

Abstract
Circadian clock confers temporal control in metabolism, with its disruption leading to the development of insulin resistance. Metabolic substrate utilization in skeletal muscle is coordinated with diurnal nutrient cycles. However, whether the molecular clock is involved in this coordination is largely unknown. Using a myocyte-selective genetic ablation mouse model of the essential clock activator Bmal1, here we identify muscle-intrinsic clock as a sensor of feeding cues to orchestrate skeletal muscle oxidation required for global nutrient flux. Bmal1 in skeletal muscle responds robustly to feeding in vivo and insulin induces its expression. Muscle Bmal1 deficiency impaired the transcriptional control of glucose metabolic pathway, resulting in markedly attenuated glucose utilization and fasting hyperglycemia. Notably, the loss of Bmal1 response to feeding abolished fasting-to-feeding metabolic fuel switch from fatty acids to glucose in skeletal muscle, leading to the activation of energy-sensing pathways for fatty acid oxidation. These altered metabolic substrate oxidations in Bmal1-deficient muscle ultimately depleted circulating lipid levels that prevented hepatic steatosis. Collectively, our findings highlight the key role of the metabolic-sensing function of skeletal muscle clock in partitioning nutrient flux between muscle and liver to maintain whole-body lipid and glucose homeostasis.
Keywords: circadian clock, skeletal muscle, glucose metabolism, fatty acid metabolism, hepatic steatosis
1 |. INTRODUCTION
The circadian clock machinery confers coordinated temporal control to metabolic processes to maintain homeostasis.1–3 Key metabolic pathways in liver, pancreatic islet, adipose tissue or skeletal muscle display strong rhythmic regulations by the circadian clock.4–6 Accumulating studies indicate that circadian clock misalignment disrupts global metabolic regulation leading to the development of obesity and insulin resistance.7–9 Appropriate timing of metabolic responses may allow adaptation to environmental nutrient cues associated with feeding-fasting and sleep-wake cycles.10,11 Interestingly, temporal orchestration was recently identified as the key driver of the global coordination of metabolic processes in distinct tissues.12 However, the mechanism and signals involved in the coordination among clock-controlled metabolic pathways remain largely unknown.
The molecular clock generates ~24-hour oscillations in gene transcription, physiology, and behavior through inter-locking transcriptional-translational feedback loops.13 Brain and muscle Arnt-like 1 (Bmal1), an essential activator of the core clock transcription circuit, is highly expressed in skeletal muscle.14,15 This basic helix-loop-helix transcription factor dimerizes with circadian locomotor output cycles kaput (CLOCK) to initiate the molecular clock transcription feedback loop. CLOCK/Bmal1 activation of the negative arms of the loop involving Per and Cry repressors, in turn, leads to subsequent inhibition of the cycle. This transcriptional negative feedback loop forms the basis of clock oscillation, although post-transcriptional and post-translational mechanisms are also required to establish the circadian clock cycle.13,16 In skeletal muscle, the circadian clock exerts transcriptional regulation on diverse physiological processes. Bmal1 and its obligate partner CLOCK are required for maintaining muscle contractile function and structural integrity.17 Our previous studies reveal that Bmal1 is a key regulator of myocyte development in postnatal muscle growth and remodeling,15,18 and in contrast, its transcriptional repressor Rev-erbα suppresses myogenesis.19 As a remarkable ~35% of rhythmic transcripts in skeletal muscl ebelongs to metabolic processes and metabolites in muscle display daily oscillations, circadian clock control is pervasive in muscle metabolism.
Metabolic activities of the skeletal muscle play a crucial role in whole-body nutrient homeostasis, accounting for greater than 80% of glucose disposal in insulin-stimulated state and a major site for lipid oxidation during fasting.20,21 Muscle fuel oxidation occurs in accordance with the nutrient flux induced by fasting-feeding cycles and the balance between diurnal nutrient availability and oxidation determines partitioning in muscle, fat, and liver.2,3 Given that oxidative metabolism in muscle is dependent on diurnal substrate availability,22 this coordination may require a functional muscle-resident clock. In line with this notion, restricted feeding has been shown to be a dominant entrainment cue for peripheral clock systems.10,23 Bmal1 regulates diverse metabolic pathways in distinct tissues, including lipid metabolism and gluconeogenesis in liver,24 insulin secretory rhythm in pancreatic islets,4,25 and adipogenic cascades in white and brown adipocytes.26,27 These metabolic regulations of Bmal1 may collectively contribute to nutrient homeostasis,28 with its global ablation or CLOCK mutants display markedly impaired glucose and lipid metabolism.17,29 Based on the pervasive temporal control involved in muscle metabolism, we generated a mouse model of myocyte-selective Bmal1 inactivation to dissect the metabolic regulations of the muscle-intrinsic clock and its specific contribution to global nutrient homeostasis.
2 |. MATERIALS AND METHODS
2.1 |. Animals
Mice were maintained in City of Hope main campus vivarium under a constant 12:12 light-dark cycle, with lights on at 7:00 AM (ZT0) and lights off at 7:00 PM (ZT12). All experiments were approved by the IACUC committee of the Beckman Research Institute of City of Hope. Bmal1fl/fl (Stock #007668) and MCK-Cre transgenic mice (Stock# 006475)14 were purchased from the Jackson Laboratory.
2.2 |. RNA extraction and quantitative reverse-transcriptase PCR analysis
TRIzol (Invitrogen) and RNeasy miniprep kits (Qiagen) were used to isolate total RNA from snap-frozen muscle tissues and cells, respectively. cDNA was generated using q-Script cDNA kit (Quanta Biosciences) and quantitative PCR was performed using a Roche 480 Light Cycler with SYBR Green (Quanta Biosciences). Relative mRNA expression was determined using the comparative Ct method to normalize target genes 36B4 as internal controls. Primers are designed using PrimerBank experimentally validated sequences.
2.3 |. Immunoblot analysis
About 20–40 μg of total protein was resolved on the SDS-PAGE gel, transferred to the nitrocellulose membrane, and used for immunoblotting. Immunoblots were developed by the chemiluminescence kit (Pierce Biotechnology). Source and dilution information of primary antibodies are shown in Supporting Table S1. Appropriate specific secondary antibodies were used at a dilution of 1:3000.
2.4 |. Chromatin immunoprecipitation-qPCR
Immunoprecipitation was performed using a specific Bmal1 antibody (Abcam AB93806)15 or control rabbit IgG with Magnetic Protein A/G beads (Magna ChIP A/G kit, Millipore), as described previously.15,26 Differentiated C2C12 myotubes chromatin was sonicated and purified following formaldehyde fixation. Real-time PCR was carried out in triplicate using purified chromatin with specific primers for predicted Bmal1 binding E or E’-box elements identified within the gene regulatory regions. Primers flanking known Bmal1 E-box in Rev-erbα promoter were used as a positive control and TBP first exon primers as a negative control. Chromatin immunoprecipitation (ChIP) primer sequences are shown in Supporting Table S2. Fold enrichment over IgG was expressed normalized to 1% of input.
2.5 |. Immunofluorescence staining
Muscles were snap-frozen in liquid-nitrogen cooled isopentane and embedded in OCT, as described.18 Cryosections of the middle region of TA were fixed, permeabilized, and endogenous IgG blocked prior to primary antibodies for immunofluorescence (Table S1).
2.6 |. Glucose tolerance test, insulin sensitivity, and serum metabolite analysis
Glucose tolerance test and insulin sensitivity assay were conducted as described previously.30,31 For the glucose tolerance test, 10 to 12-week-old mice were fasted overnight before the injection of a bolus dose of D-glucose 2 g/kg on chow diet and 1.5 g/kg after 3 months on high-fat diet. Tail vein bleeds were used to obtain plasma at 0, 15, 30, 60, and 120 minutes following glucose injection. Insulin signaling was performed on tissues, including muscle, fat, and liver, collected 20 minutes after 1 U/Kg insulin IP injection. Plasma free fatty acids (WAKO), glucose, cholesterol, and triglyceride levels were measured by respective commercial kits (Thermo Scientific).
2.7 |. NAD+ and NADH assay
Intracellular levels of NADH and total NAD (NAD+ + NADH) were measured using the NAD/NADH Quantitation Kit (Sigma) according to the manufacturer’s instructions. In brief, 200 mg of frozen quadricep was homogenized with NADH/NAD extraction buffer. For the detection of NADH, NAD in the extracted supernatant was first decomposed by heating at 60°C for 30 minutes. The total NAD level (NAD+ + NADH) was assayed by converting NAD+ to NADH, and NADH was subsequently measured by the colorimetric assay. The amount of total NAD or NADH was calculated according to the NADH standard and normalized by the protein concentration. The NAD+ value was calculated by subtracting NADH from total NAD.
2.8 |. Hepatic triglyceride content analysis
Hepatic triglyceride content analysis was performed as described previously.31 Briefly, homogenized frozen liver tissue was used for lipid extraction with aliquot for protein determination. The homogenate was mixed with chloroform, methanol, and water with 2:1:1 ratio, thoroughly vortexed, centrifuged, and the organic phase collected. The lipid extract was dried overnight and resuspended in 0.2% Triton X-100 for triglyceride assay (Sigma).
