

Abstract
Lung cancer encompasses a heterogeneous group of malignancies. Here, we discuss how the remarkable diversity of major lung cancer subtypes is manifested in their transforming cell of origin, oncogenic dependencies, phenotypic plasticity, metastatic competence, and response to therapy. More specifically, we review the increasing evidence that potentially links this biological heterogeneity to the de-regulation of cell lineage specific pathways and the transcription factors that ultimately control them. As determinants of pulmonary epithelial differentiation, these poorly characterized transcriptional networks may underlie the etiology and biological progression of distinct lung cancers, while providing insight into innovative therapeutic strategies.
Keywords: Lung cancer, metastasis, cell of origin, transcription factor
Pathologists have long been familiar with the existence of various lung cancer histological subtypes and the challenges in accurately classifying them. Thoracic cancer can be divided into two major histotypes: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). Most lung cancer patients are diagnosed with NSCLC, which can be further subclassified into adenocarcinoma (LUAD), squamous cell carcinoma (LUSC) and large cell carcinoma (LCC). Updated histological sub-classes of LUAD have been proposed for diagnosis and treatment1. As revealed by recent genome-wide studies, individual lung tumors also possess highly diverse genomes, which further underscore the biological complexity of these diseases. Untangling the relationship between lung tumor histotype, phenotype and molecular heterogeneity represents a critical barrier to improving the clinical outcome of thoracic malignancies, which collectively account for most cancer-related deaths to date2.
Lung cancer: pleiotropic origins and drivers
Current views on the cellular origins of different lung cancers are shaped by our understanding of normal pulmonary development and homeostasis. The lung epithelium arises from the ventral side of the anterior foregut endoderm, where primary lung buds are formed3. Following extensive branching of the proximal conducting airways including the trachea, bronchi and bronchioles, cells at the distal branch tips differentiate into alveolar type I (AT1) and II (AT2) cells which constitute the gas-exchanging alveoli. In the developing and adult lungs, multiple regional epithelial cell types can serve as pools of progenitor cells. In the trachea and main bronchi, basal cells give rise to secretory and ciliated cells of the luminal layer, whereas in bronchiolar epithelium, club cells (formerly known as Clara cells) can self-renew and generate ciliated cells. In the distal airway, AT1 and AT2 cells arise directly from a bipotent progenitor during embryogenesis4. A recent single-cell transcriptome analysis identified several maturation intermediates during this process5. In the post-natal lungs, AT2 cells also acquire progenitor-like functions to generate AT1 cells4, 6. Following severe injury and inflammation, distal epithelial regeneration can also occur from putative lineage negative stem cells7, 8.
Having compiled a working catalogue of pulmonary epithelial stem/progenitor lineages, recent studies have attempted to determine which of these cells are targets for transformation in various lung cancers by using genetically engineered mouse models (GEMMs)9. Several GEMMs exploit cell type specific promoters, such as surfactant protein C (SPC) in AT2 cells, club cell 10-kDa protein (CC10) in club cells and calcitonin gene-related peptide (CGRP) in pulmonary neuroendocrine cells (PNECs), to express mutations commonly seen in lung cancer patients, in a lineage specific manner10. In sum, both the oncogenic mutation and epithelial cell type in which it is expressed can influence which lung cancer subtypes will form in mice. This topic has been extensively reviewed elsewhere11–14, and recent updates are discussed here (Figure 1).
Figure 1.
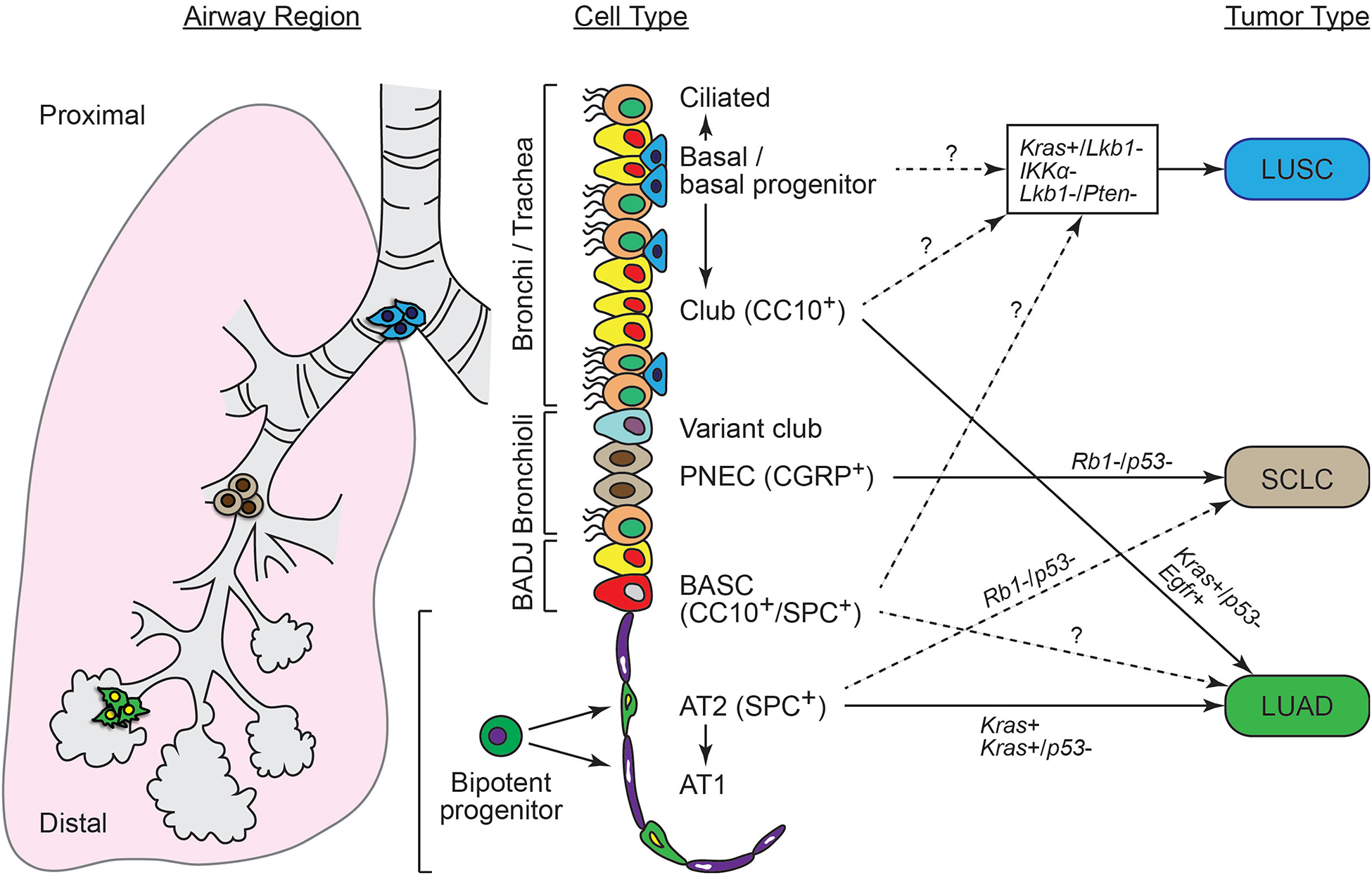
Known and predicted influence of the cell of origin vs. oncogenic mutation in the formation of different lung cancer subtypes.
Small cell lung cancer (SCLC)
Because SCLC arises in the central airways and expresses neuroendocrine markers, it has long been postulated that this lung cancer type originates from PNECs. These progenitors are rare and usually cluster as neuroendocrine bodies in the bronchioles15. Most human SCLCs harbor inactivating mutations in the prototypical tumor suppressors TP53 and the retinoblastoma 1 (RB1) genes16. By using cell-type specific conditional knockouts of these two tumor suppressors17, independent groups have confirmed that PNECs are the predominant cells of origin of SCLC18–20. Interestingly, inactivation of Tp53 and Rb1 in a subset of AT2 cells, but not in club cells, may also lead to SCLC at reduced efficiency18. The progression of Tp53/Rb1 null SCLC can be accelerated by the additional loss of Pten, which has recently been proposed as a frequent, but late genetic event during SCLC tumorigenesis20, 21.
Lung squamous cell carcinoma (LUSC)
LUSC expresses basal cell markers (including KRT5, p63 and SOX2) and frequently occurs in the proximal airway. Therefore, it has been proposed that LUSC arises from basal progenitors. Until recently, the lack of LUSC GEMM has precluded the identification of its cellular origin, and several attempts have been made to model this malignancy. Although rarely mutated in human LUSC, inactivation of the tumor suppressor Stk11 (also known as Lkb1) in Kras mutant mice can generate LUSC, but also creates a wide spectrum of NSCLCs including mixed adenosquamous carcinoma and LCC22. In addition, IKKα kinase inactivation in mice generates spontaneous LUSC exclusively, albeit at a relatively low efficiency23. The comprehensive catalog of genomic alterations in LUSC identified recurrent PTEN mutations24. Concomitant inactivation of Lkb1 and Pten leads to LUSC in GEMM with 100% penetrance25. Notably, the resulting tumors resemble many histological and transcriptional features of the human LUSC basal subtype. Given this result, it is expected that the cellular origin(s) of LUSC will soon be identified using conditional knockouts of Lkb1 and Pten in GEMM.
