

Abstract
Cooperative enzyme catalysis in nature has long inspired the application of engineered multi-enzyme assemblies for industrial biocatalysis. Despite considerable interest, efforts to harness the activity of cell-surface displayed multi-enzyme assemblies have been based on trial and error rather than rational design due to a lack of quantitative tools. In this study, we developed a quantitative approach to whole-cell biocatalyst characterization enabling a comprehensive study of how yeast-surface displayed multi-enzyme assemblies form. Here we show that the multi-enzyme assembly efficiency is limited by molecular crowding on the yeast cell surface, and that maximizing enzyme density is the most important parameter for enhancing cellulose hydrolytic performance. Interestingly, we also observed that proximity effects are only synergistic when the average inter-enzyme distance is > ~130 nm. The findings and the quantitative approach developed in this work should help to advance the field of biocatalyst engineering from trial and error to rational design.
Graphical Abstract
Cooperative catalysis is highly efficient in biology1 thanks to the ability of cells to colocalize2 or compartmentalize3 functionally related enzymes. One naturally occurring biocatalytic scheme with significant industrial potential is the hydrolysis of lignocellulosic biomass by microbes. Lignocellulosic biomass is among the most abundant carbon sources on Earth and represents a sustainable feedstock for the production of materials, value-added chemicals, and biofuels.4–12 The primary component of lignocellulosic biomass is cellulose, a polysaccharide that locks β−1,4-linked glucosyl units in a highly compact crystalline structure.13 Complete hydrolysis of cellulose to glucose requires three major types of enzymes: endoglucanases, exoglucanases (or cellobiohydrolases), and β-glucosidases,13–15 which exhibit enzyme-enzyme synergy through their cooperative activities.16–18 Despite enzyme-enzyme synergy, crystalline cellulose is highly recalcitrant, and efficient hydrolysis requires large amounts of cellulase enzymes, posing a significant challenge to the development of biomass-based refineries.13,16
To improve the efficiency of cellulose hydrolysis, microbes in nature organize cellulases into multi-enzyme assemblies termed a cellulosome.19 The cellulosome system has inspired researchers to similarly cluster different types of cellulases onto a single scaffold protein to form a soluble supramolecular complex termed a minicellulosome.19 By taking advantage of the species specificity of the dockerin-cohesin interaction, chimeric scaffold proteins composed of cohesins from multiple origins have been synthesized to direct the assembly of cellulases to form designer minicellulosomes. Among dozens of different soluble minicellulosomes and designer minicellulosomes (hereafter referred to as single-scaffolded enzyme assemblies, or sSEAs for simplicity), the most significant improvement over free enzymes was reported for a three-enzyme (trifunctional) sSEA, showing a ~5.2-fold increase in hydrolysis of straw.20 The improved cellulose hydrolysis of the sSEAs in this and other studies has been attributed to enzyme proximity; however, why only some combinations of enzymes and cellulosic substrates seem to benefit from close proximity is not clear.21–23
Yeast whole-cell biocatalysts directly displaying sSEAs represent an attractive alternative to soluble sSEAs, as whole-cell systems have the ability to convert cellulose to ethanol directly.24,25 In addition, yeast-surface displayed sSEAs have the potential to form cellulose-enzyme-microbe (CEM) complexes, which have been shown to improve the conversion of cellulose to the target product.26–28 A number of yeast-surface displayed sSEAs have been designed,29–31 including a trifunctional assembly that showed a ~1.6-fold increase in hydrolytic activity over the display of the same enzymes on separate scaffolds.29 Such enhancement is often attributed to the same proximity effects observed with soluble sSEAs; however, the enzyme density (i.e. number of enzymes per cell) and their spatial proximity are not reported, complicating conclusions and comparisons.
Building on yeast whole-cell biocatalysts displaying sSEAs, multi-scaffolded enzyme assemblies (mSEAs) have been designed to create even larger cellulase clusters, with the premise of enhancing cellulose hydrolysis through increased enzyme density and greater enzyme proximity.32 mSEAs are formed by assembling multiple primary scaffold proteins (pScafs) on a single yeast-surface displayed anchor scaffold protein (aScaf), with each pScaf binding various cellulase enzymes.33,34 Yeast displaying bifunctional mSEAs containing two pScafs exhibited ~4.2 fold greater cellulolytic activity compared to an equivalent free enzyme cocktail.33 In a more complex design, yeast displaying trifunctional mSEAs containing two pScafs achieved a greater (~1.2 fold) ethanol titer from crystalline cellulose than amorphous cellulose,34 and modestly outperformed sSEAs on both substrates,34 albeit with a much lower ethanol titer compared to other studies.29,31,35 However, the same study also reported that expanding the mSEA to additional pScafs (i.e. three and four) resulted in reduced ethanol titer, suggesting that mSEA complexity alone does not necessarily translate to improved cellulose hydrolysis.
Harnessing the full potential of cell-surface displayed mSEAs will require a better understanding of how mSEAs assemble. For example, the extent to which pScafs and cellulases saturate their respective binding sites in a surface-displayed mSEA is not reported due to a lack of quantitative tools. Therefore, comparing one mSEA design to another is difficult because one design may have considerably higher assembly efficiency or enzyme density than another. Additionally, because there are no established quantitative tools for biocatalyst characterization, decoupling the contribution of parameters like metabolic burden, aScaf display level, enzyme density, and the spatial organization of cellulases in an mSEA is a significant challenge. These challenges are by no means limited to mSEAs and instead highlight the difficulty in benchmarking the performance of whole-cell biocatalysts in general.36,37 Unlike conventional catalysis in which the type and amount of catalyst used in a reaction can be well-characterized,37 whole-cell biocatalysts are biological systems with inherent variability.
