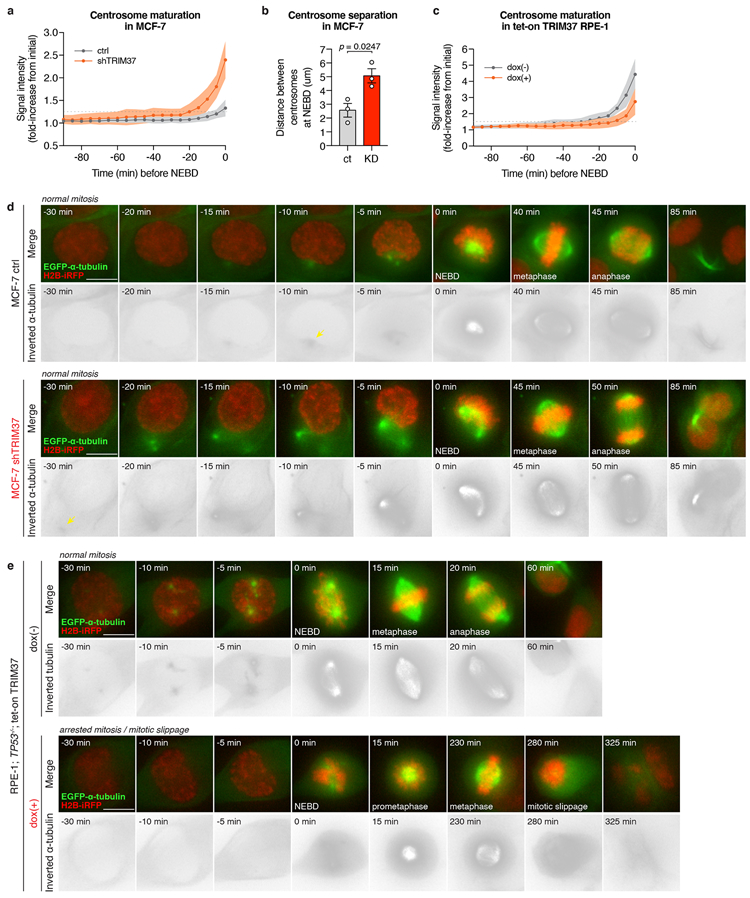
(A) Quantification of centrosomal α-tubulin intensity from time-lapse movies of dividing MCF-7 cells expressing either control or TRIM37-targeting shRNA. Quantitation of >20 cells. Mean ± s.e.m.
(B) Quantification of the distance between the two centrosomes at NEBD in MCF-7 cells. n = 3, biological replicates, each comprising >12 cells. P values, unpaired two-tailed t-test. Mean ± s.e.m.
(C) Quantification of centrosomal α-tubulin intensity from time-lapse movies of dividing RPE-1 tet-on TRIM37 cells. Quantitation of >20 cells. Mean ± s.e.m.
(D) Representative time-lapse images of centrosome maturation in MCF-7 cells. n = 3, biological replicates. Scale bars, 5 μm.
(E) Representative time-lapse images of centrosome maturation in RPE-1 tet-on TRIM37 cells. n = 3, biological replicates. Scale bars, 5 μm.
| Ranked # | Identified Proteins (579) | Gene Symbol |
Reported on BioGRID |
Accession Number |
Alternate ID | Molecular Weight |
1_Ctr_Int | 2_Ctr_Mit | 3_Trim37_I nt |
4_Trim37_ Mit |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PREDICTED: centrosomal protein of 170 kDa isoform X6 [Homo sapiens] | CEP170 | XP_011542643.1 (+34) | 176 kDa | 15 | 24 | 142 | 76 | ||
| 2 | E3 ubiquitin-protein ligase TRIM37 isoform a [Homo sapiens] | TRIM37 | x | NP_001005207.1 (+14) | 108 kDa | 0 | 0 | 115 | 66 | |
| 3 | prolow-density lipoprotein receptor-related protein 1 preproprotein [Homo sapiens] | LRP1 | NP_002323.2 (+1) | 505 kDa | 7 | 0 | 52 | 21 | ||
| 4 | tight junction protein ZO-1 isoform g [Homo sapiens] | TJP1 | NP_001341942.1 (+18) | 197 kDa | 0 | 0 | 52 | 13 | ||
| 5 | nuclear receptor corepressor 2 isoform 2 [Homo sapiens] | NCOR2 | NP_001070729.2 (+2) | 268 kDa | 0 | 1 | 41 | 5 | ||
| 6 | centrosome-associated protein 350 [Homo sapiens] | CEP350 | NP_055625.4 (+5) | 351 kDa | 3 | 4 | 28 | 17 | ||
| 7 | PREDICTED: microtubule-associated protein 4 isoform X7 [Homo sapiens] | MAP4 | XP_005265190.1 (+22) | 247 kDa | 0 | 0 | 31 | 19 | ||
| 8 | 5'-3' exoribonuclease 1 isoform c [Homo sapiens] | XRN1 | NP_001269786.1 (+7) | 193 kDa | 0 | 2 | 23 | 19 | ||
| 9 | centrosomal protein of 192 kDa [Homo sapiens] | CEP192 | x | NP_115518.3 (+8) | 279 kDa | 0 | 0 | 29 | 5 | |
| 10 | probable JmjC domain-containing histone demethylation protein 2C isoform c [Homo sapiens] | JMJD1C | NP_001269877.1 (+15) | 263 kDa | 0 | 1 | 28 | 2 | ||
| 11 | signal-induced proliferation-associated 1-like protein 3 [Homo sapiens] | SIPA1L3 | NP_055888.1 (+4) | 195 kDa | 2 | 2 | 25 | 4 | ||
| 12 | enhancer of mRNA-decapping protein 3 isoform 1 [Homo sapiens] | EDC3 | NP_001135915.1 (+3) | 56 kDa | 2 | 4 | 5 | 10 | ||
| 13 | trinucleotide repeat-containing gene 6B protein isoform 2 [Homo sapiens] | TNRC6B | NP_055903.2 (+1) | 183 kDa | 0 | 1 | 20 | 7 | ||
| 14 | rho GTPase-activating protein 29 isoform a [Homo sapiens] | ARHGAP29 | NP_001315593.1 (+5) | 142 kDa | 1 | 2 | 14 | 4 | ||
| 15 | zinc finger protein 281 isoform 1 [Homo sapiens] | ZNF281 | NP_001268222.1 (+4) | 97 kDa | 3 | 0 | 12 | 7 | ||
| 16 | splicing factor 3B subunit 3 [Homo sapiens] | SF3B3 | NP_036558.3 | 136 kDa | 0 | 0 | 12 | 8 | ||
| 17 | PREDICTED: nuclear receptor corepressor 1 isoform X1 [Homo sapiens] | NCOR1 | XP_005256923.1 (+41) | 279 kDa | 0 | 0 | 19 | 2 | ||
| 18 | cellular nucleic acid-binding protein isoform 6 [Homo sapiens] | CNBP | NP_001120668.1 (+5) | 19 kDa | 3 | 1 | 7 | 7 | ||
| 19 | SNW domain-containing protein 1 isoform 2 [Homo sapiens] | SNW1 | NP_036377.1 | 61 kDa | 0 | 0 | 1 | 10 | ||
| 20 | centrosomal protein of 152 kDa isoform 1 [Homo sapiens] | CEP152 | NP_001181927.1 (+6) | 196 kDa | 0 | 0 | 13 | 3 | ||