Lung adenocarcinoma (LUAD)
Among different lung cancer subtypes, the cellular origins of LUAD are relatively well studied owing to the availability of multiple GEMMs. Several of these rely on the spatial and temporal induction of an oncogenic Kras mutant allele. In humans, KRAS is most frequently mutated in tissues of endodermal origin, including the lung epithelium26. Independent groups have shown that Kras-driven murine LUAD preferentially arises from AT2 cells, consistent with its predominant site of diagnosis at the distal airway and the immunohistochemical staining of alveolar markers in early stage patient tumors27–30. In contrast, Kras activation driven by the CC10 promoter, presumably in club cells, bronchioalveolar stem cells (BASCs) and a small percentage of AT2 cells, leads to hyperplasia at the bronchoalveolar duct junction (BADJ), but not frank LUAD28. In principle, tumor progression in the terminal bronchioles can also occur if the Tp53 tumor suppressor gene is concomitantly deleted in CC10+ cells30. Inflammatory responses may also enhance the overall transforming effects of associated genotypes/cell phenotypes27. It is also known that mutant Egfr expression in CC10+ cells can give rise to LUAD that rarely metastasizes31. Moreover, the cell surface antigenic profile of tumor-initiating cells varies when comparing LUADs driven by Egfr and Kras mutations32. Altogether, the experimental evidence to date implicates a combination of unique genetic and environmental contexts within the distal airway that is required for LUAD initiation. The function of other driver mutations in LUAD might be equally variable across regional epithelial cell lineages, contributing even further to the heterogeneous nature of this NSCLC subtype.
Switching paths to progress
Following tumor initiation from specific cell types, lung cancers can seemingly adopt various aberrant differentiation states. In solid epithelial tumors, one of the most extensively studied manifestations of this phenomenon is epithelial to mesenchymal transition (EMT). EMT is a developmental process that is not only linked to tumor cell invasion, but also to oncogenesis and tumor initiation33. In human lung cancers, clinical evidence for EMT is seen in a fraction of LUADs that have acquired resistance to tyrosine kinase inhibitors (TKIs)34–36. Independent of EMT, lung cancer histopathological variations may correlate with cell lineage states that are unique or selective for the airways. For instance, at the genomic level, resected human NSCLCs can be distinguished by gene expression profiles associated with different stages of pulmonary development37. Despite arising from the alveolar epithelium, LUADs can convert to SCLCs, the latter being a neuroendocrine cancer more typically located in the central lungs38. LUAD conversion to an “SCLC-like” phenotype is associated with drug resistance35, 39–41. Furthermore, a significant proportion of high-grade LUADs histologically resembles LUSCs, which express markers of proximal airway basal cells. Mixed adenosquamous tumors retain epithelial markers yet are more invasive and have a worse prognosis42–44. Within the LUAD subtype specifically, a gene module of normal alveolar differentiation stratifies tumors with distinct grades, biological properties and clinical outcome45. The transcriptomic portrait of lung epithelial specification is ultimately reflected in the nomenclature adopted by The Cancer Genome Atlas (TCGA) to sub-classify human LUADs and LUSCs24, 46. This remarkable molecular heterogeneity seen between and within human lung cancer subtypes may reflect the relative abundance of various progenitor cell lineages in a tumor mass. Instead of being fixed over time, tumor histopathology can also deviate due to the capacity of a tumor-initiating cell population to dedifferentiate or transdifferentiate relative to its original lineage. Significantly, molecular aberrations or extracellular signals can directly drive this phenotypic plasticity.
In GEMMs, lineage-tracing experiments have demonstrated that Kras transformed AT2 cells self-renew without additional changes in cell fate, at least up until the formation of adenomas4. By contrast, Kras-mutant CC10+ hyperplasias can switch to a CC10−/SPC+ like phenotype to develop into LUAD in the terminal bronchioles27. This may be induced by local inflammation or oncogenic RAS-mediated AT2 differentiation, a lineage state that is more conducive to LUAD progression. Phenotypic conversion of AT2-derived LUADs is seen when mutating the tumor suppressor Stk11 in conjunction with Kras, which generates tumors that transdifferentiate into LUSCs22, 47. LUSC metaplasia from LUADs is regulated by conserved developmental signals including the Hippo/Yap48 and transforming growth factor/SMAD pathways49. Another developmental program required for respiration is the canonical WNT/β-catenin pathway50–52. Mutations in components of this pathway such as APC, GSK3B and CTNNB1 are detected in 15% of human LUADs53. Multiple studies report even more frequent overexpression of various WNT pathway components and epigenetic silencing of WNT pathway antagonists in human LUADs54. In GEMM, lung specific expression of a constitutively active mutant β-catenin is not sufficient to initiate LUAD, but may increase the pool of progenitor cells available for transformation55, 56. Deletion of the WNT antagonist APC cooperates with Kras to induce dedifferentiated invasive LUADs57. Moreover, transcriptional activation of the WNT/TCF effector LEF1 is a marker and mediator of LUAD metastasis to the brain58, 59. The mechanism by which WNT/TCF promotes LUAD may involve both the expansion of the cell of origin and epigenetic reprograming of transformed cells into a primitive state that has increased metastatic potential.
Lineage transcription factors in context
Underlying the diverse pathways, molecular perturbations and differentiation states in lung cancers are transcription factors (TFs) and co-factors whose expression and activity can be restricted for certain cell lineages3, 60. As will be discussed below, the genes encoding several such lineage TFs can be directly mutated, activated or silenced in particular lung cancer subtypes, suggesting that they are context dependent drivers of tumor progression and phenotypic heterogeneity.
Neuroendocrine factors in SCLC
Inactivating mutations in RB1 are ubiquitous to all SCLCs16. The extended pRb family of pocket proteins interacts with a myriad of DNA binding transcription factors and co-factors required for cell cycle progression, self-renewal, apoptosis, senescence and cellular differentiation61. Although components of the pRB pathway are frequently inactivated across many cancers, the strong selection for mutations in RB1 exclusively within SCLC suggests that pRb itself is a lineage-specific tumor suppressor. Remarkably, in the murine lungs, Rb is required for neuroendocrine cell fate, while the specification of other airway cell types can occur in the absence of Rb due to compensation from other pocket proteins62. This selective requirement for RB1 during mammalian lung development is consistent with the neuroendocrine origins of SCLC and the potent SCLC phenotype generated via Rb and p53 mutations in GEMMs.
Basal factors in LUSC
In LUSCs, Sry-related HMG box 2 (SOX2) is one of the most frequently amplified and overexpressed genes24, 63–65. SOX2 encodes for a TF best known for its ability to reprogram embryonic stem cells. During normal development however, SOX2 is essential for basal cell commitment in the proximal airways66. As an oncogene, SOX2 is required for the survival of LUSC cells, but its aberrant expression alone is not sufficient for transformation65. A combination of genetic alterations, such as the loss of Lkb167, might be required for Sox2-driven LUSC in mice. SOX2 is usually co-amplified with the basal lineage TF p63 at chromosome 3q2663. Their interaction and co-localization at target gene loci support oncogenesis instead of pluripotency68. Moreover, within the same amplicon as SOX2 is PRKCI, which encodes for protein kinase Cl that phosphorylates SOX2, leading to the activation of Sonic hedgehog (Shh) signaling and LUSC self-renewal69. In contrast to its role in promoting LUSCs, high levels of Sox2 in CC10+ cells restrict the formation of LUAD in the bronchioles by suppressing Notch activation70.
Alveolar factors in LUAD
In distal epithelial cells, a different SOX family member, SOX9, marks the branch tips of early lungs and functions downstream of receptor tyrosine kinase signaling to suppress premature alveolar differentiation71, 72. SOX9 is overexpressed in human LUAD, and its expression correlates with poor patient survival73, 74. Induction of Sox9 expression has been observed in murine LUADs harboring activating mutations in Kras and β-catenin and this over-expression is associated with high grade tumors56, 57. Moreover, SOX9 is preferentially upregulated in human KRAS mutant adenomas and is also induced by Notch activation75.
NK2 homeobox 1 (NKX2–1), also known as thyroid transcription factor 1 (TTF-1), is expressed in AT2 cells and a subset of bronchiolar cells76. NKX2–1 is essential for lung morphogenesis and alveolar cell differentiation77. TTF-1 protein is a biomarker of thymic cancers and LUADs. About 15% of LUADs harbor NKX2–1 amplification78–80, which correlates with poor outcome81, 82 and is required for tumor cell viability80, 83. NKX2–1 can support pro-tumorigenic signaling downstream of mutant EGFR84 and is required for EGFR mediated transformation in vivo85. Paradoxically, NKX2–1 expression also correlates with better outcome since its expression is frequently suppressed in high-grade LUADs with wild-type/non-amplified NKX2–181, 82, 86, 87. Importantly, in murine LUAD models, wild-type Nkx2–1 inhibits LUAD progression driven by Kras85, 88. This tumor-suppressive effect is closely linked to epithelial differentiation states, since Nkx2–1 haploinsufficiency promotes mucinous LUAD85 and activation of enteric lineage markers that are normally repressed in alveolar cells88. Repression of Nkx2–1 is also required for stochastic dissemination and metastasis by tumors with Kras and p53 mutations86.