In this study, we aimed to address these challenges by developing and validating a quantitative approach to whole-cell biocatalyst characterization. Using this approach, we characterized a series of yeast whole-cell biocatalysts displaying tetrafunctional mSEAs of varying complexity (i.e. aScaf1, aScaf2, aScaf3) and at varying display levels. Interestingly, we observed that aScaf-pScaf assembly is limited by molecular crowding of aScaf on the yeast cell surface, and the significance of this crowding phenomenon is dependent on aScaf size. In addition, we found that mSEA complexity (i.e. enzyme proximity) does not enhance the overall performance of yeast whole-cell biocatalysts, as increasing complexity from aScaf1 to aScaf3 only reduces the average inter-enzyme distance by ~10 nm. We further found that synergistic enzyme proximity effects are only appreciable on a length scale where the average inter-enzyme distance is > ~130 nm. Therefore, enzyme density on a whole-cell biocatalyst surface is the most important parameter for cellulose hydrolysis. We believe the quantitative approach underlying these observations is broadly applicable and may serve as a model for benchmarking whole-cell biocatalyst performance in the future.
RESULTS
Design of Yeast Whole-cell Biocatalyst Displaying mSEAs
Yeast whole-cell biocatalysts were engineered to express the anchor scaffold protein (aScaf) as a C-terminal fusion to the a-agglutinin subunit Aga2 for yeast surface display29,30,38 (Figure 1a). aScafs of varying complexity were constructed, composed of one (aScaf1), two (aScaf2), or three (aScaf3) type II cohesins derived from Clostridium thermocellum,39 with each aScaf accommodating a corresponding number of primary scaffold proteins (pScafs). The aScaf was also fused to a C-terminal V5 tag for detection and quantification.
Figure 1.

Schematic of multi-scaffolded enzyme assemblies (mSEAs) (a) Yeast were designed to display anchor scaffold proteins (aScafs) accommodating one (aScaf1), two (aScaf2) and three (aScaf3) primary scaffold proteins (pScafs) through type II cohesin-dockerin binding. (b) Representation of aScaf3-mSEA. Chimeric enzymes were assembled on the pScaf through species-specific type I cohesin-dockerin interactions, as indicated by color-coding. The carbohydrate binding module (CBM) on pScaf between ScaB and CipA cohesins is not shown for simplicity. The sequences of V5, 6×His and c-Myc epitope tags are GKPIPNPLLGLDST, HHHHHH and EQKLISEEDL, respectively. AGA1 represents the anchorage subunit of a-agglutinin of yeast cells and AGA2 the adhesion subunit. Four cellulases were used including β-glucosidase (BGL), reducing-end-cleaving cellobiohydrolase (CBHI), endoglucanase (EG), and non-reducing-end-cleaving cellobiohydrolase (CBHII).
The pScaf was designed to function as an adaptor scaffold protein that binds the aScaf and four unique cellulase enzymes through its type II dockerin and four type I cohesins, respectively (Figure 1b). Each of the four type I cohesins was derived from a scaffoldin protein of a different cellulolytic microbe: CipA of C. thermocellum, ScaB of Ruminococcus flavefaciens, CipC of C. cellulolyticum, and CbpA of C. cellulovorans (Supplementary Table 1). Meanwhile, four cellulase enzymes including a non-reducing-end-cleaving cellobiohydrolase (CBHII),40 an endoglucanase (EG),41 a reducing-end-cleaving cellobiohydrolase (CBHI)42 and a β-glucosidase (BGL)43 were fused with a type I dockerin from the corresponding species (see Supplementary Table 1 and Supplementary Table 2 for details). The species specificity of these four type I cohesin-dockerin interactions allows for the assembly of four unique enzymes by each pScaf. Through the aforementioned aScaf-pScaf-enzyme interactions, tetrafunctional mSEAs could be assembled and displayed on the surface of the yeast whole-cell biocatalyst (Figure 1b). The pScaf also included a carbohydrate binding module (CBM) between the CipA and ScaB cohesins to enhance the interaction of the whole-cell biocatalysts with the insoluble cellulose substrate.44,45 The pScaf and enzymes were also fused with a c-Myc or a His tag, respectively, for their quantification.
Quantitative Analysis of aScaf-pScaf Assembly
Successful mSEA formation hinges on the surface display of aScaf, which was first evaluated qualitatively using confocal microscopy. Yeast cells expressing aScaf1, aScaf2 or aScaf3 were stained with Alexa Fluor 647 (AF647) labeled anti-V5 antibody. As shown in Figure 2a, aScaf display was detected for all three constructs and appeared uniform across the entire cell surface. To quantify aScaf display level, the average number of aScafs per cell was evaluated using quantitative flow cytometry (Supplementary Figure 1). Quantitative flow cytometry has been routinely used to quantify the surface density of mammalian proteins by staining the target protein with fluorescently labeled monoclonal antibodies and comparing the median fluorescence intensity (MFI) to that of standard beads with known numbers of molecules of equivalent soluble fluorophores (MESF).46–48 Based on the same principle, we uniquely epitope-tagged each of the mSEA components (Figure 1b) and stained with fluorescently labeled antibodies recognizing each epitope tag. The display level of each mSEA component was then quantified by comparing its MFI to that of standard beads (see methods and Supplementary Figure 1). We observed that aScaf display level was inversely related to aScaf size, ranging from approximately 140,000 per cell for aScaf1 to approximately 70,000 per cell for aScaf3 (Figure 2b). These results suggest that aScaf surface display is limited by protein transport or metabolic burden associated with aScaf size.
Figure 2.

Characterizing aScaf-pScaf assembly on yeast cell surface. (a) Confocal microscope images of four yeast constructs stained for aScaf (green, anti-V5-AF647) and pScaf (red, anti-c-Myc-PE). Vector control (AGA2-V5) included a V5 epitope tag but no type II cohesins. Scale bar represents 10 μm. Quantification of the number of aScaf (b) and the number of pScaf (c) present on the four yeast constructs. (d) The number of pScaf assembled by aScaf1, aScaf2, and aScaf3. (e) The aScaf-pScaf assembly efficiency for aScaf1-pScaf, aScaf2-pScaf and aScaf3-pScaf. Data are represented as the mean of two independent experiments (n = 2) and error bars signify standard deviation.