| 21 | spermatogenesis-associated protein 2 [Homo sapiens] | SPATA2 | NP_001129245.1 (+3) | 58 kDa | 1 | 4 | 3 | 9 | ||
| 22 | low-density lipoprotein receptor isoform 1 precursor [Homo sapiens] | LDLR | NP_000518.1 (+5) | 95 kDa | 0 | 1 | 5 | 10 | ||
| 23 | metallothionein-2 [Homo sapiens] | MT2A | NP_005944.1 (+3) | 6 kDa | 0 | 3 | 3 | 12 | ||
| 24 | heat shock cognate 71 kDa protein isoform 1 [Homo sapiens] | HSPA8 | NP_006588.1 (+8) | 71 kDa | 2 | 2 | 6 | 6 | ||
| 25 | propionyl-CoA carboxylase beta chain, mitochondrial isoform 1 precursor [Homo sapiens] | PCCB | NP_000523.2 (+1) | 58 kDa | 2 | 0 | 8 | 3 | ||
| 26 | hornerin [Homo sapiens] | HRNR | NP_001009931.1 | 282 kDa | 0 | 1 | 1 | 7 | ||
| 27 | fibrillin-1 preproprotein [Homo sapiens] | FBN1 | NP_000129.3 | 312 kDa | 0 | 0 | 8 | 5 | ||
| 28 | neurogenic locus notch homolog protein 2 isoform 1 preproprotein [Homo sapiens] | NOTCH2 | NP_077719.2 (+5) | 265 kDa | 0 | 0 | 3 | 8 | ||
| 29 | serum albumin preproprotein [Homo sapiens] | ALB | NP_000468.1 | 69 kDa | 0 | 0 | 0 | 13 | ||
| 30 | pericentriolar material 1 protein isoform 2 [Homo sapiens] | PCM1 | NP_001302436.1 (+52) | 228 kDa | 1 | 0 | 10 | 3 | ||
| 31 | protein fantom isoform c [Homo sapiens] | RPGRIP1L | x | NP_001295263.1 (+7) | 147 kDa | 0 | 0 | 8 | 4 | |
| 32 | cell cycle and apoptosis regulator protein 2 [Homo sapiens] | CCAR2 | NP_066997.3 (+3) | 103 kDa | 2 | 0 | 6 | 2 | ||
| 33 | 60S ribosomal protein L7 [Homo sapiens] | RPL7 | NP_000962.2 (+1) | 29 kDa | 0 | 1 | 2 | 5 | ||
| 34 | zinc finger protein 318 [Homo sapiens] | ZNF318 | NP_055160.2 (+2) | 251 kDa | 1 | 0 | 8 | 2 | ||
| 35 | 60S ribosomal protein L6 isoform 1 [Homo sapiens] | RPL6 | NP_000961.2 (+9) | 33 kDa | 0 | 1 | 3 | 3 | ||
| 36 | mitotic checkpoint serine/threonine-protein kinase BUB1 beta [Homo sapiens] | BUB1B | NP_001202.4 | 120 kDa | 0 | 2 | 2 | 7 | ||
| 37 | disks large homolog 5 [Homo sapiens] | DLG5 | NP_004738.3 (+12) | 214 kDa | 1 | 0 | 7 | 0 | ||
| 38 | dermcidin isoform 2 preproprotein [Homo sapiens] | DCD | NP_001287783.1 (+1) | 12 kDa | 1 | 2 | 3 | 4 | ||
| 39 | granulins precursor [Homo sapiens] | GRN | NP_002078.1 (+1) | 64 kDa | 1 | 0 | 3 | 4 | ||
| 40 | G2 and S phase-expressed protein 1 [Homo sapiens] | GTSE1 | NP_057510.4 (+5) | 78 kDa | 0 | 2 | 1 | 6 | ||
| 41 | centrosomal protein of 85 kDa isoform 2 [Homo sapiens] | CEP85 | NP_001268446.1 (+2) | 80 kDa | 0 | 0 | 4 | 3 | ||
| 42 | nck-associated protein 5-like [Homo sapiens] | NCKAP5L | NP_001032895.2 (+4) | 139 kDa | 0 | 0 | 8 | 0 | ||
| 43 | cysteine-rich motor neuron 1 protein precursor [Homo sapiens] | CRIM1 | NP_057525.1 (+7) | 114 kDa | 0 | 0 | 4 | 4 | ||
| 44 | coatomer subunit beta' [Homo sapiens] | COPB2 | NP_004757.1 (+2) | 102 kDa | 0 | 0 | 2 | 4 | ||
| 45 | E3 ubiquitin-protein ligase RNF169 [Homo sapiens] | RNF169 | NP_001092108.1 (+1) | 77 kDa | 0 | 0 | 2 | 1 | ||
| 46 | Alstrom syndrome protein 1 [Homo sapiens] | ALMS1 | NP_055935.4 | 461 kDa | 0 | 0 | 4 | 0 | ||
| 47 | neuron navigator 1 isoform 2 [Homo sapiens] | NAV1 | NP_001161210.1 (+12) | 161 kDa | 0 | 0 | 5 | 0 | ||
| 48 | trinucleotide repeat-containing gene 6A protein isoform 2 [Homo sapiens] | TNRC6A | NP_001317449.1 (+11) | 205 kDa | 0 | 0 | 5 | 0 | ||
| 49 | PREDICTED: antigen KI-67 isoform X1 [Homo sapiens] | MKI67 | XP_011538120.1 | 321 kDa | 0 | 0 | 0 | 4 | ||
| 50 | double-stranded RNA-binding protein Staufen homolog 1 isoform c [Homo sapiens] | STAU1 | NP_001032405.1 (+17) | 56 kDa | 0 | 0 | 1 | 0 | ||
