

Summary
Rheumatoid arthritis (RA) is a chronic autoimmune disease which causes degradation of cartilage and bone. It is well appreciated that the pathogenic hallmark of RA is the mass influx of inflammatory cells into the joint. However, the role that dendritic cells (DC) may play in this inflammatory milieu is still relatively unexplored. Moreover, the contribution this unique synovial microenvironment has on DC maturation is still unknown. Using monocyte‐derived DC (MoDC), we established an in‐vitro model to recapitulate the synovial microenvironment to explore DC maturation. MoDC treated with conditioned media from ex‐vivo synovial tissue biopsy cultures [explant‐conditioned media (ECM)] have increased expression of proinflammatory cytokines, chemokines and adhesion molecules. ECM DC have increased expression of CD83 and CC‐chemokine receptor (CCR)7 and decreased expression of CCR5 and phagocytic capacity, suggestive of heightened DC maturation. ECM‐induced maturation is concomitant with altered cellular bioenergetics, whereby increased expression of glycolytic genes and increased glucose uptake are observed in ECM DC. Collectively, this results in a metabolic shift in DC metabolism in favour of glycolysis. These adaptations are in‐part mediated via signal transducer and activator of transcription‐3 (STAT‐3), as demonstrated by decreased expression of proinflammatory cytokines and glycolytic genes in ECM DC in response to STAT‐3 inhibition. Finally, to translate these data to a more in‐vivo clinically relevant setting, RNA‐seq was performed on RA synovial fluid and peripheral blood. We identified enhanced expression of a number of glycolytic genes in synovial CD1c+ DC compared to CD1c+ DC in circulation. Collectively, our data suggest that the synovial microenvironment in RA contributes to DC maturation and metabolic reprogramming.
Keywords: dendritic cell, JAK–STAT, metabolism, rheumatoid arthritis
This study demonstrates that the unique RA synovial microenvironment can induce DC maturation in vitro in addition to altering cellular bioenergetics in favour of glycolysis. We demonstrate a role for STAT3 in mediating these synovial‐induced changes at both the maturation and metabolic level and provide evidence that these changes are reflected in vivo in synovial DC.

Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease principally affecting synovial joints. It is characterized by dysregulated angiogenesis, leucocyte infiltration and lining layer proliferation, which transforms the synovium into an invasive tumour‐like pannus [1, 2]. Immune cells such as monocytes, lymphocytes and others are actively attracted into the synovial tissue, where they undergo further activation and differentiation. Monocytes differentiate into macrophages or dendritic cells (DC) under the influence of tissue‐specific factors. DC are central players in the control of both the innate and adaptive immune responses as a result of their antigen‐presenting capabilities. Distinct populations of DC have been identified, including those in peripheral blood – myeloid DC (mDC) and plasmacytoid DC (pDC) – while a population of tissue‐resident DC have also been identified in synovial tissue. The synovial microvasculature is highly dysregulated; therefore, despite increased blood flow, the increased metabolic requirements outpace the vascular supply, thus resulting in a profoundly hypoxic microenvironment [3]. Hypoxia itself drives further cytokine production, immune cell activation and proliferation, thus the increased metabolic requirements are associated with mitochondrial dysfunction and altered cellular bioenergetics, characterized by a shift towards glycolysis within the joint [4, 5].
Previous studies have demonstrated enhanced glycolysis in DC following Toll‐like receptor (TLR) stimulation [6, 7]. TLR‐activated DC undergo transcriptional reprogramming characterized by increased expression of co‐stimulatory markers and proinflammatory cytokines, increased chemotaxis and the ability to present antigen to T cells. Enhanced glycolysis is required for these DC effects, which is achieved through the rapid generation of adenosine triphosphate (ATP) and biosynthetic intermediates, thus facilitating their effector functions [8, 9]. Evidence of altered cellular bioenergetics has also been demonstrated in the context of the inflamed RA synovium, where increased expression of glycolytic enzymes and glucose transporters has been reported in RA synovial tissue [10, 11, 12]. Moreover, evidence of mitochondrial dysfunction in the RA synovium has also been reported, thus supporting a metabolic switch to glycolysis [13, 14].
The role of DC in RA has remained a relatively under‐explored avenue in human translational research. We previously reported the first description of the myeloid DC (mDC) subset, CD141+ DC in the RA synovium, that are transcriptionally and functionally distinct from their peripheral blood counterparts [15]. Moreover, synovial DC are enriched at the site of inflammation and are more mature than DC found in the circulation. This is consistent with studies reporting decreased mDC in the peripheral blood of patients with inflammatory arthritis (IA), with a concomitant increase in mDC within synovial fluid, further suggesting accumulation of DC within the inflamed synovium [16]. Furthermore, numerous studies have reported DC with elevated levels of maturation markers within the RA joint compared to circulating DCs [17, 18, 19].
These studies reinforce the hypothesis that the synovial microenvironment itself may induce phenotypical and maturation‐related adaptations in DC within the synovium. However, work in this area is challenging, due to the difficulty in obtaining acceptable numbers of cells to perform downstream functional analysis. Therefore, to further investigate the metabolic and activation pathways utilized by RA synovial DC, we established an in‐vitro monocyte‐derived DC model, whereby conditioned media from ex‐vivo RA synovial tissue explant cultures [termed explant‐conditioned media (ECM)] was used to treat healthy MoDC to recapitulate the synovial microenvironment. We hypothesized that the RA synovial microenvironment could induce DC maturation, in addition to altering DC cellular bioenergetics.
Methods
Patient recruitment, arthroscopies and ex‐vivo synovial explant culture
Patients with active RA were recruited from out‐patient clinics at the Department of Rheumatology, St Vincent’s University Hospital. Arthroscopy of the inflamed knee was performed under local anaesthetic, using a 2·7‐mm needle arthroscope (Richard Wolf, Vernon Hills, IL, USA), and macroscopic synovitis was scored. Macroscopic synovitis was assessed under direct visualization at video arthroscopy by the clinician performing the procedure. Macroscopic synovitis was scored using a well‐established, validated visual analogue scale 1–100 mm, as described previously [20].
Synovial biopsies were obtained from RA patients under direct visualization and cultured in complete media [Dulbecco’s RPMI‐1640 + 10% fetal calf serum (FCS)] within 15 min for 24 h. We have previously shown that the explants closely reflect the in‐vivo joint microenvironment, where the architecture and cell–cell contact remain intact and viable, and they spontaneously secrete proinflammatory mediators [21, 22]. Following 24‐h culture, supernatants were harvested and subsequently used as ECM for additional downstream experiments. The levels of proinflammatory and proangiogenic mediators were also measured in synovial biopsy supernatants by a meso scale discovery (MSD) proinflammatory and proangiogenic multiplex panel, according to the manufacturer’s instructions, and read using MSD Sector Imager 2400.
Cell isolation
Peripheral blood mononuclear cells (PBMC) were isolated from healthy control blood using a density gradient separation (Lymphoprep; Axis‐Shield poC, Oslo, Norway). CD14+ monocytes were subsequently purified using CD14 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), as per the manufacturer’s instructions. CD14+ monocytes were differentiated to monocyte‐derived dendritic cells (MoDC) over 7 days in the presence of recombinant granulocyte–macrophage colony‐stimulated factor (GM‐CSF) (70 ng/ml) and interleukin (IL)‐4 (50 ng/ml) (Peprotech, London, UK) in complete media (Dulbecco’s RPMI‐1640 + 10% FCS). On day 7, MoDC were harvested and plated for specific ECM experiments and left overnight to adhere in either 96‐well plates (flow cytometry or Seahorse) or 24‐well plates (RT–PCR); 1 × 105 cells were plated for flow cytometry experiments, while 3 × 105 cells were plated for RT–PCR experiments. Media were subsequently replaced with 10% ECM/media for the indicated times. MoDC, which were greater than 95% positive for the DC marker CD11c, were considered pure and used for subsequent experiments.