To mitigate complications in protein transport and to ease yeast metabolic burden, we loaded saturating concentrations of purified pScaf followed by purified enzymes in vitro. The purity and size of pScaf purified from E. coli were verified using SDS-PAGE (Supplementary Figure 2a) prior to loading onto yeast cells displaying the aScafs. The aScaf-pScaf assembly was then assessed by staining the cells using anti-V5-AF647 and anti-c-Myc-PE antibodies. Confocal microscopy imaging showed that pScaf was detected on all three yeast displaying the aScaf but not on that displaying the Aga2-V5 fusion lacking aScaf, indicating that aScaf-pScaf assembly is specific and aScaf-dependent (Figure 2a). Quantitative flow cytometry further revealed that the number of pScafs per cell increased modestly as the aScaf size increased from ~82,000 on aScaf1 to ~96,000 on aScaf3 (Figure 2c, Supplementary Figure 3). Based on these quantitative data, we observed that the number of pScaf per aScaf increased as the number of the pScaf binding sites (i.e. type II cohesins) in the aScaf increased: 0.60 ± 0.02, 0.85 ± 0.01 and 1.40 ± 0.10 for aScaf1, aScaf2 and aScaf3, respectively (Figure 2d). However, when comparing this observed aScaf-pScaf assembly efficiency with respect to its theoretical maximum, we observed that aScaf-pScaf assembly for all three constructs was incomplete, despite pScaf being loaded in excess. Given the relatively high display level of the aScaf proteins in this study, we hypothesized that the incomplete aScaf-pScaf assembly might be a consequence of steric hindrance caused by aScaf surface crowding. The fact that aScaf-pScaf assembly efficiency for aScaf1 (58%) was significantly higher than both aScaf2 (42%) and aScaf3 (46%) (Figure 2e) further suggests that the significance of this crowding phenomenon may also be affected by aScaf size (Supplementary Table 3).
Modeling aScaf-pScaf Assembly
To test our hypothesis that aScaf-pScaf assembly efficiency is limited by aScaf crowding on the yeast cell surface, we measured aScaf-pScaf assembly efficiency over a range of aScaf display levels. This was achieved by treating yeast displaying each aScaf construct with varying concentrations of Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), which reduces varying fractions of the disulfide bonds anchoring the surface-displayed aScafs (Supplementary Figure 4a). TCEP- treated yeast were then qualitatively analyzed by confocal microscopy to confirm that the aScaf display level of each construct decreased and to ensure that aScaf surface distribution remained random (Supplementary Figure 4b–c).
TCEP-treated yeast displaying each aScaf construct were then loaded with pScaf, and aScaf-pScaf assembly efficiency was systematically characterized using quantitative flow cytometry. Because the sensitivity of quantitative flow cytometry is low at very low fluorescence signal levels (i.e. very low aScaf display level), aScaf-pScaf assembly efficiency was normalized to 100% for the highest aScaf-pScaf assembly observed for each aScaf construct. Supporting our surface-crowding hypothesis, the aScaf-pScaf assembly efficiency for each construct increased when the cell surface became less crowded with aScaf protein (Figure 3a). In addition, the distinct aScaf-pScaf assembly efficiency curves suggest that aScaf size influences its sensitivity to surface crowding, with aScaf3 being the most sensitive and aScaf1 the least sensitive (Figure 3a).
Figure 3:

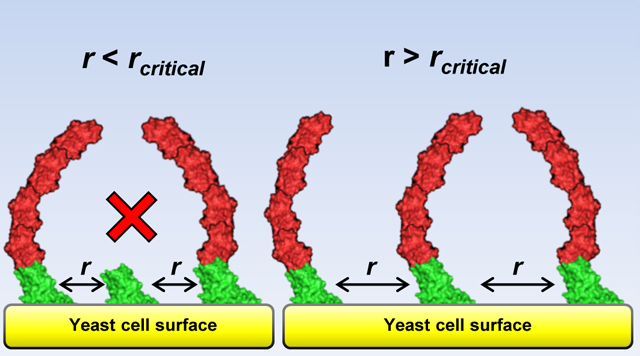
Modeling aScaf distribution and aScaf-pScaf assembly on yeast cell surface. (a) aScaf-pScaf assembly efficiency plotted as a function of aScaf display level (b) Theoretical random distribution representing the fraction of displayed aScaf plotted with respect to the distance to nearest neighbor for various aScaf display levels as calculated by Equation 2 (6.25k – 100k per cell). (c) Visual representation of aScaf-pScaf assembly inefficiency caused by aScaf surface crowding. pScaf-binding does not occur if the distance between neighboring aScafs (r) is less than the critical distance (rcrit). (d) pScaf assembly plotted with respect to the number of aScaf per cell. Solid line represents the theoretical pScaf assembly as a function of aScaf per cell based on surface crowding and rcrit values found using the random or Poisson-process distribution. Dashed lines represent the standard error of the regression. (e) The minimum distance to nearest neighbor (rcrit) required for aScaf-pScaf assembly predicted by the random distribution. Experimental data are represented as the mean of two independent experiments (n = 2) and error bars signify standard deviation unless otherwise noted. Statistical significance was evaluated from paired regression z-scores where ns (i.e. not significant) signifies p-value > 0.05, * signifies that p-value < 0.05, ** signifies p-value < 0.01, and *** signifies p-value < 0.001.
To further characterize the role of aScaf surface crowding on aScaf-pScaf assembly efficiency, the aScaf spatial distribution on the yeast cell surface was modeled based upon a Poisson process.49 This distribution was selected based on our observation that the aScaf surface distribution appears random (Figure 2a, Supplementary Figure 4b), which is further supported by the gradual decrease in aScaf-pScaf assembly efficiency with increasing aScaf display level (Figure 3a). Assuming this random process, it follows that the probability (P) that one aScaf has a distance to nearest neighbor aScaf of r is:
| (1) |
where ρ represents the aScaf display level per cell surface area. To calculate cell surface area, yeast cells were assumed to be spheres with a radius of 2 μm based on confocal imaging (Figure 2a, Supplementary Figure 4b) and previous studies.48,50 Integrating this distribution allowed the percentage of surface-displayed aScaf (faScaf) to be plotted with respect to its distance to nearest neighbor (Equation 2):
| (2) |
The integrated random distribution (Figure 3b) indicates that when the aScaf display level is high (e.g. 100,000 per cell), the distribution of aScaf distance to nearest neighbor is narrow, and the median distance between neighboring aScafs is relatively small. In contrast, when the aScaf display level is low (e.g. 6,250 per cell), the distribution of aScaf distance to nearest neighbor is broad, and the median distance between neighboring aScafs is greater.