| 51 | suprabasin isoform 1 precursor [Homo sapiens] | SBSN | NP_001159506.1 (+3) | 61 kDa | 0 | 0 | 1 | 1 | ||
| 52 | protein moonraker isoform 1 [Homo sapiens] | KIAA753 | NP_055619.2 (+5) | 109 kDa | 0 | 0 | 1 | 3 | ||
| 53 | spindle and centriole-associated protein 1 isoform a [Homo sapiens] | SPICE1 | x | NP_001318007.1 (+5) | 96 kDa | 0 | 0 | 3 | 2 | |
| 54 | zinc finger protein 609 [Homo sapiens] | ZNF69 | NP_055857.1 (+2) | 151 kDa | 0 | 0 | 5 | 0 | ||
| 55 | proline-rich protein 12 [Homo sapiens] | PRR12 | NP_065770.1 | 211 kDa | 0 | 0 | 6 | 0 | ||
| 56 | lymphokine-activated killer T-cell-originated protein kinase isoform 2 [Homo sapiens] | PBK | x | NP_001265874.1 (+2) | 37 kDa | 0 | 0 | 0 | 5 | |
| 57 | macrophage erythroblast attacher isoform 1 [Homo sapiens] | MAEA | NP_001017405.1 (+1) | 45 kDa | 0 | 0 | 0 | 4 | ||
| 58 | MAP7 domain-containing protein 3 isoform 2 [Homo sapiens] | MAP7D3 | NP_001166987.1 (+4) | 97 kDa | 0 | 0 | 2 | 1 | ||
| 59 | protein transport protein Sec24B isoform b [Homo sapiens] | SEC24B | NP_001036199.1 (+12) | 134 kDa | 0 | 0 | 1 | 0 | ||
| 60 | ribonucleoprotein PTB-binding 1 [Homo sapiens] | RAVER1 | NP_597709.2 | 80 kDa | 0 | 1 | 1 | 2 | ||
| 61 | GRB10-interacting GYF protein 2 isoform b [Homo sapiens] | GIGYF2 | NP_001096616.1 (+3) | 150 kDa | 0 | 0 | 3 | 1 | ||
| 62 | SLAIN motif-containing protein 2 [Homo sapiens] | SLAIN2 | NP_065897.1 (+1) | 63 kDa | 1 | 0 | 3 | 0 | ||
| 63 | TRAF family member-associated NF-kappa-B activator isoform a [Homo sapiens] | TANK | NP_001186064.1 (+8) | 48 kDa | 0 | 0 | 2 | 0 | ||
| 64 | rho guanine nucleotide exchange factor 18 isoform b [Homo sapiens] | ARHGEF18 | NP_001124427.1 (+11) | 131 kDa | 0 | 0 | 1 | 1 | ||
| 65 | oral-facial-digital syndrome 1 protein isoform 2 [Homo sapiens] | OFD1 | NP_001317138.1 (+8) | 112 kDa | 0 | 0 | 2 | 0 | ||
| 66 | transcription factor 12 isoform a [Homo sapiens] | TCF12 | NP_001309080.1 (+19) | 76 kDa | 0 | 0 | 0 | 1 | ||
| 67 | zinc finger E-box-binding homeobox 2 isoform 2 [Homo sapiens] | ZEB2 | NP_001165124.1 (+6) | 134 kDa | 0 | 0 | 2 | 0 | ||
| 68 | transcription factor 20 isoform 1 [Homo sapiens] | TCF2 | NP_005641.1 (+5) | 212 kDa | 0 | 0 | 2 | 0 | ||
| 69 | 60S ribosomal protein L36a-like [Homo sapiens] | RPL36AL | NP_000992.1 (+2) | 12 kDa | 0 | 1 | 1 | 2 | ||
| 70 | centrosomal protein of 131 kDa isoform b [Homo sapiens] | CEP131 | NP_001009811.2 (+11) | 117 kDa | 0 | 0 | 1 | 3 | ||
| 71 | centrobin isoform beta [Homo sapiens] | CNTROB | x | NP_001032221.1 (+22) | 104 kDa | 0 | 0 | 1 | 2 | |
| 72 | transcriptional repressor p66-beta [Homo sapiens] | GATAD2B | NP_065750.1 (+1) | 65 kDa | 1 | 0 | 2 | 0 | ||
| 73 | palladin isoform 1 [Homo sapiens] | PALLD | NP_001159580.1 (+14) | 124 kDa | 0 | 0 | 0 | 2 | ||
| 74 | sequestosome-1 isoform 2 [Homo sapiens] | SQSTM1 | NP_001135770.1 (+3) | 39 kDa | 0 | 0 | 3 | 0 | ||
| 75 | coiled-coil domain-containing protein 102A [Homo sapiens] | CCDC12A | NP_149989.2 (+1) | 63 kDa | 0 | 0 | 2 | 0 | ||
| 76 | centrosomal protein of 170 kDa protein B isoform 1 [Homo sapiens] | CEP17B | NP_001106197.1 (+6) | 168 kDa | 0 | 0 | 3 | 0 | ||
| 77 | protein FAM193A isoform 2 [Homo sapiens] | FAM193A | NP_001243595.1 (+8) | 140 kDa | 0 | 0 | 4 | 0 | ||
| 78 | phosphatidylinositol 4-phosphate 3-kinase C2 domain-containing subunit alpha isoform 1 [Homo sapiens] | PIK3C2A | NP_001308307.1 (+3) | 191 kDa | 0 | 0 | 4 | 0 | ||
| 79 | desmoplakin isoform I [Homo sapiens] | DSP | NP_004406.2 | 332 kDa | 0 | 0 | 0 | 3 | ||
| 80 | HAUS augmin-like complex subunit 6 isoform 2 [Homo sapiens] | HAUS6 | NP_001257819.1 (+1) | 104 kDa | 0 | 0 | 2 | 0 | ||