The contradictory role of certain lineage TFs likely depends on the epigenetic context in which they function. One determinant of such contexts is the relative abundance and accessibility of different co-factors in a given progenitor cell. The levels and dependencies of co-factors can be initially dictated by cell lineage states (e.g. tumor cell of origin or stage) and further dysregulated by somatic alterations (e.g. mutation, gene amplification, DNA methylation). NKX2–1 for instance can interact with multiple DNA binding transcriptional repressors or activators to expand or limit the range of target genes89. Wild-type levels of NKX2–1 maintain alveolar differentiation and inhibit proliferation by restricting the activity and genomic target loci of FOXA1/288 and AP1 factors85 respectively. In this regulatory network, decreases in NKX2–1 causes de-repression of genes that mediate cell proliferation90, EMT91, motility92, and resistance to anoikis86. Conversely, NKX2–1 amplification is often accompanied by overexpression of the lineage TF FOXA193. In this latter context which includes aberrantly high levels of NKX2–1 and FOXA1, both TFs can cooperatively access and transactivate de novo genes required for cancer cell survival93. Thus, genes that would otherwise not be targeted by physiological levels of a given lineage TF can be co-opted for malignant progression under aberrant epigenetic settings (Figure 2).
Figure 2.
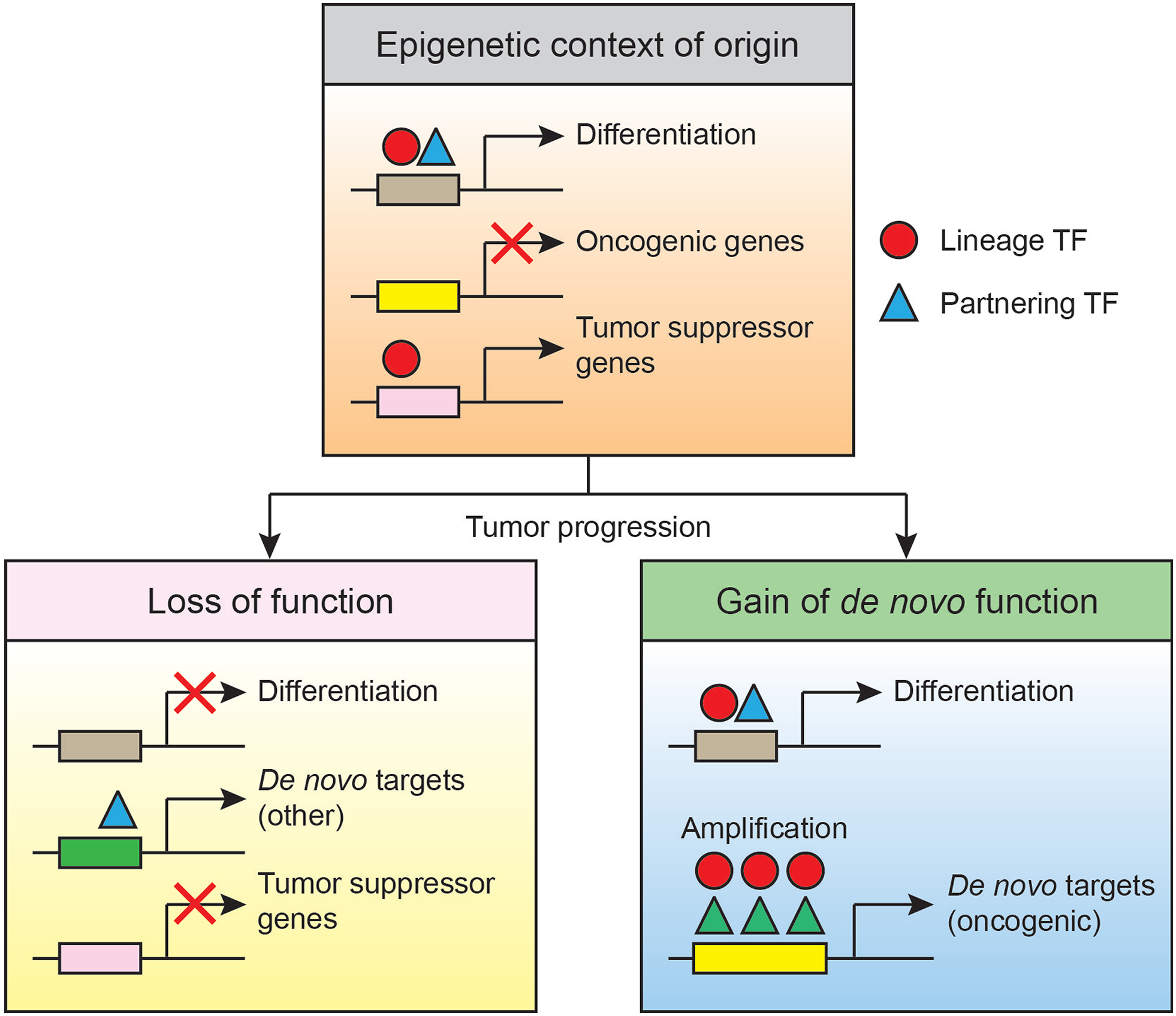
Epigenetic and cellular context dictates the effect of lineage TFs in lung cancer. Given a specific cellular origin, lung cancer progression can be driven by repression of a lineage TF and its tumor suppressive gene targets. Conversely, amplification of a lineage TF may activate oncogenic genes following de novo TF complex formation at novel target promoters. The tumor suppressive and oncogenic activities may be adopted by the same TF in different epigenetic contexts. These contexts are defined by the overall abundance and availability of TFs and their partnering co-factors. TF thresholds may be dependent on cooperating mutations and different cellular origins (not depicted).
An expanding network of alveologenic tumor suppressors
In addition to NKX2–1, several other alveolar specifying TFs can limit LUAD progression. Many of these factors genetically and physically cooperate with one another to regulate the expression of prototypical alveolar differentiation markers, including surfactant proteins. This suggests that the transcriptional network that controls alveologenesis is broadly linked to tumor suppression in the LUAD subtype.
The GATA family proteins are DNA binding TFs that can prime target promoters for repression or activation in specific tissues94. GATA6 is abundantly expressed in the distal lungs where it is required for lung branching morphogenesis and alveolar specification and it restricts the expansion of adult progenitor cells at the BADJ95–97. Gata6 directly binds to Nkx2–1 and induces the expression of surfactant proteins in AT2 cells98–100. Similar to NKX2–1, GATA6 has both oncogenic101–105 and tumor suppressive effects106, 107 in a variety of cancers. The oncogenic function of GATA6 may depend on its amplification or other cooperating gene amplifications103 and availability of interacting TF partners, such as SP1105, KLF5 and GATA4104. Elevated levels of various GATAs correlate with EMT in a murine LUAD model108. On the other hand, expression of GATAs is generally silenced in a significant proportion of human LUADs109,46. In a subset of high grade LUADs, GATA6 is co-repressed with other alveolar TFs with whom it may cooperate to inhibit LUAD metastasis45. Hypermethylation and mutation of GATA6 itself are uncommon in LUADs109, but, as in other cancers, alternative mechanisms may contribute to its down-regulation110.
The homeodomain only protein X (HOPX), reported as a marker of AT1 cells5, 6, is an atypical homedomain protein that is essential for lung maturation. Although Hopx lacks a DNA binding domain, it acts downstream of Nkx2–1 and Gata6 to modulate alveolar specific gene expression in a histone-deacetylase-dependent manner111. HOPX exhibits tumor inhibitory effects in a wide range of cancers112–116. In LUADs, HOPX expression correlates tightly with NKX2–193, and it can restrain tumor cell invasion and metastasis45. Moreover, ectopic HOPX expression can induce senescence and is preferentially silenced via DNA methylation in the LUAD subtype, where decreased HOPX levels correlate with poor clinical outcome117. Reducing both HOPX and GATA6 in LUAD cells not only reverses alveolar identity but also activates the expression of squamous basal cell markers and pro-invasive genes45. Thus, HOPX and its cooperating TFs may function similarly to NKX2–1, by controlling latent differentiation programs that limit overall metastatic competence118.
CCAAT enhancer binding protein α (C/EBPα) is a family member of basic leucine zipper TFs. C/EBPα is required for perinatal AT2 differentiation and surfactant gene expression119, 120. In the adult lungs, loss of C/EBPα impairs epithelial regeneration121. Although rarely mutated in lung cancer122, C/EBPα is often suppressed by DNA hypermethylation123, and its re-expression was shown to inhibit the proliferation and survival of human NSCLC cells124. In mice, Cebpa is dispensable for spontaneous LUAD initiation, however its loss accelerates stress-induced LUAD formation, indicative of a tumor suppressive role in response to environmental stimuli125. Further underlying the complexity of respiratory TF networks, C/EBPα binds to NKX2–1126 and Cebpa expression is also regulated downstream of Nkx2–1 during development119.
FOXA1 and FOXA2, previously termed hepatocyte nuclear factor 3α/β (HNF3 α/β), belong to the subfamily of forkhead box TFs. FOXA1/2 have similar temporal and spatial expression patterns in developing lungs. With partially overlapping functions, they play critical roles in lung morphogenesis and alveolar differentiation127, 128. Both FOXA1/2 are binding partners of NKX2–1 in LUADs88, 129, and they can activate latent epithelial differentiation genes when NKX2–1 is suppressed88. FOXA2 also cooperates with GATA6 to regulate the expression of cell adhesion genes in the developing lung epithelium130. In both normal epithelial cells and lung cancer cells, FOXA2 induces the expression of C/EBPα119, and vice versa131. Similar to other alveolar TFs, FOXA2 silencing is observed in LUADs through promoter hypermethylation132. Finally, overexpression of FOXA2 in human NSCLC cells inhibits cell growth, invasion and EMT131, 133.
