

Abstract
Background.
Autoinflammatory diseases are distinct from autoimmune diseases. Whereas autoinflammatory diseases are due to dysfunctional T-cells and B-cells, autoinflammatory diseases are due to overproduction of macrophage cytokines particularly interleukin-1 beta (IL-1β). A causative role for IL-1 in autoinflammatory diseases is derived from clinical studies blocking the IL-1 receptor or neutralizing monoclonal antibodies or soluble receptors.
Methods.
A review was performed of clinical trials in autoinflammatory diseases using the IL-1 receptor antagonist (anakinra), the soluble IL-1 receptor (rilonacept), antibodies to IL-1β (canakinumab, gevokizumab) and anti-IL-1α (xilonix).
Findings.
Anakinra blocks the IL-1 Receptor type 1 (IL-1R1) and therefore blocks the activities of both IL-1α and IL-1β. Off-label use of anakinra is common for a broad spectrum of inflammatory diseases. Neutralization of IL-1β is used to treat hereditary autoinflammatory diseases but also atherosclerosis. Rilonacept reduces arterial wall inflammation in patients with chronic kidney disease. Neutralization of IL-1α has prolonged life in patients with advanced metastatic colorectal cancer. Compared to other cytokine blocking therapies, reducing the activities of IL-1 has an excellent safety record.
Conclusions.
Blocking IL-1 therapies can be used to treat a wide-spectrum of acute and chronic inflammatory diseases.
Keywords: innate immunity, anakinra, canakinumab, xilonix, rilonacept, cancer
Introduction
The interleukin-1 (IL-1) family consists of 11 cytokines and 10 receptors. The IL-1 family is divided into three subfamilies (Figure 1). Of these, IL-1β and IL-1α are the most inflammatory members and are studied for their role in autoinflammatory diseases. IL-18, a member of the family, also plays a role in autoinflammatory inflammatory diseases but because IL-18 induces IFNγ, IL-18 is also studied for its role in autoimmune diseases. Monotherapy specifically blocking IL-1α, IL-1β or IL-18 activities results in a rapid and sustained reduction in disease severity. Although initially studied for the treatment of rare hereditary autoinflammatory diseases, blocking IL-1 activities is also effective in treating common autoinflammatory conditions such as heart failure and acute flares of gout. Ongoing trials target a broad spectrum of new indications, including cancer, atherosclerosis and hearing loss. Three IL-1-targeted agents have been approved: the IL-1 receptor antagonist, anakinra, a soluble decoy receptor, rilonacept, and a neutralizing monoclonal anti-interleukin-1β antibody, canakinumab. In addition, a monoclonal antibody directed against the IL-1 receptor and a neutralizing anti-IL-1α are in clinical trials.
Figure 1. The 3 subfamilies of the IL-1 family.
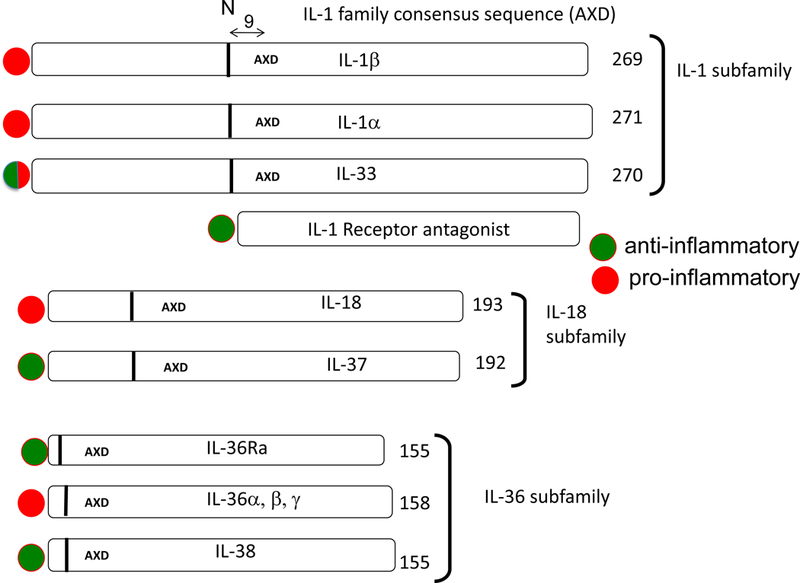
The lengths of the precursors are shown with the number of amino acids indicated at the end of each cytokine. The location of IL-1 family consensus sequence AXD is indicated in each precursor. In consensus sequence AXD, the A is an aliphatic amino acid, X is any amino acid and then D is always aspartic acid. The aspartic acid D is not the aspartic acid for the recognition of caspase-1 cleavage. Nine amino acids preceding the AXD site is a vertical bar. The vertical bar indicates the location of the optimal N-terminus. The processing enzymes of the precursors that result in an optimal N-terminus differ for each member. Some members of the IL-1 family have more than one AXD site thus creating an alternative N-terminus, for example IL-37 (137). The N-terminus affects the dimensional structure of the cytokine and therefore receptor binding and activity (138). In the case of IL-1β, nine amino acids preceding the AXD site is the caspase-1 cleavage site at amino acid 117. The IL-1 family members with proinflammatory properties are indicated by a red circle whereas a green circle represents cytokines that are anti-inflammatory. IL-1Ra is a unique member of the IL-1 family and highly homologous to IL-1β. IL-1Ra precursor has a classic signal peptide, is processed in the Golgi and readily secreted. There is an intracellular form of the IL-1Ra generated by alternate splicing. Intracellular IL-1Ra plays a role inside the cell (139). Because IL-1Ra has a signal peptide and is readily secreted, there is no AXD site.
Autoinflammatory and Autoimmune disease
Autoinflammatory diseases are associated with overactive innate inflammation and can be defined as increased inflammation mediated predominantly by IL-1, without the association with autoantibodies or autoreactive T lymphocytes (1). In autoimmune diseases, the adaptive immune system is dysfunctional. However, given the various interactions and similarities between the innate and adaptive immune systems, they cannot be viewed as completely separate entities but more as two ends of a continuum of inflammatory and immunological diseases. In autoimmune diseases, blocking TNFα is effective but blocking several cytokines of T-cell origin is also effective. In autoinflammatory diseases, neutralization of TNFα is not effective (such as in gout), or can even exacerbate the condition. In autoinflammatory diseases, the release of IL-1β from the monocyte is often elevated, but in the same cells the production of TNFα is not different from that of healthy controls.
Classic, hereditary autoinflammatory diseases
The classic autoinflammatory diseases are chronic, debilitating syndromes (2). They are rare genetic disorders, but the clinical inflammatory manifestations as well as the hematologic and metabolic abnormalities are common, characterized by recurrent episodes of systemic and local inflammation. The frequency, duration and severity of these episodes vary; in some cases, the inflammatory episodes seem almost continuous but the attacks can often resolve spontaneously only to re-occur a few weeks or several months later. Typically, patients suffer from recurrent fevers, generalized fatigue, diffuse pains in muscles and joints, frank inflammation in the linings of the lungs and peritoneal cavity, loss of appetite and poor sleep. Symptoms also include gastrointestinal disturbances and skin rashes. Neutrophilia and elevated acute phase proteins are present during the episodes.
What accounts for the inflammation? Blood monocytes from patients with autoinflammatory diseases release more IL-1β compared to cells from healthy persons (3–8). Some autoinflammatory conditions are hereditary and due to mutations in the intracellular proteins that control caspase-1, the enzyme that converts IL-1β to an active cytokine prior to release from the cell. With increased secretion of IL-1β, there is greater inflammation.
Familial Mediterranean Fever.
Familial Mediterranean Fever (FMF) is perhaps the most known autoinflammatory disease. Starting in childhood or adolescence, FMF patients suffer life of recurrent bouts of fevers and pain in linings of the abdomen. This pain mimics acute appendicitis and most patients have had abdominal surgery before the diagnosis of FMF is made. The genetic mutation was identified in the MEFV gene (after Mediterranean Fever), a previously unknown gene encoding for a protein that was given the name ‘pyrin’ from the Greek ‘pyros’ for fire (9). The term pyrexia is sometimes used for fever, as in the fever in FMF. A French consortium suggested the name ‘marenostrin’ because most patients with FMF are from Mediterranean Sea region. Wild-type pyrin is required to keep a tight control over activation of caspase-1 and the processing and release of active IL-1β whereas the mutation in pyrin results in a loss of this control with greater release of IL-1β (10).
Cryopyrin Associated Periodic Syndrome (CAPS).
In CAPS patients, single amino acid mutations are found in a protein that was originally called “cryopyrin” (11) because the clinical manifestations of the disease are triggered by exposure to cold, hence “cryo”. Following exposure to cold, even air-conditioned rooms, these patients develop fever and systemic symptoms (resembling FMF), in which the causative protein had already been called “pyrin”, hence the name “cryopyrin”. The commonly used term for the protein is NLRP3 (Nucleotide-binding domain and Leucine-rich Repeat Pyrin containing 3) but the name CAPS remains. Before mutation-analysis was available, three clinical variants of disease manifestations were recognized: Familial Cold Autoinflammatory Syndrome (FCAS), Muckle-Wells Syndrome and Neonatal Onset Multi Inflammatory Disease (NOMID, also called CINCA). It is now clear that CAPS is a continuum of diseases with severity from mildest form (FCAS) to most severe NOMID, with overlap between the clinical phenotypes. In Muckle-Wells Syndrome, the patients develop progressive loss of hearing and lose kidney function due to secondary amyloidosis. In children with NOMID there are total body rashes, destruction of the joints, hearing loss and chronic sterile inflammation in the brain. In fact, these children are thought to have learning and emotional problems, which resolve with IL-1 blockade (6, 12).