Bacterial endotoxin test
A limulus amoebocyte lysate (LAL) assay (Thermo Scientific, Fremont, CA, USA) was carried out on ECM to ensure that no bacterial endotoxin was present. An endotoxin standard stock solution was prepared and a standard curve of serial dilutions of 0·1–1 endotoxin units EU/ml was generated. ECM samples and standards were added to the plate and incubated with LAL reagent. Following this, a chromogenic substrate solution was added to the plate, resulting in the appearance of a colorimetric product which was subsequently read on a spectrophotometer at 410 nm. The levels of endotoxin in ECM were then extrapolated using the standard curve.
Quantitative RT–PCR (qRT–PCR)
Total RNA was extracted using a miRNeasy Kit (Qiagen, Valencia, CA, USA). For mRNA analysis, cDNA was synthesized with a high‐capacity cDNA reverse transcription kit (Applied Biosystems). qRT–PCR was performed with SYBR green qPCR master mix (Applied Biosystems, Foster City, CA, USA) with specific primers (Supporting information, Table S1). Relative gene expression was normalized to the expression of the housekeeping gene: ribosomal protein lateral stalk subunit P0 (RPLP0). The relative levels of each target transcript were calculated using the cycle threshold (Ct) method. mRNA in untreated control cells was adjusted to 1, serving as the basal reference value throughout experiments.
Flow cytometry
MoDC were identified using the following flow cytometry staining and gating strategy. Cells were gated based on forward‐ and side‐scatter, and dead cells and doublets were removed. Fluorescence minus one (FMO) controls were used to determine gating boundaries. Live Dead Red or Live Dead Aqua (Molecular Probes, Eugene, OR, USA) was used to eliminate dead cells. To eliminate non‐specific binding of mouse monoclonal antibodies to the Fc‐gamma receptor (FcγR) on DC, samples were blocked with a human FcγR‐binding inhibitor prior to antibody staining (eBioscience, San Diego, CA, USA). Cells were then stained as follows: human leucocyte antigen D‐related (HLA‐DR) V450, CD11c APC‐Cy7, CD83 APC, CD86 FITC and CD40 PE. For the detection of phospho STAT‐3 the BD PhosFlowTM kit (BD Biosciences, San Jose, CA, USA) with Perm Buffer III was used, according to the manufacturer’s instructions. Permeabilized cells were stained with an antibody specific for phosphorylated STAT‐3 (pY705) conjugated to PECF594. In order to adjust for spectral overlap between detectors, compensation was applied using single‐stained compensation beads (BD Biosciences). Samples were acquired using the CyAn Flow Cytometer (Beckman Coulter, Brea, CA, USA) and analysed using FlowJo software (Treestar, Inc., Ashland, OR, USA).
Antigen presentation and phagocytosis assay
DQ ovalbumin (Invitrogen, Carlsbad, CA, USA) and luciferin yellow (Sigma, St Louis, MO, USA) are model systems to examine antigen uptake and processing (DQ ovalbumin) or phagocytosis (luciferin yellow). MoDC were cultured for 24 h in the presence or absence of 10% ECM, after which cells were washed and incubated with DQ ovalbumin (25 µg/ml) for 15 min or luciferin yellow (40 µg/ml) for 120 min at 4 degrees (background) and 37 degrees. Cells were washed using cold phosphate‐buffered saline (PBS) and immediately read on the flow cytometer. Cells were excited with the 488 nm laser and fluorescence was detected using the 530/30 bandpass filter. Positive gates were set using the 4 degrees background sample (Supporting information, Fig. S3).
Glucose uptake assay
MoDC were stimulated for 6 h in the presence or absence of 10% ECM, after which cells were washed twice in glucose‐free media. MoDC were then incubated at 37°C for a further 1 h in supplemented glucose‐free media containing the fluorescently labelled glucose analogue 2NBDG (Life Technologies, Paisley, UK) at a final concentration of 100 μM before analysis using flow cytometry.
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR)–Seahorse technology
OCR and ECAR, reflecting oxidative phosphorylation (OXPHOS) and glycolysis, respectively, were measured before and after treatment with oligomycin (2 µg/ml; Seahorse Biosciences, Bristol, UK), trifluorocarbonylcyanide phenylhydrazone (FCCP) (5 μM; Seahorse Biosciences) and antimycin A (2 μM, Seahorse Biosciences) using the Seahorse XF96‐analyser (Seahorse Biosciences). MoDC were seeded at 300 000 cells per well in 96‐well XF‐microplates (Seahorse Biosciences) and allowed to adhere for 24 h. Cells were rinsed with assay medium [unbuffered Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 mM glucose, pH 7.4] before incubation with assay medium for 30 min at 37°C in a non‐CO2 incubator. Following incubation, cells were stimulated with ECM. Four baseline OCR and ECAR measurements were obtained over 28 min before injection of specific metabolic inhibitors. Moreover, to challenge the metabolic capacity of the MoDC, three OCR and ECAR measurements were obtained over 15 min following injection with oligomycin, FCCP and antimycin A.
RNA sequencing
Total RNA was extracted from CD1c+ DC RA synovial fluid and RA peripheral blood and the quality of all RNA samples was evaluated using an Agilent Bioanalyzer (Santa Clara, CA, USA). RNA samples were reverse‐transcribed, and sequencing libraries were constructed using the NuGen Ovation Universal RNA‐Seq System (Tecan Genomics, Inc., Redwood City, CA, USA; catalogue 0402‐A01), according to the manufacturer’s protocol. The resulting sequencing libraries were analyzed using the Caliper LabChip GX (Perkin Elmer, Beaconsfield, UK) and quantified using KAPA quantitative polymerase chain reaction (qPCR). Libraries were then normalized and pooled in one batch of six and one batch of five. Each pool was clustered and sequenced on an Illumina NextSeq500 instrument using 2 × 100 base pairs (bp) paired‐end reads (Illumina, San Diego, CA, USA), following the manufacturer’s protocols. The average number of reads per sample was 117 million reads, with a minimum of 90·1 million reads. Raw read quality was evaluated using FastQC. Reads were trimmed for adaptors and sequence quality. Trimmed reads were aligned to human b37.3 reference genome using the Omicsoft sequence aligner. Aligned reads were quantified using the EM algorithm with the RefSeq transcriptome model accessed on 6 June 2017 [23]. Aligned data were evaluated for quality using several quality metrics (e.g. mapping rate, coverage) and visually inspected for samples deviating from the population across multiple metrics and principle component analysis (PCA). Data are available on NCBI GEO with Accession number GSE151897. Statistical analysis of RNA‐seq data was performed in r version 3.3.1 with the Limma package version 3.28.14 [24]. The Limma Voom package (PMID, 24485249) was used to normalize and identify differentially expressed genes (DEG) from counts data. Transcripts with zero counts in more than two‐thirds of the samples were discarded from downstream analysis to reduce noise in the expression data. Gene counts were converted to log2 counts per million (cpm), quantile normalized and precision‐weighted. RNA‐seq gene features were considered differentially expressed if they satisfied a twofold change and false discovery rate (FDR) < 0·05 cut‐off. FDR control was performed with the Benjamini–Hochberg procedure. Heatmaps were generated in r with the heatmap.2 function in the gplots package version 3.0.1.