Based on this statistical model, we hypothesized that if aScaf-pScaf assembly efficiency is determined solely by aScaf surface crowding, there should be a minimum critical distance (rcrit) between neighboring aScafs required for aScaf-pScaf assembly (Figure 3c). To evaluate this critical distance for each aScaf construct, the number of pScaf per cell was plotted with respect to aScaf display level, and the integrated random distribution (Equation 2) was fit to each construct by varying rcrit (Figure 3d, Supplementary Figure 5, details in methods). This curve fitting revealed the critical distance for pScaf assembly on aScaf1, aScaf2, and aScaf3 to be 8.0 ± 0.4 nm, 11.2 ± 0.3 nm, and 13.1 ± 1.2 nm, respectively (Figure 3e). These values support our surface crowding hypothesis and demonstrate that larger aScaf proteins require greater inter-aScaf separation than smaller aScaf proteins to allow pScaf access for assembly.
Quantitative Analysis of the Overall mSEA Assembly
After characterizing aScaf-pScaf assembly on the cell surface of all three yeast constructs, we next loaded the purified enzymes (Supplementary Figure 2b) to evaluate mSEA formation. An equimolar mixture of enzymes was loaded onto aScaf1-pScaf, aScaf2-pScaf, and aScaf3-pScaf, and mSEA formation was then assessed by staining with a combination of anti-V5-AF647 (aScaf), anti-c-Myc-PE (pScaf) and/or anti-His-AF647 (enzyme) antibodies. Using quantitative flow cytometry, we found that the enzyme density increased slightly with aScaf size, ranging from ~203,000 for aScaf1 to ~228,000 for aScaf3 (Figure 4a). This result is consistent with the aScaf-pScaf binding observed in Figure 2c and suggests that enzyme density can be increased by increasing the size of the aScaf protein. When the enzyme density was compared to the number of pScaf per cell, each construct was found to display ~2.4 enzymes per pScaf, regardless of aScaf size (p values > 0.42) (Figure 4b). This corresponds to a constant pScaf-enzyme assembly efficiency of ~60%.
Figure 4.

Quantitative flow cytometric analysis of yeast cells assembling mSEAs. Quantification of (a) the enzyme density and (b) the number of enzymes per pScaf for yeast cells displaying aScaf1-pScaf, aScaf2-pScaf, and aScaf3-pScaf. (c) pScaf-enzyme assembly efficiency plotted as a function of aScaf display level. (d) Overall assembly efficiency of each mSEA incorporating both aScaf-pScaf assembly efficiency and pScaf-enzyme assembly efficiency. For all graphs, data are represented as the mean of two independent experiments (n = 2) and error bars signify standard deviation. Statistical significance was evaluated using unpaired student t tests, and ns (i.e. not significant) signifies that differences are not statistically significant (p-values > 0.05).
To determine if the relatively low (60%) pScaf-enzyme assembly efficiency was also due to aScaf surface crowding, we measured pScaf-enzyme assembly efficiency at varying aScaf display levels. Yeast displaying aScaf1, aScaf2, and aScaf3 were treated with varying concentrations of TCEP, loaded with pScaf and enzymes, and each component of the mSEA was measured using quantitative flow cytometry (Supplementary Figure 6). Interestingly, the pScaf-enzyme assembly efficiency remained ~60% regardless of aScaf display level (Figure 4c), indicating that while aScaf-pScaf assembly efficiency is dependent on aScaf display level and aScaf complexity (Figure 3a), pScaf-enzyme assembly is independent of both. This difference might be explained by the relative proximity of each assembly step with respect to the yeast cell surface: aScaf-pScaf assembly occurs close to the cell surface and is thus constrained by the spatial footprint (i.e. crowding) of neighboring aScaf proteins; in contrast, pScaf-enzyme assembly occurs further from the cell surface where there is more spatial freedom to accommodate binding. Under such an assembly scheme, aScaf-pScaf assembly efficiency is limited by aScaf surface crowding while pScaf-enzyme assembly efficiency is not.
Because pScaf-enzyme assembly efficiency was independent of both aScaf surface crowding and mSEA complexity (i.e. aScaf size), we hypothesized that its low value of 60% is simply a consequence of the disparate binding affinities of each individual enzyme dockerin to its respective cohesin on the pScaf. This was supported by the observation that BGL exhibited a much lower assembly efficiency compared to the other three enzymes when each enzyme was loaded and quantified individually (Supplementary Figure 7a). Further study of pScaf-BGL binding revealed that the dockerin fused to BGL showed a much lower binding affinity (KD=1500 nM, Supplementary Figure 7b) for its complementary cohesin than what has been reported for most other cohesin-dockerin pairs (KD ~ 1 – 25 nM).51 This unanticipated result underscores the importance of high cohesin-dockerin binding affinity for cellulosomal-based mSEA assembly. Because the overall mSEA assembly compounds inefficiencies in each assembly step (i.e. aScaf-pScaf and pScaf-enzyme), the overall mSEA assembly efficiency observed is low: 36%, 24%, and 27% for aScaf1, aScaf2, and aScaf3, respectively (Figure 4d). The low mSEA assembly efficiencies observed here are striking and suggest that conclusions drawn from previous biocatalytic systems assuming 100% assembly efficiency are probably incomplete.