TNF-Receptor Associated Periodic Syndrome.
TNF-Receptor Associated Periodic Syndrome (TRAPS) is an autosomal dominant disease caused by mutations in the TNF-receptor type 1 (13). The mutations result in an inability of the receptor to be inserted normally into the cell membrane, which is at first sight contradictory with an inflammatory phenotype. Research so far has shown that the mutated receptor accumulates inside the cells, which causes increased synthesis of IL-1 through increased reactive oxygen species activation and possibly other pathways (14, 15). Patients experience recurrent bouts of fever with local and systemic inflammation, initially thought to be due to a lack of circulating soluble TNFα receptors, but now shown to be an IL-1-mediated disease.
Hyper-IgD Syndrome.
Hyper-IgD Syndrome (HIDS) is a genetic, (autosomal recessive) autoinflammatory disorder characterized by 4–6 days of fever, muscle aches, a skin rash, painful mouth ulcers and swollen lymph nodes. The disease is caused by mutations in the enzyme termed mevalonate kinase resulting in mevalonate kinase deficiency. Several intracellular pathways of protein modification have linked mevalonate kinase deficiency to the control of IL-1 production(16), including activation of caspase-1 (17). Mevalonate kinase deficiency per se, is linked to a phenotypic continuum of disease ranging from isolated fever episodes (i.e. Hyper-IgD Syndrome) to a more severe phenotype of fever episodes in combination with cerebellar ataxia, mental retardation, anemia, liver damage and developmental delay which can result in early death.
Common Inflammatory Diseases due to Autoinflammation.
As described above, specific missense mutations in diseases such as FMF and CAPS result in periodic fevers with systemic and local inflammation. These diseases do not involve T-lymphocytes, which are characteristically the effector cells in autoimmune diseases. There are other acute and chronic inflammatory diseases such as gout, pericarditis and heart failure, which are not autoimmune diseases but rather autoinflammatory syndromes. In autoinflammatory diseases, the effector cell is a myeloid cell, characteristically a monocyte or macrophage (18). Because of the safety and relative short duration, anakinra can be used as a diagnostic as well as a treatment for patients refractory to glucocorticoid treatment with undefined autoinflammatory signs and symptoms (19).
Gout, a uniquely IL-1β mediated disease.
Worldwide, osteoarthritis dominates as the cause of painful joints followed by gouty arthritis. By comparison, rheumatoid arthritis accounts for a marginally small number of subjects with painful joints. Nevertheless, the systemic effects of rheumatoid arthritis garner considerable interest in the pathogenesis and treatment of the disease. It is often overlooked that gout is also a systemic disease but also hyperuricemia even in patients without. Gout is the tip of the iceberg for the systemic manifestation isolated hyeruricemia. Subjects with hyperuricemia are in a state of chronic inflammation. For example, there are ample studies linking hyperuricemia with morbidity and mortality associated with hypertension, atherosclerosis, chronic kidney disease and type 2 diabetes (20–22). Soluble urate suppresses the level of the IL-1 Receptor antagonist (IL-1Ra), the natural inhibitor of IL-1β. As a result, IL-1β levels increase and blood monocytes from subjects with hyperuricemia release more IL-1β compared to monocytes from subjects without elevated uric acid (23). Transcriptomic analysis of urate primed monocytes revealed broad increased mTOR signaling and decreased autophagic activity (24). Acute attacks of gouty arthritis are highly responsive to anakinra (25–32), rilonacept (33–37) or canakinumab (38, 39).
Anakinra following an acute myocardial infarction.
The first study examined the effect of anakinra in patients who had suffered a ST-elevated myocardial infarction (STEMI) and had received optimal standard of care. Standard treatment includes anti-coagulation, catherization of the affected coronary arteries and placement of stents. In that study, anakinra treatment was initiated at 100 mg subcutaneously following stent placement and continues for two weeks (40, 41). Seventy-two hours after the acute event, CRP reaches peak levels and correlates with the degree of inflammation due to the ischemic event as well as to the size of the infarcted myocardium. Anakinra treatment resulted in a significant reduction in the medium level of CRP of 256 in the placebo group to 75 mg/L in patients receiving anakinra (41). Thus, despite optimal standard of care for the acute event, anakinra administered after the event exerts a beneficial effect in reducing the progressive inflammation and damage to the myocardium. From animal studies, the infiltration of neutrophils and monocytes into the penumbra of the ischemic area likely contribute to further damage; anakinra treatment significantly reduces the infiltration (42).
Twelve weeks following the myocardial infarctions, patients were assessed for heart function such a left ventricular ejection fraction. Compared to the placebo-treated group, anakinra treated patients exhibited improved functional status, but did not reach statistical significance (41). A second trial was performed with 30 patients (41). Again, anakinra reduced CRP levels 72 hours after the myocardial infarction (p=0.002). After 10–14 weeks, cardiac function was evaluated and the reduction in CRP correlated with a reduction in left ventricular end-systolic volume (41). In both trials, the development of new onset New York Heart Association Grade III and IV heart failure was assessed. When the two trials were compared, the overall reduction in the development of heart failure was 30% in the placebo-treated groups compared to 5% in the anakinra group (p=0.035) (41).
Anakinra treatment of heart failure in patients on optimal standard of care.
Additional studies examined the effect of anakinra on heart failure in patients with poor exercise tolerance and signs of systemic inflammation. In a clinical trial, seven patients with heart failure and markers of systemic inflammation despite standard of care treatment received 100 mg of anakinra daily for 14 days. There was a statistically significant improvement in oxygen consumption compared to baseline measurements and a decrease in carbon dioxide (43). This first study established a role for treating patients with anakinra for refractory heart failure.
Patients with acute decompensated heart failure are hospitalized and present a therapeutic challenge. These patients have signs of systemic inflammation in addition to being in refractory heart failure while on standard of care. Thirty patients with acute decompensated heart failure, ejection fraction less than 40% and elevated CRP were randomized to receive either anakinra or placebo (44). Upon entering the trial, patients received either 100 mg anakinra or placebo twice daily for 3 days followed by 11 days of once daily dosing. Three days into the trial, CRP decreased by 61% in the anakinra group compared to the placebo treated group (p=0.004) (44). The authors concluded that IL-1 receptor blockade reduced the systemic inflammation in these patients but the study was not powered to determine a clinical benefit.
In these trials, patients are treated with anakinra for 14 days and although clinical and objective data indicate a functional improvement and reduced inflammation, it is possible that a more prolonged course of anakinra treatment would result in greater benefit for these patients. Patients hospitalized with an episode of acute decompensated heart failure have a high incidence for repeated hospitalizations for recurrent bouts of heart failure. Therefore, a trial was performed comparing treatment with 100 mg of daily anakinra of two and 12 weeks duration in patients discharged from the hospital following an episode of acute decompensated heart failure. In this study, a placebo treated group was also included. Patients treated with 12 weeks of anakinra exhibited improved aerobic capacity, ventilatory efficiency, quality of life and reduced readmission rates compared to placebo but also compared to patients treated with two weeks of anakinra (45).
Treating refractory idiopathic pericarditis with anakinra.
Pericarditis can be a manifestation of an inherited auto-inflammatory disorder such as TRAPS, FMF and CAPS (46, 47). Patients with Adult Onset Still Disease (AOSD) also have bouts of pericarditis (48) that respond to anakinra. However, there is no genetic association with idiopathic pericarditis, which most often develops after a viral illness. A summary of case reports that anakinra was highly effective in treating these patients was published in 2011 (49) and since that report a large number of studies have established the rapid and sustained benefit of anakinra, particularly in patients refractory to colchicine (50–62). A recent review of anakinra treatment for idiopathic pericarditis concluded that the disease is an example of IL-1-driven autoinflammation (45). Treating pericarditis with etanercept or infliximab has not been successful and in some reports anti-TNFα treatment for Crohn’s Disease has worsened the disease (63, 64).
Acute thrombotic stroke.
Anakinra was administered to patients admitted to the hospital within 6 hours of the signs of an acute thrombotic stroke (65). The dose, 2 mg/kg/hour for 72 hours, was the same dose used to treat septic shock. The trial in 34 patients was randomized and placebo controlled. Compared to placebo-treated controls, peripheral CRP, neutrophils and IL-6 levels were lower in patients treated with anakinra (65). Although the study was not powered for significant improved neurological outcome, the subgroup of patients with cortical infarcts performed better compared to the placebo group.
Intravenous anakinra was also administered to patients with subarachnoid hemorrhage due to aneurysmal rupture (66). Within 72 hours of the onset of symptoms, six patients received a bolus infusion of 500 mg of anakinra, followed by a steady infusion of 10 mg/kg/hour for 24 hours. Seven patients received placebo infusions. At 24 hours, CSF levels for IL-6 were reduced in the anakinra group compared to those patients receiving placebo at 24 hours (p=0.06) (66). In a related study, 25 patients with a cerebral ventricular drain in place as part of treatment for a subarachnoid bleed received increasing doses of intravenous anakinra (67). First, the patients received a bolus dose of anakinra (100 mg to 500 mg) followed by a four-hour infusion of anakinra from 1 to 10 mg per kilogram per hour. Plasma and CSF levels of anakinra were monitored. The objective of the study was to establish the dose regimen of peripheral anakinra that resulted in a CSF level of anakinra of 100 ng/mL within 45 minutes of the onset of the infusion. A concentration of 100 ng/mL is 100-fold greater that that measured in the CSF of children treated for 3 months with 100 mg of daily subcutaneous anakinra (6). This concentration (100 ng/mL) was deemed neuroprotective based on rats subjected to brain ischemia (68). In humans, CSF levels of 100 ng/mL were achieved with the highest regimen, that is, a bolus of 500 mg followed by 4 hours of anakinra at 10 mg/kg/hour (67, 69). The authors concluded that anakinra passively enters the brain in patients with a subarachnoid hemorrhage. Given these data, this regimen of anakinra for patients with cerebral vascular accidents such as thrombotic strokes and subarachnoid hemorrhages would provide reduced inflammation at the site of the lesion such as reduced infiltration of neutrophils and edema. Whether reduced IL-1-mediated inflammation will result in improved neurological outcomes remains to be determined in randomized, placebo-controlled blinded studies.