The runt-related transcription factors (RUNX) are required for the development of several mammalian tissues and are dysregulated in epithelial cancers134. In the murine lungs, Runx3 is expressed by distal epithelial cells including AT2 cells, and its targeted loss causes alveolar hyperplasia135, 136. RUNX3 is yet another alveolar gene noted to be methylated at a significantly high frequency in lung malignancies137. RUNX3 methylation status is an independent prognostic factor in NSCLC patients138 and is associated with chemoresistance139. Other modes of RUNX3 loss of function, such as EZH2-mediated H3K27 trimethylation140 and MDM2-mediated ubiquitination141, have been reported in other cancers, but have yet to be explored in LUAD. In GEMMs, heterozygous loss of Runx3 causes pre-neoplastic lung adenoma135, 136. Notably, Kras activation only results in non-mucinous LUADs, whereas Runx3 loss cooperates with mutant Kras to initiate both mucinous and non-mucinous LUADs142. This further suggests that deregulation of cell fate-determining TFs can drive transition to different lung tumor subtypes. The molecular connection between RUNX3 and other prototypical alveolar TFs is not yet known, but its ability to abrogate the ARF-p53 pathway by interacting with the bromodomain protein BRD2 implicates an epigenetic modulatory role142.
MYC revisited
The MYC family proto-oncogenes, which encode C-myc (Myc), N-myc and L-myc, regulate numerous biological processes. Although not restricted to pulmonary lineages, the MYC proteins are nonetheless potent transcriptional regulators of lung epithelial differentiation. N-myc is expressed at high levels in the distal airway epithelium of mice143 and is controlled by Wnt/β-catenin signaling52. Loss of N-myc and C-myc can delay lung branching morphogenesis144, 145. C-myc is amplified in human LUAD46, 146. All three MYC members are frequently amplified in SCLC16, 147, but not in LUSC24. Moreover, recent genomic studies have identified loss of function mutations in MGA and MAX, two genes that encode for MYC inhibitory proteins46, 148. In human lung cancers, alterations in MGA, MAX and MYC are mutually exclusive46, 148, indicating a more pervasive activation of the MYC pathway via mutations than was initially appreciated.
In GEMMs, expression of Myc in SPC+ or CC10+ cells induces LUAD149, 150. In addition, Myc-induced LUAD requires Kras activating mutation150, which can be substituted by Notch1 overexpression151. Myc is also required for tumor maintenance in a variety of Ras pathway initiated NSCLC models152, 153. MYC can activate or repress specific genes by recruiting different cofactors to target promoters154. Myc has also been suggested to broadly amplify gene transcription in cancer cells155. In resected human LUADs, MYC overexpression is linked to increased DNA methylation of known tumor suppressor genes46. Activation of other specific C-myc targets in human LUADs correlates with perturbations in lineage TF networks and poor clinical outcome45, 156, 157. Consistent with these in silico analysis of human tumors, Myc overexpression from SPC+ AT2 progenitors in mice is sufficient to induce macrometastases that have undergone a mixed lineage switch158. Finally, human mutations in the MYC pathway are mutually exclusive to SMARCA4 (or BRG1)148, 159, which encodes a major component of the SWI/SNF chromatin-remodeling complex160 and is a mediator of SCLC neuroendocrine differentiation148. Therefore, despite its multiple functions, the major consequence of MYC activation in lung cancers may be to epigenetically reprogram specific lineage states.
Lineage plasticity and metastasis: cause or consequence?
Epithelial lineage plasticity has been associated with cancer metastasis. As core mediators of cell fate, lineage TFs may therefore regulate genes required for lung cancer cell dissemination and distant organ colonization. However, considering the diverse functional targets of these TFs, is their capacity to regulate metastasis directly connected to the lineage state in a given cell? In lung cancer, this remains an open question, but some insight may be drawn from recent studies.
The over-expression of EMT inducing TFs in epithelial cancer cells is often interpreted as a marker of increased cell invasiveness and metastasis161. Overexpression of EMT inducing TFs has been observed in various models of metastatic LUAD162. However, this is confounded by the fact that EMT promoting TFs also target genes that are independently required for other cellular functions such as cell survival, proliferation, DNA damage responses and self-renewal163, 164. In some carcinoma models, EMT is neither sufficient nor required for cell invasion, while disseminating cancer cells that undergo the reverse process, mesenchymal to epithelial transition (MET), have a greater advantage for tumor re-initiation and metastatic colonization. Intriguingly, some studies suggest that mesenchymal carcinoma cells may not invade beyond the local tumor, but rather enhance the dissemination of other epithelial cell populations with tumor re-initiating capacity165, 166. An analogous phenomenon has been described in GEMMs of SCLCs, where a fraction of malignant cells can transition from their neuroendocrine (NE) origins into non-NE cells that express mesenchymal genes167. Although these non-NE cells are clonally related to the rest of the SCLC, they do not metastasize. On the other hand, their presence in the tumor mass enhances the ability of the NE lineage positive cells to spread. Thus, modulation of lineage states by TFs may indirectly influence metastatic potential by controlling intra-tumoral clonal dynamics, presumably via paracrine signals (Figure 3).
Figure 3.
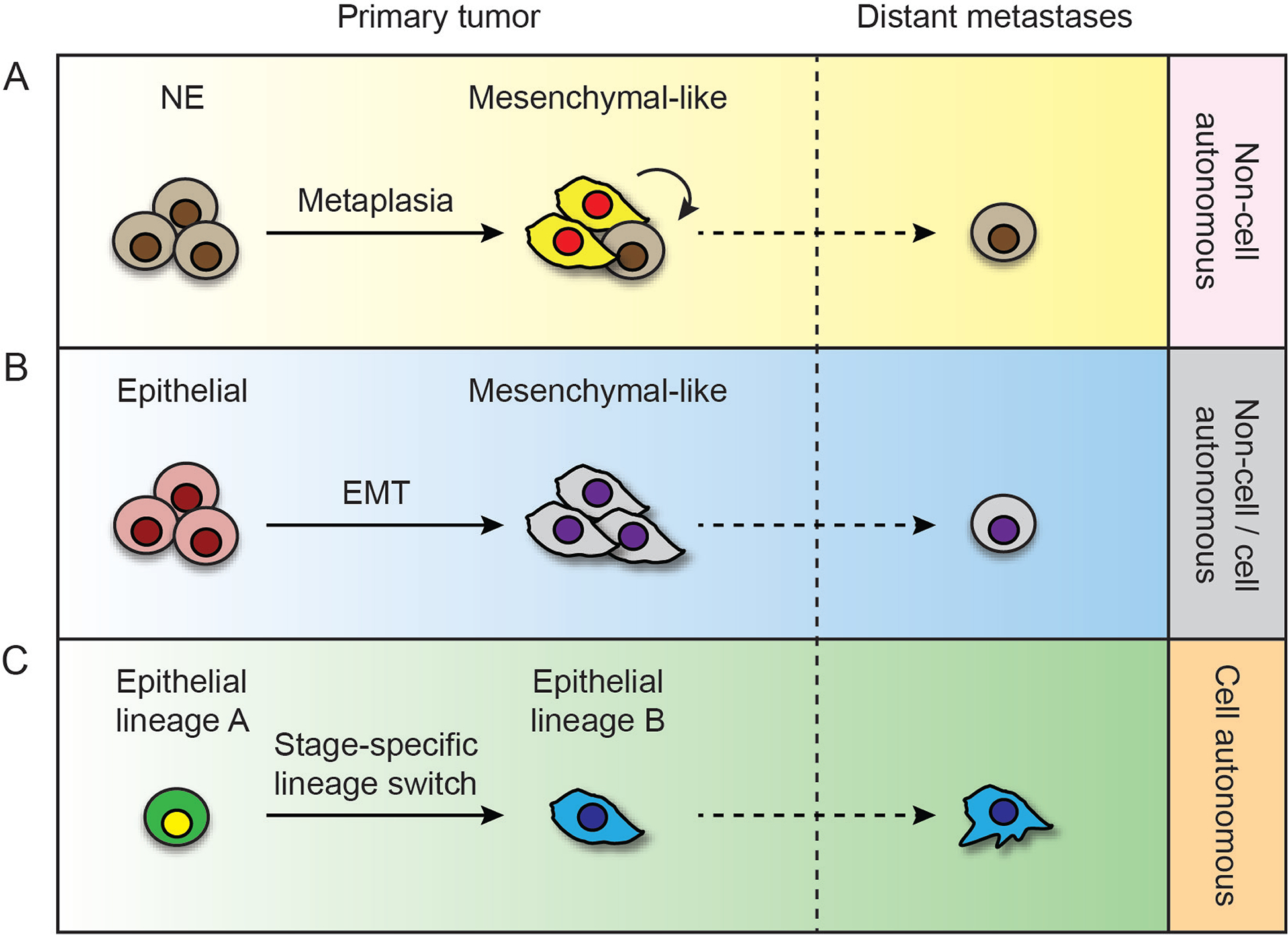
Hypothetical mechanisms by which cell lineage plasticity and heterogeneity promote lung cancer metastasis. (A) Epithelial lineage positive malignant cells, such as NE cells in SCLC, constitute the majority of distant metastasis, but their dissemination is enhanced via non-cell autonomous signals from mesenchymal transitioned tumor cells in the primary tumor. (B) LUAD cells may disseminate via a classical EMT/MET model. (C) Alternatively, LUAD cells may undergo multiple switches in the expression of airway epithelial lineage specific markers that promote metastasis via cell autonomous mechanisms.