Statistical analysis
The spss version 15 system (SPSS, Inc., Chicago, IL, USA) for Windows was used for statistical analysis. Wilcoxon’s signed‐rank test or Mann–Whitney U‐test was used for analysis of non‐parametric data. Student’s t‐test was used for parametric data analysis. P‐values of less than 0·05 (*P < 0·05) were determined as statistically significant.
Study approval
Ethical approval to conduct this study was granted by St Vincent’s Healthcare Group Medical Research and Ethics Committee, and all patients gave fully informed written consent prior to inclusion. All experiments were performed in accordance with these guidelines and regulations.
Results
Synovial ECM induces DC maturation
Representative images taken at the time of arthroscopy demonstrate macroscopic synovitis in RA patients (Fig. 1a). Synovial biopsies obtained from the site of inflammation under direct visualization, using keyhole video arthroscopy, were cultured immediately for 24 h in complete media to allow the spontaneous release of soluble mediators into the culture supernatant (Fig. 1b). Subsequently, this supernatant (ECM) was used to examine the effect of the in‐vivo synovial microenvironment on DC maturation. We have previously demonstrated that the synovial architecture and cell–cell contact of the multiple cell types within the synovium is maintained using this model [21, 22]. We have also demonstrated that the cells remain viable in culture and spontaneously secrete proinflammatory cytokines [25, 26]. Furthermore, we have demonstrated that the explant model can be utilized to examine the effect of therapeutic targets, including tofacitinib, metformin and OPN301 [21, 22, 27, 28]. Collectively, this suggests that the synovial explant ECM used throughout this study is a good model to reflect the joint microenvironment. While the ECM model reflects the joint microenvironment, only limited amounts of ECM can be obtained per synovial biopsy (approximately 200 µl). This ECM is subsequently analysed using a LAL assay and demonstrated to be free from endotoxins (Supporting information, Fig. S1). Analysis of the spontaneous release of both proinflammatory and proangiogenic mediators demonstrated very high levels of both IL‐6 and vascular endothelial growth factor (VEGF) within the ECM (Supporting information, Fig. S2). MoDC were treated for 24 h in the presence of 10% ECM, and parameters of DC maturation were assessed (Fig. 1c,d). The markers selected for analysis in this study were chosen to reflect a number of key changes which occur during DC maturation, i.e. cytokine secretion, migration, adhesion and immunological synapse formation [29, 30, 31, 32]. MoDC treated with ECM had significantly increased expression of the proinflammatory cytokines IL‐12p40 (P < 0·05), IL‐23p19 (P < 0·05) and IL‐1b (P < 0·01), the chemokine major chemoattractant protein 1 (MCP)‐1 (P < 0·05), the adhesion proteins intercellular adhesion molecule (ICAM)‐1 and ICAM‐2 (both P < 0·01) and the chemokine receptor CCR7 (P < 0·05) (Fig. 1c). Moreover, the expression of CCR5 was significantly decreased in the presence of ECM (P < 0·01) (Fig. 1c), indicative of DC maturation. The protein expression of the DC co‐stimulatory markers CD83, CD80 and CD40 were assessed by flow cytometry. and a significant up‐regulation in CD83 expression was observed in ECM‐treated DC (P < 0·05) (Fig. 1d). During DC maturation, DC lose their ability to phagocytose additional antigen. Following ECM treatment, a significant decrease in phagocytosis (P < 0·05) (Fig. 1e and Supporting information, Fig. S3) was observed, concomitant with a reduction in antigen processing (P = 0·0597) (Fig. 1e and Supporting information, Fig. S3). These data suggest that the inflamed synovial microenvironment of the RA joint induces DC maturation, as indicated by increases in cytokines, adhesion molecules, chemokines and co‐stimulatory molecules, in addition to a reduction in DC phagocytic ability.
Fig. 1.
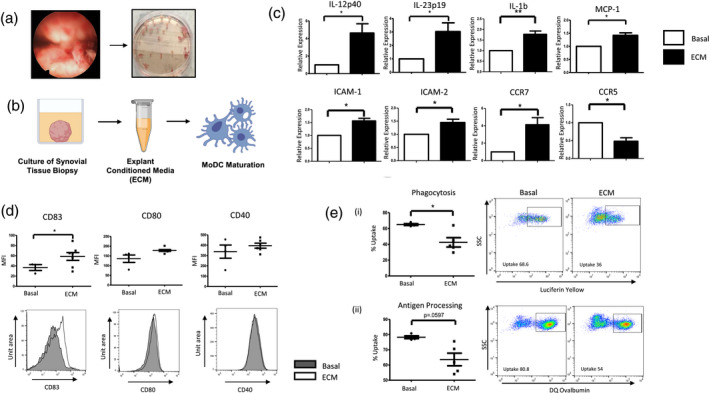
Synovial explant‐conditioned media (ECM) induces dendritic cell (DC) maturation. (a) Representative image of macroscopic synovitis in RA patients. (b) Synovial biopsies obtained from site of inflammation were cultured for 24 h in complete media. This supernatant (ECM) was used to examine the effect of the synovial microenvironment on DC maturation. (c) The relative expression of interleukin (IL)‐12p40, IL‐23p19, IL‐1b, major chemoattractant protein‐1 (MCP)‐1, intercellular adhesion molecule (ICAM)‐1, ICAM‐2, C‐C chemokine receptor (CCR)‐7 and CCR‐5 in MoDC left untreated (basal) or treated with 10% ECM for 24 h (n = 6). (d) Dot‐plots and representative histograms of CD83, CD80 and CD40 MFI on monocyte‐derived DC (MoDC) treated with 10% ECM for 24 h (n = 7). (e) Dot‐plots and representative flow cytometry plots displaying the percentage uptake of luciferin yellow (i) or DQ ovalbumin (ii) in MoDC left untreated or treated with 10% ECM for 24 h (n = 5) as a measurement of phagocytosis and antigen processing, respectively. Paired t‐test was used. *P < 0·05, **P < 0·01, significantly different from basal.
Synovial ECM induces metabolic alterations in DC
Following activation, DC require differential metabolic pathways to carry out their functions, therefore we next examined the expression of glycolytic‐related genes in DC treated with 10% ECM for 6 h. ECM‐treated DC expressed significantly higher levels of HK2 and PFKFB3 compared to basal (both P < 0·01), both of which are key glycolytic rate‐limiting enzymes. Moreover, the expression of PDK1 and PDK2, which are involved in converting pyruvate to acetyl‐coA, were both significantly increased (P < 0·05) in ECM‐treated MoDC compared to basal. The enzymes GSK3α and H6PD, which are involved in glycogen synthesis were also significantly up‐regulated (P < 0·001) in ECM‐treated MoDC compared to basal (Fig. 2a). Furthermore, DC treated with ECM had significantly higher glucose uptake compared to untreated DC, indicated by the significant increase in 2NBDG (P < 0·05, Fig. 2b). In addition to an increase in glucose uptake, ECM‐treated DC also expressed significantly higher levels of the amino acid transporter CD98 (P < 0·05) (Fig. 2b).
Fig. 2.
