

TO THE EDITOR:
Mutations in FBN1 are well known to be associated with Marfan syndrome but can also cause isolated ectopia lentis, autosomal dominant Weill–Marchesani syndrome, stiff-skin syndrome, and Shprintzen–Goldberg craniosynostosis syndrome [Barrett and Topol, 2013]. Interestingly, two patients with neonatal onset of progeroid features (a 20-year-old male and a 25-year-old female) and manifesting clinical features of Marfan syndrome, were reported to harbor novel de novo heterozygous mutations in FBN1 [Graul-Neumann et al., 2010; Goldblatt et al., 2011]. Furthermore, another 3-year and 6-month-old girl with progeroid features was reported to have a de novoFBN1 mutation with limited clinical features of Marfan syndrome fitting her young age such as downward slanting palpebral fissures, tall stature and arachnodactyly [Horn and Robinson, 2011]. Recently, another 10-year-old Japanese girl with a progeroid appearance from infancy on and overlapping features of Marfan syndrome was reported with a de novo FBN1 mutation [Takenouchi et al., 2013]. Previously, we reported two young girls as having neonatal progeroid syndrome [O’Neill et al., 2007], who on re-examination revealed several overlapping features of Marfan syndrome. Indeed, Graul-Neumann and co-authors suggested already that these patients might have a FBN1 mutation [Graul-Neumann et al., 2010]. Here we report on the whole exome sequencing in these two patients to evaluate the presence of FBN1 mutations and variants in other genes that might influence the phenotype.
Both patients and their relatives provided written informed consent to participate in the study. The study protocol was approved by the Institutional Review Board of UT Southwestern Medical Center. Detailed clinical data on these patients have been previously reported at the ages of 17 and 10 years, respectively [O’Neill et al., 2007]. We re-evaluated them again at the ages of 23 and 17 years, respectively and report additional clinical features.
Patient 1
This patient had partial colectomy at age 16 years. She has had iron deficiency anemia requiring blood transfusion likely due to poor nutritional intake as well as menorrhagia. She has thoracic and lumbar scoliosis and bilateral protrusio acetabuli. She has been noted to have thick calvarium prominent on both frontal regions on head magnetic resonance imaging. She gets recurrent episodes of nausea and vomiting and severe headaches. She complains of feelings of hypoglycemia which requires her to eat frequent meals throughout the day. She has had surgery on right foot due to upturning toes. She was noted to have ectopia lentis in the left eye requiring lensectomy and partial vitrectomy at age 17 years. She also has myopic macular degeneration. She had many features of Marfan syndrome which are listed in Table I. She was on oral contraceptives and was having regular menstrual periods. She was taking iron and vitamin D daily. Interestingly, her total body fat on dual emission X-ray absorptiometry was 28.4%. An echocardiogram at age 23 years demonstrated normal left ventricular ejection fraction, somewhat myxomatous appearing leaflets with no significant prolapse. Her aortic root diameter at the sinus of valsalva was 2.5 cm and at the sinotubular ridge was 2.0 cm (Z score: 1.56).
TABLE I.
Clinical Features of Previously Reported and Our Patients With Marfanoid Neonatal Progeroid Syndrome With Mutations in FBN1
| Feature | Graul-Neumann et al. [2010] | Goldblatt et al. [2011] | Horn and Robinson [2011] | Takenouchi et al. [2013] | Patient 1 | Patient 2 |
|---|---|---|---|---|---|---|
| Age (year] | 27 | 20 | 3.5 | 10 | 23 | 17 |
| Sex | F | M | F | F | F | F |
| Race | Caucasian | Caucasian | Caucasian | Asian | Hispanic | Caucasian |
| Height (cm; [centile]) | 170 | (50–75) | 108 | 149 | 157.5 | 176 |
| Weight (kg; [centile]) | 39.0 | (<3) | 14.5 | 21.7 | 26.4 | 41.9 |
| BMI (kg/m2) | 13.3 | NA | 12.4 | 9.8 | 10.6 | 13.5 |