An alternative mechanistic consequence of tumor metaplasia is that it may lead to the activation of lineage gene programs that directly control the metastatic capacity of a given cell. The activation of latent basal epithelial markers, such as cytokeratins (K) has been reported in more invasive tumor cells. For example, cells at the invasive border of luminal breast tumors express the basal epithelial marker K14 and basal TF p63. These invasive basal cells arise from the well-differentiated luminal cells and are responsible for collective tumor cell invasion in a manner that is independent of EMT168. In liver cancer, the biliary/progenitor marker K19 is expressed in a subset of hepatocellular carcinomas with a high incidence of microvascular invasion and metastasis169. Similarly, LUAD cells may acquire invasion potential by down-regulating alveolar differentiation genes and upregulating the squamous basal cell markers K6A/B45. Importantly, knockdown of these cytokeratins in their respective disease models inhibits tumor cell invasiveness, suggesting an unexpected direct role for basal/biliary epithelial markers in malignant invasion and metastasis.
Harnessing lineage states for therapy
The TFs discussed here are but a few examples linking the molecular control of cell lineage fate to lung cancer progression. Despite the broad biological significance of these observations, their clinical and therapeutic impacts remains underexplored. Part of the reason for this is the intrinsic technical barriers in pharmacologically manipulating pathways or TFs that are not only tissue specific, but also likely to have dosage and stage specific effects during tumor progression. Nevertheless, exploiting aberrant tumor differentiation states or lineage TFs for therapy has been shown to be clinically beneficial in leukemia and neuroblastoma170 and pre-clinically feasible in melanoma171, 172, glioma173, 174, nasopharyngeal carcinoma175, and rhabdomyosarcoma176. Driving the tumors toward more differentiated states may not only delay their growth but also potentially increase their sensitivity to commonly used cytotoxic agents.
Several novel compounds have recently been explored to target the expression or activity of TFs. For example, drugs that inhibit the chromatin-remodeling BET bromodomain proteins are being actively investigated as a strategy to target the lineage-dependent activity of TFs in multiple cancers, including LUAD177, 178. Another potential approach is to use covalent inhibitors of cyclin-dependent kinase 7, which preferentially mediates the expression of super-enhancer associated genes179, 180. Since such genes include oncogenic MYC and neuroendocrine TFs, this class of transcription targeting drugs may be particularly effective against SCLC181.
Assessing the lineage state of the bulk tumor may have more immediate clinical benefits for anticipating dynamic therapeutic responses. This is relevant in the treatment of KRAS mutant LUADs, which are highly heterogeneous. Notably, a subset of KRAS mutant LUADs that are well-differentiated and retain epithelial markers respond specifically to a combination of drugs that inhibit MAP kinase and BCL-Xl182. In contrast, LUADs that have undergone lineage transition become drug refractory, even when those treatments target their driver oncogenes. At the same time, such lineage transitions may have foreseeable shifts in signaling dependencies. For example, the survival of TKI resistant EGFR mutant tumor cells that are more mesenchymal-like requires activation of the receptor tyrosine kinase AXL34, 183, which can be inhibited with small molecules.
Given the fact that lung cancers can adopt various differentiation states, new biological insight may be revealed by comprehensively defining their underlying transcriptional circuits. These circuits may in turn modify the oncogenic dependencies of a given tumor, providing novel therapeutic opportunities. Major challenges remain in directly inducing or reverting these lineage specific pathways at different stages of disease progression. Nevertheless, several lung cancer differentiation states, defined either transcriptionally or histologically, may already be used as both prognostic and predictive biomarkers. Moving forward, a systematic search for efficacious agents tailored not only to the oncogenic drivers of various lung cancer subtypes, but also to the cell lineage of their malignant compartment, holds the promise of abating metastatic progression.
Acknowledgments
We thank all members of the Nguyen laboratory for discussions and critical review of the manuscript. We apologize for omitting some examples and primary references due to space constraints. Our research is funded by grants from Uniting Against Lung Cancer (to W.K.C.C) and the National Cancer Institute (1R01CA166376, 1R21CA170537, 1R01CA191489; to D.X.N.).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Travis WD, Brambilla E, Riely GJ. New pathologic classification of lung cancer: relevance for clinical practice and clinical trials. J Clin Oncol 2013; 31: 992–1001. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64: 9–29. [DOI] [PubMed] [Google Scholar]
- 3.Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell 2010; 18: 8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014; 507: 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014; 509: 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 2013; 123: 3025–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zuo W, Zhang T, Wu DZ, Guan SP, Liew AA, Yamamoto Y et al. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature 2015; 517: 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015; 517: 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meuwissen R, Berns A. Mouse models for human lung cancer. Genes Dev 2005; 19: 643–664. [DOI] [PubMed] [Google Scholar]
- 10.Rawlins EL, Perl AK. The a“MAZE”ing world of lung-specific transgenic mice. Am J Respir Cell Mol Biol 2012; 46: 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanpain C Tracing the cellular origin of cancer. Nat Cell Biol 2013; 15: 126–134. [DOI] [PubMed] [Google Scholar]
- 12.Hanna JM, Onaitis MW. Cell of origin of lung cancer. J Carcinog 2013; 12: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutherland KD, Berns A. Cell of origin of lung cancer. Mol Oncol 2010; 4: 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer 2014; 14: 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet 2011; 378: 1741–1755. [DOI] [PubMed] [Google Scholar]
- 16.Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012; 44: 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003; 4: 181–189. [DOI] [PubMed] [Google Scholar]
- 18.Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011; 19: 754–764. [DOI] [PubMed] [Google Scholar]
- 19.Park KS, Liang MC, Raiser DM, Zamponi R, Roach RR, Curtis SJ et al. Characterization of the cell of origin for small cell lung cancer. Cell Cycle 2011; 10: 2806–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song H, Yao E, Lin C, Gacayan R, Chen MH, Chuang PT. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc Natl Acad Sci U S A 2012; 109: 17531–17536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014; 156: 1298–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007; 448: 807–810. [DOI] [PubMed] [Google Scholar]
- 23.Xiao Z, Jiang Q, Willette-Brown J, Xi S, Zhu F, Burkett S et al. The pivotal role of IKKalpha in the development of spontaneous lung squamous cell carcinomas. Cancer Cell 2013; 23: 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489: 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 2014; 25: 590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinlan MP, Quatela SE, Philips MR, Settleman J. Activated Kras, but not Hras or Nras, may initiate tumors of endodermal origin via stem cell expansion. Mol Cell Biol 2008; 28: 2659–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mainardi S, Mijimolle N, Francoz S, Vicente-Dueñas C, Sánchez-García I, Barbacid M. Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc Natl Acad Sci U S A 2014; 111: 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu X, Rock JR, Lu Y, Futtner C, Schwab B, Guinney J et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc Natl Acad Sci U S A 2012; 109: 4910–4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin C, Song H, Huang C, Yao E, Gacayan R, Xu SM et al. Alveolar type II cells possess the capability of initiating lung tumor development. PLoS One 2012; 7: e53817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutherland KD, Song JY, Kwon MC, Proost N, Zevenhoven J, Berns A. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc Natl Acad Sci U S A 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev 2006; 20: 1496–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curtis SJ, Sinkevicius KW, Li D, Lau AN, Roach RR, Zamponi R et al. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell 2010; 7: 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 2013; 27: 2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012; 44: 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011; 3: 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uramoto H, Shimokawa H, Hanagiri T, Kuwano M, Ono M. Expression of selected gene for acquired drug resistance to EGFR-TKI in lung adenocarcinoma. Lung Cancer 2011; 73: 361–365. [DOI] [PubMed] [Google Scholar]
- 37.Borczuk AC, Gorenstein L, Walter KL, Assaad AA, Wang L, Powell CA. Non-small-cell lung cancer molecular signatures recapitulate lung developmental pathways. Am J Pathol 2003; 163: 1949–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watson WL, Berg JW. Oat cell lung cancer. Cancer 1962; 15: 759–768. [DOI] [PubMed] [Google Scholar]