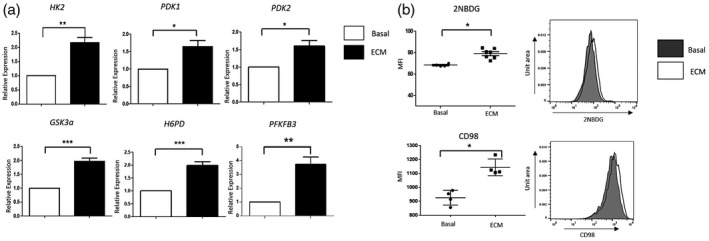
Synovial explant‐conditioned media (ECM) induces metabolic alterations in dendritic cells (DC). (a) The relative expression of hexokinase 2 (HK2), pyruvate dehydrogenase kinase (PDK)1, PDK2, glycogen synthase kinase 3 alpha (GSK3a), hexose‐6‐phosphate dehydrogenase (H6PD) and 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphatase 3 (PFKFB3) in monocyte‐derived DC (MoDC) left untreated (basal) or treated with 10% ECM for 6 h (n = 6). (b) Dot‐plots and representative histograms of 2NBDG (glucose uptake) (n = 7) and CD98 mean fluorescence intensity (MFI) (n = 4) on MoDC treated with 10% ECM for 6 h. Paired t‐test was used. *P < 0·05, **P < 0·01, ***P < 0·001 significantly different from basal.
Synovial ECM induces glycolysis in DC
To further investigate if the observed changes in the metabolic parameters results in altered cellular bioenergetics, we simultaneously measured the two major cellular energy pathways, OXPHOS and glycolysis, using the Seahorse XF‐Analyzer (Seahorse Biosciences) over a continuation of 6 h 10% ECM treatment. We demonstrated significant changes in cellular bioenergetics as early as 1 h post‐ECM exposure. Specifically, there was a significant increase in ECAR (indicative of glycolysis) 1 h post‐ECM exposure (P < 0·05). ECM‐treated DC maintained this increase in ECAR at 3 h (P < 0·01) and 6 h (P < 0·05) post‐treatment (Fig. 3a). In contrast, there was no significant change in baseline OCR (indicative of mitochondrial respiration) following ECM treatment over a time–course of 1–6 h. Moreover, the ECAR : OCR ratio was significantly increased in DC at 1 and 3 h post‐ECM exposure (both P < 0·05) (Fig. 3a). To further examine mitochondrial function in DC in response to ECM, a Mito Stress test (Seahorse Biosciences) was performed, thus allowing for multiple parameters of mitochondrial function to be assessed simultaneously in real time. Figure 3b demonstrates the average bioenergetic traces for OCR and ECAR in DCs before and after injections of the mitochondrial inhibitors oligomycin, trifluorocarbonylcyanide phenylhydrazone (FCCP) and antimycin A following 10% ECM treatment for 6 h. Oligomycin acts as an inhibitor to adenosine triphosphate (ATP) synthase, and is used to evaluate the maximal glycolytic capacity. FCCP, which acts a mitochondrial uncoupler, was injected to evaluate the maximal respiratory capacity. Maximal respiratory capacity was determined by subtracting baseline OCR from FCCP‐induced OCR and the respiratory reserve (baseline OCR subtracted from maximal respiratory capacity). Baseline glycolysis was calculated by the average of three baseline ECAR measurements obtained before injections of oligomycin FCCP and antimycin A. ECM significantly increased baseline glycolysis (P < 0·05) (Fig. 3c). Moreover, the maximal glycolytic capacity (i.e. the maximum possible rate of glucose conversion to pyruvate or lactate) was significantly increased in ECM treated DC (P < 0·05) (Fig. 3c), in addition to a significantly higher maximal respiratory capacity (P < 0·05) (Fig. 3c), highlighting the potential capability of DCs to respond to energetic demands within the synovium. Taken together, these data suggest a distinct metabolic shift towards glycolysis in ECM‐treated DCs (Fig. 3d).
Fig. 3.
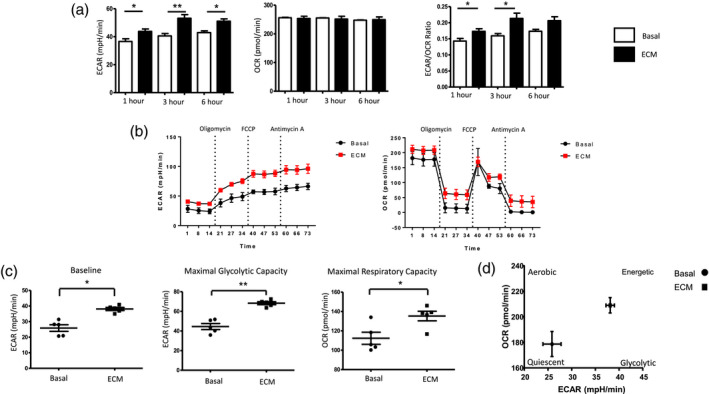
Synovial explant‐conditioned media (ECM) induces glycolysis in dendritic cells (DC). (a) Representative bar graphs demonstrating baseline extracellular acidification rate (ECAR), baseline oxygen consumption rate (OCR), the ECAR : OCR ratio after injection of ECM (10%) over 1, 3 and 6 h. (b) Average Seahorse profiles demonstrating (i) extracellular acidification rate (ECAR) (glycolysis) and (ii) OCR (oxidative phosphorylation) before and after injections of oligomycin, trifluorocarbonylcyanide phenylhydrazone (FCCP) and anti‐mycin A in monocyte‐derived DC (MoDC) treated with 10% ECM for 6 h (n = 5). (c) Representative bar graphs demonstrating baseline ECAR, maximal glycolytic capacity and maximal respiratory capacity in MoDC treated with 10% ECM for 6 h (n = 5). (D) Energy map of MoDC showing a distinct shift in ECM‐treated MoDC towards a glycolytic/energetic phase.
STAT‐3 mediates DC maturation in the synovial microenvironment
Next, we examined potential pathways activated in ECM‐treated DC in order to delineate the molecular mechanisms driving these inflammatory and metabolic changes. Given that the Janus kinase (JAK)–STAT pathway, specifically STAT‐3, is known to play a role in DC maturation [33] and glycolysis [34, 35], we analysed STAT‐3 expression following 10% ECM treatment for 6 h. We demonstrated a significant increase in STAT‐3 expression in ECM‐treated DC (P < 0·001) (Fig. 4a), in addition to a significant increase in STAT‐3 phosphorylation in response to ECM (P < 0·01) (Fig. 4a). These data suggest a possible role for STAT‐3 in mediating DC maturation and metabolism within the RA synovium.
Fig. 4.
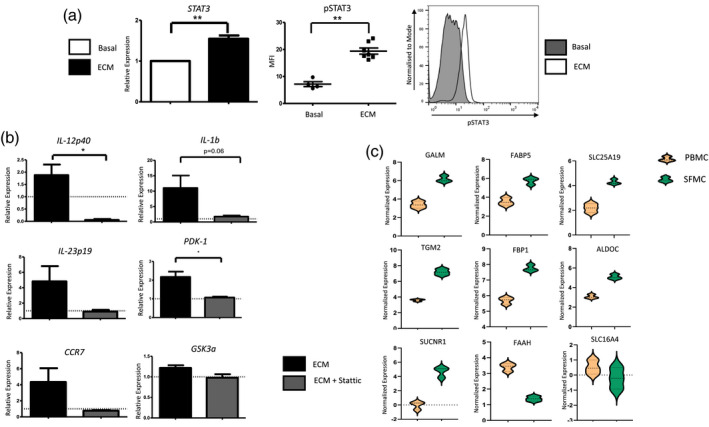
Signal transducer and activator of transcription‐3 STAT‐3 mediates dendritic cell (DC) maturation in the synovial microenvironment. [a (i)] The relative expression of STAT‐3 in monocyte‐derived DC (MoDC) treated with 10% explant‐conditioned media (ECM) for 6 h (n = 7). (ii) Dot‐plot and histogram representing mean fluorescent intensity (MFI) of pSTAT‐3 in MoDC treated with 10% ECM (n = 7). (b) Relative expression of the inflammatory genes interleukin (IL)‐12p40, IL‐1b, IL‐23p19 and CCR7 in MoDC treated with 10% ECM for 24 h in the presence or absence of Stattic (5 µM). Cells were pretreated with 6‐nitrobenzo[b]thiophene‐1,1‐dioxide (Stattic) for 1 h prior to ECM treatment. Dashed line represents basal expression of genes. (b) Relative expression of glycolytic genes PDK‐1 and GSK3a in MoDC treated with 10% ECM for 6 h in the presence or absence of Stattic (5 µM). Cells were pretreated with Stattic for 2 h prior to ECM treatment. Dashed line represents basal expression of genes. (c) Volcano plots following RNA‐Seq analysis, depicting genes involved in cellular metabolism which are differentially expressed between synovial CD1c+ DC and peripheral blood CD1c+ DC in RA patients (n = 3). Orange cluster represents genes expressed in CD1c+ DC isolated from peripheral blood, whereas the green cluster represents genes expressed in CD1c+ DC isolated from synovial fluid. Data were analysed using a paired t‐test. *P < 0·05, **P < 0·01 significantly different from basal.
