

Abstract
Following the discovery that hydroxylated derivative 3 (Fig. 1) was one of the oxidative metabolites of the original lead 1, it was found that hydroxylated compound 4 possesses higher in vitro anti-HIV potency than the corresponding non-hydroxylated compound 2. Structural hybridation of 4 with the orally available analog 5 resulted in another orally-available spirodiketopiperazine CCR5 antagonist 6a that possesses more favorable pharmaceutical profile for use as a drug candidate.
Keywords: CCR5, Chemokine, Anti-HIV
Since the United States Food and Drug Administration (US FDA) approval of the first antiretroviral drug in 1987, new agents have been added to the list of treatment options for human immunodeficiency virus type-1 (HIV-1) infection. Due to the increased incidence of drug-resistant viruses, there has been a growing need for novel treatments of HIV-1 infections. Therefore, the identification of new antiretroviral drugs that have unique mechanisms of action remains an important therapeutic objective.1 In 1996, it was revealed that one of the C–C chemokine receptors, CCR5, is utilized by HIV-1 as one of the essential co-receptors and that the endogenous ligand showed anti-HIV-1 activity in vitro.2 For such reasons, CCR5 antagonists that inhibit viral entry into the host cell are thought to be one of the most promising class of new antiviral drugs.
Since these discoveries, many pharmaceutical companies and academic institutions have been enthusiastically investigating novel CCR5 antagonists.3 Thus far Maraviroc is currently the only approved CCR5 antagonist on the market for the treatment of HIV-1 infection. Due to its new mechanism of action, CCR5 antagonists may be utilized in salvage therapy in patients who have become resistant to one or multiple classes of antiretrovirals, in combination with other classes of active antiretroviral agents.
We have previously reported the discovery of spirodiketopiperazines 1 and 2 as structurally novel CCR5 antagonists (Fig. 1).4 Both of these compounds showed potent anti-HIV activity, in vitro. After the incubation of 1 with human liver microsomes, the hydroxylated analog of 1′ position was identified as the biologically active metabolite.5 Since the proposed structure of metabolite had two chiral centers, we synthesized all four possible stereo-isomers as their optically pure form from the corresponding β-hydroxylated leucine to evaluate their biological activities. Compound 3, having a (3R,1′R)-configuration, exhibited approximately 4.5-12-fold higher potency than the other isomers in Ca assay. Based on the observation of this unexpectedly improved antagonist activity of compound 3, we applied this information to compound 2 which showed more potent in vitro activity than compound 1. Thus, it was found that the (3R, 1′R)-hydroxylated compound 4 also exhibited strong antagonist activity.5,6 Although the hydroxylated analogs 3 and 4 could not improve the bioavailability in rodents5, they showed some favorable pharmaceutical properties. For example, 4 did not show any significant inhibition of CYP 3A4 up to 30 μM (2: 9.3 μM of IC50). Furthermore, aqueous solubility (pH 6.8) of 4 was significantly improved relative to the corresponding non-hydroxylated analog (2: <0.2 μg/mL; 4: 2.7 μg/mL). Further assessment and optimization led us to the discovery of the spirodiketopiperazine-based structure 5 as an orally-available CCR5 antagonist.7 Based on the observation described above, we synthesized and evaluated 6a, which was expected to have improved potency and oral availability with favorable pharmaceutical properties due of its hybrid structure possessing both of the features derived from 4 and 5. The other possible stereoisomers 6b-d were also synthesized and evaluated.
Figure 1.
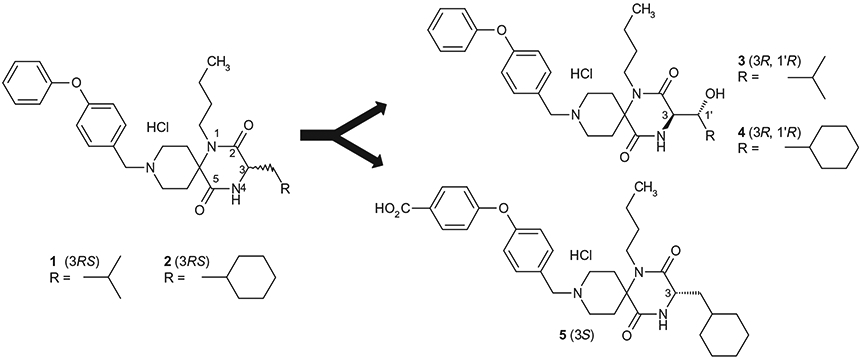
Lead compounds of CCR5 antagonists.