- 39.Alam N, Gustafson KS, Ladanyi M, Zakowski MF, Kapoor A, Truskinovsky AM et al. Small-cell carcinoma with an epidermal growth factor receptor mutation in a never-smoker with gefitinib-responsive adenocarcinoma of the lung. Clin Lung Cancer 2010; 11: E1–4. [DOI] [PubMed] [Google Scholar]
- 40.Popat S, Wotherspoon A, Nutting CM, Gonzalez D, Nicholson AG, O’Brien M. Transformation to “high grade” neuroendocrine carcinoma as an acquired drug resistance mechanism in EGFR-mutant lung adenocarcinoma. Lung Cancer 2013; 80: 1–4. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe S, Sone T, Matsui T, Yamamura K, Tani M, Okazaki A et al. Transformation to small-cell lung cancer following treatment with EGFR tyrosine kinase inhibitors in a patient with lung adenocarcinoma. Lung Cancer 2013; 82: 370–372. [DOI] [PubMed] [Google Scholar]
- 42.Filosso PL, Ruffini E, Asioli S, Giobbe R, Macri L, Bruna MC et al. Adenosquamous lung carcinomas: a histologic subtype with poor prognosis. Lung Cancer 2011; 74: 25–29. [DOI] [PubMed] [Google Scholar]
- 43.Cooke DT, Nguyen DV, Yang Y, Chen SL, Yu C, Calhoun RF. Survival comparison of adenosquamous, squamous cell, and adenocarcinoma of the lung after lobectomy. Ann Thorac Surg 2010; 90: 943–948. [DOI] [PubMed] [Google Scholar]
- 44.Nakagawa K, Yasumitu T, Fukuhara K, Shiono H, Kikui M. Poor prognosis after lung resection for patients with adenosquamous carcinoma of the lung. Ann Thorac Surg 2003; 75: 1740–1744. [DOI] [PubMed] [Google Scholar]
- 45.Cheung WK, Zhao M, Liu Z, Stevens LE, Cao PD, Fang JE et al. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell 2013; 23: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han X, Li F, Fang Z, Gao Y, Li F, Fang R et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun 2014; 5: 3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao Y, Zhang W, Han X, Li F, Wang X, Wang R et al. YAP inhibits squamous transdifferentiation of Lkb1-deficient lung adenocarcinoma through ZEB2-dependent DNp63 repression. Nat Commun 2014; 5: 4629. [DOI] [PubMed] [Google Scholar]
- 49.Ischenko I, Liu J, Petrenko O, Hayman MJ. Transforming growth factor-beta signaling network regulates plasticity and lineage commitment of lung cancer cells. Cell Death Differ 2014; 21: 1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goss AM, Tian Y, Tsukiyama T, Cohen ED, Zhou D, Lu MM et al. Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev Cell 2009; 17: 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mucenski ML, Wert SE, Nation JM, Loudy DE, Huelsken J, Birchmeier W et al. beta-Catenin is required for specification of proximal/distal cell fate during lung morphogenesis. J Biol Chem 2003; 278: 40231–40238. [DOI] [PubMed] [Google Scholar]
- 52.Shu W, Guttentag S, Wang Z, Andl T, Ballard P, Lu MM et al. Wnt/beta-catenin signaling acts upstream of N-myc, BMP4, and FGF signaling to regulate proximal-distal patterning in the lung. Dev Biol 2005; 283: 226–239. [DOI] [PubMed] [Google Scholar]
- 53.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455: 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst 2014; 106: djt356. [DOI] [PubMed] [Google Scholar]
- 55.Reynolds SD, Zemke AC, Giangreco A, Brockway BL, Teisanu RM, Drake JA et al. Conditional stabilization of beta-catenin expands the pool of lung stem cells. Stem Cells 2008; 26: 1337–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pacheco-Pinedo EC, Durham AC, Stewart KM, Goss AM, Lu MM, Demayo FJ et al. Wnt/β-catenin signaling accelerates mouse lung tumorigenesis by imposing an embryonic distal progenitor phenotype on lung epithelium. J Clin Invest 2011; 121: 1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sánchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nguyen DX, Chiang AC, Zhang XH, Kim JY, Kris MG, Ladanyi M et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 2009; 138: 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bleckmann A, Siam L, Klemm F, Rietkotter E, Wegner C, Kramer F et al. Nuclear LEF1/TCF4 correlate with poor prognosis but not with nuclear beta-catenin in cerebral metastasis of lung adenocarcinomas. Clin Exp Metastasis 2013; 30: 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maeda Y, Dave V, Whitsett JA. Transcriptional control of lung morphogenesis. Physiol Rev 2007; 87: 219–244. [DOI] [PubMed] [Google Scholar]
- 61.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 2008; 8: 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wikenheiser-Brokamp KA. Rb family proteins differentially regulate distinct cell lineages during epithelial development. Development 2004; 131: 4299–4310. [DOI] [PubMed] [Google Scholar]
- 63.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 2009; 41: 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hussenet T, Dali S, Exinger J, Monga B, Jost B, Dembele D et al. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS One 2010; 5: e8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu Y, Futtner C, Rock JR, Xu X, Whitworth W, Hogan BL et al. Evidence that SOX2 overexpression is oncogenic in the lung. PLoS One 2010; 5: e11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Que J, Luo X, Schwartz RJ, Hogan BL. Multiple roles for Sox2 in the developing and adult mouse trachea. Development 2009; 136: 1899–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mukhopadhyay A, Berrett KC, Kc U, Clair PM, Pop SM, Carr SR et al. Sox2 cooperates with Lkb1 loss in a mouse model of squamous cell lung cancer. Cell Rep 2014; 8: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watanabe H, Ma Q, Peng S, Adelmant G, Swain D, Song W et al. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest 2014; 124: 1636–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 2014; 25: 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu X, Huang L, Futtner C, Schwab B, Rampersad RR, Lu Y et al. The cell of origin and subtype of K-Ras-induced lung tumors are modified by Notch and Sox2. Genes Dev 2014; 28: 1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang DR, Martinez Alanis D, Miller RK, Ji H, Akiyama H, McCrea PD et al. Lung epithelial branching program antagonizes alveolar differentiation. Proc Natl Acad Sci U S A 2013; 110: 18042–18051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rockich BE, Hrycaj SM, Shih HP, Nagy MS, Ferguson MA, Kopp JL et al. Sox9 plays multiple roles in the lung epithelium during branching morphogenesis. Proc Natl Acad Sci U S A 2013; 110: E4456–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou CH, Ye LP, Ye SX, Li Y, Zhang XY, Xu XY et al. Clinical significance of SOX9 in human non-small cell lung cancer progression and overall patient survival. J Exp Clin Cancer Res 2012; 31: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang SS, Fang WT, Hou YH, Huang SF, Yen BL, Chang JL et al. Upregulation of SOX9 in lung adenocarcinoma and its involvement in the regulation of cell growth and tumorigenicity. Clin Cancer Res 2010; 16: 4363–4373. [DOI] [PubMed] [Google Scholar]
- 75.Capaccione KM, Hong X, Morgan KM, Liu W, Bishop JM, Liu L et al. Sox9 mediates Notch1-induced mesenchymal features in lung adenocarcinoma. Oncotarget 2014; 5: 3636–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ikeda K, Clark JC, Shaw-White JR, Stahlman MT, Boutell CJ, Whitsett JA. Gene structure and expression of human thyroid transcription factor-1 in respiratory epithelial cells. J Biol Chem 1995; 270: 8108–8114. [DOI] [PubMed] [Google Scholar]
- 77.Minoo P, Hamdan H, Bu D, Warburton D, Stepanik P, deLemos R. TTF-1 regulates lung epithelial morphogenesis. Dev Biol 1995; 172: 694–698. [DOI] [PubMed] [Google Scholar]
- 78.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 2007; 450: 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kendall J, Liu Q, Bakleh A, Krasnitz A, Nguyen KC, Lakshmi B et al. Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci U S A 2007; 104: 16663–16668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kwei KA, Kim YH, Girard L, Kao J, Pacyna-Gengelbach M, Salari K et al. Genomic profiling identifies TITF1 as a lineage-specific oncogene amplified in lung cancer. Oncogene 2008; 27: 3635–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barletta JA, Perner S, Iafrate AJ, Yeap BY, Weir BA, Johnson LA et al. Clinical significance of TTF-1 protein expression and TTF-1 gene amplification in lung adenocarcinoma. J Cell Mol Med 2009; 13: 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tang X, Kadara H, Behrens C, Liu DD, Xiao Y, Rice D et al. Abnormalities of the TITF-1 lineage-specific oncogene in NSCLC: implications in lung cancer pathogenesis and prognosis. Clin Cancer Res 2011; 17: 2434–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tanaka H, Yanagisawa K, Shinjo K, Taguchi A, Maeno K, Tomida S et al. Lineage-specific dependency of lung adenocarcinomas on the lung development regulator TTF-1. Cancer Res 2007; 67: 6007–6011. [DOI] [PubMed] [Google Scholar]
- 84.Yamaguchi T, Yanagisawa K, Sugiyama R, Hosono Y, Shimada Y, Arima C et al. NKX2–1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell 2012; 21: 348–361. [DOI] [PubMed] [Google Scholar]
- 85.Maeda Y, Tsuchiya T, Hao H, Tompkins DH, Xu Y, Mucenski ML et al. Kras(G12D) and Nkx2–1 haploinsufficiency induce mucinous adenocarcinoma of the lung. J Clin Invest 2012; 122: 4388–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM et al. Suppression of lung adenocarcinoma progression by Nkx2–1. Nature 2011; 473: 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Berghmans T, Paesmans M, Mascaux C, Martin B, Meert AP, Haller A et al. Thyroid transcription factor 1--a new prognostic factor in lung cancer: a meta-analysis. Ann Oncol 2006; 17: 1673–1676. [DOI] [PubMed] [Google Scholar]