| 81 | U2 snRNP-associated SURP motif-containing protein isoform 1 [Homo sapiens] | U2SURP | NP_001073884.1 (+2) | 118 kDa | 0 | 0 | 1 | 0 | ||
| 82 | YEATS domain-containing protein 2 isoform 3 [Homo sapiens] | YEATS2 | NP_001338298.1 (+7) | 151 kDa | 0 | 0 | 3 | 0 | ||
| 83 | TBC1 domain family member 2A isoform 1 [Homo sapiens] | TBC1D2 | NP_001254500.1 (+2) | 105 kDa | 0 | 0 | 1 | 0 | ||
| 84 | zinc finger protein 746 isoform 1 [Homo sapiens] | ZNF746 | NP_001156946.1 (+3) | 69 kDa | 0 | 0 | 2 | 1 | ||
| 85 | 60S ribosomal protein L29 [Homo sapiens] | RPL29 | NP_000983.1 | 18 kDa | 0 | 0 | 1 | 2 | ||
| 86 | G2/mitotic-specific cyclin-B1 isoform 2 [Homo sapiens] | CCNB1 | NP_001341773.1 (+1) | 44 kDa | 0 | 1 | 0 | 2 | ||
| 87 | signal-induced proliferation-associated 1-like protein 1 isoform 2 [Homo sapiens] | SIPA1L1 | NP_001271174.1 (+44) | 197 kDa | 0 | 0 | 1 | 0 | ||
| 88 | thyroid receptor-interacting protein 6 [Homo sapiens] | TRIP6 | NP_003293.2 | 50 kDa | 0 | 0 | 1 | 0 | ||
| 89 | PREDICTED: mediator of RNA polymerase II transcription subunit 23 isoform X4 [Homo sapiens] | MED23 | XP_006715675.1 (+7) | 85 kDa | 0 | 0 | 1 | 0 | ||
| 90 | kinesin-like protein KIF14 isoform 1 [Homo sapiens] | KIF14 | NP_055690.1 (+7) | 186 kDa | 0 | 0 | 1 | 0 | ||
| 91 | ataxin-1 [Homo sapiens] | ATXN1 | NP_000323.2 (+1) | 87 kDa | 0 | 0 | 1 | 0 | ||
| 92 | E3 ubiquitin-protein ligase TRIM33 isoform alpha [Homo sapiens] | TRIM33 | NP_056990.3 (+6) | 123 kDa | 0 | 0 | 1 | 0 | ||
| 93 | 60S ribosomal protein L18 isoform 1 [Homo sapiens] | RPL18 | NP_000970.1 (+1) | 22 kDa | 0 | 0 | 1 | 1 | ||
| 94 | regulation of nuclear pre-mRNA domain-containing protein 2 isoform 3 [Homo sapiens] | RPRD2 | NP_001284603.1 (+4) | 153 kDa | 0 | 0 | 1 | 0 | ||
| 95 | PREDICTED: DNA cross-link repair 1A protein isoform X1 [Homo sapiens] | DCLRE1A | XP_011538731.1 (+3) | 116 kDa | 0 | 0 | 0 | 1 | ||
| 96 | MAX gene-associated protein isoform 2 [Homo sapiens] | MGA | NP_001074010.2 (+16) | 315 kDa | 0 | 0 | 3 | 0 | ||
| 97 | nuclear receptor-interacting protein 1 [Homo sapiens] | NRIP1 | NP_003480.2 (+11) | 127 kDa | 0 | 0 | 2 | 0 | ||
| 98 | zinc finger CCCH domain-containing protein 11A isoform 1 [Homo sapiens] | ZC3H11A | NP_001306167.1 (+19) | 89 kDa | 0 | 0 | 0 | 2 | ||
| 99 | germinal-center associated nuclear protein [Homo sapiens] | MCM3AP | NP_003897.2 (+3) | 218 kDa | 0 | 0 | 2 | 0 | ||
| 100 | MDS1 and EVI1 complex locus protein EVI1 isoform a [Homo sapiens] | MECOM | NP_001098547.3 (+17) | 126 kDa | 0 | 0 | 2 | 0 | ||
| 101 | C2 domain-containing protein 3 isoform 1 [Homo sapiens] | C2CD3 | NP_001273506.1 (+5) | 260 kDa | 0 | 0 | 3 | 0 | ||
| 102 | ankyrin repeat domain-containing protein 12 isoform 2 [Homo sapiens] | ANKRD12 | NP_001077094.1 (+5) | 233 kDa | 0 | 0 | 1 | 0 | ||
| 103 | high mobility group protein HMG-I/HMG-Y isoform b [Homo sapiens] | HMGA1 | NP_001306006.1 (+4) | 11 kDa | 0 | 0 | 0 | 2 | ||
| 104 | arginase-1 isoform 2 [Homo sapiens] | ARG1 | NP_000036.2 (+2) | 35 kDa | 0 | 0 | 1 | 1 | ||
| 105 | SCL-interrupting locus protein isoform 1 [Homo sapiens] | STIL | NP_001041631.1 (+17) | 143 kDa | 0 | 0 | 1 | 1 | ||
| 106 | progesterone-induced-blocking factor 1 isoform 1 [Homo sapiens] | PIBF1 | NP_001336584.1 (+8) | 93 kDa | 0 | 0 | 1 | 1 | ||
| 107 | fibrillin-2 precursor [Homo sapiens] | FBN2 | NP_001990.2 (+1) | 315 kDa | 0 | 0 | 1 | 1 | ||
| 108 | protein SMG7 isoform 5 [Homo sapiens] | SMG7 | NP_001167532.1 (+21) | 128 kDa | 0 | 0 | 1 | 1 | ||
| 109 | rho GTPase-activating protein 22 isoform 1 [Homo sapiens] | ARHGAP22 | NP_001242953.1 (+16) | 78 kDa | 0 | 0 | 1 | 1 | ||