2.9 |. Oil-Red-O staining
Frozen liver tissues were collected in OCT, sectioned at 10 um, and air-dried. Sections were fixed with formalin and stained with a 60% oil-red-O working solution at room temperature for 15 minutes. The sections were counterstained with hematoxylin, rinsed under water, and mounted in water-soluble mounting medium and imaged.
2.10 |. Ex vivo muscle fatty acid and glucose oxidation assay
Fatty acid oxidation rate was determined by the measurement of 14C-CO2 production from 1 mmol/L 1-14C-palmitate complexed with BSA in isolated soleus muscle, as previously described.30 Briefly, soleus muscle was isolated from hindlimbs of 4-hour fasted mice and the weight measured. Each muscle strip was incubated in Krebs-Henseleit buffer at 37°C containing 14C palmitate conjugated with 4% fatty acid-free albumin for 1 hour. Oxygen was then mixed with the reaction for 10 minutes followed by shaking at 37°C for 20 minutes. Hyamine hydrochloride was added to the center of the top hanging well to trap released CO2 and incubate for 40 minutes. The reaction was stopped by 60% perchloric acid and shaken for an additional 90 minutes. 14C-CO2 radioactivity trapped in the hyamine hydrochloride filter paper was measured by beta-scintillation to calculate the rate of fatty acid oxidation, while acid-soluble metabolites were analyzed by 14C radioactivity in the supernatant. Data were normalized to homogenate total protein. The glucose oxidation method was adapted from published protocols32,33 similarly as for fatty acid oxidation using 200 μM cold glucose and 0.2 μCi/mL U-14C-glucose as a substrate.34 14C-CO2 radioactivity trapped by hyamine hydrochloride in hanging well was measured to calculate the oxidation rate with normalization to the protein content.
2.11 |. Hyperinsulinemic-euglycemic clamp
Low-dose hyperinsulinemic-euglycemic clamp was conducted as described previously.31 Mice were cannulated through the right jugular vein and hyperinsulinemic-euglycemic clamp was performed under conscious condition after 4 days of recovery. Briefly, a primed dose (10 μCi) and a constant rate of intravenous dose (0.1 μCi/min) of 3H-glucose were infused. Tail vein blood was sampled after 50 minutes of glucose infusion to determine the basal glucose production. Mice were then primed with bolus insulin followed by continuous infusion at 3 mU/kg/min. About 10% glucose was simultaneously infused at a rate adjusted to maintain a steady blood glucose level at 100–140 mg/dL. At the end of the clamp period, blood was collected to measure the hepatic glucose production and peripheral glucose disposal rates.35 For the analysis of insulin-stimulated glucose transport in individual tissues, 2-deoxy-D-[1-14C] glucose was administered as a bolus 45 minutes before the end of clamps. Following the clamp, animals were euthanized, and adipose tissue, liver, and individual muscle groups were snap-frozen and stored at −80°C. Tissue 14C-glucose uptake was assayed by scintillation counting and calculated from 14C-glucose plasma profile fitted with a double exponential curve and tissue content, as previously described.36
2.12 |. Indirect calorimetry by Comprehensive Laboratory Animal Monitoring System (CLAMS)
Mice were single housed and acclimated in metabolic cages using the Comprehensive Laboratory Animal Monitoring System (Columbus Instruments), with ad libitum food and controlled lighting for 2 days prior to metabolic recording. Metabolic parameters, including oxygen consumption, CO2 production, respiratory exchange ratio (RER), ambulatory activity, and food intake were recorded for 5 consecutive days as previously described.26
2.13 |. Statistical analysis
Data were expressed as mean ± SE. Differences between groups were examined for statistical significance using unpaired two-tailed Student’s t test or ANOVA for multiple group comparison as indicated using Prism by GraphPad. P < .05 was considered statistically significant.
3 |. RESULTS
3.1 |. Diurnal expression of clock regulators and Bmal1 induction by feeding in skeletal muscle
To interrogate metabolic functions of the Bmal1-driven clock in skeletal muscle, we first examined Bmal1 diurnal regulations in distinct myofiber types. As compared to abundant expression in liver and white adipose tissue, Bmal1 protein is relatively enriched in slow oxidative soleus muscle than the predominantly fast glycolytic tibialis anterior (TA), with highest levels in diaphragm and heart (Figure 1A). Interestingly, diurnal patterns of Bmal1 are evident in soleus, TA, and diaphragm with consistently high expression at ZT2. Pyruvate dehydrogenase kinase (PDK4), as a control, is highly expressed in oxidative muscles as expected without significant diurnal rhythm. To test whether muscle clock specifically responds to feeding or fasting cues independent of circadian regulation, we used a fasting and refeeding regimen as shown in Figure 1B with identical circadian time sample collection at 12 PM following the same fasting duration, while lighting schedules are maintained. In the fasted group, food was withheld from 8 PM to 12 PM of the following day;while the refed group was fasted from 4 PM to 8 PM the following day prior to refeeding for 4 hours from 8 AM to 12 PM. Refeeding elicited a striking Bmal1 induction in soleus muscle with high basal expression, and moderate up-regulation was detected in the liver (Figure 1C). In contrast, the clock repressor Rev-erbα was induced by fasting evident in soleus and TA, suggesting a coordinated clock response to fasting-feeding cues. Pdk4 up-regulation by fasting and Glut4 induction by refeeding demonstrate the normal metabolic switch coinciding with nutrient flux. Consistent with its protein expression, Bmal1 transcript was stimulated by ~2-fold in the refed state (Figure 1D). Using differentiated C2C12 myotubes, we next examined which feeding signals, including insulin or glucose, that Bmal1 responds to, together with fasting-mimicking signals including forskolin to stimulate cAMP and fatty acids. Consistent with Bmal1 induction by feeding, insulin markedly stimulated, while forskolin inhibited Bmal1 protein expression in myotubes (Figure 1E), while insulin induction of Bmal1 mRNA was also observed (Figure 1F). In contrast to its induction by fasting in mice, Rev-erbα transcript did not respond to either insulin or forskolin in myotube. Neither glucose or palmitate regulated Bmal1 or Rev-erbα expression (Figure 1G), and high glucose also did not affect Bmal1 or Rev-erbα protein levels (Figure 1H). Thus, Bmal1 in skeletal muscle responds robustly to feeding cues in vivo, likely by insulin stimulation but not glucose.