Traumatic brain injury.
Traumatic brain injury is a major cause of death worldwide in persons under the age of 40 years. An even greater number are left with severe disabilities, which results in a significant economic cost. A trial randomized, open-labeled trial was performed comparing 5 days of 100 mg subcutaneous anakinra to placebo in 20 patients with diffuse traumatic brain injury within the previous 24 hours. After a 6-hour monitoring period, patients received either anakinra or placebo. A central microdialysis was in placed in each patient as part of standard of care. Patients were intubated and were receiving ventilatory support. During the 6 hour monitoring period, the mean level in the CSF was 78 pg/mL but rose to 138 pg/mL 12 hours after the first dose of anakinra (70). In general, inflammatory cytokines in the CSF were lower in patients treated with anakinra; of these, macrophage-derived chemoattractant-1 (MDC-1) was significantly lower on days 1–5 (p=0.05) compared to patients treated with the placebo. On the 5th day of anakinra treatment, mean MDC-1 was 1.04 pg/mL compared to 45.4 pg/mL in the placebo group (70). The study was too small to detect clinic improvement, although the marked decrease in CSF levels MDC-1 represents a beneficial CNS effect of anakinra. Treatment earlier and longer would likely result in improved clinical outcomes.
Treatment of epilepsy with IL-1 blockade.
A role for IL-1 in seizure disorders is based on the innovative studies of Vezzani and Barfai (71). Although IL-1α is found in brain astrocytes and microglia, the data indicate that IL-1β contributes to epileptic seizures. Indeed, there was a clinical trial of an oral caspase-1 inhibitor (VX-765, NCT01048255) in subjects refractory to standard anti-epilepsy drugs. In that trial, 48 people received 900 mg of VX-765 three times each day and 12 people received placebo for six weeks followed by a six week post-treatment observation period. Although the primary endpoint was safety and tolerability, decreased seizure rates were less in the treatment arm. For example, 13–19% of the treatment group was seizure-free during the last two weeks compared to 0–9% in the placebo group. During the first two weeks of the follow-up period, there was a 19–31% decrease in the seizure rate of the treatment arm compared to 0–9% in the placebo arm.
Several studies have focused on febrile seizures since these are the most common type of seizure activity. Using an animal model for febrile seizures, an agonist role for IL-1β and an antagonist role for endogenous IL-1Ra in the hippocampus have been reported (72). Other studies have examined circulating cytokines in patients with recurrent seizures and find elevated levels of IL-6, IL-1Ra in the post-ictal period (73). In one study, elevated IL-1β has also been observed in the intracellular-ictal period in patients with recurrent temporal lobe epilepsy (74). Some studies have reported polymorphisms in IL-1α, IL-1β and IL-1Ra that are associated with subjects who develop epilepsy as adults (75–79).
Anakinra has been administered a young patient with a severe seizure disorder termed febrile infection-related epilepsy syndrome. This syndrome, which often follows an infectious encephalopathy, has a high mortality rate and there are few treatment options. The patient had recurrent seizures each day, which be came increasing less and even ceased while being treated with daily subcutaneous anakinra (80). When anakinra was stopped, seizures resumed only to decrease again upon restarting. CSF cytokines decreased with anakinra treatment.
Anakinra for MAS (Macrophage Activation Syndrome).
The pathogenesis of MAS has been the topic of increasing reports (81) and the incidence of MAS is likely underestimated. For example, there are case reports of MAS in patients with Ebola virus, parasitic and influenza infections (82, 83). The use of anakinra in MAS is primarily in patients with systemic juvenile idiopathic arthritis (sJIA). Similar to sJIA, some patients with Adult Onset Still’s Disease (AOSD) also develop MAS. There are several reports of anakinra use in sJIA and AOSD. Although canakinumab is approved for sJIA, anakinra is commonly used in these patients, possibly because anakinra also reduces the activity of IL-1α. Patients with sJIA or AOSD and treated with anakinra or canakinumab can develop MAS while on therapy. In some cases, the dose of anakinra is increased with clinical improvement.
What is the role for IL-1 in MAS? IL-1 acts at several levels related to the signs and symptoms of MAS. Fever and the increase hepatic ferritin are IL-1 mediated since IL-18 does not cause fever (84, 85), does not induce PGE2 (86) and dose not induce hepatic acute phase proteins (87). A critical role for IL-1 in MAS is the induction of IL-18. The endothelium contains constitutively preformed IL-18 precursor (88) and IL-1 induces the release of IL-18. High levels of IL-18 reported in MAS are not of myeloid origin but rather are derived from the IL-1-activated endothelium. IL-1-induced myocardial suppression is mediated by IL-18 (88, 89). IL-1 mediates the fever, hyperferritimia, coagulopathy and the production of IL-18. Also shown, IL-18 likely mediates the hypersplenism, elevated IFNγ, hypertriglyceridemia and hypotension of MAS. With high levels of IL-18-dependent IFNγ, there is macrophage activation and hemophagocytosis, which characterizes MAS.
Anakinra treatment of MAS as a manifestation with septic shock.
There were three randomized, placebo-controlled trials of anakinra to reduce all cause 28-day mortality in patients with a diagnosis of septic shock (90–92). Nearly 2,000 patients were enrolled in these trials. The first trial was dose-response open labeled trial (91). The second trial tested two doses, 1 mg/kg/hour for 72 hours compared to 2 mg/kg/hour for 72 hours (92). This trial revealed that survival increased over placebo when the patient enrolled in the trial had a greater than 24% risk of death but did not reach statistical significance. The third trial was performed in 91 centers world-wide but stopped at mid-way as the likelihood of reaching a statistically significant endpoint was low (90). A re-analysis of the data from the third trial was performed subsequently (93). The analysis revealed that patients with fever, disseminated intravascular coagulation, hepatobiliary dysfunction, cytopenias, and hyperferritinemia were features of MAS. Data were available for 763 adults from the original study cohort, randomized to receive either anakinra or placebo. Concurrent hepatobiliary dysfunction/disseminated intravascular coagulation was noted in 43 patients (5.6% of total; 18–75 years old; 47% women). Treatment with anakinra improved 28-day survival: 65.4% anakinra vs. 35.3% placebo), with hazard ratio for death 0.28 (0.11–0.71; p = 0.0071) (93).
Is atherosclerosis an autoinflammatory disease?
For at least three decades, a role for cytokine-mediated inflammation in the arteries, particularly the coronary and carotid arteries, has been the subject of basic as well as pre-clinical studies. The first clinical testing of such a concept was the use of anakinra in patients with rheumatoid arthritis and cardiovascular disease (94, 95). In those studies, anakinra treatment resulted in improved in endothelial and coronary aortic function as well as left ventricular myocardial deformation.
Background of the CANTOS trial.
The design of the CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcomes Study) trial was published in 2011 (96) and based on a reduction in circulating CRP in inflammatory diseases such as gout and several hereditary diseases upon treatment with anti-IL-1β monoclonal antibodies (reviewed in (18)). However, anakinra treatment had already been used in a randomized, placebo-controlled study in Type 2 diabetics; this trial was published in the New England Journal of Medicine in 2007. In that trial, after 4 and 13 weeks, anakinra significantly reduced CRP, IL-6 and the level of glycated hemoglobin (97). In many ways, the data from the anakinra trial served as a rationale for the CANTOS trial because Type 2 diabetics are comprise a large number of the high-risk population for a second cardiovascular event. Thus, a Phase 2b trial was performed using a dose-response of canakinumab of 5, 15, 50 and 150 mg monthly for 4 months in 556 Type 2 diabetics being treated with standard of care doses of statins but with CRP >2 mg/L. The median percentage reductions in CRP were 52, 66, 69, and 72, respectively, compared to 2.9 in the placebo group (98). As anticipated, IL-6 levels were also significantly reduced in the study. In many, ways the data from Phase 2b canakinumab study was similar to those of the 2007 anakinra trial (97). Another trial in Type 2 diabetic patients specifically used a neutralizing antibody to IL-1β, gevokizumab (99). That trial similarly reported the reductions in CRP. Therefore, at the time of the initiation of CANTOS trial, blocking IL-1 with anakinra or specifically IL-1β in Type 2 patients at risk for a second cardiovascular event was established.
CANTOS achieves its primary and secondary endpoints.