| 110 | SEC23-interacting protein [Homo sapiens] | SEC23IP | NP_009121.1 (+3) | 111 kDa | 0 | 0 | 1 | 0 | ||
| 111 | NP_001278761.1-DECOY | ERFE | NP_001278761.1-DECOY | ? | 0 | 0 | 0 | 1 | ||
| 112 | protein CYR61 precursor [Homo sapiens] | CYR61 | NP_001545.2 | 42 kDa | 0 | 0 | 1 | 0 | ||
| 113 | protein-glutamine gamma-glutamyltransferase E [Homo sapiens] | TGM3 | NP_003236.3 | 77 kDa | 0 | 0 | 1 | 0 | ||
| 114 | PREDICTED: RNA-binding protein 10 isoform X5 [Homo sapiens] | RBM10 | XP_016885374.1 (+10) | 80 kDa | 0 | 0 | 0 | 1 | ||
| 115 | ankyrin repeat and LEM domain-containing protein 2 [Homo sapiens] | ANKLE2 | NP_055929.1 (+3) | 104 kDa | 0 | 0 | 1 | 0 | ||
| 116 | neurogenic locus notch homolog protein 1 preproprotein [Homo sapiens] | NOTCH1 | NP_060087.3 (+1) | 272 kDa | 0 | 0 | 1 | 0 | ||
| 117 | BCL-6 corepressor isoform a [Homo sapiens] | BCOR | NP_001116855.1 (+13) | 188 kDa | 0 | 0 | 2 | 0 | ||
| 118 | pleckstrin homology-like domain family B member 2 isoform b [Homo sapiens] | PHLDB2 | NP_001127909.1 (+3) | 140 kDa | 0 | 0 | 2 | 0 | ||
| 119 | smoothelin isoform d [Homo sapiens] | SMTN | NP_001193946.1 (+20) | 108 kDa | 0 | 0 | 0 | 2 | ||
| 120 | desmoglein-1 preproprotein [Homo sapiens] | DSG1 | NP_001933.2 | 114 kDa | 0 | 0 | 2 | 0 | ||
| 121 | msx2-interacting protein [Homo sapiens] | SPEN | NP_055816.2 | 402 kDa | 0 | 0 | 2 | 0 | ||
| 122 | BLOC-1-related complex subunit 6 [Homo sapiens] | BORCS6 | NP_060092.2 | 37 kDa | 0 | 0 | 2 | 0 | ||
| 123 | interferon regulatory factor 2-binding protein 1 [Homo sapiens] | IRF2BP1 | NP_056464.1 | 62 kDa | 0 | 0 | 1 | 0 | ||
| 124 | ubinuclein-1 isoform a [Homo sapiens] | UBN1 | NP_001072982.1 (+9) | 122 kDa | 0 | 0 | 2 | 0 | ||
| 125 | supervillin isoform 3 [Homo sapiens] | SVIL | NP_001310528.1 (+24) | 214 kDa | 0 | 0 | 1 | 0 | ||
| 126 | coiled-coil domain-containing protein 14 isoform 2 [Homo sapiens] | CCDC14 | x | NP_001295246.1 (+13) | 84 kDa | 0 | 0 | 1 | 0 | |
| 127 | transcription intermediary factor 1-alpha isoform b [Homo sapiens] | TRIM24 | NP_003843.3 (+1) | 113 kDa | 0 | 0 | 1 | 0 | ||
| 128 | nephrocystin-4 isoform b [Homo sapiens] | NPHP4 | NP_001278522.1 (+15) | 101 kDa | 0 | 0 | 0 | 1 | ||
| 129 | ubiquitin-like protein fubi and ribosomal protein S30 precursor [Homo sapiens] | FAU | NP_001988.1 | 14 kDa | 0 | 0 | 0 | 2 | ||
| 130 | calmodulin-regulated spectrin-associated protein 1 [Homo sapiens] | CAMSAP1 | NP_056262.3 (+7) | 178 kDa | 0 | 0 | 2 | 0 | ||
| 131 | NP_001010982.2-DECOY | AFMID | NP_001010982.2-DECOY (+5) | ? | 0 | 0 | 0 | 2 | ||
| 132 | actin, alpha skeletal muscle [Homo sapiens] | ACTA1 | NP_001091.1 (+1) | 42 kDa | 0 | 0 | 1 | 0 | ||
| 133 | metallothionein-1E [Homo sapiens] | MT1E | NP_783316.2 | 6 kDa | 0 | 0 | 0 | 1 | ||
| 134 | AP-3 complex subunit sigma-1 isoform 2 [Homo sapiens] | AP3S1 | NP_001002924.2 (+3) | 19 kDa | 0 | 0 | 0 | 1 | ||
| 135 | 40S ribosomal protein S8 [Homo sapiens] | RPS8 | NP_001003.1 | 24 kDa | 0 | 0 | 0 | 1 | ||
| 136 | UPF0705 protein C11orf49 isoform 1 [Homo sapiens] | C11orf49 | NP_001003676.1 (+6) | 31 kDa | 0 | 0 | 0 | 1 | ||
| 137 | 40S ribosomal protein S28 [Homo sapiens] | RPS28 | NP_001022.1 | 8 kDa | 0 | 0 | 0 | 1 | ||
| 138 | nudC domain-containing protein 1 isoform 2 [Homo sapiens] | NUDCD1 | NP_001121683.1 (+3) | 64 kDa | 0 | 0 | 0 | 1 | ||
| 139 | protein SGT1 homolog isoform B [Homo sapiens] | SUGT1 | NP_001124384.1 (+8) | 41 kDa | 0 | 0 | 0 | 1 | ||
| 140 | insulin-like growth factor-binding protein 7 isoform 2 precursor [Homo sapiens] | IGFBP7 | NP_001240764.1 (+1) | 29 kDa | 0 | 0 | 0 | 1 | ||