FIGURE 1.
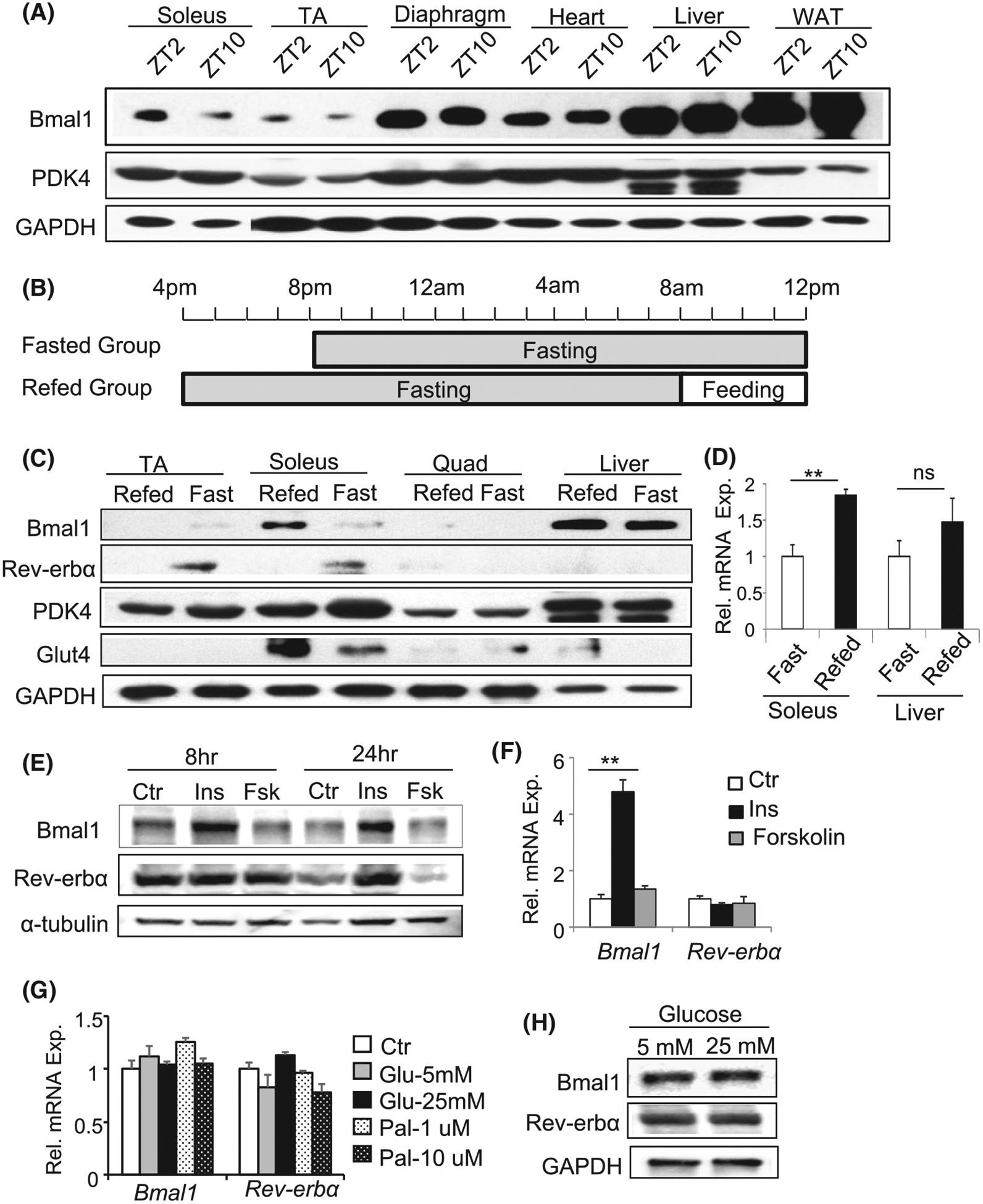
Circadian expression of Bmal1 protein and response to feeding-fasting signals in skeletal muscle. A, Immunoblot analysis of Bmal1 protein together with PDK4 in different muscle types Tibialis Anterior (TA), Soleus, diaphragm, and heart, liver, white adipose tissue (WAT) in normal mice. Pooled protein samples of n = 5–6 mice/group were used. B, Fasting and refeeding regimen with sample collection at the same circadian time of the day at 12 PM. Fasted group of mice was withheld food from 8 to 12 PM the following day and the refed group was fasted from 4 PM to 8 AM the next day followed by refed for 4 hours from 8 AM to 12 PM. The time of lighting was maintained with lights on at 7 AM (ZT0) and off at 7 PM (ZT12). C, Bmal1 protein level in distinct tissues in response to fasting and refeeding. D, Bmal1 mRNA expression during fasting and refeeding in TA muscle and liver as analyzed by RT-qPCR (n = 5–6/group). E and F, Bmal1 and Rev-erbα protein (E) or mRNA level (F) in response to insulin (10 ug/mL), forskolin (10 uM) treatment using differentiated myotubes (n = 3). G and H, Bmal1 and Rev-erbα mRNA (G) or protein level (H) in response to glucose or BSA-conjugated palmitate treatment in myotubes at an indicated concentration (n = 3)
3.2 |. Bmal1 promotes skeletal muscle glucose oxidation through direct transcriptional control of glucose metabolic pathway
As our previous studies found key roles of Bmal1 in myogenesis and muscle growth,15,18 we generated mice with selective ablation of Bmal1 in post-mitotic myocytes to determine its metabolic function in skeletal muscle using floxed Bmal1 (BMfl/fl) and MCK-Cre transgenic mice.37 In myocyte-specific Bmal1-deficient MCK-Cre+/BMfl/fl (mBMKO) mice muscle, Bmal1 protein was efficiently deleted, including TA, quadriceps, and soleus as compared to littermate MCK-Cre−/BMfl/fl (mBMCtr) controls, while its expression in the liver remains intact (Figure 2A). Residual Bmal1 was detected in soleus, possibly due to non-myocyte expression in vasculature or satellite cells. Myofiber-specific Bmal1 ablation did not affect total muscle mass (Figure 2B), gastrocnemius muscle weight (Figure 2C) or myofiber size (Figure S1A), ruling out potential muscle growth-related effects in this model. In addition, loss of Bmal1 in mature myocyte did not affect myogenesis, as mBMKO primary myoblasts display normal myogenic progression as indicated by comparable morphological differentiation (Figure S1B), MyHC immunostaining (Figure S1C), and myogenic gene expression (Figure S1D) to that of mBMCtr cells.
FIGURE 2.
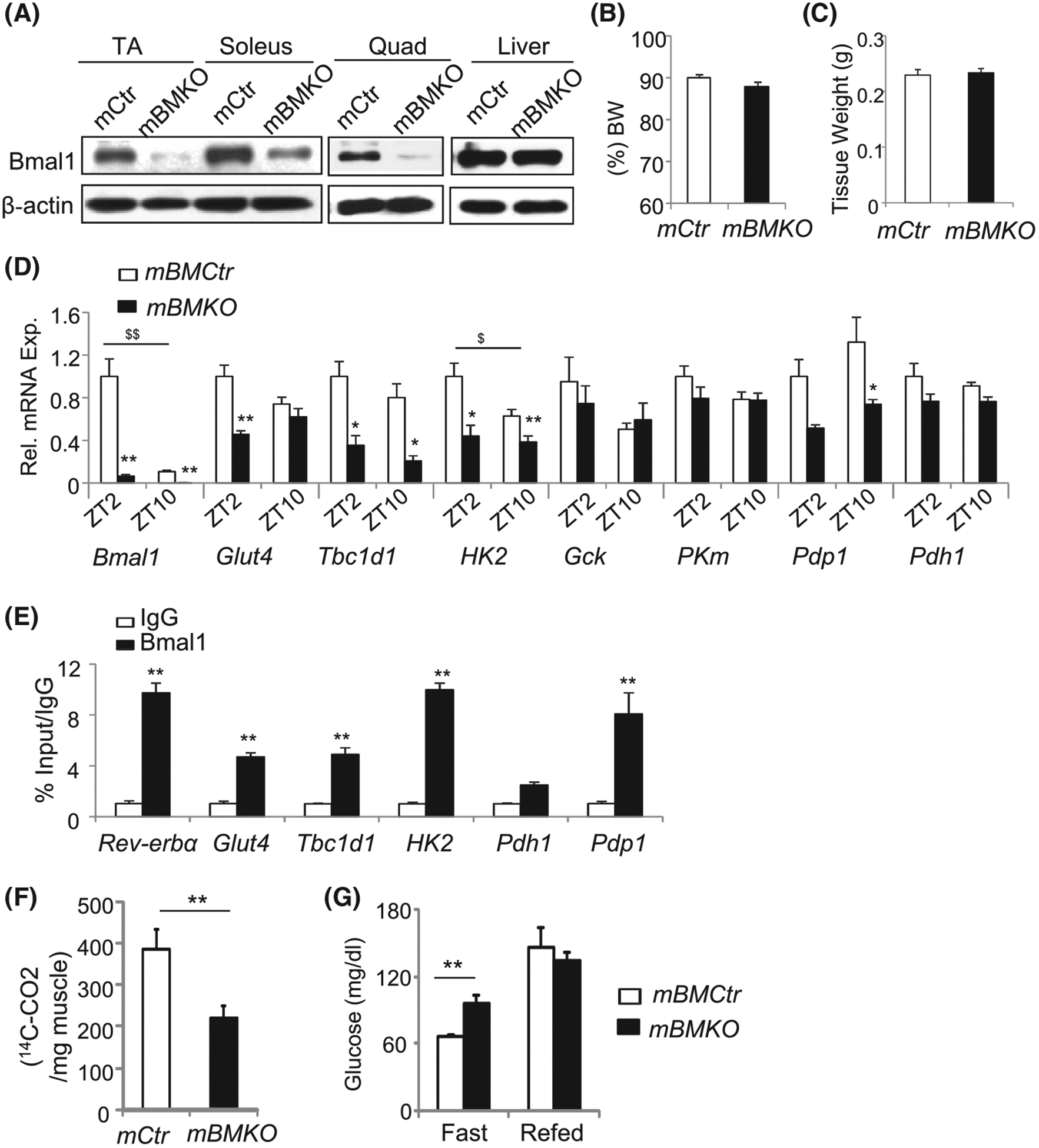
Regulation of muscle Bmal1 on glucose metabolism. A, Bmal1 ablation in muscle but not the liver in the mBMKO mice. Pooled samples (n = 4–5 mice/group) were collected at ZT2. B and C, NMR analysis of lean muscle mass content (B), and isolated gastrocnemius (GN) muscle tissue weight (C) in 12-week-old control and mBMKO mice (n = 7 mCtr and n = 8 mBMKO). D, RT-qPCR analysis of glucose metabolic pathway gene expression in TA of control and mBMKO mice (N = 5–6). *,**P < .05 or .01 mBMKO vs mBMCtr, $,$ $P < .05 or .01 ZT2 vs ZT10 by two-way ANOVA. E, ChIP-qPCR analysis of Bmal1 chromatin occupancy of the E-box element in the regulatory regions of glucose metabolic genes in C2C12 differentiated myotube (n = 4). The fold of enrichment is expressed as a percent of 1% input normalized to IgG. F, Glucose oxidation rate as measured in soleus muscle of control and mBMKO mice using C-14 glucose under insulin-stimulated condition. n = 6/group. G, Plasma glucose levels under fasting or refed state in control and mBMKO mice (n = 7 mBMCtr and n = 9 mBMKO)
Based on Bmal1 induction by feeding and insulin, we examined its potential involvement in glucose metabolism. Genes involved in insulin-responsive glucose transport (Glut4), Glut4 membrane translocation (Tbc1d1), intracellular glucose metabolism (Hk2), and pyruvate dehydrogenase phosphatase (Pdp1) were markedly down-regulated in Bmal1-deficient muscle (Figure 2D), suggesting attenuated glucose utilization. Interestingly, genes responsible for mitochondrial pyruvate oxidation (PKm, Pdh1) were not affected. We next determined whether altered glucose metabolic gene expression is mediated by Bmal1 transcriptional control, by examining Bmal1 chromatin occupancy on identified E- or E’-box in gene regulatory regions, as previously described.15 Robust Bmal1 association of Glut4, Tbc1d1, Hk2, and Pdp1 promoters were detected with enrichments comparable to that of a known Bmal1 target gene, Rev-erba (Figure 2E). Direct analysis of ex vivo glucose oxidation in isolated mBMKO soleus reveals that the loss of Bmal1 significantly attenuated C14-glucose oxidation by ~46% as compared to controls (Figure 2F). In line with the impaired glucose utilization in Bmal1-deficient muscle, fasting plasma glucose level in these mice was significantly elevated than mBMCtr (Figure 2G), indicating the impact of muscle Bmal1 deficiency on systemic glucose metabolism.