The role of low density lipoprotein (LDL) in the pathogenesis of atherosclerosis has been the basis for drug development for several decades. Lowering plasma LDL with statins has proven efficacy in reducing myocardial infarctions and deaths. However, the atherosclerotic process includes a significant role for cytokine-mediated arterial inflammation and IL-1β has been the most studied cytokine for its detrimental role in the formation of the atherosclerotic lesion (100). The CANTOS trial was a world-wide study in 10,000 patients who had survived a previous myocardial infarction or stroke but despite standard of care with statin therapy, continued to have a level of CRP greater than 2 mg/L; this level of CRP is a biomarker for risk of a second cardiovascular event (101). The primary endpoint of the trial was the first occurrence of a fatal or nonfatal myocardial infarction or non-fatal stroke. This is also defined as a three-point MACE. The first secondary end points included hospitalization for unstable angina that required rapid revascularization. There was another secondary endpoint; did IL-1β neutralization reduce the incidence of new-onset type 2 diabetes in patients with prediabetes at the time of randomization? Thus the CANTOS trial tested the hypothesis that IL-1β was causative in the atherosclerotic process, which resulted in a cardiovascular event.
Three doses were used; 50 mg, 150 mg, and 300 mg of canakinumab or Placebo administered 4 times a year for 4 years. The decrease in CRP was dose dependent. Importantly, canakinumab did not reduce lipid levels and this observation provides the conclusion that inflammation, and particularly IL-1β-induced inflammation, is as important as lowering LDL in reducing cardiovascular events. The reduction in a composite of cardiovascular events met the primary endpoint. The hazard ratios as compared with placebo were 0.85 (0.74 to 0.98; P=0.021) in subjects receiving 150 mg and 0.86 (0.75 to 0.99; P=0.031) in the 300 mg group. The reduction in the 50 mg dose was not statistically significant. The secondary endpoint of hospitalization for unstable angina and rapid angioplasty was reduced compared to placebo treated patients (Hazard Ratio of 0.83; 95% confidence level 0.73 to 0.95; P=0.005).
Is hearing loss as an autoinflammatory syndrome.
Sensorineural deafness is a prominent characteristic of persons with Muckle-Wells syndrome and persons with mutations in NLRP3 (102). The first reports of efficacy of anakinra to improve hearing in were in patients with Muckle-Wells syndrome (103) and several other reports subsequently followed (47, 104–111). In general, the reversal in sensorineural deafness with anakinra treatment was unexpected and signified the concept that hearing loss in autoinflammatory diseases was due to a reversible chronic inflammatory response and not due to loss of neuronal function. Thus early treatment is more likely to be beneficial (112). The autoinflammatory syndromes are gain-of-function mutations of NLRP3 and are autosomal dominant diseases. A recent study reported tow families with a missense mutation (Arg918Gln) in NLRP3 as the cause of autosomal-dominant sensorineural hearing loss (113). Treatment of one family member with IL-1β blockade reversed the systemic inflammation. However, treatment of the other family revealed that hearing loss was present without systemic inflammation (113). Thus local macrophages in the cochlea may release IL-1β, which leads to the hearing loss. It is also likely that somatic mutation in NLRP3 may account for other hearing-lose disorders.
Autoinflammation in Cancer: the role of IL-1 in the progression of cancer
IL-1 and the tumor microenvironment.
In the tumor microenvironment, one considers the role of IL-1 from the infiltrating myeloid cells as well as IL-1 production from the tumor cell or both. In addition, the production of IL-1Ra from the infiltrating myeloid cells is of considerable relevance. It is also possible that tumors also produce IL-1Ra. In health, the IL-1α precursor is found in nearly all epithelial cells such as the entire gastrointestinal tract, the epithelial cells of the lung, the ductal epithelium of the breast, prostatic cells, the hepatocyte and lining of the bladder and the renal epithelium. Therefore, tumors originating from these cells will contain the IL-1α precursor. Melanoma cells, B –cells of lymphomas, Hodgkin’s cells and the plasma cell of multiple myeloma contain the IL-1β precursor as reviewed in (114). In general, the level of IL-1α or IL-1β often correlates with poor outcomes (115). The level of IL-1β in the tumor microenvironment may be from the tumor cell itself but more likely to be from infiltrating myeloid cells, such as monocytes, neutrophils, dendritic and Natural Killer (NK) cells. T-cells are often present in the tumor and can be a source of either IL-1α or IL-1β. Regardless of whether IL-1 is of tumor cell origin or from infiltrating bone marrow-derived cells, IL-1 plays a major role in inducing angiogenesis (116–118). As the tumor increases in size due to neovascularization, IL-1 induction of matrix metalloproteinases facilitates the entry of tumor cells into the circulation and result in local or distant metastasis. IL-1 itself is a growth factor for malignant cells but IL-1 also induces growth factor production from resident stromal cells. For example, in multiple myeloma, IL-1β from the plasma cell in the bone marrow induces IL-6 from the stromal cells; IL-6 is a growth factor for plasma cells.
IL-1 in the systemic response to cancer.
Although levels of circulating IL-1β or IL-1α are low and often below the level of quantification in cancer (119), the systemic response to cancer reveals that the inflammation of cancer is due, in part, to IL-1. In two studies treating patients with metastatic colorectal cancer with a neutralizing antibody to IL-1α, the levels of IL-6 fall consistently as do the platelet counts (120, 121). Not unexpectedly, the inflammation of cancer increases the levels of circulating of IL-1Ra (114). In several studies, the levels of IL-1Ra reflect IL-1 driven inflammation, as is the case with non-cancer inflammation such a patients with coronary artery inflammation (122). However, in some cancers, the expected elevated levels of IL-1Ra are low, as reviewed in (114)
Anakinra to treat pre-multiple myeloma.
The first study to use a IL-1 blocking therapy to treat cancer is in patients with a diagnosis of pre-myeloma stage. Anakinra treatment was assessed in patients with smoldering or indolent myeloma, and first reported by Lust and co-workers in 2009 (123). The treatment was the standard dose of 100 mg daily of anakinra plus a weekly low dose of 20 mg of oral dexamethasone. The rationale for using anakinra in these patients is to reduce progression to overt myeloma, a consistently fatal disease even with advanced chemotherapeutics and bone marrow transplantation. The mechanism of action of IL-1 blockade is based on data that IL-1β released from bone marrow plasma cell induces IL-6 production from marrow stromal cells; futhermore, IL-6 is a growth factor for the malignant plasma cell (124). Since the amount of IL-1β released from malignant plasma cells is low, anakinra would reduce IL-6 with greater efficacy compared to neutralizing IL-6. In addition, experimental data revealed that although anakinra was highly effective in reducing IL-1β induced IL-6 in vitro, adding dexamethasone resulted in the death of the plasma cells (123–125).
A follow-up assessment of the treated cohort of patients has recently been reported (126). A reduction in serum C-reactive protein (CRP) level of 40 or greater percent from baseline was used as a biomarker of response to anakinra. Of the 47 patients, 22 patients did not have this a 40% fall in CRP; the median progression free survival was 11 months. However, in 25 patients with a fall in CRP of 40% or greater, the median progression free survival was 104 months (p<0.001). Moreover, the median overall survival of the responders has not been reached whereas the overall survival of those patients without a 40% reduction in CRP was 7.9 years p<0.001).
The clinical assessment of patients with smoldering myeloma includes a therapeutic intervention or a “wait and see” approach. Since the progression to active myeloma in many patients is 6 months to one year, blocking IL-1 with anakinra is a strategy for preventing progression of the disease. Each of the 47 patients in the study met established criteria for smoldering myeloma or indolent myeloma (126). Patients who did progress had statistically significant evidence of more active disease at time of enrollment. The standard of care for active multiple myeloma often is autologous bone marrow transplantation, but not in older patients. Therefore, when considering the overall survival in the 47 patients, the data include those with transplantation. In the 25 patients with a CRP decrease of 40% or greater and without bone marrow transplantation, many remain without progression to active disease. As such, a universally fatal disease is held at bay by daily anakinra and a weekly low dose of dexamethasone. It is possible that anakinra therapy in those without a 40% fall in CRP (considered non-responders) began anakinra therapy at a time when progression had already advanced. In contrast, anakinra when anakinra was started at an earlier stage of the disease, the Progression Free Survival and as well as Overall Survival was significantly greater.
Anakinra for solid tumors.
Despite a considerable number of reports in various animal models of solid tumors, there has been limited use of anakinra to treat solid tumors. Models of breast cancer are of particular interest based on data that the level of IL-1β in biopsies and excised tumors from patients with hormone receptor negative breast cancer correlate with poor prognosis (127). A polymorphism in the IL-1β gene (rs1143627) has been proposed to relate to increased breast cancer risk but a meta analysis of several studies reveals the polymorphisms is not a risk factor (128).
How much of anakinra therapy is blockade of IL-1α activity?
Because IL-1α and IL-1β can play a role in augmenting immune recognition, a prevailing, but misunderstood concept, is that blocking IL-1β or IL-1α would contribute to the immunosuppression of cancer and be contraindicated. However, reversing the immunosuppression of cancer has been validated with blocking CTLA4 and PD-1. Therefore, there is less reluctance to neutralize an immunostimulatory cytokine such as IL-1α. In what may be landmark milestone studies, patients with refractory, endstage cancer and losing weight received a course of neutralizing human anti-human IL-1α(129). Within the treated population, a significant number of patients responded with an increase in lean body mass (LBM), decreased constitutional symptoms and extended life compared to non-responders.