| 141 | RNA-binding motif protein, X chromosome isoform 1 [Homo sapiens] | RBMX | NP_002130.2 | 42 kDa | 0 | 0 | 0 | 1 | ||
| 142 | histone H1.2 [Homo sapiens] | HIST1H1C | NP_005310.1 (+4) | 21 kDa | 0 | 0 | 0 | 1 | ||
| 143 | A-kinase anchor protein 8 [Homo sapiens] | AKAP8 | NP_005849.1 (+2) | 76 kDa | 0 | 0 | 0 | 1 | ||
| 144 | DNA-binding protein RFX5 [Homo sapiens] | RFX5 | NP_000440.1 (+9) | 65 kDa | 0 | 0 | 1 | 0 | ||
| 145 | 60S ribosomal protein L28 isoform 2 [Homo sapiens] | RPL28 | NP_000982.2 (+4) | 16 kDa | 0 | 0 | 1 | 0 | ||
| 146 | protein diaphanous homolog 3 isoform a [Homo sapiens] | DIAPH3 | NP_001035982.1 (+8) | 137 kDa | 0 | 0 | 1 | 0 | ||
| 147 | interferon regulatory factor 2-binding protein 2 isoform B [Homo sapiens] | IRF2BP2 | NP_001070865.1 (+1) | 59 kDa | 0 | 0 | 1 | 0 | ||
| 148 | BEN domain-containing protein 3 [Homo sapiens] | BEND3 | NP_001073919.1 (+4) | 94 kDa | 0 | 0 | 1 | 0 | ||
| 149 | nuclear factor related to kappa-B-binding protein isoform 1 [Homo sapiens] | NFRKB | NP_001137307.1 (+15) | 139 kDa | 0 | 0 | 1 | 0 | ||
| 150 | U3 small nucleolar RNA-associated protein 14 homolog A isoform 2 [Homo sapiens] | UTP14A | NP_001159693.1 (+3) | 82 kDa | 0 | 0 | 1 | 0 | ||
| 151 | C7orf55-LUC7L2 protein [Homo sapiens] | LUC7L2 | NP_001231513.1 (+3) | 54 kDa | 0 | 0 | 1 | 0 | ||
| 152 | myocyte-specific enhancer factor 2D isoform 2 [Homo sapiens] | MEF2D | NP_001258558.1 (+10) | 55 kDa | 0 | 0 | 1 | 0 | ||
| 153 | protein transport protein Sec16A isoform 2 [Homo sapiens] | SEC16A | NP_001263347.1 (+21) | 249 kDa | 0 | 0 | 1 | 0 | ||
| 154 | E3 ubiquitin-protein ligase UBR5 isoform 2 [Homo sapiens] | UBR5 | NP_001269802.1 (+6) | 309 kDa | 0 | 0 | 1 | 0 | ||
| 155 | protein KRBA1 isoform 2 [Homo sapiens] | KRBA1 | NP_001277116.1 (+9) | 112 kDa | 0 | 0 | 1 | 0 | ||
| 156 | next to BRCA1 gene 1 protein isoform b [Homo sapiens] | NBR1 | NP_001278500.1 (+8) | 104 kDa | 0 | 0 | 1 | 0 | ||
| 157 | calmodulin-regulated spectrin-associated protein 2 isoform 1 [Homo sapiens] | CAMSAP2 | NP_001284636.1 (+4) | 168 kDa | 0 | 0 | 1 | 0 | ||
| 158 | cleavage and polyadenylation specificity factor subunit 6 isoform 2 [Homo sapiens] | CPSF6 | NP_001287876.1 (+3) | 63 kDa | 0 | 0 | 1 | 0 | ||
| 159 | SCY1-like protein 2 isoform 1 [Homo sapiens] | SCYL2 | NP_001304713.1 (+4) | 104 kDa | 0 | 0 | 1 | 0 | ||
| 160 | leucine zipper putative tumor suppressor 2 isoform a [Homo sapiens] | LZTS2 | NP_001305028.1 (+6) | 73 kDa | 0 | 0 | 1 | 0 | ||
| 161 | serine/threonine-protein phosphatase 1 regulatory subunit 10 [Homo sapiens] | PPP1R1 | NP_002705.2 (+3) | 99 kDa | 0 | 0 | 1 | 0 | ||
| 162 | sodium channel protein type 9 subunit alpha [Homo sapiens] | SCN9A | NP_002968.1 (+6) | 225 kDa | 0 | 0 | 1 | 0 | ||
| 163 | histone-lysine N-methyltransferase 2D [Homo sapiens] | KMT2D | NP_003473.3 (+9) | 593 kDa | 0 | 0 | 1 | 0 | ||
| 164 | ras GTPase-activating protein nGAP isoform 1 [Homo sapiens] | RASAL2 | NP_004832.1 (+11) | 129 kDa | 0 | 0 | 1 | 0 | ||
| 165 | zinc finger protein 217 [Homo sapiens] | ZNF217 | NP_006517.1 (+4) | 115 kDa | 0 | 0 | 1 | 0 | ||
| 166 | E3 ubiquitin-protein ligase RBBP6 isoform 1 [Homo sapiens] | RBBP6 | NP_008841.2 (+1) | 202 kDa | 0 | 0 | 1 | 0 | ||
| 167 | zinc finger protein 592 [Homo sapiens] | ZNF592 | NP_055445.2 (+4) | 138 kDa | 0 | 0 | 1 | 0 | ||
| 168 | tubulin polyglutamylase TTLL5 [Homo sapiens] | TTLL5 | NP_055887.3 | 144 kDa | 0 | 0 | 1 | 0 | ||
| 169 | TBC1 domain family member 2B isoform b [Homo sapiens] | TBC1D2B | NP_055894.6 (+3) | 104 kDa | 0 | 0 | 1 | 0 | ||
| 170 | synemin isoform B [Homo sapiens] | SYNM | NP_056101.5 (+2) | 140 kDa | 0 | 0 | 1 | 0 | ||