3.3 |. Bmal1-driven muscle clock is required for global glucose homeostasis
We next performed the glucose tolerance test (GTT) and low-dose hyperinsulinemic-euglycemic clamp to determine the contribution of muscle clock to systemic glucose homeostasis. Consistently higher fasting glucose was observed in mBMKO mice than controls on a regular chow diet (Figure 3A). Furthermore, the glucose level at 2 hours post glucose injection during GTT was significantly elevated (Figure 3A), suggesting glucose intolerance. Fasting blood glucose on a high-fat diet was also increased in mBMKO than mBMCtr mice when both cohorts developed significant insulin resistance (Figure 3B), although excursion curves showed a tendency of higher glucose levels in mBMKO mice. Surprisingly, under a steady-state low-dose hyperinsulinemic-euglycemic clamp, the overall glucose infusion rate or disposal rate did not differ significantly between mBMKO mice and controls (Figure 3C). However, glucose uptake in mBMKO TA and EDL muscles was markedly reduced (Figure 3D), consistent with ex vivo glucose oxidation rate. The ability of muscle Bmal1-deficient mice to maintain normal glucose disposal rate despite significant defective muscle uptake suggests potential compensation by other tissues. Indeed, mBMKO mice display 30%−50% higher glucose uptake in fat depots than controls, including both epidydimal white adipose tissue (eWAT) and inguinal adipose tissue (iWAT), (Figure 3E). Surprisingly, under the low-dose insulin clamp when hepatic glucose production was reduced to 1/3 of basal level, there was a lack of suppression of hepatic glucose production from mBMKO (Figure 3F). These data indicate that attenuated muscle glucose disposal in muscle Bmal1-deficient mice was largely compensated by adipose tissue uptake, and lack of suppression of hepatic glucose production leads to fasting hyperglycemia. In addition, we determined insulin signaling activity in these tissues. In agreement with the findings of impaired glucose uptake in skeletal muscle, insulin-stimulated Akt phosphorylation was significantly impaired in Bmal1-deficient muscle as compared to the robust response of the controls (Figure 3G). In contrast, there was ~3-fold higher insulin-induced Akt signaling was found in eWAT of mBMKO mice (Figure 3H), suggesting a compensatory mechanism of augmented insulin sensitivity in mBMKO fat depot to maintain systemic glucose metabolism.
FIGURE 3.
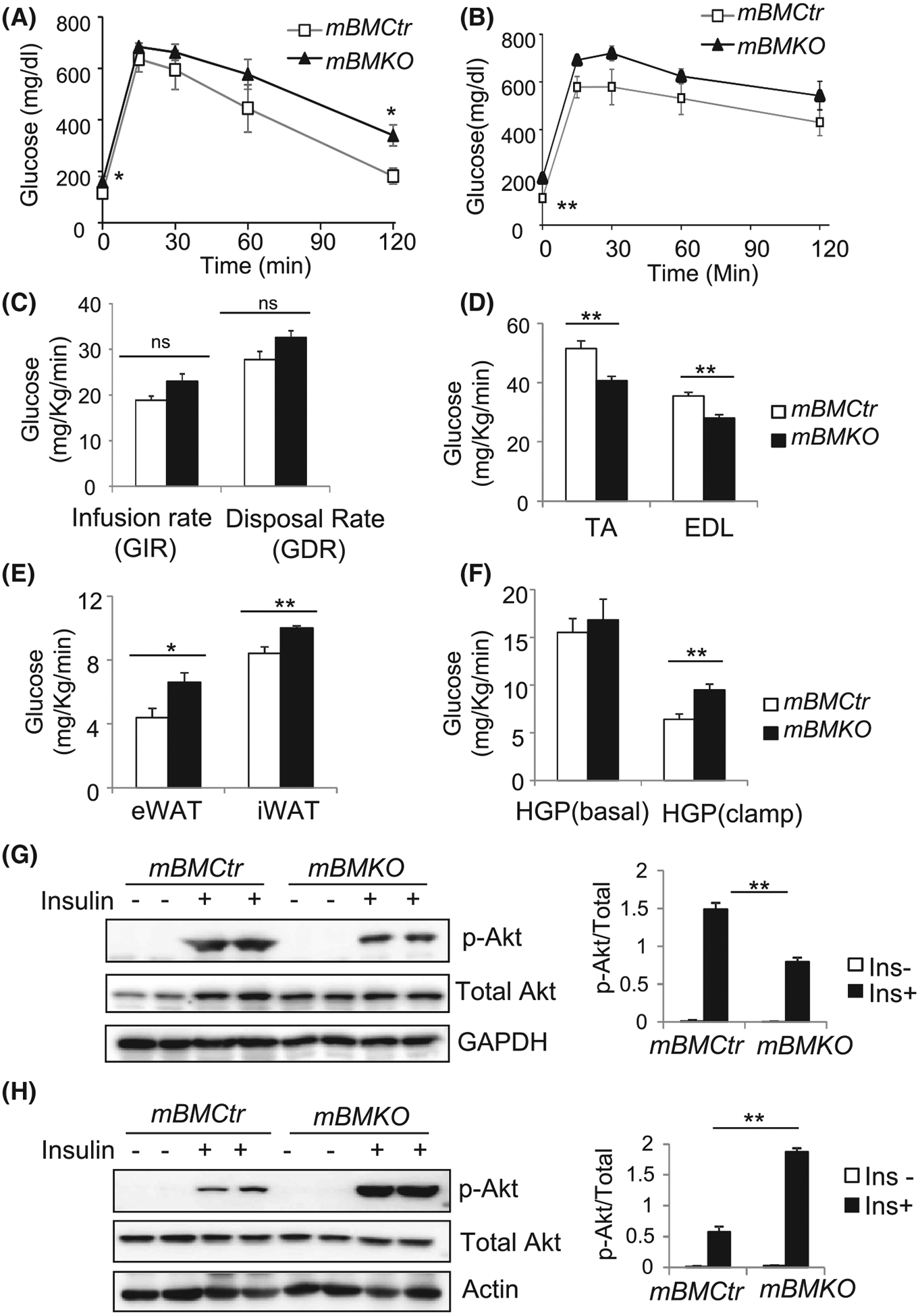
Glucose homeostasis in mice with muscle Bmal1 ablation. A and B, Glucose level during the IP glucose tolerance test of mice on regular chow diet (A), or after 12 weeks of 45% high-fat diet (B) in mBMKO (n = 9) vs mBMCtr (n = 10) mice. C-F, Low-dose hyperinsulinemic-euglycemic clamp study in regular chow-fed 12-weeks old mBMKO vs mBMCtr mice (n = 6/group). Steady-state glucose infusion rate and disposal rate (C), glucose uptake in TA and extensor digitalis longus (EDL) muscle (D), glucose uptake in visceral epididymal white adipose tissue (eWAT) and inguinal subcutaneous white adipose tissue (iWAT) (E), and hepatic glucose production under basal and low-dose clamp condition (F). *, **P < .05 or .01 mBMKO vs mBMCtr by unpaired Student’s t test. G and H, Insulin signaling activity in skeletal muscle (G) and eWAT (H) as assessed by immunoblot of Akt phosphorylation 20 minutes after IP insulin injection (5 mg/Kg). Pooled protein samples of n = 3/lane
3.4 |. Activation of energy-sensing pathways and impaired fasting to feeding metabolic fuel transition in Bmal1-deficient skeletal muscle
As Bmal1 is highly induced by feeding signals, we postulate that the loss of this sensing function may disrupt metabolic adaptation required for fasting to feeding transition. We first determined whether cellular energy status as reflected by NAD+/NADH levels are altered between fasting and refeeding in Bmal1-deficient muscle. Interestingly, despite a similar total level of NAD+ and NADH to mBMCtr mice at either fed or fasted state (Figure 4A), the increase in the NADH level upon feeding observed in mBMCtr is lost due to significantly lower levels upon refeeding (Figure 4B). In addition, a fasting-driven increase in the NAD+/NADH ratio was markedly attenuated in the mBMKO muscle (Figure 4C). Thus, Bmal1-deficient muscles exhibit a persistent energy-depleting state and energy-sensing pathways driving the fasting to feeding switch also display significantly impaired response (Figure D,E). PGC1α, the master regulator of fasting-driven muscle oxidative metabolism,38 was markedly induced in Bmal1-deficient muscle under the fasted condition and remained elevated after refeeding as compared to controls (Figure 4D). Similarly, the phosphorylation of AMP kinase (AMPK), a kinase activated by energy depletion,22 was persistently high with significantly attenuated feeding-induced inhibition (Figure 4E). In Bmal1-deficient muscle, analysis of the major insulin-sensitive glucose transporter Glut4 protein revealed near-complete loss of induction by feeding in soleus as compared to controls (Figure 4F). In contrast, PDK4, the key enzymatic switch to promote fat utilization during fasting,39–41 lost its fasting-induced up-regulation as seen in mBMCtr mice in both TA and soleus leading to persistent expression. Gene expression profile analysis of metabolic enzymes involved in glucose and fat oxidation demonstrated impaired induction glucose metabolic pathway with preferential fat oxidation program (Figure 4G,H). Glucose metabolic genes identified as Bmal1 transcriptional targets (Figure 2E), including Glut4, Tbc1d1, and Hk2, display significantly reduced expression levels with complete loss of feeding-induced induction of Tbc1d1 in mBMKO muscle (Figure 4G). Consistent with its protein levels, mRNA of Pdk4 was markedly induced in fasted Bmal1-null TA muscle as compared to controls (Figure 4H), together with the up-regulation of fatty acid oxidation genes medium-chain acyl-CoA dehydrogenase (Acadm) and carnitine palmitoyl-transferase Cpt2. Together these results reveal an energy-depleted state resembling persistent fasting in skeletal muscle lacking Bmal1, with severe loss of fasting to feeding metabolic fuel adaptation.