Test compounds 6a-d listed in Table 1 were synthesized as outlined in Scheme 1. As shown in Scheme 1, the Ugi four component coupling reaction of N-benzylpiperizine-4-one, n-butyl amine, an appropriate optically-active amino acid among 7a–d and 2-(4-morpholinyl)ethyl isonitrile followed by deprotection of N-Boc group and cyclization under acetic acid condition afforded the 9-N-benzyl spirodiketopiperazines is represented by the general formula 8 in 60–90% yield from 7a–d. The stereochemical purity of the Ugi products was confirmed by NMR. Catalytic hydrogenation of 8 followed by reductive 9-N-alkylation with 4-(4-formylphenoxy)benzoic acid resulted in 6a–d in 50–90% yield. Optically-active amino acids 7a–d were prepared using Sharpless asymmetric epoxidation as previously reported.8 The synthetic procedures of amino acid 7a (2R, 3R) and 7c (2S, 3R) are described as a representative example in Scheme 2. According to the same procedure as described for preparation of 7a from 13a, the amino acid 7d (2S, 3S) was synthesized from the enantiomer of 13a, which was prepared from 12 by Sharpless asymmetric epoxidation using another enantiomeric diethyl tartrate. According to the same procedure as described for the preparation of 7c from 13a, the amino acid 7b (2R, 3S) was prepared from the enantiomer of 13a.
Table 1.
Activity profiles of stereoisomers 6a–d
| Compound | Ca assay IC50 (nM) | Fusion assay pIC50 | |
|---|---|---|---|
| 1 | 94 | 6.07 | |
| 2 | 28 | NDa | |
| 3 | 84 | 6.64 | |
| 4 | 53 | 8.11 | |
| 5 | 13 | 7.06 | |
| 6a(3R, 1’R) | 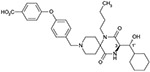 |
34 | 8.25 |
| 6b(3R, 1’S) | 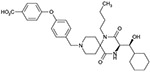 |
44 | 6.74 |
| 6c(3S, 1’R) | 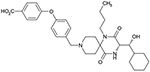 |
470 | 6.42 |
| 6d(3S, 1’S) | 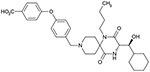 |
51 | 6.35 |
ND : Not determined.
Scheme 1.
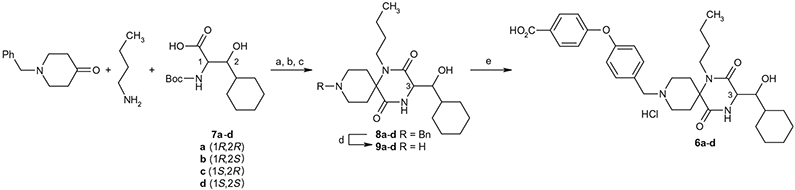
Synthesis of spirodiketopiperazines 6a–d. Reagents and conditions: (a) 2-(4-morpholinyl)-ethyl isonitrile, MeOH, 55 °C; (b) concd HCl, 55 °C; (c) AcOH/toluene, 80 °C; (d) H2, Pd(OH)2/C, EtOH, 50 °C, then 4 N HCl/AcOEt, 60–90% yield in four-steps; (e) 4-(4-formylphenoxy)benzoic acid, NaBH(OAc)3, AcOH, DMF and then 4 N HCl/AcOEt, 50–90% yield.
Scheme 2.