| Body fat (%] | 20.5 | NA | NA | NA | 28.4 | 27.7 |
| Gestational age at birth (week) | 36 | 28 | 32 | 34.4 | 32 | 32 |
| Birth weight (g; [centile or SD]) | 1,780 (−2.31 SD) | 1,040 (<3) | 1,185 (−1.5 SD) | 1,427 (−2.3 SD) | 1,190 (10) | 1,172 (10) |
| Birth length (cm; [centile or SD]) | 41.5 (−2.56 SD) | NA | 40 (−0.8 SD) | 40 (−1.8 SD) | 40 (39) | 40.75 (39) |
| Birth head circumference (cm; [centile or SD]) | 32 (−2.96 SD) | NA | 29 (−0.4 SD) | 30.6 (−0.3 SD) | 29 (30) | 28.5 (30) |
| Arm span/heighta | 0.99 | Normal | 0.94 | 0.98 | ||
| Upper/lower segment | 0.95 | Normal | 0.99 | 0.80 | ||
| Myopia | Y | Y | N | Y | Y | Y |
| Proptosis | Y | Y | Y | Y | Y | Y |
| Lens subluxation | Bilateral | Bilateral | N | N | Left eye | N |
| Xerophthalmia | Y | N | Y | Y | ||
| Retrognathia | Y | Y | Y | Y | Y | Y |
| High palate | Y | Y | Y | Y | N | |
| Dental crowding | NA | Y | Y | N | ||
| Pectus excavatum | NA | Y | N | Y | ||
| Wrist sign | Y | Y | Y | Y | ||
| Thumb sign | Y | N | Y | Y | ||
| Elbow contracture | NA | Y | Y | Y | ||
| Pes planus | NA | Y | N | Y | ||
| Joint hypermobility | Mild | Y | Y | Y | Y | |
| Bruises | Y | Y | N | N | ||
| Thick frontal bone | NA | Y | Y | NA | ||
| Aortic bulb dilatation | Y | N | N | N | N | Y |
| MVPS | Y | N | Y | N | N | Y |
| Other features | Kyphosis, pigmented naevi, lumbosacral dural ectasia | Failed orchidopexy, arrested hydrocephalous | Arrested hydrocephalous, motor retardation, deep set eyes, down slanting palpebral fissures, pes valgus | Arachnodactyly, arrested hydrocephalous, dural ectasia, hydronephrosis, hypertension, scaphocephaly | Scoliosis | Scoliosis |
| Menarche age (year) | 14 | NA | NA | 12 | 16 |
MVPS, mitral valve prolapsed syndrome; Y, yes; N, no; NA, not available.
Due to elbow contractures, the arm span may be longer than measured. The presence of dural ectasia was not determined in our patients.
Patient 2
She reported mild strabismus of the left eye. She had weekly episodes of severe headaches. She also had some anxiety, depression, and attention deficit disorder and received medications for this. She had scoliosis and had pain in right hip. She also had mild hearing loss in left ear. She had menarche at age 16 years and was having regular menstrual periods. She had several features of Marfan syndrome which are listed in Table I. Her total body fat on dual emission X-ray absorptiometry was 27.7%. On ophthalmologic examination, she had extreme myopia and iridodonesis but there were no lens dislocation. A recent echocardiogram demonstrated a widened aortic root (diameter at the sinus of valsalva 3.1 cm and at the sinotubular ridge 2.4 cm; Z score: 2.29).
DNA was extracted from peripheral blood leucocytes from the patients and their relatives. Whole exome sequencing of the two patients was performed by Knome Inc. (Boston, MA) using the SureSelect Target Enrichment System for Illumina Pair-End Sequencing Library protocol. The mean depth of coverage of targeted regions for Patients 1 and 2 were 76.5 and 77.5, respectively. A total of 28,991 and 28,529 autosomal variants passing the GATK quality filters were called for Patients 1 and 2, respectively.
We filtered their variants by function, conservation and novelty, and then examined their intersection (Table II). After filtering by the predetermined criteria for potentially pathogenic variants, 105 remained in Patient 1 and 64 remained in Patient 2. There was not a single variant shared by both patients, but there were two genes, KLHL35 and FBN1, harboring novel heterozygous missense variants in both of them (Table III). Sanger sequencing of c.225C>A (p.Pro75Thr) variant in KLHL35 in Patient 1 and her parents revealed that the unaffected father also harbored the same variant and of c.1439G>T (p. Arg480Leu) variant in Patient 2 and her parents revealed her unaffected mother as a carrier of the variant, thus excluding KLHL35 variants as disease-causing.
TABLE II.