Modeling Overall mSEA Assembly
Using the constant 60% pScaf-enzyme assembly efficiency observed in Figure 4b–c and the critical distance values calculated in Figure 3e, the theoretical enzyme density was plotted as a function of aScaf display level for each construct (Figure 5a). Based on crowding-limited and aScaf-size dependent aScaf-pScaf assembly, each construct has a distinct optimum aScaf display level that maximizes enzyme density (arrows in Figure 5a). Of the three constructs tested, aScaf3 is predicted to provide the greatest enzyme density (~246,000 per cell) at an optimum aScaf3 display level of ~101,000 per cell. Interestingly, under crowding-limited assembly our model indicates that the optimum aScaf display level for each construct is associated with the same aScaf-pScaf assembly efficiency value of 1/e (or ~37%) (Figure 5b). If the aScaf display level is lower than this optimum, assembly is occurring in an aScaf display-limited (DL) regime. In the DL assembly regime, more aScafs could be added to the surface without crowding effects dominating, increasing the number of possible pScafs and enzymes assembled per cell, but at the expense of mSEA assembly efficiency (Figure 5c). If the aScaf display level is greater than this optimum, assembly is occurring in a crowding-limited (CL) regime. In the CL assembly regime, not only will the additional aScafs be unable to bind pScaf, but they will also crowd out previously accessible aScafs and make them unavailable for pScaf binding. This crowding produces a net reduction in the number of possible pScafs and thus enzymes assembled per cell. These results highlight a clear tradeoff that arises from aScaf crowding: increasing aScaf display level (from 0 to optimum) causes an increase in enzyme density while reducing enzyme proximity.
Figure 5:

Modeling theoretical mSEA assembly (a) The theoretical enzyme density plotted as a function of aScaf display level. Data shown on right is a zoomed-in view across a physiologically relevant range of aScaf display levels and with experimental data overlaid. Experimental data are represented as the mean of two independent experiments (n = 2) and error bars signify standard deviation (b) Predicted enzyme density as a percent of the theoretical maximum plotted with respect to aScaf-pScaf assembly efficiency. (c) Tradeoff between the theoretical number of enzymes per mSEA (dashed lines) and the percent of theoretical maximum enzyme density (solid lines) plotted as a function of aScaf display level for aScaf1, aScaf2, and aScaf3. Theoretical mSEA assembly was determined assuming 60% pScaf-enzyme assembly efficiency and rcrit values found using the Poisson-process distribution. DL represents the aScaf display-limited regime and CL represents the aScaf crowding-limited regime.
Nevertheless, at typical yeast-surface display levels (0 – 150,000 proteins per cell), our model indicates that at the same aScaf display level, increasing the size of the aScaf protein will allow for greater enzyme density (Figure 5a, right panel). This insight might explain in part why larger and more complex cellulosomal structures tend to outperform smaller and simpler structures in some studies.29 However, because larger aScaf constructs are associated with greater critical distances for pScaf assembly, the apparent advantages of such constructs diminish rapidly as aScaf display level increases beyond ~150,000 per cell (Figure 5a, left panel).
Role of Enzyme Density and Proximity on Cellulose Hydrolysis
While a number of studies have reported the importance of enzyme density and enzyme proximity for cellulose hydrolysis,33,52 there have been few attempts to quantify and decouple the contribution of each to cellulolytic activity. Further, those studies that have isolated the importance of enzyme proximity on cellulose hydrolysis have reported mixed results depending on the type of cellulosic substrate.23 Using the quantitative approach developed in this study, we aimed to evaluate the significance of both enzyme density and enzyme proximity on cellulose hydrolysis.
Because the enzymes used here were fused with heterologous dockerins and/or His-tags (Figure 1), the activity of each was first verified by hydrolyzing 0.5% phosphoric acid swollen cellulose (PASC) (Supplementary Figure 8a–d). Additionally, the activity of yeast cells displaying aScaf1-mSEA, aScaf2-mSEA, and aScaf3-mSEA was measured at varying concentrations of PASC substrate to evaluate the half-saturating concentration of substrate and limiting rate of the reaction after 24 h (Supplementary Figure 9a–b). After confirming the activity and synergy of all four enzymes, yeast cells displaying aScaf were treated with varying concentrations of TCEP to produce 18 populations for each, spanning a wide range of aScaf display levels (Supplementary Figure 10). These yeast populations were then loaded with pScaf and enzymes and the aScaf display level, enzyme density, and the number of enzymes per mSEA were quantified as described in Figures 4 and 5. The enzyme density and the number of enzymes per mSEA were then plotted with respect to the average inter-aScaf distance () (Figure 6a), which was calculated by integrating the random distribution using Equation 3:
| (3) |
The resulting relationships demonstrate that larger inter-aScaf distance allows a greater number of enzymes per mSEA, but at the expense of enzyme density (Figure 6a). While the number of enzymes per mSEA represents a qualitative measure of enzyme proximity, to better quantitatively evaluate enzyme proximity, we further calculated average inter-enzyme distance using and the number of enzymes per mSEA (see methods for details). As shown in Figure 6b, aScaf3 provides the greatest enzyme proximity across all enzyme densities, followed by aScaf2, and aScaf1, respectively (Figure 6b). However, the maximum reduction in average inter-enzyme distance at a given enzyme density when increasing mSEA complexity from aScaf1 to aScaf3 was less than 10 nm.
Figure 6:

Structure-performance relationship of multi-enzyme assemblies (a) Enzyme density (circle) and the number of enzyme per mSEA (triangle) plotted as a function of inter-aScaf distance for aScaf1, aScaf2, and aScaf3. (b) Average inter-enzyme distance plotted as a function of enzyme density for aScaf1 (blue), aScaf2 (red), and aScaf3 (green). Average inter-enzyme distance was calculated using parameters determined in (a). (c) Glucose released from 0.3% phosphoric acid swollen cellulose (PASC) by yeast whole-cell biocatalysts displaying mSEAs on aScaf1, aScaf2, and aScaf3. Low, medium, and high corresponds to enzyme density of ~50,000 per cell, ~100,000 per cell, and ~150,000 per cell, respectively. (d) Glucose released from 0.3% PASC by an equimolar mixture of free enzymes (open circles) or an equivalent amount of enzyme assembled on soluble pScaf proteins (sSEAs, filled circles), plotted as a function of inter-enzyme distance. (e) Proximity enhancement of sSEAs over soluble enzymes on PASC hydrolysis plotted as a function of inter-enzyme distance. Error bars on data represent standard deviation from the mean of two independent experiments (n = 2) Statistical significance was evaluated using unpaired student t tests where ns (i.e. not significant) signifies p-value > 0.05, * signifies that p-value < 0.05, ** signifies p-value < 0.01, and *** signifies p-value < 0.001.