The study, albeit small, is the first to specifically neutralize IL-1α, a highly inflammatory member of the IL-1 family. It is a unique contribution for many reasons, particularly in end stage cancer patients. Since blocking the IL-1 receptor with anakinra or neutralizing IL-1β with canakinumab or rilonacept (reviewed in (18)) is without symptom side-effects, it was not unexpected that there were no symptom side-effects with anti-IL-1α. Importantly, the study evokes an examination of likely mechanisms, which could account for the objective and constitutional endpoints. First, the data demonstrate that treatment reduces systemic inflammation since a fall in circulating IL-6 levels remains one of the most consistent observations of blocking IL-1 (18). The source of the inflammatory trigger is likely the tumor itself, as all cancer cells of epithelial cell origin contain IL-1α in its precursor form. Inflammation also is due to invading stromal cells into the tumor microenvironment. As tumors out grow their vascular supply, they become necrotic, the IL-1α precursor is readily released and triggers local production of chemokines, which facilitate an influx of neutrophils and monocytes(18). Unlike precursor of IL-1β, the IL-1α precursor is fully active(130). Neutralization of local IL-1α likely reduces the infiltration of tumor-associated macrophages and myeloid derived suppressor cells, which contribute to the immunosuppression cancer mediated by inflammation (131).
At some point, this local inflammation must become systemic in order to account for one of the clinical endpoints of the study, the association of increased survival (19.3 months) with increased LBM in patients with colorectal cancer compared to 6.6 months in those who failed to increase LBM. The association of non-cancer chronic inflammation with loss of LBM is well established and IL-1 can directly induce muscle protein breakdown (132). The study also reports a reduction in fatigue, which is consistent with the use of anakinra in patients with inflammatory diseases unrelated to cancer (reviewed in(18)). In the early 1990’s either IL-1β or IL-1α was administered to patients with suppressed bone-marrow due to chemotherapy in order to stimulate hematopoiesis; although effective, picomolar concentrations of IL-1α was toxic with fever, severe fatigue, loss of appetite, myalgias and frank hypotension (133). Since picomolar concentrations of IL-1α induce these symptoms, demonstrating elevated circulating IL-1α should not be a criterion for the rationale of blocking IL-1α in cancer. IL-1α is also present on platelets and may account for the systemic effects such as elevated IL-6. In fact, the longstanding reports of platelet involvement in metastasis, which would include platelet-endothelial cell interaction (134), may now be, in part, understood by neutralization of IL-1α. This study is the first clinical evidence that endogenous IL-1α-induced IL-6 contributes to the thrombocytosis in cancer (133).
Additional possible mechanisms of action with neutralization of IL-1α include decreased angiogenesis (116) and decreased immunosuppression (135). IL-1α neutralization also includes direct anti-tumor properties by inhibition of tumor growth. With IL-1α presence in non-cancerous as well as cancerous cells and given the broad inflammatory properties of IL-1α, no one mechanism accounts for the study’s observations (129). We are left questions. Is refractory endstage cancer with progressive loss in LBM sufficient to initiate treatment with IL-1α neutralization? Given the near total lack of side-effects, the long-term safety of IL-1 blockade (18) and the efficacy of MABp1, there are few reasons to withhold treatment in this population. Would neutralizing IL-1α exhibit a greater efficacy if used earlier as an adjunct during chemotherapy? For example, would neutralizing IL-1α potentiate the anti-tumor effect of tyrosine kinase inhibitors? Since we know that IL-1 induces myeloid suppressor cells(131), would neutralizing IL-1α be beneficial if used in combination with anti-CTLA4 or anti-PD-1?
CANTOS reveals a role for IL-1β in the progression of cancer.
Patients considered for entry into the CANTOS trial with a known cancer were not enrolled. The 10,061 patients were randomized to receive 50, 150, 300 mg of canakinumab or Placebo every 3 months. However, during the 4 year duration, these subjects were evaluated for a diagnosis of cancer by oncologists blinded to the whether the patients were treated with canakinumab or the Placebo. The first observation was remarkable; there were 196 cancer deaths in the study but in patients receiving the 300 mg dose, there was 51% reduction in death from any cancer (p=0.0009) (136). The baseline CRP levels in those patients who were determined to have lung cancer was 6.0 mg/L compared to 4.2 mg/L in patients with no CRP (p<0.0001). Similarly, the baseline IL-6 in the cancer group was 3.2 pg/ml compared to 2.6 pg/mL in those without a diagnosis of cancer (p<0.0001). The incidence of lung cancer diagnosis was reduced by 67% in patients receiving the 300 mg dose (P=0.00008). In this group of patients with lung cancer, treatment with the 300 mg dose of canakinumab reduced fatal lung cancer by 77% (0.0002).
Can autoinflammation explain cancer progression?
The reduction in the incidence and survival of cancer demonstrated in the CANTOS trial reveals the role of IL-1 driven inflammation and the progression of cancer. As listed in Table 1, there are several known inflammatory properties of IL-1, which mediate cancer progression. A fundamental concept in autoinflammation is that regardless of the initiating event, as inflammation increases the production of IL-1 also increases and contributes to more inflammation. From Table 1, cancer progression from its early stages of vascularization and local spread to invasion into the circulation and lymphatics suggest autoinflammatory mechanisms independent of T or B cell function. The suppression of acquired immune responses in cancer is specific for the neoantigens of the tumor but the IL-1-driven immunosuppression is non-specific and therefore due to autoinflammation. The data from the CANTOS trial as well as anti-IL-1α treatment of advanced metastatic cancer support the concept that IL-1 blockade a safe, non-toxic, durable checkpoint inhibitor, reversing the immunosuppression of cancer-induced inflammation Would reversing the immunosuppression of cancer by the toxic, checkpoint inhibitors be better served when used in combination with IL-1 blockade? Would treating very early cancer with IL-1 blockade monotherapy prevent cancer progression?
Table 1.
IL-1-mediated Mechanisms of Cancer Promotion
| Angiogenesis, IL-1-dependent VEGF production, increased VEGFR |
| Generation of Myeloid-derived Suppressor Cells (MDSC) |
| Elevated IL-8 and chemokines, including monocyte chemotaxis (for MDSC) |
| Migration of endothelial precursors into tumors |
| Increased basic FGF, PDGF, IL-6 and other stromal growth factors |
| Neutrophil accumulation in tumor |
| Growth factor production (GM-CSF, FGF, IL-6, IL-1, chemokines) |
| Thrombocytosis, platelet-derived IL-1α |
| Induction of matrix metalloproteinases (MMP) for local spread |
| Cyclooxygenase (increased PGE2) for immunosuppression |
| Immunosuppression (increased PD-l ligand and CTLA-4) |
| Low IL-1Ra circulating levels due to high uric acid levels |
| Increased endothelial tissue factor for coagulopathy |
| Increased ICAM-1, increased VCAM-1 for local and metastatic tumor spread |
Conclusions.
Blocking IL-1 therapies can be used to treat a wide-spectrum of acute and chronic inflammatory diseases, including cancer, due to autoinflammation.
Acknowledgments
Supported by NIH Grants AI-15614
Footnotes
The author declares no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell 2010;140(6):784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 2009;27:621–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drenth JP, van der Meer JW, Kushner I. Unstimulated peripheral blood mononuclear cells from patients with the hyper-IgD syndrome produce cytokines capable of potent induction of C-reactive protein and serum amyloid A in Hep3B cells. J Immunol 1996;157(1):400–4. [PubMed] [Google Scholar]
- 4.Gattorno M, Piccini A, Lasiglie D, Tassi S, Brisca G, Carta S, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum 2008;58(5):1505–15. [DOI] [PubMed] [Google Scholar]
- 5.Gattorno M, Tassi S, Carta S, Delfino L, Ferlito F, Pelagatti MA, et al. Pattern of interleukin-1beta secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum 2007;56(9):3138–48. [DOI] [PubMed] [Google Scholar]
- 6.Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med 2006;355(6):581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med 2005;201(9):1479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colina M, Pizzirani C, Khodeir M, Falzoni S, Bruschi M, Trotta F, et al. Dysregulation of P2X7 receptor-inflammasome axis in SAPHO syndrome: successful treatment with anakinra. Rheumatology (Oxford) 2010;49(7):1416–8. [DOI] [PubMed] [Google Scholar]
- 9.Mansfield E, Chae JJ, Komarow HD, Brotz TM, Frucht DM, Aksentijevich I, et al. The familial Mediterranean fever protein, pyrin, associates with microtubules and colocalizes with actin filaments. Blood 2001;98(3):851–9. [DOI] [PubMed] [Google Scholar]