Exome Variants Filtering of Two Marfanoid Neonatal Progeroid Syndrome Patients
| Number of variants | ||
|---|---|---|
| Filter | Patient 1 | Patient 2 |
| Total | 28,991 | 28,529 |
| Functiona | 9,010 | 8,911 |
| Frequencyb | 161 | 95 |
| Conservationc | 105 | 64 |
| Gened | 2 | 3 |
Missense, nonsense, splice site, and frameshift variants.
Variants not present in the 1,000 Genome Project database, 69 Complete Genomic genomes, dbSNP 134, or the NHLBI Exome Sequencing Project database.
Variants with GERP++ conservation score >2.0.
Variants present in the same gene.
TABLE III.
Two Genes Harbored Five Rare Variants in the Two Marfanoid Neonatal Progeroid Syndrome Patients
| Gene | Genomic location (hg19) | Variant | Functional predictiona | Patient 1 | Patient 2 |
|---|---|---|---|---|---|
| KLHL35 | chr11:75134860 | c.1439G>T | Possibly damaging | G/G | G/T |
| chr11:75141452 | c.225C>A | Probably damaging | C/A | A/A | |
| FBN1 | chr15:48704765 | c.8226+1G>T | - | G/G | G/T |
| chr15:48704770 | c.8222T>C | Benign | T/T | T/C | |
| chr15:48704785 | c.8206_8027insA | - | C/CA | C/C |
By PolyPhen-2: http://genetics.bwh.harvard.edu/pph2/.
Patient 1 had heterozygousc.8206_8027insA(p.Thr2736Asnfs*1) mutation and Patient 2 had two heterozygous variants in close proximity to each other, that is, c.8222T>C (p.Ile2741Thr) and c.8226+1G>T (c.IVS64+1G>T; predicted to result in p. Glu2742Glufs*43). These variants were confirmed upon Sanger sequencing (Fig. 1A–D). Furthermore, none of their parents and none of the two siblings of Patient 1 and the five siblings of Patient 2 harbored any of these variants suggesting that these mutations were de novo. Interestingly, both the heterozygous variants in Patient 2 were on the same allele (Fig. 1E). There was no other variant in FBN1 present in either patient.
FIG. 1.
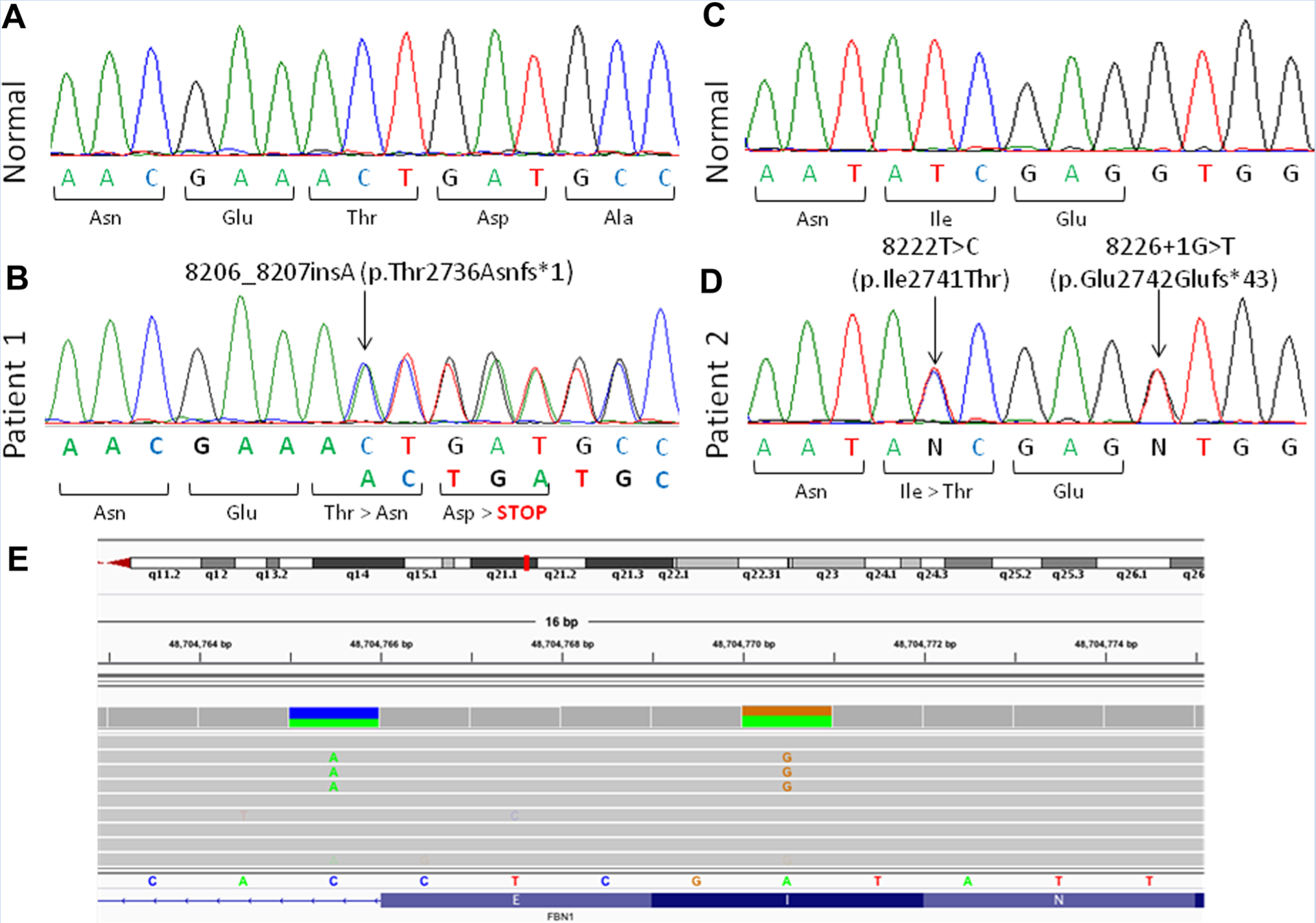
Chromatograms from our Patients with progeroid fibrillinopathy. A: Electropherogram for normal sequence of a part of exon 65 of FBN1 and B: the sequence from Patient 1 showing heterozygous c.8206_8027insA mutation. C: Normal sequence electropherogram of a part of exon 65 of FBN1 and D: the sequence from Patient 2 showing heterozygous c.8222T>C and heterozygous c.8226+1G>T mutations. The amino acid substitution is shown below the electropherogram. E: Data from whole exome sequencing of Patient 2 in the region of exon 65 of FBN1 showing that both the heterozygous variants c.8222T>C and c.8226+1G>T are on the same allele.