| 171 | ras-related protein Rab-10 [Homo sapiens] | RAB1 | NP_057215.3 | 23 kDa | 0 | 0 | 1 | 0 | ||
| 172 | RNA-binding protein 27 [Homo sapiens] | RBM27 | NP_061862.1 (+7) | 119 kDa | 0 | 0 | 1 | 0 | ||
| 173 | ANKHD1-EIF4EBP3 protein [Homo sapiens] | ANKHD1 | NP_065741.3 | 277 kDa | 0 | 0 | 1 | 0 | ||
| 174 | rho GTPase-activating protein 31 [Homo sapiens] | ARHGAP31 | NP_065805.2 (+2) | 157 kDa | 0 | 0 | 1 | 0 | ||
| 175 | GRB10-interacting GYF protein 1 [Homo sapiens] | GIGYF1 | NP_072096.2 (+9) | 115 kDa | 0 | 0 | 1 | 0 | ||
| 176 | deubiquitinating protein VCIP135 [Homo sapiens] | VCPIP1 | NP_079330.2 | 134 kDa | 0 | 0 | 1 | 0 | ||
| 177 | PREDICTED: nuclear pore complex protein Nup88 isoform X2 [Homo sapiens] | NUP88 | XP_016880183.1 (+3) | 69 kDa | 0 | 0 | 1 | 0 | ||
| 178 | biotin--protein ligase isoform 1 [Homo sapiens] | HLCS | NP_000402.3 (+17) | 81 kDa | 0 | 0 | 0 | 1 | ||
| 179 | microtubule-associated protein 1A [Homo sapiens] | MAP1A | NP_002364.5 (+1) | 305 kDa | 0 | 0 | 1 | 0 | ||
| 180 | NP_036475.3-DECOY | NNT | NP_036475.3-DECOY (+4) | ? | 0 | 0 | 0 | 1 | ||
| 181 | four and a half LIM domains protein 2 isoform a [Homo sapiens] | FHL2 | NP_001034581.1 (+10) | 32 kDa | 0 | 0 | 0 | 1 | ||
| 182 | zinc finger protein 776 isoform 1 [Homo sapiens] | ZNF776 | NP_775903.3 | 60 kDa | 0 | 0 | 0 | 1 | ||
| 183 | protein SCAF8 isoform a [Homo sapiens] | SCAF8 | NP_001273117.1 (+4) | 149 kDa | 0 | 0 | 1 | 0 | ||
| 184 | 60S ribosomal protein L4 [Homo sapiens] | RPL4 | NP_000959.2 | 48 kDa | 0 | 0 | 0 | 1 | ||
Supplementary Material
Acknowledgements
This work was supported by a Cancer Research UK Career Development Fellowship C52690/A19270 (to J.R.C.), and by National Institutes of Health grants R01GM114119 and R01GM133897, an American Cancer Society Scholar grant RSG-16-156-01-CCG, and an American Cancer Society Mission Boost Grant MBG-19-173-01-MBG (to A.J.H). Z.Y.Y. is supported by the National Science Scholarship from A*STAR, Singapore. The Wellcome Centre for Human Genetics is supported by Wellcome grant 090532/Z/09/Z. C.J.L./A.N.J.T laboratories are funded by NC3Rs (NC/P001262/1), Breast Cancer Now funding to the Breast Cancer Now Toby Robins Research Centre (CTR-Q4-Y2), private donations to the ICR Development Office, and NHS funding to the NIHR Biomedical Research Centres at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London, and the Royal Marsden Hospital. We also thank R. Peat (Francis Crick Institute) for breast cancer cell lines.
Footnotes
Reprints and permissions information is available at www.nature.com/reprints.
Competing interests: C.J.L. makes the following disclosures that are not directly relevant to work published or discussed in this manuscript: received research funding from: AstraZeneca, Merck KGaA, Artios. Received consultancy, SAB membership or honoraria payments from: Syncona, Sun Pharma, Gerson Lehrman Group, Merck KGaA, Vertex, AstraZeneca, Tango, 3rd Rock, Ono Pharma, Artios. Has stock in: Tango, Ovibio. C.J.L. is also a named inventor on patents describing the use of DNA repair inhibitors and stands to gain from the development as part of the ICR “Rewards to Inventors” scheme. A.N.J.T makes the following disclosures that are not directly relevant to work published or discussed in this manuscript: received research funding from: AstraZeneca and Merck KGaA. A.N.J.T. also receives “Rewards to Inventors” scheme payments from the Institute of Cancer Research (ICR) associated with patents describing the use of DNA repair inhibitors and stands to gain from their development.