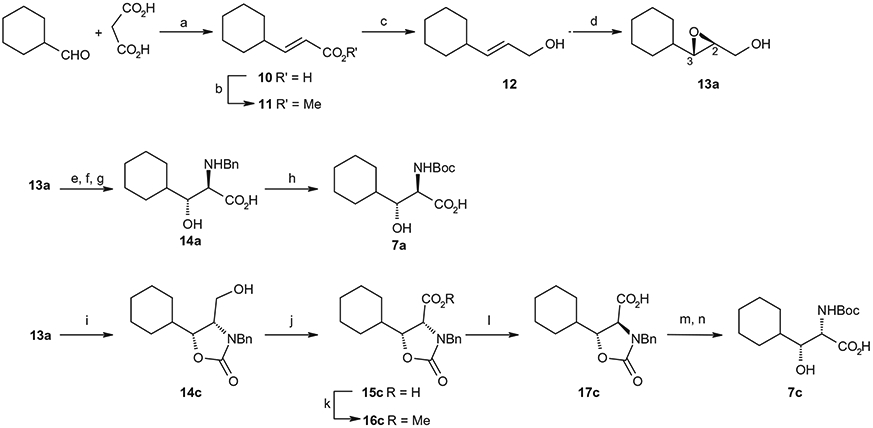
Synthetic method of optically-active β-hydroxy-N-Boc-α-amino acids 7a and 7c. Reagents and conditions: (a) piperidine, pyridine, 97%; (b) concd H2SO4, MeOH, 87%; (c) DIBAL, THF, 90%; (d) (−)-diethyl-D-tartrate, Ti(Oi-Pr)4, cumene hydroperoxide, MS4A, CH2Cl2, 85%; (e) SO3.pyridine, Et3N, DMSO; (f) 2-methyl-2-butene, NaClO2, NaH2PO4, MeCN, H2O; (g) BnNH2, 5.0 M NaOH, H2O, 35% from 13; h) H2, Pd(OH)2/C, MeOH, then Boc2O, 1.0 M NaOH, 97%; (i) benzyl isocyanate, NaH, THF, 28%; (j) Jones reagent, acetone; (k) TMS-diazomethane, diethyl ether, 62% from 14c; (l) KOH, EtOH, 98%; (m) 2.0 M KOH; (n) H2, Pd(OH)2/C then Boc2O, 84% from 17c.
Compounds 6a–d listed in Table 1 were evaluated for their inhibitory activities against calcium mobilization of human CCR5 over-expressed CHO cell (hCCR5/CHO) stimulated by MIP-1α (Ca assay) 6a and for their inhibitory activity of cell-cell fusion reaction between target cells expressing CD4/CCR5 and effector cells expressing the envelope protein of HIV-1.9
As shown in Table 1, we evaluated 6a-d to investigate the structure–activity relationship (SAR) of the four stereoisomers. In our previous paper,5 we reported that the (3R, 1′R)-isomer 4 was identified to show the most potent in vitro activities among the four possible stereoisomers. Based on the SAR of 4 and its possible three stereoisomers, 6a was predicted to show the most potent antagonistic activity. Unexpectedly, however, isomers 6a, 6b and 6d showed nearly equipotent antagonistic activity, while 6c (3S, 1′R) showed nearly 10-fold less antagonistic activity. Regarding the 1′R-isomers, the 3R-isomer 6a showed more potency than the corresponding 3S-isomer 6c. Regarding the 1′S-isomers, both the 3R-isomer 6b and 3S-isomer 6d exhibited nearly equipotent activity. This specific SAR was consistent with the SAR of 4 and its three stereoisomers.5
According to the results of the fusion assay, 6a showed nearly two orders of magnitude more potency in the pIC50 values relative to those of other three stereoisomers 6b–d. This lack of correlation between anti-chemokine activity and anti-HIV activity has been previously reported, and is attributed to the allosteric nature of CCR5 inhibition.10 Such a discrepancy observed between the calcium assay and the fusion assay was considered to be because of the following reasons. The calcium assay (receptor antagonist activity) was conducted using the natural ligand (MIP-1α), while the fusion assay (inhibitory effect on HIV envelope glycoprotein-mediated cell–cell fusion) was conducted using envelope glycoprotein. To more extensively estimate the activity of these compounds, the inhibition of virus entry mediated by membrane fusion and/or the HIV replication inhibition should be evaluated in addition to the receptor antagonist activity. The HIV replication inhibition results are described further in the article.