Our data support previous observations that de novo FBN1 null mutations in patients presenting with manifestations fitting Marfan syndrome but also progeroid features are disease-causing [Graul-Neumann et al., 2010; Goldblatt et al., 2011; Horn and Robinson, 2011; Takenouchi et al., 2013]. Our study excluded the possibility of disease-causing variants in other genes (both de novo variants and homozygous or compound heterozygous variants) and thus provides firm evidence linking FBN1 variants with this syndrome. Furthermore, the variants in FBN1 in our patients and those reported previously are in close proximity. The splice site mutation c.8226+1G>T was previously reported [Horn and Robinson, 2011] and the frameshift insertion c.8206_8027insA seemed to be deposited into dbSNP 136 (rs193922241). All the variants are null variants either due to a nucleotide insertion, deletion or splice site alteration. This suggests clustering of null variants near exon 65 of FBN1 causing a phenotype strongly resembling Marfan syndrome with additional progeroid features. We suggest calling it progeroid fibrillinopathy. The pathogenic significance of the de novo heterozygous missense variant, c.8222T>C (p.Ile2741Thr), which is on the same allele as the other heterozygous null c.8226+1G>T variant in Patient 2, is not clear. The c.8222T>C variant is not a known polymorphism; however, PolyPhen-2 in silico prediction of its function was benign. Previously, a few individuals with other syndromes harboring two nearby de novo missense mutations on the same allele have been reported [Tessitore et al., 1999; Rump et al., 2006; Margraf et al., 2012].
FBN1 contains 66 exons and encodes for fibrillin-1, a 350-KDa, 2871-amino acid glycoprotein [Jensen et al., 2012]. Although collagens, laminins, and fibronectin are the main protein components of the extracellular matrix, fibrillins act as major structural components as 10–12 nm calcium-binding extensible microfibrils. The microfibrils provide a scaffold for elastin in elastic issues. While Marfan syndrome patients have mutations in FBN1 scattered all throughout the gene [Detaint et al., 2010], clustering of mutations in and around exon 65 have been responsible for the progeroid fibrillinopathy until now (Fig.2). Takenouchi etal.[2013] proposed that the presence of an extremely charged “ETEKHKRN” protein motif in the carboxyltermini of the truncated FBN1 proteins may be responsible for this severe phenotype. However, mutated FBN1 variants in both our patients lack this “ETEKHKRN” motif and thus our data do not support this hypothesis.