To support the hypothesis that STAT‐3 mediates DC maturation and bioenergetics within the synovial microenvironment, MoDC were pretreated for 1 h with the STAT‐3 inhibitor, Stattic (6‐nitrobenzo[b]thiophene‐1,1‐dioxide), before 10% ECM treatment for 24 and 6 h. STAT‐3 inhibition significantly abrogated ECM‐induced IL‐12p40 expression (P < 0·05) (Fig. 4a), in addition to decreasing the expression of IL‐1b, IL‐23p19 and CCR7 (Fig. 4a). Moreover, STAT‐3 inhibition significantly abrogated ECM‐induced PDK‐1 expression (P < 0·05) (Fig. 4a), suggesting that STAT‐3 may play a partial role in DC maturation and metabolism. Our data support the hypothesis that the RA synovial microenvironment induces DC maturation and function, which is consistent with our previous study demonstrating that DC within the RA synovium are indeed more mature [15]. Moreover, our data also suggest that the RA synovial microenvironment may induce glycolytic reprogramming of DC. To support this hypothesis in vivo, we performed RNA sequencing on both peripheral blood and synovial fluid CD1c+ DC from RA patients. Reactome analysis [36] revealed that a number of genes within the cellular metabolism pathway were differentially regulated between synovial fluid and peripheral blood CD1c+ DC. Specifically, we demonstrated an increase in the expression of genes involved in glucose, galactose and fatty acid metabolism (GALM, FABP5, FBP1, TGM2, ALDOC), the TCA cycle (SUCNR1) and metabolic transporters (SLC25A19) in RA synovial CD1c+ DC compared to DC in the circulation (Fig. 4c and Supporting information, Fig. S4). Furthermore, the expression of FAAH (involved in the breakdown of fatty acid amides) and SLC16A4 (which facilitates the transport of lactate and pyruvate) were significantly decreased in synovial DC compared to peripheral blood DC. Collectively, these in‐vivo data suggest that metabolic dysregulation may occur within synovial DC in RA.
Discussion
In this study, we report an induction of DC maturation concomitant with altered cellular bioenergetics in response to the RA synovial microenvironment. MoDC exposed to ECM have increased expression of proinflammatory cytokines, co‐stimulatory markers, chemokines and adhesion molecules, in addition to a reduction in their ability to phagocytose and process antigen. ECM DC have altered cellular bioenergetics with increased expression of glycolytic genes, glucose uptake and increased expression of the amino acid transporter CD98, ultimately leading to a more glycolytic metabolic profile. We report a potential role for STAT‐3 in partially mediating these effects, whereby ECM‐treated DC up‐regulated STAT‐3 and pharmacological inhibition of STAT‐3 prevented ECM‐induced cytokine and glycolytic gene expression. Finally, we identified a number of differentially expressed genes involved in cellular metabolism in synovial fluid and peripheral blood CD1c+ DC in RA patients, suggesting that the metabolic alterations observed in MoDC in vitro may reflect the metabolic adaptations of synovial cells in vivo – specifically CD1c+ DC.
It is well appreciated that the synovial microenvironment is abundant in cytokines, chemokines, metabolites and matrix‐degrading enzymes, in addition to a complex network of immune cells. However, the effect of this unique microenvironment on immune cell function has been relatively unexplored. Previous studies have shown that MoDC can transdifferentiate into osteoclasts in the presence of RA synovial fluid [37], while others have shown that synovial fluid can induce apoptosis and cell death of chondrocytes [38]. Moreover, Carsons et al. report that RA synovial fluid can differentiate CD34+ myeloid progenitor cells into DC [39]. While previous studies have examined the role of the synovial microenvironment on cellular functions using synovial fluid as a model system, to the best of our knowledge the effect of ECM on immune cell maturation has not previously been reported. ECM induces expression of the cytokines IL‐12p40, IL‐23p19 and IL‐1. IL‐23 (comprising IL‐12p40 and IL‐23p19) is one of the essential factors required for the survival and maintenance of T helper type 17 (Th17) cells. While the IL‐23/IL‐17 axis has been shown to be more relevant in the context of psoriatic arthritis rather than that of RA, exTh17 cells have been shown to accumulate in RA synovial fluid suggestive of a role for IL‐23 early in disease [40]. In addition to mediating the production of cytokines, ECM‐treated MoDC also express significantly higher levels of the chemokine MCP‐1, a known regulator of monocyte, T cell and natural killer (NK) cell infiltration [41], levels of which are significantly higher within the RA synovium compared to OA [42]. We also report a significant increase in ICAM‐1, ICAM‐2 and CCR7, all of which are necessary for the movement of DC to lymph nodes where interactions with antigen‐specific T cells occur. Moreover, studies also suggest that these markers, in addition to facilitating movement, may also be involved in the immunological synapse, thus facilitating T cell activation. Furthermore, Moschovakis et al. recently demonstrated that CCR7‐deficient mice were completely resistant to collagen‐induced arthritis (CIA), while selective CCR7 expression on DC restored arthritis severity.
ECM‐treated MoDC express significantly higher surface levels of CD83. While CD83 signalling on DC is known to result in an up‐regulation of major histocompatibility complex (MHC)‐II and CD86 (both required for T cell activation), its expression is also one of the best markers to date to distinguish stably mature DC [43]. This is consistent with previous data, which have reported DC within the synovium to be more mature than their peripheral blood counterparts [15, 16, 44]. While CD83 expression acts as a marker of DC maturation, its precise function, or indeed its ligand, is not yet known. This contrasts with other, more established, co‐stimulatory markers such as CD40 and CD80, which bind CD40L and CD28, respectively, on T cells to drive T cell activation and proliferation. While ECM significantly induces CD83 expression, it does not affect the expression of CD40 and CD80. This may suggest that while ECM can induce MoDC maturation, additional signals, i.e. contact‐dependent signals, may be required to induce the expression of specific co‐stimulatory markers. Moreover, while our model examines the effect of synovial tissue‐derived soluble mediators on MoDC maturation, antigen uptake and subsequent antigen‐specific DC changes will also shape the activation and maturation status of DC in RA. An important unanswered question that remains is: do soluble mediators in the synovial microenvironment alone induce T cell proliferation and/or differentiation or are additional cues also needed to functionally affect T cell responses? Future studies in this area can aid in our understanding of synovial DC : T cell interactions.
We demonstrate that ECM‐treated DC have significantly less phagocytic and antigen‐processing capabilities indicative of their increased maturation status. Taken together, these data suggest that the synovial microenvironment may induce DC maturation.