The cellulolytic performance of each population was then assessed using 0.3% PASC as a substrate in the presence of the glucose metabolism inhibitor, methylglyoxal. The concentration of glucose released was plotted with respect to the enzyme density for each aScaf construct (Supplementary Figure 11a). Interestingly, when compared at similar enzyme densities, the whole-cell biocatalyst performance appeared independent of mSEA complexity (i.e. aScaf1, aScaf2, or aScaf3) and instead depended solely on enzyme density (Figure 6c). These results indicate that the relatively modest increase in enzyme proximity (i.e. 1 – 10 nm as shown in Figure 6b) caused by greater mSEA complexity is insufficient to produce synergistic proximity effects.
Given the limited range of inter-enzyme distances observed with surface-displayed mSEAs in Figure 6b (i.e. 6 nm – 17 nm), we hypothesized that any performance enhancement from proximity effects will only be measurable on a longer length scale. To test this hypothesis, 0.3% PASC was hydrolyzed by free enzyme cocktails, spanning inter-enzyme distances of ~50 nm to ~250 nm (i.e. 12.8 μM to 0.1 μM in Supplementary Figure 11b). The average inter-enzyme distance in solution (d) was calculated from the molar enzyme concentration (C):
| (4), ref53 |
Proximity effects were evaluated by comparing the hydrolytic performance of soluble sSEAs (i.e., enzymes assembled on soluble pScaf protein) to an equivalent concentration of free enzymes over the range of inter-enzyme distances.
At lower inter-enzyme distances, the amount of glucose released by free enzymes and sSEAs was similar. However, at greater inter-enzyme distances, the sSEAs released significantly more glucose than the free enzymes (Figure 6d). The proximity enhancement, defined as the ratio of the glucose released by the sSEAs to that by free enzymes, was then plotted separately as a function of inter-enzyme distance in solution (Figure 6e). Consistent with our hypothesis and our results on the yeast cell surface, no proximity effect was detected for inter-enzyme distances less than < 100 nm. However, at inter-enzyme distances >130 nm, the sSEAs introduced proximity effects that enhanced hydrolytic performance up to 2.0-fold. Taken together, these data suggest that enzyme proximity can enhance cellulolytic performance, but this enhancement is only significant when the equivalent distance between free enzymes is relatively large (>130 nm). In contrast, when inter-enzyme distances are less than ~100 nm, additional increases in enzyme proximity do not appear to be associated with further synergistic proximity effects on the PASC hydrolysis.
Direct Fermentation of Cellulose to Ethanol
Next, we evaluated the ability of the yeast whole-cell biocatalyst displaying aScaf3-mSEA to directly ferment cellulose to ethanol. 1% or 2% PASC was used as the substrate and ethanol production was sampled over 168 hours (Figure 7). Within 48 hours, the ethanol titer from 1% PASC had reached 3.49 g/L. After 168 h of fermentation, ethanol titer achieved from 1% PASC was 4.22 g/L, corresponding to 78% theoretical ethanol yield. Using 2% PASC as substrate, ethanol titer reached 5.13 g/L within 48 h and 7.17 g/L after 168 hours. However, the ethanol titer from 2% PASC after 168 h corresponded to just 65% theoretical ethanol yield. The ethanol titers achieved from both 1% and 2% PASC are among the highest reported to date (Table 1), which is likely a consequence of the high enzyme density of aScaf3-mSEA. Further, the aScaf3-mSEA biocatalyst also exhibited greater ethanol productivity than previously reported when using PASC (Table 1).
Figure 7:

Time course of ethanol production from PASC by aScaf3-mSEA. Ethanol production from 1% PASC (blue) and 2% PASC (red), were recorded over 168 hours. Yeast displaying cells without enzyme loaded were used as a control. Data are represented as the mean of two independent experiments (n = 2) and error bars signify standard deviation.
Table 1.
Cellulosic ethanol production using yeast whole-cell biocatalysts
| Carbon source | Cell density | Ethanola (g/L) | Productivityb (g/L/h) | Source |
|---|---|---|---|---|
| 1% PASC | NA | 3.5 | 0.073 | ref31 |
| OD600 50 | 1.8 | 0.034 | ref29 | |
| OD600 50 | 1.091 | 0.020 | ref34 | |
| OD600 50 | 2.7 | 0.031 | ref30 | |
| OD600 50 | 4.22 | 0.088 | This study | |
| 2% PASC | 150 g wet cells/L | 6.7 | 0.149 | ref8 |
| OD600 50 | 7.17 | 0.133 | This study |
Highest titer reported in each study from the literature and this study.
Productivity was calculated after 48 hours of the fermentation reactions.
DISCUSSION
A wide variety of biocatalytic systems have been engineered based on multi-enzyme assembly.54–57 Previous studies have shown that assembling mSEAs on whole-cell biocatalyst surfaces promotes greater cellulose hydrolysis.33,34 However, the molecular principles that govern how these mSEAs form and the mechanisms by which they improve cellulolytic activity are currently not well understood due to a lack of quantitative tools. Here, we developed experimental and theoretical approaches to quantify each step of mSEA assembly and thereby provide insights into what parameters affect assembly efficiency and biocatalyst performance.