FIG. 2.
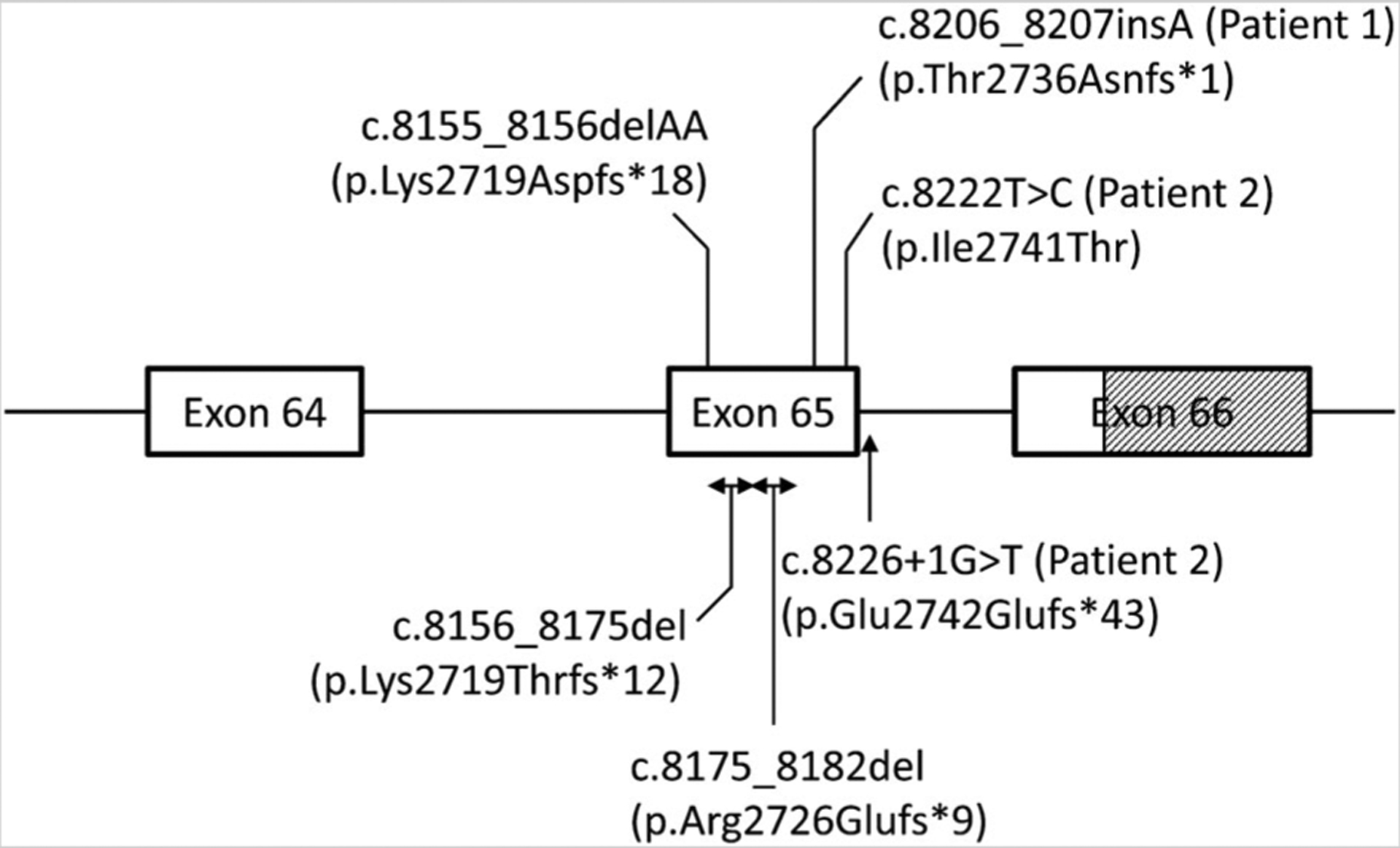
The gene structure of FBN1 at the 3′ end and clustering of the reported de novo variants in and around exon 65 in patients with progeroid fibrillinopathy. Boxes denote exons and the lines between the two boxes denote introns. Shaded area denotes untranslated region of the exon. The c.8155_8156delAA mutation was previously reported by Graul-Neumann et al. [2010], c. 8156_8175del mutation by Goldblatt et al. [2011], c.8226+1G>T mutation by Horn and Robinson [2011], and c.8175_8182del mutation by Takenouchi et al. [2013]. The total number of exons in the FBN1 transcript is 66 and the first exon is noncoding.