FIGURE 4.
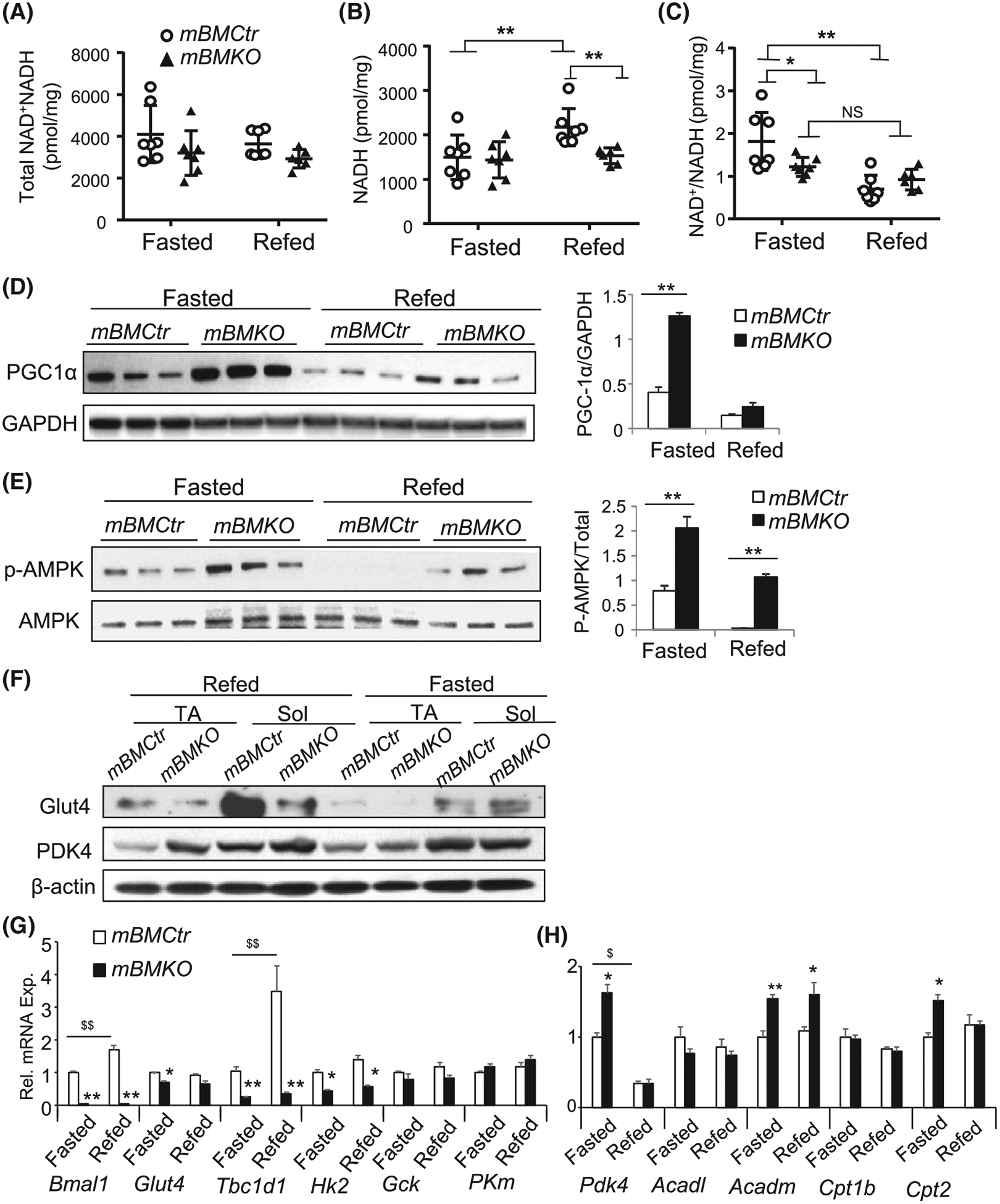
Activation of energy-sensing signaling pathways and impaired fasting-to-feeding metabolic transition in Bmal1-deficient muscle. A-C, Skeletal muscle total NAD++NADH (A), NADH levels (B) and NAD+/NADH ratio (C) in TA muscle of mBMCtr and mBMKO mice in response to feeding and fasting (n = 6–7/group). D and E, Immunoblot analysis of PGC-1a protein expression (D), and AMPK phosphorylation (E) under fasting or refeeding conditions in mBMCtr and mBMKO mice (n = 3/group). F, Immunoblot analysis of Glut4 and PDK4 protein regulations, and (G, H) RT-qPCR analysis of genes involved in fat oxidation (G) and glucose metabolism (H) in response to fasting and refeeding in mBMCtr (n = 6) and mBMKO (n = 5) mice. *,**P < .05 or .01 mBMKO vs mBMCtr; $,$ $P < .05 & .01 fasted vs refed by two-way ANOVA
3.5 |. Loss of Bmal1 in muscle induced fatty acid oxidation
Based on elevated PGC1α and AMPK phosphorylation in Bmal1-deficient muscle, we next determined whether activation of these energy-sensing pathways induces fatty acid oxidation to potentially compensate for the attenuated glucose utilization. Acetyl-CoA carboxylase (ACC) is a direct downstream target of AMPK resulting in its inactivation, which consequently inhibits fatty acid synthesis cascade, while promoting oxidation.42,43 In comparison to mBMCtr, ACC phosphorylation was increased in fasted mBMKO muscle whereas fatty acid synthase (FASN) protein expression was markedly reduced (Figure 5A). Furthermore, direct ex vivo measurement of fatty acid oxidation in soleus muscle using C14-palmitate revealed a ~2.2-fold higher rate in mBMKO mice than the controls (Figure 5B), while both can be stimulated similarly by epinephrine and inhibited by Rotenone. Analysis of the fat oxidation gene program reveals persistently elevated Pdk4 transcript at ZT2 and ZT10 in Bmal1-deficient muscle with the loss of its normal diurnal expression pattern as seen in mBMCtr mice (Figure 5C). In addition, concerted up-regulation of various steps involved in fatty acid metabolism, including transport (Lpl, Cd36 Cpt-1b, Cpt2) and oxidation (Acox1, Acadl, Acads) may underlie the increased fat oxidation rate (Figure 5D). We postulated that the persistent fat oxidation over glucose metabolism in mBMKO muscle may lead to oxidative muscle fiber type switching. Indeed, a shift toward oxidative fiber-type with more abundant oxidative myosin isoform Type IIA but diminished glycolytic Type IIB fibers was detected by immunostaining in TA muscle (Figure 5E) and further validated by total protein analysis (Figure 5F). Consistent with preferential fat oxidation in Bmal1-deficient muscle, a moderate yet significantly reduced respiratory exchange ratio (RER) in the post-absorptive dark phase (9 PM −3 AM), but not in the light phase, was found in mBMKO mice by indirect calorimetry (Figure 5G,H). Interestingly, the RER peak in mBMKO was delayed as compared to that of the controls, potentially due to the impaired glucose oxidation leading to a slower rate of substrate switching from fat to glucose. Moreover, overall oxygen consumption, activity levels, and food intake (Figure S2) did not differ from controls with comparable light and dark phase partitioning.
FIGURE 5.