Using quantitative flow cytometry, we observed that aScaf surface crowding limits aScaf-pScaf assembly, but not pScaf-enzyme assembly. This result suggests aScafs must be separated from their nearest neighbor by a minimum critical distance rcrit to allow pScaf access, and rcrit is a function of aScaf size. The relationship between aScaf size and critical distance could be a consequence of outside-in aScaf-pScaf assembly, in which cohesins on aScafs closest to the bulk solution likely bind pScafs before cohesins closer to the cell surface. As more of the distal cohesins are occupied by pScafs, neighboring partial assemblies will obstruct the diffusion of soluble pScafs from the bulk to interior cohesins, resulting in additional aScaf-pScaf assembly inefficiency. As a result, one would expect larger aScaf constructs to require greater inter-aScaf spacing so that soluble pScafs may access internal cohesins by diffusing between bulky neighboring assemblies. Based on this assembly mechanism, the critical distance associated with each aScaf construct should also be a function of pScaf size. It is important to note that our crowding model assumes aScaf-pScaf assembly is a binary event (i.e. pScaf binds all sites on a particular aScaf or none at all). In reality, pScaf binding can be intermediate resulting in partial aScaf-pScaf assemblies, which should also be affected by aScaf surface crowding. In addition, our model fails to include secondary and tertiary crowding effects from neighboring aScafs that are not nearest. Nevertheless, the remarkable agreement with our experimental data (Figure 3d, Supplementary Figure 5), suggests that our relatively simple crowding model captures some of this additional complexity, without attempting to explain it from first principles.
Interestingly, aScaf-pScaf assembly inefficiency caused by surface crowding is not an impediment to achieving a high enzyme density. Instead, the mathematical model developed here predicts that enzyme density is maximized when surface crowding allows only 37% of expressed aScaf to bind pScaf (Figure 5b). This result may seem counterintuitive, as one might assume that high assembly efficiency correlates with high enzyme density. However, because aScaf crowding limits aScaf-pScaf assembly efficiency, high assembly efficiencies are only possible at relatively low aScaf display levels. While increasing aScaf display level causes a reduction in aScaf-pScaf assembly efficiency (Figure 3a), the additional aScafs (up to an optimum) provide more pScaf binding sites than are lost by inefficient assembly (Figure 5c). The resulting tradeoff between enzyme density and mSEA assembly efficiency demonstrates how a quantitative approach to whole-cell biocatalyst characterization can reveal unexpected insights into stochastic protein assembly. Even when aScaf crowding limits aScaf-pScaf assembly, the enzyme density can potentially be improved through protein engineering and the rational design of mSEA components. For example, it might be possible to reduce the critical distances for aScaf-pScaf assembly by modulating cohesin orientation and/or binding modes58 to allow more efficient packing of assemblies on the cell surface.
In addition to characterizing aScaf-pScaf and pScaf-enzyme assembly, the quantitative approach used in this study allowed us to explore how parameters (e.g. enzyme density and enzyme proximity) affect the overall cellulolytic performance of whole-cell biocatalysts. Using in vitro assembly, we revealed that the enzyme density and not mSEA complexity (i.e. enzyme proximity) is the most important parameter for hydrolyzing PASC. In addition, we found that while aScaf3 generally provided the greatest enzyme proximity, the difference in average inter-enzyme distance between aScaf1 and aScaf3 was relatively small (< 10 nm, Figure 6b), and thus insufficient to produce any synergistic proximity effects. Using soluble sSEAs and free enzymes, we determined that enzyme proximity effects on PASC are only significant when the average distance between equivalent amounts of free enzymes is > 130 nm. Because most mSEAs on whole-cell biocatalysts have inter-enzyme distances well below this 130 nm threshold, enzyme proximity is not likely to play an important role in the hydrolytic performance.
While there is great interest in improving cellulosome-inspired enzyme systems for biomass degradation, many fundamental questions concerning how these systems assemble and what aspects of the assembly (e.g. assembly efficiency, enzyme density, and enzyme proximity) are most important for catalytic activity remain unanswered. As a result, biocatalyst design has generally been empirically driven based on trial and error. Here we developed and utilized both quantitative and theoretical approaches to understand the assembly and performance of a series of yeast whole-cell biocatalysts of varying complexity. This work presents a quantitative study of cellulosomal-structure assembly and revealed insights that could provide guidance for future designs of cell-surface bound protein assemblies. Moreover, we believe that the quantitative techniques described here could provide a standardized approach to whole-cell biocatalyst benchmarking. Such an approach should allow more meaningful comparisons between whole-cell biocatalytic systems from different labs and help shift the field of whole-cell biocatalysis from trial and error towards rational design.
METHODS
Strains, Media and Reagents
The yeast S. cerevisiae strain EBY100 (Invitrogen, Carlsbad, CA) was used for mSEA surface display. The E. coli strains (Invitrogen) Mach1 and BL21 (DE3) were used for recombinant DNA manipulation and protein expression, respectively. All recombinant yeast and E. coli strains are summarized in Supplementary Table 1. C. thermocellum DSM1237, C. cellulovorans, C. cellulolyticum and R. flavefaciens were purchased from ATCC (Manassas, VA) and cultured anaerobically following ATCC protocols. Recombinant EBY100 cells were cultured using SC-Trp medium: 1.67 g/L yeast nitrogen base without amino acids, 5 g/L ammonium sulfate (Difco Laboratories, Detroit, MI), 20 g/L glucose, 15 g/L adenine hemisulfate, and 0.64 g/L complete supplement mixture without tryptophan (MP Biomedicals, Solon, OH). Induction of aScaf display on yeast surface was performed in YPG media (1% yeast extract, 2% peptone, 2% galactose). E. coli was cultured in Luria-Bertani (LB) medium containing 50 μg/mL kanamycin. Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Protein Design and Purification
The expression cassettes of aScafs and pScaf were cloned in pYD1 (Invitrogen, Carlsbad CA) and pET28a (Novagen, WI, USA), respectively. The chimeric enzymes EG, CBHI, CBHII and BGL fused to respective dockerin were cloned in pET28a. Sources of all protein sequences and primers used in this study are listed in Supplementary Table 1 and Supplementary Table 2, respectively. All plasmids were constructed using either homologous recombination in EBY100 or ligation in Mach1. Detailed plasmid construction steps are provided in supplementary information. EBY100 strains transformed with pYD1-Aga2-V5, pYD1-aScaf1, pYD1-aScaf2, pYD1-aScaf3 were induced in YPG for 60 hours at 20 °C. BL21 (DE3) strains transformed with pET28a-pScaf, pET28a-EG, pET28a-CBHI, pET28a-CBHII or pET28a-BGL were induced in LB with 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) overnight at 16 °C. pScaf and enzymes were then purified from BL21 (DE3) cell lysate using fast protein liquid chromatography (FPLC) with a HisTrap HP column (GE healthcare, Fairfield, CT). The His tag on pScaf was cleaved using thrombin (EMD Millipore, Billerica, MA) according to the manufacturer’s protocol following purification.