- 88.Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, Wang DG et al. Nkx2–1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol Cell 2013; 50: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yamaguchi T, Hosono Y, Yanagisawa K, Takahashi T. NKX2–1/TTF-1: an enigmatic oncogene that functions as a double-edged sword for cancer cell survival and progression. Cancer Cell 2013; 23: 718–723. [DOI] [PubMed] [Google Scholar]
- 90.Kim IJ, Quigley D, To MD, Pham P, Lin K, Jo B et al. Rewiring of human lung cell lineage and mitotic networks in lung adenocarcinomas. Nat Commun 2013; 4: 1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saito RA, Watabe T, Horiguchi K, Kohyama T, Saitoh M, Nagase T et al. Thyroid transcription factor-1 inhibits transforming growth factor-beta-mediated epithelial-to-mesenchymal transition in lung adenocarcinoma cells. Cancer Res 2009; 69: 2783–2791. [DOI] [PubMed] [Google Scholar]
- 92.Hosono Y, Yamaguchi T, Mizutani E, Yanagisawa K, Arima C, Tomida S et al. MYBPH, a transcriptional target of TTF-1, inhibits ROCK1, and reduces cell motility and metastasis. EMBO J 2012; 31: 481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Watanabe H, Francis JM, Woo MS, Etemad B, Lin W, Fries DF et al. Integrated cistromic and expression analysis of amplified NKX2–1 in lung adenocarcinoma identifies LMO3 as a functional transcriptional target. Genes Dev 2013; 27: 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Molkentin JD. The zinc finger-containing transcription factors GATA-4, −5, and −6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem 2000; 275: 38949–38952. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Y, Goss AM, Cohen ED, Kadzik R, Lepore JJ, Muthukumaraswamy K et al. A Gata6-Wnt pathway required for epithelial stem cell development and airway regeneration. Nat Genet 2008; 40: 862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang H, Lu MM, Zhang L, Whitsett JA, Morrisey EE. GATA6 regulates differentiation of distal lung epithelium. Development 2002; 129: 2233–2246. [DOI] [PubMed] [Google Scholar]
- 97.Keijzer R, van Tuyl M, Meijers C, Post M, Tibboel D, Grosveld F et al. The transcription factor GATA6 is essential for branching morphogenesis and epithelial cell differentiation during fetal pulmonary development. Development 2001; 128: 503–511. [DOI] [PubMed] [Google Scholar]
- 98.Shaw-White JR, Bruno MD, Whitsett JA. GATA-6 activates transcription of thyroid transcription factor-1. J Biol Chem 1999; 274: 2658–2664. [DOI] [PubMed] [Google Scholar]
- 99.Liu C, Glasser SW, Wan H, Whitsett JA. GATA-6 and thyroid transcription factor-1 directly interact and regulate surfactant protein-C gene expression. J Biol Chem 2002; 277: 4519–4525. [DOI] [PubMed] [Google Scholar]
- 100.Bruno MD, Korfhagen TR, Liu C, Morrisey EE, Whitsett JA. GATA-6 activates transcription of surfactant protein A. J Biol Chem 2000; 275: 1043–1049. [DOI] [PubMed] [Google Scholar]
- 101.Kwei KA, Bashyam MD, Kao J, Ratheesh R, Reddy EC, Kim YH et al. Genomic profiling identifies GATA6 as a candidate oncogene amplified in pancreatobiliary cancer. PLoS Genet 2008; 4: e1000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhong Y, Wang Z, Fu B, Pan F, Yachida S, Dhara M et al. GATA6 activates Wnt signaling in pancreatic cancer by negatively regulating the Wnt antagonist Dickkopf-1. PLoS One 2011; 6: e22129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin L, Bass AJ, Lockwood WW, Wang Z, Silvers AL, Thomas DG et al. Activation of GATA binding protein 6 (GATA6) sustains oncogenic lineage-survival in esophageal adenocarcinoma. Proc Natl Acad Sci U S A 2012; 109: 4251–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chia NY, Deng N, Das K, Huang D, Hu L, Zhu Y et al. Regulatory crosstalk between lineage-survival oncogenes KLF5, GATA4 and GATA6 cooperatively promotes gastric cancer development. Gut 2014. [DOI] [PubMed] [Google Scholar]
- 105.Belaguli NS, Aftab M, Rigi M, Zhang M, Albo D, Berger DH. GATA6 promotes colon cancer cell invasion by regulating urokinase plasminogen activator gene expression. Neoplasia 2010; 12: 856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Capo-chichi CD, Roland IH, Vanderveer L, Bao R, Yamagata T, Hirai H et al. Anomalous expression of epithelial differentiation-determining GATA factors in ovarian tumorigenesis. Cancer Res 2003; 63: 4967–4977. [PubMed] [Google Scholar]
- 107.Kamnasaran D, Qian B, Hawkins C, Stanford WL, Guha A. GATA6 is an astrocytoma tumor suppressor gene identified by gene trapping of mouse glioma model. Proc Natl Acad Sci U S A 2007; 104: 8053–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang Y, Ahn YH, Gibbons DL, Zang Y, Lin W, Thilaganathan N et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest 2011; 121: 1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guo M, Akiyama Y, House MG, Hooker CM, Heath E, Gabrielson E et al. Hypermethylation of the GATA genes in lung cancer. Clin Cancer Res 2004; 10: 7917–7924. [DOI] [PubMed] [Google Scholar]
- 110.Caslini C, Capo-chichi CD, Roland IH, Nicolas E, Yeung AT, Xu XX. Histone modifications silence the GATA transcription factor genes in ovarian cancer. Oncogene 2006; 25: 5446–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yin Z, Gonzales L, Kolla V, Rath N, Zhang Y, Lu MM et al. Hop functions downstream of Nkx2.1 and GATA6 to mediate HDAC-dependent negative regulation of pulmonary gene expression. Am J Physiol Lung Cell Mol Physiol 2006; 291: L191–199. [DOI] [PubMed] [Google Scholar]
- 112.Yamashita K, Kim MS, Park HL, Tokumaru Y, Osada M, Inoue H et al. HOP/OB1/NECC1 promoter DNA is frequently hypermethylated and involved in tumorigenic ability in esophageal squamous cell carcinoma. Mol Cancer Res 2008; 6: 31–41. [DOI] [PubMed] [Google Scholar]
- 113.Yamaguchi S, Asanoma K, Takao T, Kato K, Wake N. Homeobox gene HOPX is epigenetically silenced in human uterine endometrial cancer and suppresses estrogen-stimulated proliferation of cancer cells by inhibiting serum response factor. Int J Cancer 2009; 124: 2577–2588. [DOI] [PubMed] [Google Scholar]
- 114.Waraya M, Yamashita K, Katoh H, Ooki A, Kawamata H, Nishimiya H et al. Cancer specific promoter CpG Islands hypermethylation of HOP homeobox (HOPX) gene and its potential tumor suppressive role in pancreatic carcinogenesis. BMC Cancer 2012; 12: 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ooki A, Yamashita K, Kikuchi S, Sakuramoto S, Katada N, Kokubo K et al. Potential utility of HOP homeobox gene promoter methylation as a marker of tumor aggressiveness in gastric cancer. Oncogene 2010; 29: 3263–3275. [DOI] [PubMed] [Google Scholar]
- 116.Katoh H, Yamashita K, Waraya M, Margalit O, Ooki A, Tamaki H et al. Epigenetic silencing of HOPX promotes cancer progression in colorectal cancer. Neoplasia 2012; 14: 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen Y, Yang L, Cui T, Pacyna-Gengelbach M, Petersen I. HOPX is methylated and exerts tumour suppressive function through Ras-induced senescence in human lung cancer. J Pathol 2014. [DOI] [PubMed] [Google Scholar]
- 118.Watanabe H, Meyerson M. Hopping between differentiation states in lung adenocarcinoma. Cancer Cell 2013; 23: 707–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martis PC, Whitsett JA, Xu Y, Perl AK, Wan H, Ikegami M. C/EBPalpha is required for lung maturation at birth. Development 2006; 133: 1155–1164. [DOI] [PubMed] [Google Scholar]
- 120.Basseres DS, Levantini E, Ji H, Monti S, Elf S, Dayaram T et al. Respiratory failure due to differentiation arrest and expansion of alveolar cells following lung-specific loss of the transcription factor C/EBPalpha in mice. Mol Cell Biol 2006; 26: 1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sato A, Xu Y, Whitsett JA, Ikegami M. CCAAT/enhancer binding protein-α regulates the protease/antiprotease balance required for bronchiolar epithelium regeneration. Am J Respir Cell Mol Biol 2012; 47: 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Costa DB, Dayaram T, D’Alo F, Wouters BJ, Tenen DG, Meyerson M et al. C/EBP alpha mutations in lung cancer. Lung Cancer 2006; 53: 253–254. [DOI] [PubMed] [Google Scholar]
- 123.Tada Y, Brena RM, Hackanson B, Morrison C, Otterson GA, Plass C. Epigenetic modulation of tumor suppressor CCAAT/enhancer binding protein alpha activity in lung cancer. J Natl Cancer Inst 2006; 98: 396–406. [DOI] [PubMed] [Google Scholar]
- 124.Halmos B, Huettner CS, Kocher O, Ferenczi K, Karp DD, Tenen DG. Down-regulation and antiproliferative role of C/EBPalpha in lung cancer. Cancer Res 2002; 62: 528–534. [PubMed] [Google Scholar]
- 125.Sato A, Yamada N, Ogawa Y, Ikegami M. CCAAT/enhancer-binding protein-α suppresses lung tumor development in mice through the p38Į MAP kinase pathway. PLoS One 2013; 8: e57013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tomita T, Kido T, Kurotani R, Iemura S, Sterneck E, Natsume T et al. CAATT/enhancer-binding proteins alpha and delta interact with NKX2–1 to synergistically activate mouse secretoglobin 3A2 gene expression. J Biol Chem 2008; 283: 25617–25627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wan H, Kaestner KH, Ang SL, Ikegami M, Finkelman FD, Stahlman MT et al. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development 2004; 131: 953–964. [DOI] [PubMed] [Google Scholar]