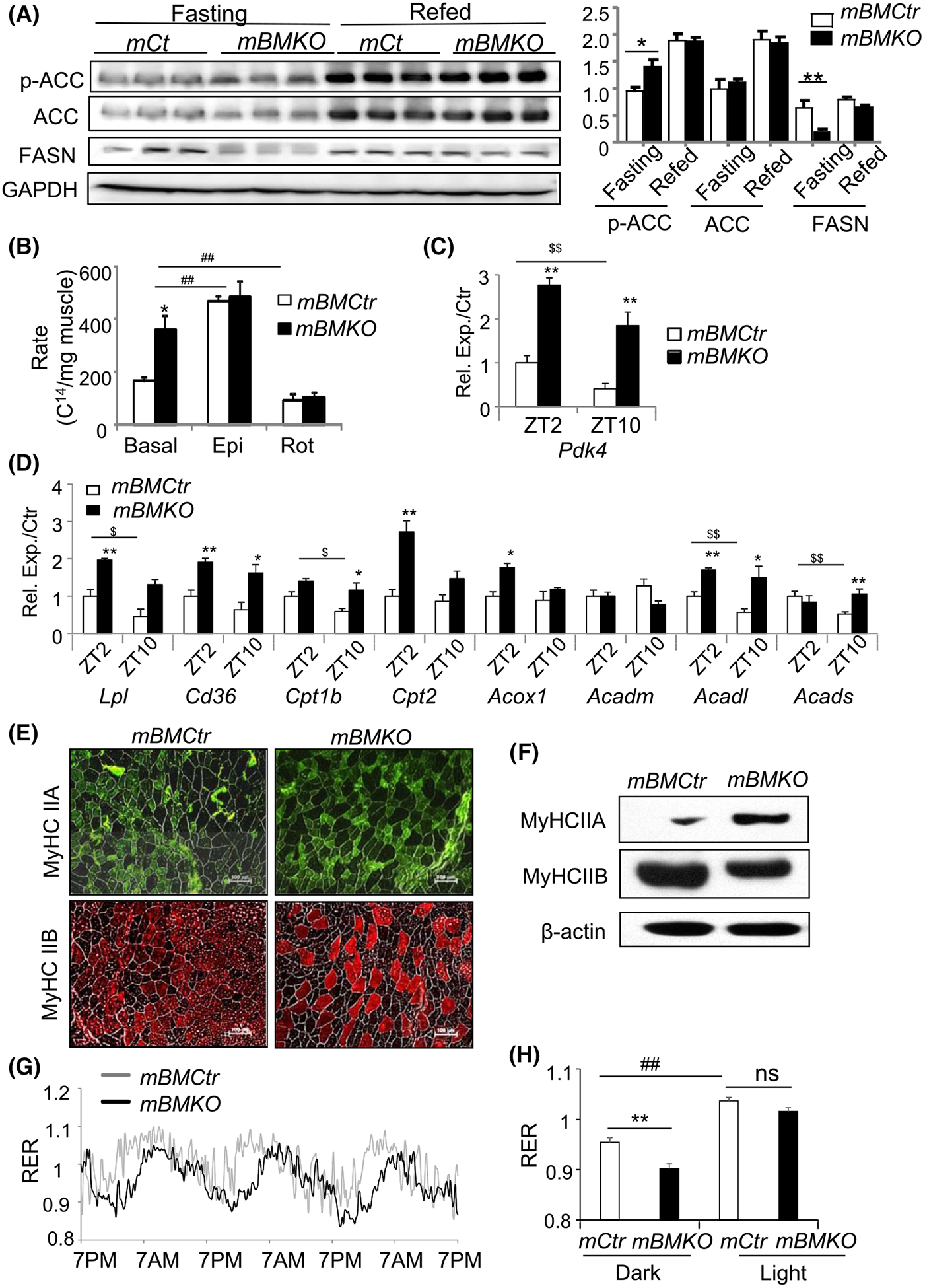
Enhanced fatty acid metabolism in Bmal1-deficinet muscle. A, Immunoblot analysis of phosphorylation of acetyl-coA carboxylase (ACC) and fatty acid synthetase (FASN) protein expression in fasting and refed mBMCtr and mBMKO mice (n = 3/group). B, Ex vivo fatty acid β-oxidation rate of soleus muscle using C14-palmitic acid substrate under basal non-stimulated, epinephrin-stimulated states and rotenone inhibition (n = 6/group). **P < .01 mBMKO vs mBMCtr; ##P < .05 & .01 treatment vs basal by two-way ANOVA. C and D, RT-qPCR analysis of Pdk4 and additional genes involved in lipid metabolism in TA muscle of mBMKO (n = 5) vs mBMCtr (n = 6) at ZT2 and ZT10. *,**P < .01 mBMKO vs mBMCtr; $,$ $P < .05 & .01 ZT2 vs ZT10 by ANOVA. E and F, Representative images of immunofluorescence staining of myosin heavy chain (MyHC) isoforms Type IIA and IIB fiber types (E), and immunoblot analysis (F) of pooled samples in TA of mBMCtr (n = 5) and mBMKO (n = 4) mice. G and H, Representative average tracing of 3-day respiratory exchange ratio (RER) monitoring by CLAMS calorimetry (G), and quantification of dark (9 PM-3 AM) and light period (9 AM-3 PM) (H) in mBMCtr and mBMKO mice (n = 6/group). **P < .01 mBMKO vs mBMCtr, and ##<.01 dark vs light period by unpaired Student’s t test
3.6 |. Induction of fat oxidation in mBMKO muscle reduces systemic lipid levels and prevents hepatic steatosis
Metabolic activities of skeletal muscle critically influence systemic metabolic homeostasis.20,41 We tested whether augmented fat oxidation in muscle Bmal1 loss can drive global lipid homeostasis. Strikingly, muscle Bmal1-deficient mice display nearly 40%−50% reductions of circulating lipid levels, including free fatty acid, triglyceride, and glycerol (Figure 6A). These effects on lipids are most evident in the fasted state but not significantly altered following refeeding, while serum cholesterol does not differ between mBMKO and controls. The reduction of circulating lipids indicates that persistent fat oxidation in mBMKO muscle reduces global lipid flux, and we postulate this may prevent its hepatic accumulation. Following overnight fasting, in comparison to the normal lipid-laden appearance in mBMCtr liver due to fat mobilization to the liver for secretion (Figure 6B), mBMKO liver retains a strikingly reddish color, suggesting lack of lipid accumulation. Oil-red-O staining confirmed the marked reduction of lipids (Figure 6C). Quantitative analysis revealed a ~58% lower hepatic triglyceride content in mBMKO liver at fasting but not at the refed state (Figure 6D). In addition, a diurnal rhythm of hepatic lipid accumulation that is significantly lower at ZT10 than ZT2 was detected in normal controls, likely due to a fasting state at ZT2 following early evening feeding. This normal rhythm in hepatic lipid storage was abolished in muscle Bmal1-deficient mice with persistent low lipid content (Figure 6E), and reduced lipid staining at ZT2 further confirmed this finding (Figure 6G, upper panel). When mBMKO mice were challenged by high-fat diet for 10 weeks, in comparison to an expected robust ~2-fold increase in hepatic triglyceride content in normal controls as compared to the chow diet, they were significantly protected against steatosis with ~30% less lipid accumulation at either ZT2 or ZT10 examined as shown by the analysis of total content (Figure 6F) or staining (Figure 6G, lower panel). The findings of our study are summarized in a working model illustrated in Figure 6H, which in aggregate, demonstrates a key function of the muscle Bmal1-driven clock as a metabolic sensor required for the orchestration of glucose and lipid metabolism to maintain homeostasis.
FIGURE 6.