- 10.Chae JJ, Aksentijevich I, Kastner DL. Advances in the understanding of familial Mediterranean fever and possibilities for targeted therapy. Br J Haematol 2009;146(5):467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001;29(3):301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldbach-Mansky R Current status of understanding the pathogenesis and management of patients with NOMID/CINCA. Curr Rheumatol Rep 2011;13(2):123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999;97(1):133–44. [DOI] [PubMed] [Google Scholar]
- 14.Simon A, Park H, Maddipati R, Lobito AA, Bulua AC, Jackson AJ, et al. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc Natl Acad Sci U S A 2010;107(21):9801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J Exp Med 2011;208(3):519–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoffels M, Simon A. Hyper-IgD syndrome or mevalonate kinase deficiency. Curr Opin Rheumatol 2011;23(5):419–23. [DOI] [PubMed] [Google Scholar]
- 17.Gosavi S, Chavez LL, Jennings PA, Onuchic JN. Topological frustration and the folding of interleukin-1 beta. J Mol Biol 2006;357(3):986–96. [DOI] [PubMed] [Google Scholar]
- 18.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 2012;11(8):633–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrison SR, McGonagle D, Nizam S, Jarrett S, van der Hilst J, McDermott MF, et al. Anakinra as a diagnostic challenge and treatment option for systemic autoinflammatory disorders of undefined etiology. JCI Insight 2016;1(6):e86336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cicero AF, Salvi P, D’Addato S, Rosticci M, Borghi C, Brisighella Heart Study g. Association between serum uric acid, hypertension, vascular stiffness and subclinical atherosclerosis: data from the Brisighella Heart Study. J Hypertens 2014;32(1):57–64. [DOI] [PubMed] [Google Scholar]
- 21.Athyros VG, Mikhailidis DP. Uric acid, chronic kidney disease and type 2 diabetes: a cluster of vascular risk factors. J Diabetes Complications 2014;28(2):122–3. [DOI] [PubMed] [Google Scholar]
- 22.Gustafsson D, Unwin R. The pathophysiology of hyperuricaemia and its possible relationship to cardiovascular disease, morbidity and mortality. BMC Nephrol 2013;14:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crisan TO, Cleophas MC, Oosting M, Lemmers H, Toenhake-Dijkstra H, Netea MG, et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann Rheum Dis 2016;75(4):755–62. [DOI] [PubMed] [Google Scholar]
- 24.Crisan TO, Cleophas MCP, Novakovic B, Erler K, van de Veerdonk FL, Stunnenberg HG, et al. Uric acid priming in human monocytes is driven by the AKT-PRAS40 autophagy pathway. Proc Natl Acad Sci U S A 2017;114(21):5485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 2007;9(2):R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Announ N, Palmer G, Guerne PA, Gabay C. Anakinra is a possible alternative in the treatment and prevention of acute attacks of pseudogout in end-stage renal failure. Joint Bone Spine 2009;76(4):424–6. [DOI] [PubMed] [Google Scholar]
- 27.Cronstein BN, Sunkureddi P. Mechanistic aspects of inflammation and clinical management of inflammation in acute gouty arthritis. J Clin Rheumatol 2013;19(1):19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dinarello CA, van der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol 2013;25(6):469–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh P, Cho M, Rawat G, Simkin PA, Gardner GC. Treatment of acute gouty arthritis in complex hospitalized patients with anakinra. Arthritis Care Res (Hoboken) 2013;65(8):1381–4. [DOI] [PubMed] [Google Scholar]
- 30.Lopalco G, Rigante D, Giannini M, Galeazzi M, Lapadula G, Iannone F, et al. Safety profile of anakinra in the management of rheumatologic, metabolic and autoinflammatory disorders. Clin Exp Rheumatol 2016;34(3):531–8. [PubMed] [Google Scholar]
- 31.McGonagle D, Tan AL, Shankaranarayana S, Madden J, Emery P, McDermott MF. Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra. Ann Rheum Dis 2007;66(12):1683–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossi-Semerano L, Fautrel B, Wendling D, Hachulla E, Galeotti C, Semerano L, et al. Tolerance and efficacy of off-label anti-interleukin-1 treatments in France: a nationwide survey. Orphanet J Rare Dis 2015;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gillespie J, Mathews R, McDermott MF. Rilonacept in the management of cryopyrin-associated periodic syndromes (CAPS). J Inflamm Res 2010;3:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitha E, Schumacher HR, Fouche L, Luo SF, Weinstein SP, Yancopoulos GD, et al. Rilonacept for gout flare prevention during initiation of uric acid-lowering therapy: results from the PRESURGE-2 international, phase 3, randomized, placebo-controlled trial. Rheumatology (Oxford) 2013;52(7):1285–92. [DOI] [PubMed] [Google Scholar]
- 35.Schumacher HR Jr., Evans RR, Saag KG, Clower J, Jennings W, Weinstein SP, et al. Rilonacept (interleukin-1 trap) for prevention of gout flares during initiation of uric acid-lowering therapy: results from a phase III randomized, double-blind, placebo-controlled, confirmatory efficacy study. Arthritis Care Res (Hoboken) 2012;64(10):1462–70. [DOI] [PubMed] [Google Scholar]
- 36.Terkeltaub R, Sundy JS, Schumacher HR, Murphy F, Bookbinder S, Biedermann S, et al. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis 2009;68(10):1613–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Terkeltaub RA, Schumacher HR, Carter JD, Baraf HS, Evans RR, Wang J, et al. Rilonacept in the treatment of acute gouty arthritis: a randomized, controlled clinical trial using indomethacin as the active comparator. Arthritis Res Ther 2013;15(1):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlesinger N, Mysler E, Lin HY, De Meulemeester M, Rovensky J, Arulmani U, et al. Canakinumab reduces the risk of acute gouty arthritis flares during initiation of allopurinol treatment: results of a double-blind, randomised study. Ann Rheum Dis 2011;70(7):1264–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlesinger N, Alten RE, Bardin T, Schumacher HR, Bloch M, Gimona A, et al. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann Rheum Dis 2012;71(11):1839–48. [DOI] [PubMed] [Google Scholar]
- 40.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am J Cardiol 2010;105(10):1371–7 e1. [DOI] [PubMed] [Google Scholar]
- 41.Abbate A, Canada JM, Van Tassell BW, Wise CM, Dinarello CA. Interleukin-1 blockade in rheumatoid arthritis and heart failure: a missed opportunity? Int J Cardiol 2014;171(3):e125–6. [DOI] [PubMed] [Google Scholar]
- 42.Toldo S, Mezzaroma E, Van Tassell BW, Farkas D, Marchetti C, Voelkel NF, et al. Interleukin-1beta blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol 2013;98(3):734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abbate A, Van Tassell BW, Biondi-Zoccai GG. Blocking interleukin-1 as a novel therapeutic strategy for secondary prevention of cardiovascular events. BioDrugs 2012;26(4):217–33. [DOI] [PubMed] [Google Scholar]
- 44.Van Tassell BW, Abouzaki NA, Oddi Erdle C, Carbone S, Trankle CR, Melchior RD, et al. Interleukin-1 blockade in acute decompensated heart failure: A randomized, double-blinded, placebo-controlled pilot study. J Cardiovasc Pharmacol 2016;67(6):544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buckley L, Viscusi MM, Van Tassell B, Abbate A. Interleukin-1 blockade for the treatment of pericarditis. Eur Heart J Cardiovasc Pharmacother 2017;in press. [DOI] [PMC free article] [PubMed]
- 46.Cantarini L, Lucherini OM, Cimaz R, Galeazzi M. Recurrent pericarditis caused by a rare mutation in the TNFRSF1A gene and with excellent response to anakinra treatment. Clin Exp Rheumatol 2010;28(5):802. [PubMed] [Google Scholar]
- 47.Kuemmerle-Deschner JB, Lohse P, Koetter I, Dannecker GE, Reess F, Ummenhofer K, et al. NLRP3 E311K mutation in a large family with Muckle-Wells syndrome - description of a heterogeneous phenotype and response to treatment. Arthritis Res Ther 2011;13(6):R196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerfaud-Valentin M, Maucort-Boulch D, Hot A, Iwaz J, Ninet J, Durieu I, et al. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore) 2014;93(2):91–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scott IC, Vijay Hajela V, Hawkins PN, Lachmann HJ. A case series and systematic literature review of anakinra and immunosuppression in idiopathic recurrent pericarditis. J Cardiology Cases 2011;4:e93–e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scardapane A, Brucato A, Chiarelli F, Breda L. Efficacy of an interleukin-1beta receptor antagonist (anakinra) in idiopathic recurrent pericarditis. Pediatr Cardiol 2013;34(8):1989–91. [DOI] [PubMed] [Google Scholar]
- 51.Vassilopoulos D, Lazaros G, Tsioufis C, Vasileiou P, Stefanadis C, Pectasides D. Successful treatment of adult patients with idiopathic recurrent pericarditis with an interleukin-1 receptor antagonist (anakinra). Int J Cardiol 2012;160(1):66–8. [DOI] [PubMed] [Google Scholar]
- 52.Camacho-Lovillo M, Mendez-Santos A. Successful treatment of idiopathic recurrent pericarditis with interleukin-1 receptor antagonist (Anakinra). Pediatr Cardiol 2013;34(5):1293–4. [DOI] [PubMed] [Google Scholar]
- 53.Finetti M, Insalaco A, Cantarini L, Meini A, Breda L, Alessio M, et al. Long-term efficacy of interleukin-1 receptor antagonist (anakinra) in corticosteroid-dependent and colchicine-resistant recurrent pericarditis. J Pediatr 2014;164(6):1425–31 e1. [DOI] [PubMed] [Google Scholar]
- 54.Imazio M Idiopathic recurrent pericarditis as an immune-mediated disease: current insights into pathogenesis and emerging treatment options. Expert Rev Clin Immunol 2014;10(11):1487–92. [DOI] [PubMed] [Google Scholar]