Isomers 6a-d were investigated for their inhibitory activity on cytochrome P450 (2C9, 2D6 and 3A4 isomers) as shown in Table 2. For comparative purposes, the profiles of 1-5 were included to demonstrate the improvement shown with 6a-d. The initial leads 1 and 2 showed P450 inhibition on all the enzymes tested, while the hydroxylated analogs 3 and 4 showed no inhibition on 3A4 up to 30 μM. Analog 5 which also possessed the p-carboxylic acid functional group still showed inhibitory activity on 2C9 and 3A4, while it did not show inhibitory activity on 2D6 up to 30 μM. The analog 6a that possesses the best potential as a drug candidate did not show inhibitory activity on all the tested enzymes up to 30 μM. As described above, introduction of the hydrophilic functions such as hydroxyl and/or carboxylic acid functions was effective to remove or reduce these inhibitory activities. Among the tested compounds, only (3R, 1′S)-isomer 6b showed inhibitory activity against 2C9. While this result was not able to be explained rationally, (3R, 1′S)-configuration was estimated to have a tendency to show relatively more affinity to the enzyme 2C9 than the other three configurations.
Table 2.
P450 inhibition of 1–5 and 6a–d
| Compound | P450 inhibition IC50 (μM) |
||
|---|---|---|---|
| 2C9 | 2D6 | 3A4 | |
| 1 | 5.9 | 4.6 | 19.7 |
| 2 | 5.5 | 7.8 | 9.3 |
| 3 | >30 | >30 | >30 |
| 4 | 3.7 | 9.8 | >30 |
| 5 | 8.8 | >30 | 24.9 |
| 6a | >30 | >30 | >30 |
| 6b | 18 | >30 | >30 |
| 6c | >30 | >30 | >30 |
| 6d | >30 | >30 | >30 |
The (3R, 1′R)-configuration of 6a was identified as possessing the most optimized stereochemistry regarding the biological potency, while the p-carboxylic acid function was found to be necessary for oral bioavailability, as illustrated by 5. For the simultaneous optimization of the biological potency and oral bioavailability, the (3R, 1′R)-configuration and the p-carboxylic acid function are both needed, as indicated by the area under the curve (AUC) values (6a > 6b–d) after oral dosing described in Table 3.
Table 3.
Pharmacokinetic profiles of 6a–d
| Compound | 10 mg/kg, po |
3 mg/kg, iv |
||||||
|---|---|---|---|---|---|---|---|---|
| Cmax (ng/mL) | T1/2 (min) | AUC (ng h/mL) | BA (%) | AUC (ng h/mL) | T1/2 (min) | CL (mL/min/kg) | Vss (mL/kg) | |
| 1 | 33.3a | 75.7 | 96.7a | 1.3 | 372 | 13 | 137 | 2349 |
| 2 | 5.6a | 103 | 24.8a | 1.9 | 400 | 19.9 | 113 | 2542 |
| 5 | 2400 | 48.4 | 10532 | 34 | 3091 | 11.1 | 16 | 145 |
| 6a (3R, 1′R) | 2360 | 120 | 3422 | 23 | 4402 | 21.8 | 11.3 | 132 |
| 6b (3R, 1′S) | 70 | 35.2 | 67.5 | NDb | NDb | NDb | NDb | NDb |
| 6c (3S, 1′R) | 280 | 14.4 | 126 | NDb | NDb | NDb | NDb | NDb |
| 6d (3S, 1′S) | 260 | 19.2 | 213 | 6.5 | 988 | 16.2 | 50.9 | 836 |
Cmax and AUC are normalized to a dose of 10 mg/kg.
ND: Not determined.
Analogs 6a-d were evaluated for their pharmacokinetics (PK) values after their single dose oral administration and intravenous administration in rats. These data are summarized in Table 3. Among the four tested compounds, 6a showed the best oral exposure (AUC = 3422 ng h/mL) and maximum plasma concentration (Cmax = 2360 ng/mL). A marked improvement of the AUC value of 6a relative to the chemical lead 1 (Table 5) after oral dosing was estimated to be due to the marked reduction of the clearance (CL) and tissue distribution (Vss) after intravenous dosing. Thus, stereochemistry of position-3 and position-1′ of 6a (3R, 1′R) was thought to be involved as one of the factors for the improvement of PK values as illustrated by the lower Cmax and AUC values after oral dosing of 6b (3R, 1′S), 6c (3S, 1′R), and 6d (3S, 1′S). Stability of these test compounds in rat liver microsomes was investigated but all isomers 6a-d did not show significant differences between each other in in vitro metabolic stability, Caco2 permeability, and solubility, as shown in Table 4. In vitro metabolic stability and in vivo clearance of these test compounds did not show a strong relationship. Therefore, one cannot rule out other clearance processes in addition to metabolic ones to explain the mismatch in Cmax/AUC across the 4 isomers. The differences in their susceptibility to the systemic metabolism, transporter recognition and/or the changes in Vss may suggest additional processes.