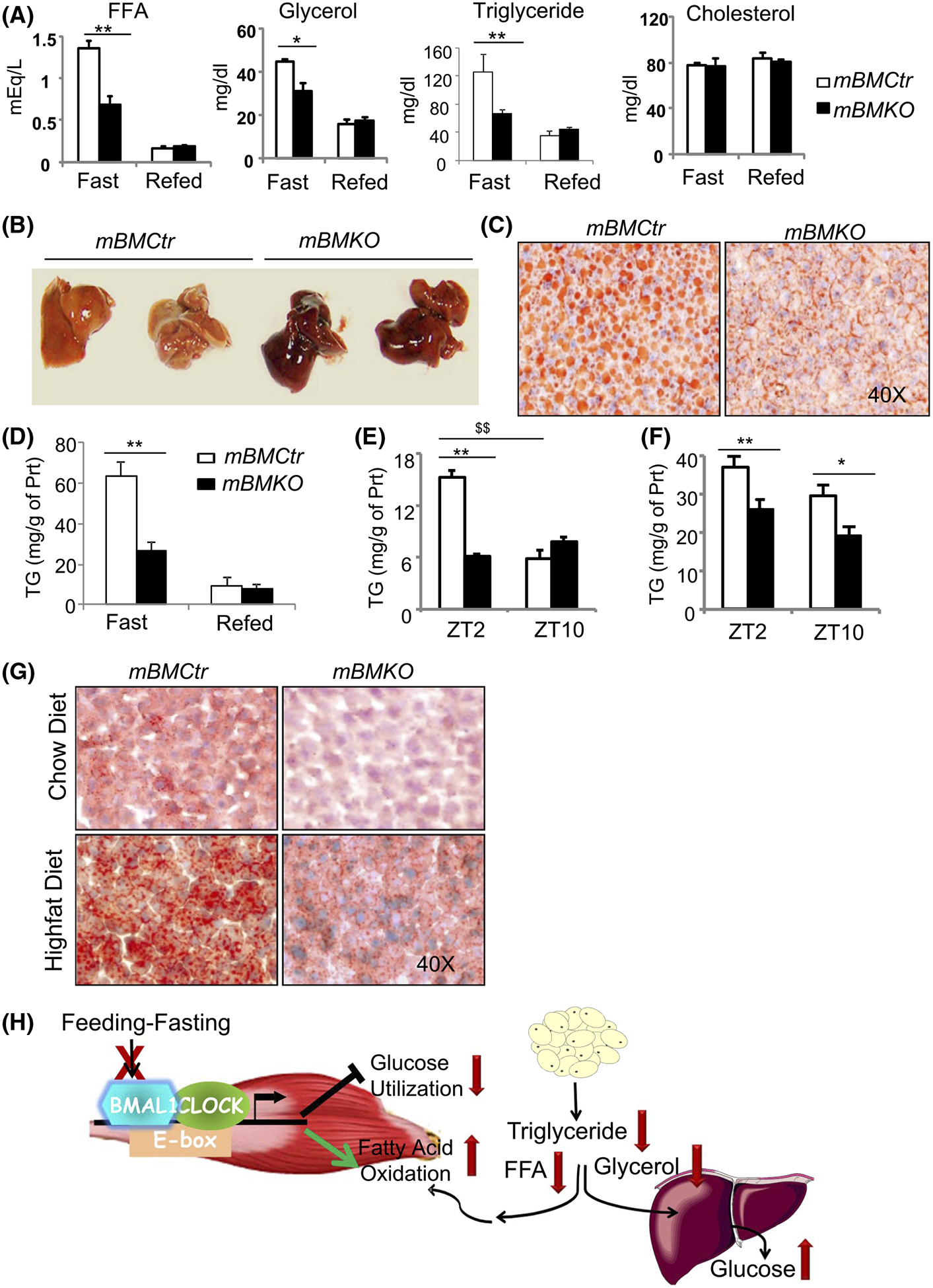
Muscle Bmal1 ablation reduces circulating lipid levels and hepatic accumulation. A, The impact of muscle Bmal1 deficiency on systemic lipids levels, including free fatty acid (FAA), glycerol, triglyceride, and cholesterol in mBMCtr (n = 8) and mBMKO mice (n = 10) under fasted or refed conditions. B and C, Representative images of gross appearance (B), and oil-red-O staining (C) of mBMCtr and mBMKO mice after 16 hour overnight fasting. D, Hepatic triglyceride content analysis of mBMCtr (n = 6) and mBMKO mice (n = 8) during fasting and refeeding. E and F, Hepatic triglyceride content at ZT2 and ZT10 under normal chow (E), or after high-fat diet for 10 weeks (F), in mBMCtr (n = 7) and mBMKO mice (n = 6), with representative images of oil-red-O staining at ZT2 (G). H, Proposed working model of skeletal muscle Bmal1-driven clock in the regulation of glucose and lipid homeostasis. Bmal1 is induced by feeding-associated insulin stimulation. Loss of Bmal1 in myocytes impairs response to feeding with reduced glucose utilization and induction of fat oxidation. Altered metabolic oxidation in the Bmal1-deficient muscle significantly attenuates global glucose disposal but depletes circulating lipids and its hepatic storage
4 |. DISCUSSION
Metabolic adaptation in accordance with daily feeding and fasting cues in skeletal muscle is essential for nutrient homeostasis. However, whether the tissue-intrinsic muscle clock is involved in coordinating this response is largely unknown. Using a model with selective clock ablation in mature myocytes, our study demonstrated a critical function of the Bmal1-driven clock in sensing nutrient flux to orchestrate global glucose and lipid partitioning. As accumulating evidence suggests that clock dysregulation impairs metabolic homeostasis leading to the development of insulin resistance,7–9 our findings implicate perturbation of the skeletal muscle tissue clock as a significant contributor to clock disruption-associated metabolic disorders. Our findings, as summarized in Figure 6H, demonstrate that muscle Bmal1-driven clock is capable of sensing feeding cues to promote glucose utilization through direct transcriptional control of the glucose metabolic pathway. Disruption of this sensing mechanism in muscle attenuates glucose metabolism with a profound metabolic shift toward FA oxidation, resembling a persistent fasting state that augments hepatic glucose output but depletes hepatic lipid storage.
As an evolutionarily conserved mechanism to anticipate and adapt to environmental changes, feeding and its associated metabolic signals entrain the circadian clock system in peripheral tissues outside the central clock.10 Recent transcriptomic and metabolomic analyses reveal the global temporal coordination of metabolism in distinct tissues that involve clock circuits.12 However, the precise mechanisms underlying clock orchestration of metabolic processes to adapt to feeding-fasting cycles remain unclear. We found that Bmal1 was robustly induced by feeding in muscle and responds to insulin whereas clock repressor Rev-erbα was stimulated upon fasting, suggesting that the clock machinery may constitute, at least in part, a sensing mechanism for nutrient oscillation during feeding-fasting transitions. Corroborating this notion, the loss of Bmal1 in muscle impairs the transition from fatty acid metabolism to glucose oxidation requisite for adaptation to feeding, displaying the persistent up-regulation of fatty acid oxidative gene program even at the fed state. The nutrient-sensing function of Bmal1 in skeletal muscle is in keeping with food as an entrainment cue for peripheral clocks,10,23 although the precise signals mediating this response warrants further investigation. Insulin induced by feeding could be a major clock input signal with its downstream signaling potentially modulating clock gene transcription, as our data suggest. Meng et al recently discovered an important direct glucose-sensing mechanism in muscle mediated by a HDAC5-Baf60c-Deptor-Akt cascade.44 Our findings of insulin-responsive Bmal1 induction may function synergistically with this pathway, in parallel or through cross-talk between Bmal1 and Baf60c-mediated transcription programs, to facilitate glucose disposal. In addition, metabolic cues induced by fasting may also help entrain the muscle clock,45 although palmitate we tested does not induce clock gene expression. These intriguing possibilities warrant future investigations. A clock metabolic sensing mechanism may also apply to nutrient adaptations in many tissues such as the pancreatic β-cells, in which the lack of clock regulation severely blunts glucose-responsive insulin secretion.4,25
A large body of epidemiological and experimental studies has established that circadian disruption impairs metabolic homeostasis.7–9 However, our current understanding of the specific contributions of tissue-resident clocks to whole-body metabolic regulation is limited. We found surprisingly that the effects of the myocyte loss of Bmal1 are not confined to defective glucose metabolism with persistent lipid oxidation in the muscle. The loss of clock function in muscle led to striking reductions of circulating lipid levels and its hepatic storage, highlighting an indispensable metabolic-sensing role of the muscle-resident clock to maintain inter-organ nutrient flux and homeostasis. It is possible that muscle clock ablation on lipid homeostasis may involve partitioning in adipose tissue, which we will explore in our immediate future studies. Our findings in aggregate suggest a new concept of tissue-intrinsic clock as metabolic sensors to orchestrate tissue-specific metabolic processes required for global homeostasis. It is also conceivable that impaired metabolic-sensing mechanism due to tissue clock dys-synchrony in circadian misalignment may contribute to the overall metabolic dysregulations that predispose to the development of obesity and diabetes. Specific circadian etiologies and mechanisms underlying metabolic diseases, particularly a muscle-liver nutrient partitioning abnormality as suggested by our study, warrants further investigations.
Several previous studies using distinct Cre transgenic lines to induce the loss of muscle clock yield largely similar findings of attenuated glucose oxidation in skeletal muscle,46–49 highlighting the reproducibility of our current findings. These studies are mostly limited to glucose metabolism in skeletal muscle46,47 or focused on global transcriptional and metabolomic profiling.48,49 We identify the new function of muscle clock in metabolic-sensing beyond glucose metabolism that is required for muscle-liver nutrient flux and global homeostasis, with its loss profoundly altering metabolic substrate adaptation, lipid homeostasis and its storage in the liver. In addition, although Dyar et al46 initially reported the disruption of glucose metabolic pathway regulation with muscle fiber type switch in the Bmal1-deficient mouse model using MLC-Cre-mediated deletion, the underlying mechanism was not addressed. Our results reveal that Bmal1 direct transcription control of key steps of glucose metabolism mediates its regulation on glucose disposal. Furthermore, hyperinsulinemic-euglycemic clamp performed at steady-state determined the impact of muscle clock deficiency on other insulin-sensitive tissues, which uncover a key compensatory mechanism of enhanced adipose depot uptake for impaired muscle glucose utilization. This mechanism may account for moderate changes in glucose homeostasis observed in distinct muscle Bmal1-deficient models,46–49 potentially providing new insight into etiologies underlying circadian misalignment-induced metabolic disorders. Similar adipose tissue compensation for the loss of muscle glucose disposal has been well-documented in mice lacking the insulin receptor in skeletal muscle.37,50 The clamp study also revealed enhanced hepatic glucose output that may contribute to, at least in part, fasting hyperglycemia in muscle Bmal1-deficient mice. Most importantly, by exploring the muscle clock response to metabolic stimuli, particularly through the fasting-refeeding regimen, we uncovered the muscle clock function in sensing feeding cues that coordinates global nutrient partitioning. With the prevalence of frequent circadian misalignment in a modern society, these mechanistic insights implicate potential contributions of tissue-intrinsic clock circuits to the global metabolic dysregulations in shiftwork or “social jet-leg”.51
In summary, our study uncovered a key role of the skeletal muscle-intrinsic clock in sensing metabolic signals to orchestrate whole-body nutrient homeostasis. Elucidation of muscle clock-controlled metabolic pathways that impacts global metabolic homeostasis sheds lights on tissue-intrinsic clock mechanisms mediating clock disruption-induced metabolic disorders, which may ultimately lead to the discovery of new targets for therapeutic interventions toward circadian etiologies in metabolic diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Metabolic Phenotyping Core Laboratory at Baylor College of Medicine for its expert technical support in insulin clamp study.