- 128.Wan H, Dingle S, Xu Y, Besnard V, Kaestner KH, Ang SL et al. Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J Biol Chem 2005; 280: 13809–13816. [DOI] [PubMed] [Google Scholar]
- 129.Minoo P, Hu L, Xing Y, Zhu NL, Chen H, Li M et al. Physical and functional interactions between homeodomain NKX2.1 and winged helix/forkhead FOXA1 in lung epithelial cells. Mol Cell Biol 2007; 27: 2155–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jimenez FR, Lewis JB, Belgique ST, Wood TT, Reynolds PR. Developmental lung expression and transcriptional regulation of claudin-6 by TTF-1, Gata-6, and FoxA2. Respir Res 2014; 15: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Halmos B, Bassères DS, Monti S, D’Aló F, Dayaram T, Ferenczi K et al. A transcriptional profiling study of CCAAT/enhancer binding protein targets identifies hepatocyte nuclear factor 3 beta as a novel tumor suppressor in lung cancer. Cancer Res 2004; 64: 4137–4147. [DOI] [PubMed] [Google Scholar]
- 132.Basseres DS, D’Alò F, Yeap BY, Löwenberg EC, Gonzalez DA, Yasuda H et al. Frequent downregulation of the transcription factor Foxa2 in lung cancer through epigenetic silencing. Lung Cancer 2012; 77: 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tang Y, Shu G, Yuan X, Jing N, Song J. FOXA2 functions as a suppressor of tumor metastasis by inhibition of epithelial-to-mesenchymal transition in human lung cancers. Cell Res 2011; 21: 316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ito Y, Bae SC, Chuang LS. The RUNX family: developmental regulators in cancer. Nat Rev Cancer 2015; 15: 81–95. [DOI] [PubMed] [Google Scholar]
- 135.Lee KS, Lee YS, Lee JM, Ito K, Cinghu S, Kim JH et al. Runx3 is required for the differentiation of lung epithelial cells and suppression of lung cancer. Oncogene 2010; 29: 3349–3361. [DOI] [PubMed] [Google Scholar]
- 136.Lee JM, Shin JO, Cho KW, Hosoya A, Cho SW, Lee YS et al. Runx3 is a crucial regulator of alveolar differentiation and lung tumorigenesis in mice. Differentiation 2011; 81: 261–268. [DOI] [PubMed] [Google Scholar]
- 137.Liang Y, He L, Yuan H, Jin Y, Yao Y. Association between RUNX3 promoter methylation and non-small cell lung cancer: a meta-analysis. J Thorac Dis 2014; 6: 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yanagawa N, Tamura G, Oizumi H, Kanauchi N, Endoh M, Sadahiro M et al. Promoter hypermethylation of RASSF1A and RUNX3 genes as an independent prognostic prediction marker in surgically resected non-small cell lung cancers. Lung Cancer 2007; 58: 131–138. [DOI] [PubMed] [Google Scholar]
- 139.Zheng Y, Wang R, Song HZ, Pan BZ, Zhang YW, Chen LB. Epigenetic downregulation of RUNX3 by DNA methylation induces docetaxel chemoresistance in human lung adenocarcinoma cells by activation of the AKT pathway. Int J Biochem Cell Biol 2013; 45: 2369–2378. [DOI] [PubMed] [Google Scholar]
- 140.Fujii S, Ito K, Ito Y, Ochiai A. Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by increasing histone H3 methylation. J Biol Chem 2008; 283: 17324–17332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chi XZ, Kim J, Lee YH, Lee JW, Lee KS, Wee H et al. Runt-related transcription factor RUNX3 is a target of MDM2-mediated ubiquitination. Cancer Res 2009; 69: 8111–8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee YS, Lee JW, Jang JW, Chi XZ, Kim JH, Li YH et al. Runx3 inactivation is a crucial early event in the development of lung adenocarcinoma. Cancer Cell 2013; 24: 603–616. [DOI] [PubMed] [Google Scholar]
- 143.Okubo T, Knoepfler PS, Eisenman RN, Hogan BL. Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development 2005; 132: 1363–1374. [DOI] [PubMed] [Google Scholar]
- 144.Moens CB, Auerbach AB, Conlon RA, Joyner AL, Rossant J. A targeted mutation reveals a role for N-myc in branching morphogenesis in the embryonic mouse lung. Genes Dev 1992; 6: 691–704. [DOI] [PubMed] [Google Scholar]
- 145.Cohen JC, Scott DK, Miller J, Zhang J, Zhou P, Larson JE. Transient in utero knockout (TIUKO) of C-MYC affects late lung and intestinal development in the mouse. BMC Dev Biol 2004; 4: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Iwakawa R, Kohno T, Kato M, Shiraishi K, Tsuta K, Noguchi M et al. MYC amplification as a prognostic marker of early-stage lung adenocarcinoma identified by whole genome copy number analysis. Clin Cancer Res 2011; 17: 1481–1489. [DOI] [PubMed] [Google Scholar]
- 147.Iwakawa R, Takenaka M, Kohno T, Shimada Y, Totoki Y, Shibata T et al. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes Cancer 2013; 52: 802–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Romero OA, Torres-Diz M, Pros E, Savola S, Gomez A, Moran S et al. MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov 2014; 4: 292–303. [DOI] [PubMed] [Google Scholar]
- 149.Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J et al. Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS One 2008; 3: e2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Allen TD, Zhu CQ, Jones KD, Yanagawa N, Tsao MS, Bishop JM. Interaction between MYC and MCL1 in the genesis and outcome of non-small-cell lung cancer. Cancer Res 2011; 71: 2212–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Allen TD, Rodriguez EM, Jones KD, Bishop JM. Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Res 2011; 71: 6010–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Soucek L, Whitfield JR, Sodir NM, Massó-Vallés D, Serrano E, Karnezis AN et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev 2013; 27: 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Juan J, Muraguchi T, Iezza G, Sears RC, McMahon M. Diminished WNT -> β-catenin -> c-MYC signaling is a barrier for malignant progression of BRAFV600E-induced lung tumors. Genes Dev 2014; 28: 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 2005; 6: 635–645. [DOI] [PubMed] [Google Scholar]
- 155.Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012; 151: 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sinha S, Adler AS, Field Y, Chang HY, Segal E. Systematic functional characterization of cis-regulatory motifs in human core promoters. Genome Res 2008; 18: 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008; 2: 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rapp UR, Korn C, Ceteci F, Karreman C, Luetkenhaus K, Serafin V et al. MYC is a metastasis gene for non-small-cell lung cancer. PLoS One 2009; 4: e6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat 2008; 29: 617–622. [DOI] [PubMed] [Google Scholar]
- 160.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011; 11: 481–492. [DOI] [PubMed] [Google Scholar]
- 161.Brabletz T To differentiate or not--routes towards metastasis. Nat Rev Cancer 2012; 12: 425–436. [DOI] [PubMed] [Google Scholar]
- 162.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev 2009; 23: 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Brabletz T EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell 2012; 22: 699–701. [DOI] [PubMed] [Google Scholar]
- 164.Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol 2014; 16: 488–494. [DOI] [PubMed] [Google Scholar]
- 165.Tsuji T, Ibaragi S, Hu GF. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res 2009; 69: 7135–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Celià-Terrassa T, Meca-Cortés O, Mateo F, de Paz AM, Rubio N, Arnal-Estapé A et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest 2012; 122: 1849–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Calbo J, van Montfort E, Proost N, van Drunen E, Beverloo HB, Meuwissen R et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 2011; 19: 244–256. [DOI] [PubMed] [Google Scholar]
- 168.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013; 155: 1639–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Govaere O, Komuta M, Berkers J, Spee B, Janssen C, de Luca F et al. Keratin 19: a key role player in the invasion of human hepatocellular carcinomas. Gut 2014; 63: 674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Reynolds CP, Lemons RS. Retinoid therapy of childhood cancer. Hematol Oncol Clin North Am 2001; 15: 867–910. [DOI] [PubMed] [Google Scholar]
- 171.Saez-Ayala M, Montenegro MF, Sanchez-Del-Campo L, Fernandez-Perez MP, Chazarra S, Freter R et al. Directed phenotype switching as an effective antimelanoma strategy. Cancer Cell 2013; 24: 105–119. [DOI] [PubMed] [Google Scholar]
- 172.Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 2014; 4: 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013; 340: 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Campos B, Wan F, Farhadi M, Ernst A, Zeppernick F, Tagscherer KE et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin Cancer Res 2010; 16: 2715–2728. [DOI] [PubMed] [Google Scholar]
- 175.Yan M, Zhang Y, He B, Xiang J, Wang ZF, Zheng FM et al. IKKalpha restoration via EZH2 suppression induces nasopharyngeal carcinoma differentiation. Nat Commun 2014; 5: 3661. [DOI] [PubMed] [Google Scholar]
- 176.Tremblay AM, Missiaglia E, Galli GG, Hettmer S, Urcia R, Carrara M et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 2014; 26: 273–287. [DOI] [PubMed] [Google Scholar]
- 177.Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A 2012; 109: 19408–19413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin Cancer Res 2013; 19: 6183–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014; 511: 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014; 159: 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014; 26: 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 2013; 23: 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013; 19: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]