Seminal studies in the last decade have reinforced the hypothesis that immune cells adapt their metabolic and biosynthetic needs according to their function. Several studies have demonstrated a metabolic switch within the RA joint. Specifically, the presence of dysfunctional mitochondria, in addition to increased expression of glucose transporters and glycolytic enzymes have all been demonstrated in the RA synovium [12, 14, 45]. To date, however, the role of the synovial microenvironment on DC bioenergetics or the potential metabolic requirements of synovial DC themselves for survival and function within the joint are not understood. Our data suggest that soluble mediators within the RA joint can significantly increase glycolytic genes, glucose uptake and the expression of amino acid transporters, ultimately leading to a more glycolytic phenotype. This is consistent with previous data demonstrating that RA synovial cells have increased glycolytic capacities and metabolically switch to produce ATP via glycolysis, even in the presence of O2 (i.e. Warburg metabolism) [12, 46, 47].
While the bioenergetic requirements of synovial DC have yet to be explored, our data suggest that the RA synovial microenvironment induces a metabolic shift in DC to a more glycolytic phenotype. Previous studies have demonstrated a diverse spectrum of metabolic requirements between resting immature DC and activated mature DC. Resting DC predominantly utilize OXPHOS to generate ATP which sustains their immature phenotype [48]. However, upon exposure to antigen or TLR‐stimulation, glycolysis is increased and OXPHOS is maintained, while long‐term activated DCs have elevated glycolysis concomitant with a cessation in OXPHOS [6, 49]. These metabolic requirements underpin DC maturation, whereby metabolic inhibition results in an inability of DC to become functionally mature even in the presence of inflammatory signals [8].
We also demonstrate a role for STAT‐3 in mediating synovial‐induced DC maturation and metabolic reprogramming. Inhibition of STAT‐3 using the pharmacological inhibitor, Stattic, inhibits ECM‐mediated induction of IL‐12p40, IL‐1b and PDK‐1. Previous studies have illustrated that pSTAT‐3 is significantly increased within the RA synovium [50, 51]. Moreover, in RA synovial explant cultures, JAK2 inhibition decreased IL‐6, IL‐8 and matrix metallopeptidase 3 (MMP3) secretion while inducing IL‐10 [35]. STAT‐3 expression in RA also correlates with synovitis [52]. Furthermore, Tofacitinib, a reversible, competitive inhibitor of JAK1 and JAK3, has been approved in a wide variety of countries for the treatment of RA and has previously been demonstrated to inhibit glycolysis while concomitantly inducing OXPHOS in RA synovial fibroblasts [21]. Collectively, this reinforces the suggestion that STAT‐3 may contribute to RA pathogenesis. Specifically, we demonstrate that this effect may in part be via DC maturation. Synovial biopsies spontaneously produce a variety of proinflammatory and proangiogenic mediators such as IL‐6, IL‐1β, TNF‐α, basic fibroblast growth factor (bFGF) and VEGF (Supporting information, Fig. S2). While it is unclear which component of the ECM drives STAT‐3 activation in MoDC, the observed excessive production of IL‐6 from RA synovial explants suggests that ECM‐derived IL‐6 may be responsible. While VEGF is also produced at high levels, it does not directly activate STAT‐3. However, it may do so indirectly by regulating NOTCH, which can subsequently regulate STAT‐3 [53, 54]. Moreover, additional factors may directly or indirectly regulate STAT‐3 via IFN‐γ or TNF‐α, respectively [28, 55]. Finally, it has also been shown that STAT‐3 is regulated by signalling components in cellular metabolism, which may also be implicated in the synovial STAT‐3 response [56]. While outside the scope of this manuscript, future experiments could directly examine IL‐6 blockade in addition to other potential stimulants within ECM‐treated MoDC to determine microenvironment and cytokine‐specific effects on DC responses.
While previous studies have demonstrated a role for STAT‐3 mediated DC maturation, to the best of our knowledge this is the first study to highlight a potential role for STAT‐3 specifically in MoDC in response to the RA synovial microenvironment. Additional studies examining the effect of STAT‐3 inhibition on glucose uptake, glucose transporters and real‐time glycolytic capacity would strengthen our understanding of the role of STAT‐3 in DC metabolism in RA. Moreover, an additional unanswered question remains as to whether the in‐vitro role of STAT‐3 in MoDC maturation described in this study is reflected in vivo in RA synovial DC. While previous studies have explored the role of STAT‐3 in DC differentiation, maturation, inflammation and in disease settings such as cancer [21, 35, 57, 58, 59, 60], additional studies on STAT‐3 signalling specifically in RA synovial DC are needed to fully understand the role STAT‐3 may play in DC maturation in RA.
Throughout this study, ECM was used to recapitulate the RA synovial microenvironment. Therefore, one unavoidable limitation within this study is the inability to measure contact‐dependent interactions on DC maturation which no doubt also plays a pivotal role. Moreover, to carry out this work monocyte‐derived DC were used as a model of in‐vivo DC. While this model is widely used, and much of human translational DC research utilizes this system, it cannot be assumed that all effects demonstrated in MoDC will be mirrored in vivo. Therefore, to further confirm our findings in a more in‐vivo‐relevant setting, we performed RNA‐seq on CD1c+ DC isolated from the synovial fluid and peripheral blood of RA patients. We subsequently interrogated this sequencing data set for genes and pathways involved in cellular metabolism and identified several genes involved in metabolism which were significantly enriched in synovial CD1c+ DC compared to peripheral blood CD1c+ DC. These data further support our findings, demonstrating that the in‐vivo joint microenvironment alters the metabolic profile of MoDC.
In conclusion, this study demonstrates that the unique RA synovial microenvironment can induce DC maturation in vitro in addition to altering cellular bioenergetics in favour of glycolysis. We demonstrate a role for STAT‐3 in mediating these synovial‐induced changes at both the maturation and metabolic level and provide evidence that these changes are reflected in vivo in synovial DC.
Disclosures
None.
Supporting information
Fig. S1. Endotoxin levels in RA ECM. Barchart representing the levels of endotoxin in ECM (n = 10). An LAL assay was performed on RA ECM and was determined to be free from contaminating levels of endotoxins.
Fig. S2. Proinflammatory and Proangiogenic mediators in RA ECM. Barchart representing the production of IL‐6, IL‐1β, TNFα, bFGF and VEGF released from RA synovial tissue biopsies (n = 7).
Fig. S3. Gating controls for Phagocytosis and Antigen Processing Assays. Representative flow cytometry dotplots displaying the percentage uptake of Luciferin Yellow (i) or DQ Ovalbumin (ii). Gates were set using the 4 degree background control for each sample used.
Fig. S4. Differentially expressed metabolic genes in synovial fluid and peripheral blood RA CD1c+DC. Heatmap generated from RNA‐Seq analysis depicting hierarchical clustering of differentially expressed genes involved in cellular metabolism in CD1c+DC isolated from synovial fluid and peripheral blood of RA patients (n = 3).
Table S1. Oligonucleotide primers used in RT‐PCR.