Funding information
This project was supported by grants from the National Institute of Health 1R01DK112794, Muscular Dystrophy Association 381294, and American Heart Association 17GRNT33370012 to KM; and National Institute of Health grant DK097160-01 to VY
Abbreviations:
- Bmal1
brain and muscle Arnt-like protein 1
- CLOCK
circadian locomotor output cycles kaput
- MyHC
myosin heavy chain
- ROR
RAR-related orphan receptor
Footnotes
CONFLICT OF INTEREST
I certify that neither I nor my co-authors have a conflict of interest that is relevant to the subject matter or materials included in this work. No financial or material disclosers to report.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Panda S, Antoch MP, Miller BH, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. [DOI] [PubMed] [Google Scholar]
- 2.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134:728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee J, Moulik M, Fang Z, et al. Bmal1 and β-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced β-cell failure in mice. Mol Cell Biol. 2013;33:2327–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller BH, McDearmon EL, Panda S, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci U S A. 2007;104:3342–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zvonic S, Ptitsyn AA, Conrad SA, et al. Characterization of peripheral circadian clocks in adipose tissues. Diabetes. 2006;55:962–970. [DOI] [PubMed] [Google Scholar]
- 7.Buxton OM, Cain SW, O’Connor SP, et al. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci Transl Med. 2012;4:129ra143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, McEwen BS. Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc Natl Acad Sci U S A. 2011;108:1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheer FA, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A. 2009;106:4453–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14:2950–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asher G, Reinke H, Altmeyer M, Gutierrez-Arcelus M, Hottiger MO, Schibler U. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell. 2010;142:943–953. [DOI] [PubMed] [Google Scholar]
- 12.Dyar KA, Lutter D, Artati A, et al. Atlas of circadian metabolism reveals system-wide coordination and communication between clocks. Cell. 2018;174:1571–1585.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18:164–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bunger MK, Wilsbacher LD, Moran SM, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatterjee S, Nam D, Guo B, et al. Brain and muscle Arnt-like 1 is a key regulator of myogenesis. J Cell Sci. 2013;126:2213–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 2012;35:445–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews JL, Zhang X, McCarthy JJ, et al. CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proc Natl Acad Sci U S A. 2010;107:19090–19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chatterjee S, Yin H, Nam D, Li Y, Ma K. Brain and muscle Arnt-like 1 promotes skeletal muscle regeneration through satellite cell expansion. Exp Cell Res. 2014. [DOI] [PubMed] [Google Scholar]
- 19.Chatterjee S, Yin H, Li W, Lee J, Yechoor VK, Ma K. The nuclear receptor and clock repressor Rev-erbα suppresses myogenesis. Sci Rep. 2019;9:4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zurlo F, Larson K, Bogardus C, Ravussin E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Investig. 1990;86:1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007. [DOI] [PubMed] [Google Scholar]
- 22.de Lange P, Moreno M, Silvestri E, Lombardi A, Goglia F, Lanni A. Fuel economy in food-deprived skeletal muscle: signaling pathways and regulatory mechanisms. FASEB Journal. 2007;21:3431–3441. [DOI] [PubMed] [Google Scholar]
- 23.Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M. Entrainment of the circadian clock in the liver by feeding. Science 2001;291:490–493. [DOI] [PubMed] [Google Scholar]
- 24.Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A. 2008;105:15172–15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcheva B, Ramsey KM, Buhr ED, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo B, Chatterjee S, Li L, et al. The clock gene, brain and muscle Arnt-like 1, regulates adipogenesis via Wnt signaling pathway. FASEB J. 2012;26:3453–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nam D, Guo B, Chatterjee S, et al. The adipocyte clock controls brown adipogenesis through the TGF-β and BMP signaling pathways. J Cell Sci. 2015;128:1835–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDearmon EL, Patel KN, Ko CH, et al. Dissecting the functions of the mammalian clock protein BMAL1 by tissue-specific rescue in mice. Science 2006;314:1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woldt E, Sebti Y, Solt LA, et al. Rev-erb-α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat Med. 2013;19:1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma K, Cabrero A, Saha PK, et al. Increased β-oxidation but no insulin resistance or glucose intolerance in mice lacking adiponectin. J Biol Chem. 2002;277:34658–34661. [DOI] [PubMed] [Google Scholar]
- 31.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Investig. 2006;116:1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wende AR, Huss JM, Schaeffer PJ, Giguere V, Kelly DP. PGC-1alpha coactivates PDK4 gene expression via the orphan nuclear receptor ERRα: a mechanism for transcriptional control of muscle glucose metabolism. Mol Cell Biol. 2005;25:10684–10694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caruso M, Miele C, Formisano P, et al. In skeletal muscle, glucose storage and oxidation are differentially impaired by the IR1152 mutant receptor. The Journal of biological chemistry. 1997;272:7290–7297. [DOI] [PubMed] [Google Scholar]
- 34.Xu W, Zhou H, Xuan H, Saha P, Wang G, Chen W. Novel metabolic disorders in skeletal muscle of Lipodystrophic Bscl2/Seipin deficient mice. Mol Cell Endocrinol. 2019;482:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujita Y, Kojima H, Hidaka H, Fujimiya M, Kashiwagi A, Kikkawa R. Increased intestinal glucose absorption and postprandial hyperglycaemia at the early step of glucose intolerance in Otsuka Long-Evans Tokushima Fatty rats. Diabetologia. 1998;41:1459–1466. [DOI] [PubMed] [Google Scholar]
- 36.Kraegen EW, James DE, Jenkins AB, Chisholm DJ. Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. The American journal of physiology. 1985;248:E353–362. [DOI] [PubMed] [Google Scholar]
- 37.Bruning JC, Michael MD, Winnay JN, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2:559–569. [DOI] [PubMed] [Google Scholar]
- 38.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Investig. 2006;116:615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roche TE, Hiromasa Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol Life Sci. 2007;64:830–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugden MC, Holness MJ. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem. 2006;112:139–149. [DOI] [PubMed] [Google Scholar]
- 41.Sugden MC, Bulmer K, Holness MJ. Fuel-sensing mechanisms integrating lipid and carbohydrate utilization. Biochem Soc Trans. 2001;29:272–278. [DOI] [PubMed] [Google Scholar]
- 42.Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science, 2001;291:2613–2616. [DOI] [PubMed] [Google Scholar]
- 43.Glund S, Schoelch C, Thomas L, et al. Inhibition of acetyl-CoA carboxylase 2 enhances skeletal muscle fatty acid oxidation and improves whole-body glucose homeostasis in db/db mice. Diabetologia. 2012;55:2044–2053. [DOI] [PubMed] [Google Scholar]
- 44.Meng ZX, Gong J, Chen Z, et al. Glucose Sensing by Skeletal Myocytes Couples Nutrient Signaling to Systemic Homeostasis. Mol Cell. 2017;66:332–344.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kinouchi K, Magnan C, Ceglia N, et al. Fasting imparts a switch to alternative daily pathways in liver and muscle. Cell Rep. 2018;25:3299–3314.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dyar KA, Ciciliot S, Wright LE, et al. Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Mol Metab. 2014;3:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harfmann BD, Schroder EA, Kachman MT, Hodge BA, Zhang X, Esser KA. Muscle-specific loss of Bmal1 leads to disrupted tissue glucose metabolism and systemic glucose homeostasis. Skelet Muscle. 2016;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dyar KA, Hubert MJ, Mir AA, et al. Transcriptional programming of lipid and amino acid metabolism by the skeletal muscle circadian clock. PLoS Biol. 2018;16:e2005886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hodge BA, Wen Y, Riley LA, et al. The endogenous molecular clock orchestrates the temporal separation of substrate metabolism in skeletal muscle. Skelet Muscle. 2015;5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim JK, Michael MD, Previs SF, et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Investig. 2000;105:1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stenvers DJ, Scheer F, Schrauwen P, la Fleur SE, Kalsbeek A. Circadian clocks and insulin resistance. Nat Rev Endocrinol. 2019;15:75–89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.