ACKNOWLEDGMENTS
The authors acknowledge Knome Inc. for providing exome analysis of the patient samples and interpretation of the data, Benjamin Levine, M.D. and Elizabeth Brickner, M.D. for echocardiography of Patient 2 and its interpretation, Anil Agarwal, Ph.D., for help with designing primers for Sanger sequencing; and Pei-Yun Tseng, Mary Tunison, and Shourya Kumar for technical assistance. This work was supported by the National Institutes of Health grants R01-DK54387, M01-RR00633 and UL1-RR-024982, and by the Southwestern Medical Foundation.
REFERENCES
- Barrett PM, Topol EJ. 2013. The fibrillin-1 gene: Unlocking new therapeutic pathways in cardiovascular disease. Heart 99:83–90. [DOI] [PubMed] [Google Scholar]
- Detaint D, Faivre L, Collod-Beroud G, Child AH, Loeys BL, Binquet C, Gautier E, Arbustini E, Mayer K, Arslan-Kirchner M, Stheneur C, Halliday D, Beroud C, Bonithon-Kopp C, Claustres M, Plauchu H, Robinson PN, Kiotsekoglou A, De Backer J, Ades L, Francke U, De Paepe A, Boileau C, Jondeau G. 2010. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur Heart J 31:2223–2229. [DOI] [PubMed] [Google Scholar]
- Goldblatt J, Hyatt J, Edwards C, Walpole I. 2011. Further evidence for a marfanoid syndrome with neonatal progeroid features and severe generalized lipodystrophy due to frameshift mutations near the 3′ end of the FBN1 gene. Am J Med Genet A 155A:717–720. [DOI] [PubMed] [Google Scholar]
- Graul-Neumann LM, Kienitz T, Robinson PN, Baasanjav S, Karow B, Gillessen-Kaesbach G, Fahsold R, Schmidt H, Hoffmann K, Passarge E. 2010. Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3′ terminus of the FBN1-gene. Am J Med Genet A 152A:2749–2755. [DOI] [PubMed] [Google Scholar]
- Horn D, Robinson PN. 2011. Progeroid facial features and lipodystrophy associated with a novel splice site mutation in the final intron of the FBN1 gene. Am J Med Genet A 155A:721–724. [DOI] [PubMed] [Google Scholar]
- Jensen SA, Robertson IB, Handford PA. 2012. Dissecting the fibrillin microfibril: Structural insights into organization and function. Structure 20:215–225. [DOI] [PubMed] [Google Scholar]
- Margraf RL, Krautscheid PMF, Pattison DC, Voelkerding KV, Mao R. 2012. Novel p.C620L RET mutation detected in a patient with medullary thyroid carcinoma. Int J Clin Med 3:498–501. [Google Scholar]
- O’Neill B, Simha V, Kotha V, Garg A. 2007. Body fat distribution and metabolic variables in patients with neonatal progeroid syndrome. Am J Med Genet A 143:1421–1430. [DOI] [PubMed] [Google Scholar]
- Rump P, Letteboer TG, Gille JJ, Torringa MJ, Baerts W, van Gestel JP, Verheij JB, van Essen AJ. 2006. Severe complications in a child with achondroplasia and two FGFR3 mutations on the same allele. Am J Med Genet A 140:284–290. [DOI] [PubMed] [Google Scholar]
- Takenouchi T, Hida M, Sakamoto Y, Torii C, Kosaki R, Takahashi T, Kosaki K. 2013. Severe congenital lipodystrophy and a progeroid appearance: Mutation in the penultimate exon of FBN1 causing a recognizable phenotype. Am J Med Genet A 161:3057–3062. [DOI] [PubMed] [Google Scholar]
- Tessitore A, Sinisi AA, Pasquali D, Cardone M, Vitale D, Bellastella A, Colantuoni V. 1999. A novel case of multiple endocrine neoplasia type 2A associated with two de novo mutations of the RET protooncogene. J Clin Endocrinol Metab 84:3522–3527. [DOI] [PubMed] [Google Scholar]