References
- 1. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med [internet] 2011. [cited 2019 Jun 19]; 365:2205–19. Available at: http://www.nejm.org/doi/abs/10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 2. Kennedy A, Ng CT, Biniecka M et al Angiogenesis and blood vessel stability in inflammatory arthritis. Arthritis Rheum 2010; 62:711–21. [DOI] [PubMed] [Google Scholar]
- 3. Koch AE. Angiogenesis as a target in rheumatoid arthritis. Ann Rheum Dis 2003; 62:60ii–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biniecka M, Fox E, Gao W et al Hypoxia induces mitochondrial mutagenesis and dysfunction in inflammatory arthritis. Arthritis Rheum [internet] 2011. [cited 2019 Jun 19]; 63:2172–82. Available at: http://doi.wiley.com/10.1002/art.30395. [DOI] [PubMed] [Google Scholar]
- 5. Gaber T, Dziurla R, Tripmacher R, Burmester GR, Buttgereit F. Hypoxia inducible factor (HIF) in rheumatology: low O2! See what HIF can do!. Ann Rheum Dis 2005. 10.1136/ard.2004.031641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krawczyk CM, Holowka T, Sun J et al Toll‐like receptor‐induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010; 115:4742–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jantsch J, Chakravortty D, Turza N et al Hypoxia and hypoxia‐inducible factor‐1α modulate lipopolysaccharide‐induced dendritic cell activation and function. J Immunol [internet] 2008. [cited 2020 Feb 25]; 180:4697–705. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18354193. [DOI] [PubMed] [Google Scholar]
- 8. Everts B, Amiel E, Huang SCC et al TLR‐driven early glycolytic reprogramming via the kinases TBK1‐IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol 2014; 15:323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Everts B, Amiel E, Van Der Windt GJW et al Commitment to glycolysis sustains survival of NO‐producing inflammatory dendritic cells. Blood 2012; 120:1422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bustamante MF, Oliveira PG, Garcia‐Carbonell R et al Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann Rheum Dis 2018; 77:1636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garcia‐Carbonell R, Divakaruni AS, Lodi A et al Critical role of glucose metabolism in rheumatoid arthritis fibroblast‐like synoviocytes. Arthritis Rheum 2016; 68:1614–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biniecka M, Canavan M, McGarry T et al Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheum Dis [internet] 2016. [cited 2017 Dec 13]; 75:2192–200. Available at: http://www.ncbi.nlm.nih.gov/pubmed/27013493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moodley D, Mody G, Patel N, Chuturgoon AA. Mitochondrial depolarisation and oxidative stress in rheumatoid arthritis patients. Clin Biochem 2008; 41:1396–401. [DOI] [PubMed] [Google Scholar]
- 14. Harty LC, Biniecka M, O’Sullivan J et al Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann Rheum Dis [internet] 2012. [cited 2019 Jun 19]; 71:582–8. Available at: http://ard.bmj.com/lookup/doi/10.1136/annrheumdis‐2011‐200245. [DOI] [PubMed] [Google Scholar]
- 15. Canavan M, Walsh AM, Bhargava V et al Enriched Cd141+ DCs in the joint are transcriptionally distinct, activated, and contribute to joint pathogenesis. JCI Insight [internet] 2018. [cited 2019 Jun 19]; 3 Available at: https://insight.jci.org/articles/view/95228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jongbloed SL, Lebre MC, Fraser AR et al Enumeration and phenotypical analysis of distinct dendritic cell subsets in psoriatic arthritis and rheumatoid arthritis. Arthritis Res Ther [internet] 2006. [cited 2017 May 17]; 8:R15 Available at: http://www.ncbi.nlm.nih.gov/pubmed/16507115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Page G, Lebecque S, Miossec P. Anatomic localization of immature and mature dendritic cells in an ectopic lymphoid organ: correlation with selective chemokine expression in rheumatoid synovium. J Immunol [internet] 2002. [cited 2017 May 17]; 168:5333–41. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11994492. [DOI] [PubMed] [Google Scholar]
- 18. Segura E, Touzot M, Bohineust A et al Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity [internet] 2013. [cited 2017 May 17]; 38:336–48. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23352235. [DOI] [PubMed] [Google Scholar]
- 19. Pettit AR, MacDonald KP, O’Sullivan B, Thomas R. Differentiated dendritic cells expressing nuclear RelB are predominantly located in rheumatoid synovial tissue perivascular mononuclear cell aggregates. Arthritis Rheum [internet] 2000. [cited 2017 May 17]; 43:791–800. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10765923. [DOI] [PubMed] [Google Scholar]
- 20. Veale DJ, Reece RJ, Parsons W et al Intra‐articular primatised anti‐CD4: efficacy in resistant rheumatoid knees. A study of combined arthroscopy, magnetic resonance imaging, and histology. Ann Rheum Dis [internet] 1999 [cited 2017 Dec 13]; 58:342–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10340958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McGarry T, Orr C, Wade S et al JAK/STAT blockade alters synovial bioenergetics, mitochondrial function, and proinflammatory mediators in rheumatoid arthritis. Arthritis Rheum [internet] 2018 [cited 2020 Mar 17]; 70:1959–70. Available at: https://onlinelibrary.wiley.com/doi/abs/10.1002/art.40569. [DOI] [PubMed] [Google Scholar]
- 22. Gao W, McGarry T, Orr C, McCormick J, Veale DJ, Fearon U. Tofacitinib regulates synovial inflammation in psoriatic arthritis, inhibiting STAT activation and induction of negative feedback inhibitors. Ann Rheum Dis 2016; 75:311–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA‐Seq gene expression estimation with read mapping uncertainty. Bioinformatics [internet] 2010. [cited 2017 May 17]; 26:493–500. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20022975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ritchie ME, Phipson B, Wu D et al Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res [internet] 2015. [cited 2017 May 17]; 43:e47 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25605792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Connolly M, Mullan RH, McCormick J et al Acute‐phase serum amyloid A regulates tumor necrosis factor alpha and matrix turnover and predicts disease progression in patients with inflammatory arthritis before and after biologic therapy. Arthritis Rheum [internet] 2012. [cited 2020 May 13]; 64:1035–45. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22076945. [DOI] [PubMed] [Google Scholar]
- 26. McGarry T, Veale DJ, Gao W, Orr C, Fearon U, Connolly M. Toll‐like receptor 2 (TLR2) induces migration and invasive mechanisms in rheumatoid arthritis. Arthritis Res Ther [internet] 2015. [cited 2020 May 13]; 17:153 Available at: http://www.ncbi.nlm.nih.gov/pubmed/26055925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gallagher L, Cregan S, Biniecka M et al Insulin resistant pathways are associated with disease activity in rheumatoid arthritis and are subject to disease modification through metabolic reprogramming; a potential novel therapeutic approach. Arthritis Rheum 2019. 10.1002/art.41190. [DOI] [PubMed] [Google Scholar]
- 28. Nic An Ultaigh S, Saber TP, McCormick J et al Blockade of Toll‐like receptor 2 prevents spontaneous cytokine release from rheumatoid arthritis ex vivo synovial explant cultures. Arthritis Res Ther 2011; 13:R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gardella S, Andrei C, Costigliolo S, Olcese L, Zocchi MR, Rubartelli A. Secretion of bioactive interleukin‐1β by dendritic cells is modulated by interaction with antigen specific T cells. Blood 2000; 95:3809–15. [PubMed] [Google Scholar]
- 30. Sallusto F, Schaerli P, Loetscher P et al Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol 1998; 28:2760–9. [DOI] [PubMed] [Google Scholar]