Data availability statement:
All source data for graphs and gels in Fig. 1-4 and Extended Data Fig. 1-10 are available as .xslx tables and Supplementary Information within the manuscript. Other data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Burrell RA, McGranahan N, Bartek J & Swanton C The causes and consequences of genetic heterogeneity in cancer evolution. Nature 501, 338–345, doi: 10.1038/nature12625 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Kalimutho M et al. Patterns of Genomic Instability in Breast Cancer. Trends in pharmacological sciences 40, 198–211, doi: 10.1016/j.tips.2019.01.005 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Lord CJ & Ashworth A PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158, doi: 10.1126/science.aam7344 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curtis C et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346, doi: 10.1038/nature10983 https://www.nature.com/articles/nature10983#supplementary-information (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dawson S-J, Rueda OM, Aparicio S & Caldas C A new genome-driven integrated classification of breast cancer and its implications. The EMBO journal 32, 617–628, doi: 10.1038/emboj.2013.19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali HR et al. Genome-driven integrated classification of breast cancer validated in over 7,500 samples. Genome biology 15, 431, doi: 10.1186/s13059-014-0431-1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong YL et al. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 348, 1155–1160, doi: 10.1126/science.aaa5111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moyer TC, Clutario KM, Lambrus BG, Daggubati V & Holland AJ Binding of STIL to Plk4 activates kinase activity to promote centriole assembly. J Cell Biol 209, 863–878, doi: 10.1083/jcb.201502088 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lambrus BG et al. p53 protects against genome instability following centriole duplication failure. J Cell Biol 210, 63–77, doi: 10.1083/jcb.201502089 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuella-Martin R et al. 53BP1 Integrates DNA Repair and p53-Dependent Cell Fate Decisions via Distinct Mechanisms. Mol Cell 64, 51–64, doi: 10.1016/j.molcel.2016.08.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meitinger F et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. The Journal of Cell Biology 214, 155–166, doi: 10.1083/jcb.201604081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong CS et al. 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. eLife 5, e16270, doi: 10.7554/eLife.16270 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambrus BG et al. A USP28–53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 214, 143–153, doi: 10.1083/jcb.201604054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ciriello G et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519, doi: 10.1016/j.cell.2015.09.033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pereira B et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature communications 7, 11479, doi: 10.1038/ncomms11479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monni O et al. Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proceedings of the National Academy of Sciences of the United States of America 98, 5711–5716, doi: 10.1073/pnas.091582298 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parssinen J, Kuukasjarvi T, Karhu R & Kallioniemi A High-level amplification at 17q23 leads to coordinated overexpression of multiple adjacent genes in breast cancer. Br J Cancer 96, 1258–1264, doi: 10.1038/sj.bjc.6603692 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinclair CS, Rowley M, Naderi A & Couch FJ The 17q23 amplicon and breast cancer. Breast Cancer Res Treat 78, 313–322 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Meitinger F et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol 214, 155–166, doi: 10.1083/jcb.201604081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balestra FR, Strnad P, Fluckiger I & Gonczy P Discovering regulators of centriole biogenesis through siRNA-based functional genomics in human cells. Dev Cell 25, 555–571, doi: 10.1016/j.devcel.2013.05.016 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Oegema K, Davis RL, Lara-Gonzalez P, Desai A & Shiau AK CFI-400945 is not a selective cellular PLK4 inhibitor. Proceedings of the National Academy of Sciences 115, E10808, doi: 10.1073/pnas.1813310115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mason JM et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 26, 163–176, doi: 10.1016/j.ccr.2014.05.006 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Kawakami M et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc Natl Acad Sci U S A 115, 1913–1918, doi: 10.1073/pnas.1719760115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong YL et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 348, 1155–1160, doi: 10.1126/science.aaa5111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monni O et al. Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proc Natl Acad Sci U S A 98, 5711–5716, doi: 10.1073/pnas.091582298 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhatnagar S et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 516, 116–120, doi: 10.1038/nature13955 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Branon TC et al. Efficient proximity labeling in living cells and organisms with TurboID. Nature biotechnology 36, 880, doi: 10.1038/nbt.4201 https://www.nature.com/articles/nbt.4201#supplementary-information (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roux KJ, Kim DI, Raida M & Burke B A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. The Journal of Cell Biology 196, 801, doi: 10.1083/jcb.201112098 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oughtred R et al. The BioGRID interaction database: 2019 update. Nucleic acids research 47, D529–d541, doi: 10.1093/nar/gky1079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhatnagar S et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 516, 116, doi: 10.1038/nature13955 https://www.nature.com/articles/nature13955#supplementary-information (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang W, Xia Z-J, Farré J-C & Subramani S TRIM37, a novel E3 ligase for PEX5-mediated peroxisomal matrix protein import. The Journal of Cell Biology 216, 2843, doi: 10.1083/jcb.201611170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Densham RM et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol 23, 647–655, doi: 10.1038/nsmb.3236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ben-David U et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330, doi: 10.1038/s41586-018-0409-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silkworth WT, Nardi IK, Paul R, Mogilner A & Cimini D Timing of centrosome separation is important for accurate chromosome segregation. Molecular Biology of the Cell 23, 401–411, doi: 10.1091/mbc.e11-02-0095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaseda K, McAinsh AD & Cross RA Dual pathway spindle assembly increases both the speed and the fidelity of mitosis. Biol Open 1, 12–18, doi: 10.1242/bio.2011012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y et al. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J Clin Invest 122, 4362–4374, doi: 10.1172/JCI63084 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meitinger F et al. TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature (2020). [DOI] [PMC free article] [PubMed]
- 38.Di Z et al. Ultra high content image analysis and phenotype profiling of 3D cultured micro-tissues. PLoS One 9, e109688, doi: 10.1371/journal.pone.0109688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Booij TH et al. Development of a 3D Tissue Culture-Based High-Content Screening Platform That Uses Phenotypic Profiling to Discriminate Selective Inhibitors of Receptor Tyrosine Kinases. J Biomol Screen 21, 912–922, doi: 10.1177/1087057116657269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandercock AM et al. Identification of anti-tumour biologics using primary tumour models, 3-D phenotypic screening and image-based multi-parametric profiling. Mol Cancer 14, 147, doi: 10.1186/s12943-015-0415-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Wang S & Li W RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185, doi: 10.1093/bioinformatics/bts356 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, doi: 10.1093/bioinformatics/btp616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Firat-Karalar EN & Stearns T in Methods in Cell Biology Vol. 129 (eds Renata Basto & Karen Oegema) 153–170 (Academic Press, 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mi H et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic acids research 45, D183–d189, doi: 10.1093/nar/gkw1138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mi H, Muruganujan A, Casagrande JT & Thomas PD Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 8, 1551–1566, doi: 10.1038/nprot.2013.092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.