Table 5.
Anti-HIV activity of the representative compounds
| Compound | Anti-HIV-1 activity IC50 (nM)a |
|
|---|---|---|
| HIV-1Ba-L (R5) | HIV-1MM (R5MDR) | |
| 6a | 0.4 ± 0.3 | 0.6 ± 0.2 |
| TAK-779 | 28 ± 32 | 14 ± 8 |
| SCH-351125 | 4 ± 2 | 3 ± 0.5 |
| Zidovudineb | 7 ± 4 | 250 ± 98 |
| Nelfinavirc | 12 ± 8 | >1000 |
IC50 values are based on the inhibition of HIV p24 antigen expression in PBMC.
Zidovudine is a reverse transcriptase inhibitor.
Nerfinavir is a HIV-1 protease inhibitor
Table 4.
In vitro metabolic stability, Caco2 permeability, and solubility of 6a–d
| Compound | % Remaininga |
Caco2 × 10−6 (cm/s) | Solubilityd (μM) | |
|---|---|---|---|---|
| HLMb | RLMc | |||
| 6a (3R, 1′R) | 36 | 39 | 3.8 | 70 |
| 6b (3R, 1′S) | 90.1 | 76.5 | 1.6 | 81 |
| 6c (3S, 1′R) | 88.1 | 74.8 | 1.6 | 85 |
| 6d (3S, 1′S) | 69.8 | 52.7 | 2.1 | 80 |
The data show the remaining% 15 min after incubating with the 0.5 mg/mL liver microsomes.
Human liver microsomes
Rat liver microsomes
Kinetic solubility
Non-hydroxylated analog 2, hydroxylated analog 4, and hydroxylated benzoic acid analog 6a were evaluated for their anti-HIV activity using CCR5-tropic (R5) HIV-1 and multidrug-resistant HIV-1 virus, as shown in Tables 1 and 5. Analogs 4 and 6a showed potent anti-HIV activity as indicated by a significant decrease in virus p24 production relative to 2. These compounds potently inhibited not only the replication of laboratory and primary HIV-1 R5 strains, but also that of various multidrug-resistant monocyte/macrophage tropic (R5) HIV-1.6b These compounds were inactive against T-cell-tropic (X4) HIV-1. These data support the hypothesis that spirodiketopiperazines, such as 2, 4, and 6a, show potent anti-HIV activity through their antagonistic effects on CCR5.
In summary, the design and synthesis of the structurally novel spirodiketopiperazines for use as orally-available CCR5 antagonists was performed based on the information previously reported by our group. Structural hybridization of the hydroxylated derivative 4 possessing more favorable pharmaceutical profiles and the orally-available non-hydroxylated derivative 5 resulted in the discovery of 6a, which shows potential as an orally-available anti-HIV drug candidate. Compound 6a possessing (3R, 1′R)-configuration exhibited potent CCR5 antagonist activity and the most potent activity by fusion assay among the tested four stereoisomers. For its biological potency and oral availability, both the optimized (3R, 1′R)-configuration and the p-carboxylic acid function on the terminal phenoxy moiety were found to be required. More detailed SAR including PK data will be reported in due course.