- 31. Comrie WA, Li S, Boyle S, Burkhardt JK. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM‐1 mobility. J Cell Biol 2015; 208:457–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Foti M, Granucci F, Aggujaro D et al Upon dendritic cell (DC) activation chemokines and chemokine receptor expression are rapidly regulated for recruitment and maintenance of DC at the inflammatory site. Int Immunol [internet] 1999; 11: 979–86 [cited 2020 May 12]. Available at: https://academic.oup.com/intimm/article‐abstract/11/6/979/707790. [DOI] [PubMed] [Google Scholar]
- 33. Kawakami Y, Inagaki N, Salek‐Ardakani S et al Regulation of dendritic cell maturation and function by Bruton’s tyrosine kinase via IL‐10 and Stat3. Proc Natl Acad Sci USA 2006; 103:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li M, Jin R, Wang W et al STAT3 regulates glycolysis via targeting hexokinase 2 in hepatocellular carcinoma cells. Oncotarget 2017; 8:24777–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gao W, McCormick J, Connolly M, Balogh E, Veale DJ, Fearon U. Hypoxia and STAT3 signalling interactions regulate pro‐inflammatory pathways in rheumatoid arthritis. Ann Rheum Dis [internet] 2015. [cited 2020 Feb 24]; 74:1275–83. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24525913. [DOI] [PubMed] [Google Scholar]
- 36. Wu G, Haw R. Functional interaction network construction and analysis for disease discovery In: Methods in molecular biology. Clifton, NJ: Humana Press Inc.; 2017:235–53. [DOI] [PubMed] [Google Scholar]
- 37. Rivollier A, Mazzorana M, Tebib J et al Immature dendritic cell transdifferentiation into osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood 2004; 104:4029–37. [DOI] [PubMed] [Google Scholar]
- 38. Röhner E, Matziolis G, Perka C et al Inflammatory synovial fluid microenvironment drives primary human chondrocytes to actively take part in inflammatory joint diseases. Immunol Res 2012; 52:169–75. [DOI] [PubMed] [Google Scholar]
- 39. Santiago‐Schwarz F, Anand P, Liu S, Carsons SE. Dendritic cells (DCs) in rheumatoid arthritis (RA): progenitor cells and soluble factors contained in RA synovial fluid yield a subset of myeloid DCs that preferentially activate Th1 inflammatory‐type responses. J Immunol 2001; 167:1758–68. [DOI] [PubMed] [Google Scholar]
- 40. Basdeo SA, Cluxton D, Sulaimani J et al Ex‐Th17 (nonclassical Th1) cells are functionally distinct from classical Th1 and Th17 cells and are not constrained by regulatory T cells. J Immunol 2017; 198:2249–2259. [DOI] [PubMed] [Google Scholar]
- 41. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein‐1 (MCP‐1): an overview. J Interferon Cytokine Res 2009; 29:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koch AE, Kunkel SL, Harlow LA et al Enhanced production of monocyte chemoattractant protein‐1 in rheumatoid arthritis. J Clin Invest 1992; 90:772–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cao W, Lee SH, Lu J. CD83 is preformed inside monocytes, macrophages and dendritic cells, but it is only stably expressed on activated dendritic cells. Biochem J 2005; 385:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lebre MC, Jongbloed SL, Tas SW, Smeets TJ, McInnes IB, Tak PP. Rheumatoid arthritis synovium contains two subsets of CD83–DC‐LAMP– dendritic cells with distinct cytokine profiles. Am J Pathol [internet] 2008. [cited 2017 May 17]; 172:940–50. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18292234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Valcárcel‐Ares MN, Riveiro‐Naveira RR, Vaamonde‐García C et al Mitochondrial dysfunction promotes and aggravates the inflammatory response in normal human synoviocytes. Rheumatology [cited 2020 Feb 28]; 2014; 53:1332–1343. Available at: https://academic.oup.com/rheumatology/article‐abstract/53/7/1332/1794504. [DOI] [PubMed] [Google Scholar]
- 46. McGarry T, Biniecka M, Gao W et al Resolution of TLR2‐induced inflammation through manipulation of metabolic pathways in rheumatoid arthritis. Sci Rep 2017; 7:43165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Balogh E, Veale DJ, McGarry T et al Oxidative stress impairs energy metabolism in primary cells and synovial tissue of patients with rheumatoid arthritis. Arthritis Res Ther [internet] 2018. [cited 2020 Feb 28]; 20:95 Available at: https://arthritis‐research.biomedcentral.com/articles/10.1186/s13075‐018‐1592‐1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol [internet] 2015. [cited 2020 Feb 28]; 15:18–29. Available at: 10.1038/nri3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guak H, Al Habyan S, Ma EH et al Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun 2018; 9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oike T, Sato Y, Kobayashi T et al Stat3 as a potential therapeutic target for rheumatoid arthritis. Sci Rep 2017; 7:10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De Hooge ASK, Van De Loo FAJ, Koenders MI et al Local activation of STAT‐1 and STAT‐3 in the inflamed synovium during zymosan‐induced arthritis: Exacerbation of joint inflammation in STAT‐1 gene‐knockout mice. Arthritis Rheum [internet] 2004. [cited 2020 Feb 28]; 50:2014–23. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15188379. [DOI] [PubMed] [Google Scholar]
- 52. Ju JH, Heo YJ, La Cho M et al Modulation of STAT‐3 in rheumatoid synovial T cells suppresses Th17 differentiation and increases the proportion of Treg cells. Arthritis Rheum 2012; 64:3543–3552. [DOI] [PubMed] [Google Scholar]
- 53. Gao W, Sweeney C, Walsh C et al Notch signalling pathways mediate synovial angiogenesis in response to vascular endothelial growth factor and angiopoietin 2. Ann Rheum Dis [internet] 2013. [cited 2020 May 13]; 72:1080–8. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23161900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee JH, Suk J, Park J et al Notch signal activates hypoxia pathway through HES1‐dependent SRC/signal transducers and activators of transcription 3 pathway. Mol Cancer Res 2009; 7:1663–71. [cited 2020 May 13]; Available at: http://mcr.aacrjournals.org/. [DOI] [PubMed] [Google Scholar]
- 55. Rosengren S, Corr M, Firestein GS, Boyle DL. The JAK inhibitor CP‐690,550 (tofacitinib) inhibits TNF‐induced chemokine expression in fibroblast‐like synoviocytes: autocrine role of type I interferon. Ann Rheum Dis [internet] 2012. [cited 2020 May 13]; 71:440–7. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22121136. [DOI] [PubMed] [Google Scholar]
- 56. Demaria M, Poli V. PKM2, STAT3 and HIF‐1α. JAK–STAT [internet] 2012. [cited 2020 Mar 17]; 1:194–6. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24058770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3L‐dependent dendritic cell differentiation. Immunity 2003;19:903–12. [DOI] [PubMed] [Google Scholar]
- 58. Jung ID, Noh KT, Lee CM et al Oncostatin M induces dendritic cell maturation and Th1 polarization. Biochem Biophys Res Commun 2010; 394:272–8. [DOI] [PubMed] [Google Scholar]
- 59. Nefedova Y, Cheng P, Gilkes D et al Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol 2005; 175:4338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li R, Fang F, Jiang M et al STAT3 and NF‐κB are simultaneously suppressed in dendritic cells in lung cancer. Sci Rep 2017; 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Endotoxin levels in RA ECM. Barchart representing the levels of endotoxin in ECM (n = 10). An LAL assay was performed on RA ECM and was determined to be free from contaminating levels of endotoxins.
Fig. S2. Proinflammatory and Proangiogenic mediators in RA ECM. Barchart representing the production of IL‐6, IL‐1β, TNFα, bFGF and VEGF released from RA synovial tissue biopsies (n = 7).
Fig. S3. Gating controls for Phagocytosis and Antigen Processing Assays. Representative flow cytometry dotplots displaying the percentage uptake of Luciferin Yellow (i) or DQ Ovalbumin (ii). Gates were set using the 4 degree background control for each sample used.
Fig. S4. Differentially expressed metabolic genes in synovial fluid and peripheral blood RA CD1c+DC. Heatmap generated from RNA‐Seq analysis depicting hierarchical clustering of differentially expressed genes involved in cellular metabolism in CD1c+DC isolated from synovial fluid and peripheral blood of RA patients (n = 3).
Table S1. Oligonucleotide primers used in RT‐PCR.