- 55.Lazaros G, Vasileiou P, Koutsianas C, Antonatou K, Stefanadis C, Pectasides D, et al. Anakinra for the management of resistant idiopathic recurrent pericarditis. Initial experience in 10 adult cases. Ann Rheum Dis 2014;73(12):2215–7. [DOI] [PubMed] [Google Scholar]
- 56.Cantarini L, Lopalco G, Selmi C, Napodano S, De Rosa G, Caso F, et al. Autoimmunity and autoinflammation as the yin and yang of idiopathic recurrent acute pericarditis. Autoimmun Rev 2015;14(2):90–7. [DOI] [PubMed] [Google Scholar]
- 57.D’Elia E, Brucato A, Pedrotti P, Valenti A, De Amici M, Fiocca L, et al. Successful treatment of subacute constrictive pericarditis with interleukin-1beta receptor antagonist (anakinra). Clin Exp Rheumatol 2015;33(2):294–5. [PubMed] [Google Scholar]
- 58.Jain S, Thongprayoon C, Espinosa RE, Hayes SN, Klarich KW, Cooper LT, et al. Effectiveness and safety of anakinra for management of refractory pericarditis. Am J Cardiol 2015;116(8):1277–9. [DOI] [PubMed] [Google Scholar]
- 59.Baskar S, Klein AL, Zeft A. The use of IL-1 Receptor antagonist (anakinra) in idiopathic recurrent pericarditis: a narrative review. Cardiol Res Pract 2016;2016:7840724. [DOI] [PMC free article] [PubMed]
- 60.Lazaros G, Imazio M, Brucato A, Vassilopoulos D, Vasileiou P, Gattorno M, et al. Anakinra: an emerging option for refractory idiopathic recurrent pericarditis: a systematic review of published evidence. J Cardiovasc Med (Hagerstown) 2016;17(4):256–62. [DOI] [PubMed] [Google Scholar]
- 61.Brucato A, Imazio M, Gattorno M, Lazaros G, Maestroni S, Carraro M, et al. Effect of anakinra on recurrent pericarditis among patients with colchicine resistance and corticosteroid dependence: The AIRTRIP randomized clinical Trial. JAMA 2016;316(18):1906–12. [DOI] [PubMed] [Google Scholar]
- 62.Imazio M, Brucato A, Pluymaekers N, Breda L, Calabri G, Cantarini L, et al. Recurrent pericarditis in children and adolescents: a multicentre cohort study. J Cardiovasc Med (Hagerstown) 2016;17(9):707–12. [DOI] [PubMed] [Google Scholar]
- 63.Ambrose NL, O’Connell PG. Anti-TNF alpha therapy does not always protect rheumatoid arthritis patients against developing pericarditis. Clin Exp Rheumatol 2007;25(4):660. [PubMed] [Google Scholar]
- 64.Devasahayam J, Pillai U, Lacasse A. A rare case of pericarditis, complication of infliximab treatment for Crohn’s disease. J Crohns Colitis 2012;6(6):730–1. [DOI] [PubMed] [Google Scholar]
- 65.Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, et al. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry 2005;76(10):1366–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singh N, Hopkins SJ, Hulme S, Galea JP, Hoadley M, Vail A, et al. The effect of intravenous interleukin-1 receptor antagonist on inflammatory mediators in cerebrospinal fluid after subarachnoid haemorrhage: a phase II randomised controlled trial. J Neuroinflammation 2014;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galea J, Ogungbenro K, Hulme S, Greenhalgh A, Aarons L, Scarth S, et al. Intravenous anakinra can achieve experimentally effective concentrations in the central nervous system within a therapeutic time window: results of a dose-ranging study. J Cereb Blood Flow Metab 2011;31(2):439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clark SR, McMahon CJ, Gueorguieva I, Rowland M, Scarth S, Georgiou R, et al. Interleukin-1 receptor antagonist penetrates human brain at experimentally therapeutic concentrations. J Cereb Blood Flow Metab 2008;28(2):387–94. [DOI] [PubMed] [Google Scholar]
- 69.Ogungbenro K, Hulme S, Rothwell N, Hopkins S, Tyrrell P, Galea J. Study design and population pharmacokinetic analysis of a phase II dose-ranging study of interleukin-1 receptor antagonist. J Pharmacokinet Pharmacodyn 2016;43(1):1–12. [DOI] [PubMed] [Google Scholar]
- 70.Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab 2014;34(5):845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun 2011;25(7):1281–9. [DOI] [PubMed] [Google Scholar]
- 72.Heida JG, Moshe SL, Pittman QJ. The role of interleukin-1beta in febrile seizures. Brain Dev 2009;31(5):388–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uludag IF, Bilgin S, Zorlu Y, Tuna G, Kirkali G. Interleukin-6, interleukin-1 beta and interleukin-1 receptor antagonist levels in epileptic seizures. Seizure 2013;22(6):457–61. [DOI] [PubMed] [Google Scholar]
- 74.Uludag IF, Duksal T, Tiftikcioglu BI, Zorlu Y, Ozkaya F, Kirkali G. IL-1beta, IL-6 and IL1Ra levels in temporal lobe epilepsy. Seizure 2015;26:22–5. [DOI] [PubMed] [Google Scholar]
- 75.Chou IC, Lin WD, Wang CH, Tsai CH, Li TC, Tsai FJ. Interleukin (IL)-1beta, IL-1 receptor antagonist, IL-6, IL-8, IL-10, and tumor necrosis factor alpha gene polymorphisms in patients with febrile seizures. J Clin Lab Anal 2010;24(3):154–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haspolat S, Baysal Y, Duman O, Coskun M, Tosun O, Yegin O. Interleukin-1alpha, interleukin-1beta, and interleukin-1Ra polymorphisms in febrile seizures. J Child Neurol 2005;20(7):565–8. [DOI] [PubMed] [Google Scholar]
- 77.Kanemoto K, Kawasaki J, Miyamoto T, Obayashi H, Nishimura M. Interleukin (IL)1beta, IL-1alpha, and IL-1 receptor antagonist gene polymorphisms in patients with temporal lobe epilepsy. Ann Neurol 2000;47(5):571–4. [PubMed] [Google Scholar]
- 78.Nakayama J, Arinami T. Molecular genetics of febrile seizures. Epilepsy Res 2006;70 Suppl 1:S190–8. [DOI] [PubMed] [Google Scholar]
- 79.Serdaroglu G, Alpman A, Tosun A, Pehlivan S, Ozkinay F, Tekgul H, et al. Febrile seizures: interleukin 1beta and interleukin-1 receptor antagonist polymorphisms. Pediatr Neurol 2009;40(2):113–6. [DOI] [PubMed] [Google Scholar]
- 80.Kenney-Jung DL, Vezzani A, Kahoud RJ, LaFrance-Corey RG, Ho ML, Muskardin TW, et al. Febrile infection-related epilepsy syndrome treated with anakinra. Ann Neurol 2016;80(6):939–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schulert GS, Grom AA. Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies. Annu Rev Med 2015;66:145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van der Ven AJ, Netea MG, van der Meer JW, de Mast Q. Ebola virus disease has features of hemophagocytic lymphohistiocytosis syndrome. Front Med 2015;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kumar N, Goyal J, Goel A, Shakoory B, Chatham W. Macrophage activation syndrome secondary to human monocytic ehrlichiosis. Indian J Hematol Blood Transfus 2014;30(Suppl 1):145–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gatti S, Beck J, Fantuzzi G, Bartfai T, Dinarello CA. Effect of interleukin-18 on mouse core body temperature. Am J Physiol Regul Integr Comp Physiol 2002;282(3):R702–9. [DOI] [PubMed] [Google Scholar]
- 85.Robertson MJ, Mier JW, Logan T, Atkins M, Koon H, Koch KM, et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin Cancer Res 2006;12(14 Pt 1):4265–73. [DOI] [PubMed] [Google Scholar]
- 86.Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL, Dinarello CA. Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A 2004;101(23):8815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stuyt RJ, Netea MG, Verschueren I, Dinarello CA, Kullberg BJ, van der Meer JW. Interleukin-18 does not modulate the acute-phase response. J Endotoxin Res 2005;11(2):85–8. [DOI] [PubMed] [Google Scholar]
- 88.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci U S A 2001;98(5):2871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abbate A, Van Tassell BW, Christopher S, Abouzaki NA, Sonnino C, Oddi C, et al. Effects of Prolastin C (Plasma-Derived Alpha-1 Antitrypsin) on the acute inflammatory response in patients with ST-segment elevation myocardial infarction (from the VCU-alpha 1-RT pilot study). Am J Cardiol 2015;115(1):8–12. [DOI] [PubMed] [Google Scholar]
- 90.Opal SM, Fisher CJJ, Dhainaut JF, Vincent J-L, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial. Crit Care Med 1997;25:1115–24. [DOI] [PubMed] [Google Scholar]
- 91.Fisher CJJ, Slotman GJ, Opal SM, Pribble J, Bone RC, Emmanuel G, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med 1994;22:12–21. [DOI] [PubMed] [Google Scholar]
- 92.Fisher CJ Jr., Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA 1994;271(23):1836–43. [PubMed] [Google Scholar]
- 93.Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med 2016;44(2):275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ikonomidis I, Tzortzis S, Lekakis J, Paraskevaidis I, Andreadou I, Nikolaou M, et al. Lowering interleukin-1 activity with anakinra improves myocardial deformation in rheumatoid arthritis. Heart 2009;95(18):1502–7. [DOI] [PubMed] [Google Scholar]
- 95.Ikonomidis I, Tzortzis S, Andreadou I, Paraskevaidis I, Katseli C, Katsimbri P, et al. Increased benefit of interleukin-1 inhibition on vascular function, myocardial deformation, and twisting in patients with coronary artery disease and coexisting rheumatoid arthritis. Circ Cardiovasc Imaging 2014;7(4):619–28. [DOI] [PubMed] [Google Scholar]
- 96.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J 2011;162(4):597–605. [DOI] [PubMed] [Google Scholar]
- 97.Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007;356(15):1517–26. [DOI] [PubMed] [Google Scholar]
- 98.Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, et al. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation 2012;126(23):2739–48. [DOI] [PubMed] [Google Scholar]