References and notes
- 1.(a) Mitsuya H; Erickson J In Textbook of AIDS Medicine, 2nd ed.; Williams & Wilkins, 2001; pp 751–780; [Google Scholar]; (b) Hoffmann C; Mulcahy F Overview of Antiretroviral Agents In HIV Medicine 2006; Hoffmann C, Rockstroh JK, Kamps BS, Eds.; Flying Publisher, 2006; p 94; [Google Scholar]; (c) Mehellou Y; Clercq ED J. Med. Chem. 2010, 53, 521. [DOI] [PubMed] [Google Scholar]
- 2.(a) Feng Y; Broder CC; Kennedy PE; Berger EA Science 1996, 272, 872;8629022 [Google Scholar]; (b) Deng HK; Liu R; Ellmeier W; Choe S; Unutmaz D; Burkhart M; Marzio PD; Marmon S; Sutton RE; Hill CM; Davis CB; Peiper SC; Schall TJ; Littman DR; Landau NR Nature 1996, 381, 661; [DOI] [PubMed] [Google Scholar]; (c) Dragic T; Litwin V; Allaway GP; Martin SR; Huang Y; Nagashima KA; Cayanan C; Maddon PJ; Koup RA; Moore JP; Paxton WA Nature 1996, 381, 667; [DOI] [PubMed] [Google Scholar]; (d) Alkhatib G; Combadiere C; Broder CC; Feng Y; Kennedy PE; Murphy PM; Berger EA Science 1996, 272, 1955; [DOI] [PubMed] [Google Scholar]; (e) Berger EA; Murphy PM; Farber JM Annu. Rev. Immunol 1999, 17, 657; [DOI] [PubMed] [Google Scholar]; (f) Caldwell DJ; Evans JD, Expert Opin. Pharmacother 2008, 9, 3231. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kazmierski WM; Gudmundsson KS; Piscitelli SC Annu. Rep. Med. Chem 2007, 42, 301; [Google Scholar]; (b) Lemoine RC; Wanner J Curr. Top. Med. Chem 2010, 10, 1299. [DOI] [PubMed] [Google Scholar]
- 4.Habashita H; Kokubo M; Hamano S; Hamanaka N; Toda M; Shibayama S; Tada H; Sagawa K; Fukushima D; Maeda K; Mitsuya HJ Med. Chem 2006, 49, 4140. [DOI] [PubMed] [Google Scholar]
- 5.Nishizawa R; Nishiyama T; Hisaichi K; Matsunaga N; Minamoto C; Habashita H; Takaoka Y; Toda M; Shibayama S; Tada H; Sagawa K; Fukushima D; Maeda K; Mitsuya H Bioorg. Med. Chem. Lett 2007, 17, 727. [DOI] [PubMed] [Google Scholar]
- 6.(a) Maeda K; Yoshimura K; Shibayama S; Habashita H; Tada H; Sagawa K; Miyakawa T; Aoki M; Fukushima D; Mitsuya H J. Biol. Chem 2001, 276, 35194; [DOI] [PubMed] [Google Scholar]; (b) Maeda K; Nakata H; Koh Y; Miyakawa T; Ogata H; Takaoka Y; Shibayama S; Sagawa K; Fukushima D; Moravek J; Koyanagi Y; Mitsuya H J. Virol 2004, 78, 8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Nishizawa R; Nishiyama T; Hisaichi K; Hirai K; Habashita H; Takaoka Y; Tada H; Sagawa K; Shibayama S; Maeda K; Mitsuya H; Nakai H; Fukushima D; Toda M Bioorg. Med. Chem. Lett 2010, 20, 763; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nishizawa R; Nishiyama T; Hisaichi K; Hirai K; Habashita H; Takaoka Y; Tada H; Sagawa K; Shibayama S; Maeda K; Mitsuya H; Nakai H; Fukushima D; Toda M Bioorg. Med. Chem 2010, 18, 5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Caldwell CG; Bondy SS Synthesis 1990, 34–36; [Google Scholar]; (b) Nagamitsu T; Sunazuka T; Tanaka H; Omura S; Sprengeler PA; Smith AB III J. Am. Chem. Soc 1996, 118, 3584–3590. [Google Scholar]
- 9.Jenkinson S; McCoy DC; Kerner SA; Ferris RG; Lawrence WK; Clay WC; Condreay JP; Smith CJ Biomol. Screen 2003, 8, 463. [DOI] [PubMed] [Google Scholar]
- 10.(a) Shankaran K; Donnelly KL; Shah SK; Guthikonda RN; MacCoss M; Mills SG; Gould SL; Malkowitz L; Siciliano SJ; Springer MS; Carella A; Carver G; Hazuda D; Holmes K; Kessler J; Lineberger J; Miller MD; Emini EA; Schleif WA Bioorg. Med. Chem. Lett 2004, 14, 3419; [DOI] [PubMed] [Google Scholar]; (b) Wood A; Armour D Prog. Med. Chem 2005, 43, 239. [DOI] [PubMed] [Google Scholar]