- 99.Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, et al. Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care 2012;35(8):1654–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol 2009;54(23):2129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377(12):1119–31.This paper reports the results of a large trial in 10,061 patients testing the hypothesis that neutralization of IL-1β by canakinumab reduces a second myocardial infarction or stroke in high risk subjects who have already experienced a heart attack or stroke. The subjects were high risk because despite standard of car with statins, the level of CRP in the circulation was above 2 mg/L. Subjects were randomized to receive 50, 150 or 300 mg of canakinumab or Placebo every three months for 4 years. The primary end point was a reduction in a composite of fatal or non-fatal myocardial infarctions or non-fatal stroke termed MACE. There were several secondary endpoints. Of these, a reduction in hospitalizations for acute coronary syndromes requiring rapid revascularization was determined. The study achieved primary and secondary endpoint. The dose of 150 mg was most effective, although a dose of 300 mg was also effective. The study revealed that canakinumab can reduce the incidence of cardiovascular events without affecting a change in serum lipids. Thus, a role for inflammation, in this case IL-1β, in the pathogenesis of atherosclerosis was demonstrated
- 102.Aganna E, Martinon F, Hawkins PN, Ross JB, Swan DC, Booth DR, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002;46(9):2445–52. [DOI] [PubMed] [Google Scholar]
- 103.Rynne M, Maclean C, Bybee A, McDermott MF, Emery P. Hearing improvement in a patient with variant Muckle-Wells syndrome in response to interleukin 1 receptor antagonism. Ann Rheum Dis 2006;65(4):533–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, Wittkowski H, Bialkowski A, Tzaribachev N, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum 2011;63(3):840–9. [DOI] [PubMed] [Google Scholar]
- 105.Kuemmerle-Deschner JB, Wittkowski H, Tyrrell PN, Koetter I, Lohse P, Ummenhofer K, et al. Treatment of Muckle-Wells syndrome: analysis of two IL-1-blocking regimens. Arthritis Res Ther 2013;15(3):R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stew BT, Fishpool SJ, Owens D, Quine S. Muckle-Wells syndrome: a treatable cause of congenital sensorineural hearing loss. B-ENT 2013;9(2):161–3. [PubMed] [Google Scholar]
- 107.Kitley JL, Lachmann HJ, Pinto A, Ginsberg L. Neurologic manifestations of the cryopyrin-associated periodic syndrome. Neurology 2010;74(16):1267–70. [DOI] [PubMed] [Google Scholar]
- 108.Ahmadi N, Brewer CC, Zalewski C, King KA, Butman JA, Plass N, et al. Cryopyrin-associated periodic syndromes: otolaryngologic and audiologic manifestations. Otolaryngol Head Neck Surg 2011;145(2):295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Klein AK, Horneff G. Improvement of sensoneurinal hearing loss in a patient with Muckle-Wells syndrome treated with anakinra. Klin Padiatr 2011;222(4):266–8. [DOI] [PubMed] [Google Scholar]
- 110.Eungdamrong J, Boyd KP, Meehan SA, Latkowski JA. Muckle-Wells treatment with anakinra. Dermatol Online J 2013;19(12):20720. [PubMed] [Google Scholar]
- 111.Gerard S, le Goff B, Maugars Y, Berthelot JM, Malard O. Lasting remission of a Muckle-Wells syndrome with CIAS-1 mutation using half-dose anakinra. Joint Bone Spine 2007;74(6):659. [DOI] [PubMed] [Google Scholar]
- 112.Sibley CH, Plass N, Snow J, Wiggs EA, Brewer CC, King KA, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: a cohort study to determine three- and five-year outcomes. Arthritis Rheum 2012;64(7):2375–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nakanishi H, Kawashima Y, Kurima K, Chae JJ, Ross AM, Pinto-Patarroyo G, et al. NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc Natl Acad Sci U S A 2017;114(37):E7766–E75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev 2010;29(2):317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lewis AM, Varghese S, Xu H, Alexander HR. Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J Transl Med 2006;4:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A 2003;100(5):2645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M, et al. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J Immunol 2009;183(7):4705–14. [DOI] [PubMed] [Google Scholar]
- 118.Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, Baron R, et al. The role of IL-1beta in the early tumor cell-induced angiogenic response. J Immunol 2013;190(7):3500–9. [DOI] [PubMed] [Google Scholar]
- 119.Pusztai L, Mendoza TR, Reuben JM, Martinez MM, Willey JS, Lara J, et al. Changes in plasma levels of inflammatory cytokines in response to paclitaxel chemotherapy. Cytokine 2004;25(3):94–102. [DOI] [PubMed] [Google Scholar]
- 120.Hickish T, Andre T, Wyrwicz L, Saunders M, Sarosiek T, Kocsis J, et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2017;18(2):192–201. [DOI] [PubMed] [Google Scholar]
- 121.Hong DS, Janku F, Naing A, Falchook GS, Piha-Paul S, Wheler JJ, et al. Xilonix, a novel true human antibody targeting the inflammatory cytokine interleukin-1 alpha, in non-small cell lung cancer. Invest New Drugs 2015;33(3):621–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Biasucci LM, Liuzzo G, Fantuzzi G, Caligiuri G, Rebuzzi AG, Ginnetti F, et al. Increasing levels of interleukin (IL)-1Ra and IL-6 during the first 2 days of hospitalization in unstable angina are associated with increased risk of in-hospital coronary events. Circulation 1999;99(16):2079–84. [DOI] [PubMed] [Google Scholar]
- 123.Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc 2009;84(2):114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lust JA, Donovan KA. The role of interleukin-1 beta in the pathogenesis of multiple myeloma. Hematol Oncol Clin North Am 1999;13(6):1117–25. [DOI] [PubMed] [Google Scholar]
- 125.Xiong Y, Donovan KA, Kline MP, Gornet MK, Moon-Tasson LL, Lacy MQ, et al. Identification of two groups of smoldering multiple myeloma patients who are either high or low producers of interleukin-1. J Interferon Cytokine Res 2006;26(2):83–95. [DOI] [PubMed] [Google Scholar]
- 126.Lust JA, Lacy MQ, Zeldenrust SR, Witzig TE, Moon-Tasson LL, Dinarello CA, et al. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am J Hematol 2016;91(6):571–4. [DOI] [PubMed] [Google Scholar]
- 127.O’Shaughnessy C, Young RR, Levin MK, Baisch J, Timis R, Muniz LS, et al. Safety and immunologic activity of anakinra in HER2-negative metastatic breast cancer J Clin Oncol 34, 2016 (suppl; abstr e14565) 2016;34 Suppl):e14565. [Google Scholar]
- 128.Wang Y, Wang Y, Li L. Note of clarification regarding data about the association between the interleukin-1beta −31T>C polymorphism and breast cancer risk. Breast Cancer Res Treat 2016;155(3):415–7. [DOI] [PubMed] [Google Scholar]
- 129.Hong DS, Hui D, Bruera E, Janku F, Naing A, Falchook GS, et al. MABp1, a first-in-class true human antibody targeting interleukin-1alpha in refractory cancers: an open-label, phase 1 dose-escalation and expansion study. Lancet Oncol 2014;15(6):656–66. [DOI] [PubMed] [Google Scholar]
- 130.Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, et al. The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front Immunol 2013;4:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol 2012;22(1):33–40. [DOI] [PubMed] [Google Scholar]
- 132.Baracos V, Rodemann HP, Dinarello CA, Goldberg AL. Stimulation of muscle protein degradation and prostaglandin E2 release by leukocytic pyrogen (interleukin-1). A mechanism for the increased degradation of muscle proteins during fever. N Engl J Med 1983;308(10):553–8. [DOI] [PubMed] [Google Scholar]
- 133.Smith JW 2nd, Longo DL, Alvord WG, Janik JE, Sharfman WH, Gause BL, et al. The effects of treatment with interleukin-1 alpha on platelet recovery after high-dose carboplatin. N Engl J Med 1993;328(11):756–61. [DOI] [PubMed] [Google Scholar]
- 134.Kaplanski G, Porat R, Aiura K, Erban JK, Gelfand JA, Dinarello CA. Activated platelets induce endothelial secretion of interleukin-8 in vitro via an interleukin-1-mediated event. Blood 1993;81(10):2492–5. [PubMed] [Google Scholar]
- 135.Smith CJ, Emsley HC, Udeh CT, Vail A, Hoadley ME, Rothwell NJ, et al. Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine 2012;58(3):384–9. [DOI] [PubMed] [Google Scholar]
- 136.Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017;390(10105):1833–42.This study reveals that in subjects at risk for a second cardiovascular event have a lower incidence of cancer and cancer deaths. Upon randomization into the CANTOS trial, the 10,061 subjects did not have evidence of cancer. A special judication committee reviewed each subject for the presence of cancer before and after the trial. Of the 196 subjects with cancer, there was a significant lower incidence of death from all cancers in subjcts receiving 300 mg of canakinumab every 3 months for 4 years. Deaths due to lung cancer dominated the data. The subjects with a diagnosis of cancer had higher levels of CRP upon randomization compared to levels in subjects without cancer suggesting a greater amount of inflammation associated with cancer. One explanation for the benefit of neutralizing IL-1β in these high risk patients is the reduction in IL-1β driven mechanisms of cancer promotion, such as angiogenesis, myeloid-derived suppressor cells and endothelial adhesion molecules.
- 137.Dinarello CA, Nold-Petry C, Nold M, Fujita M, Li S, Kim S, et al. Suppression of innate inflammation and immunity by interleukin-37. Eur J Immunol 2016;46(5):1067–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J Biol Chem 2011;286(49):42594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Arend WP. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev 2002;13(4–5):323–40. [DOI] [PubMed] [Google Scholar]