Quantitative Characterization of mSEAs
aScaf-pScaf assembly was performed by incubating 1.0 μM pScaf with 1.0×106 yeast displaying each aScaf type for 2 h at 30 °C or overnight at 4 °C in 50 mM PBS buffer (pH 7.4) containing 1.0% BSA to limit non-specific binding. After incubation, cells were washed and either co-stained or split, and stained with anti-V5-AF647 (Invitrogen) and anti-c-Myc-PE (Invitrogen) at 2 ng/μL of each antibody for one hour on ice. aScaf-pScaf assembly was assessed qualitatively using an Olympus FV-1200 confocal microscope (Olympus, Melville, NY) with a 60x objective. To assemble mSEAs, pScaf was loaded as described above, followed by loading of one or a combination of two, three or four cellulases, each at 0.1 μM. The yeast-cell displayed mSEAs were then stained with anti-His-AF647 (Biolegend, San Diego, CA) or anti-V5-AF647 or anti-c-Myc-AF647 (R&D Systems, Minneapolis, MN) at 2 ng/μL of each antibody for one hour on ice. Co-stained cells were then analyzed using an Applied Biosystems Attune Acoustic Focusing Flow Cytometer (Applied Biosystems, Waltham, MA), and the MFI of each component was recorded. The number of each component of the mSEA assembly on the yeast cell (i.e. aScaf, pScaf, and enzyme) was determined using quantitative flow cytometry with Quantum™ Alexa Fluor 647 and Quantum™ R-PE fluorescence quantitation beads (Bangs Laboratories, Fishers, IN). Briefly, quantitation beads with four densities of molecules of equivalent soluble fluorophore (MESF) were analyzed by flow cytometry, and the MFI corresponding to each distinct density of fluorophores was determined. Using these data, MESF (provided by the manufacturer) was plotted with respect to MFI (Supplementary Figure 1), and linear regression was performed to determine m and b:
| (5) |
The number of aScaf, pScaf and enzyme on each co-stained yeast cell was then determined by plugging the MFI for each (i.e. aScaf, pScaf, and enzyme) into the appropriate standard curve regression equation (Supplementary Figure 1).
Modeling aScaf Spatial Distribution and mSEA Assembly
aScaf spatial distribution on the yeast cell surface was modeled as a Poisson-process distribution as described in equation 1. The critical distance for aScaf1, aScaf2, and aScaf3 was determined by fitting the theoretical distribution to an experimental relationship between pScaf per cell and aScaf display level, over a range of aScaf display levels. Briefly, aScaf-pScaf assembly efficiency (ηE) was first defined as:
| (6) |
where pScaf and aScaf represent the respective number of proteins per cell and β represents the number of pScaf binding sites per aScaf (e.g. β=3 for aScaf3). The random distribution was then fit to this relationship using faScaf derived in equation 2:
| (7). |
where the coefficient A normalizes aScaf-pScaf assembly efficiency to maximum value of 100%. Importantly, ηE represents the experimentally determined aScaf-pScaf assembly efficiency and ηT represents the fraction of total aScaf with a distance to nearest neighbor less than or equal to the distance rcrit. Therefore, if our crowding hypothesis is accurate:
| (8) |
This relationship was ln-transformed to obtain slope-intercept form:
| (9) |
Where y = and x = ρ. Then was plotted with respect to ρ, and linear regression was performed on the ln-transformed set to evaluate A and rcrit:
| (10) |
The regression equation was then exponentiated and the predicted pScaf per cell was plotted as a function of aScaf per cell for each aScaf construct based on found critical distances (Figure 3d). The number of enzymes per aScaf was similarly found as a function of aScaf display level assuming 60% assembly pScaf-enzyme assembly efficiency (Figure 5a–c).
Modeling inter-enzyme Distance
The average inter-enzyme distance across the entire surface of yeast whole-cell biocatalysts was calculated as a weighted average of the distance between enzymes in a single mSEA and the distance between neighboring mSEAs. To determine this, the radius of the mSEA assemblies (rmSEA) was estimated as described:
| (11), ref53 |
where MWmSEA represents the molecular weight of the mSEA. Each mSEA was assumed roughly spherical, so mSEA volume (VmSEA) was then calculated as:
| (12) |
After determining mSEA volume, the average distance between enzymes within the same mSEA (rEmSEA) was calculated using a modified form of the Wigner-Seitz radius:
| (13) |
where EPA represents the number of enzymes per assembly. Unsurprisingly, rEmSEA was found to be relatively constant for all aScaf constructs and assembly efficiencies, as mSEA volume scales with the number of enzymes per mSEA. Next, the average distance between mSEAs was calculated using the average inter-aScaf distance (raScaf) derived in Equation 3. The average inter-enzyme distance () was then calculated as the weighted average:
| (14) |
If EPA = 1, the average inter-enzyme distance across the whole-cell will be equal to the inter-aScaf distance (raScaf).
Enzyme Activity Assay and Fermentation
Phosphoric acid-swollen cellulose (PASC) was prepared from crystalline cellulose (Avicel PH-101) as described previously.18 For activity assays involving the release of reducing-end sugars, the Somogy-Nelson method was used to quantify product formation.59 For activity assays involving TCEP treatment, yeast cells expressing aScaf were treated with various concentrations of TCEP to remove a fraction of aScaf from the surface prior to mSEA assembly. Next, yeast cells displaying aScaf-mSEAs were washed twice with hydrolysis buffer (50 mM sodium acetate, pH 5.0) and re-suspended to a concentration of 2.25×108 cells/300 μL hydrolysis buffer (OD600 = 30) containing 0.3% PASC and 100 mM methylglyoxal60. For activity assays using soluble sSEAs, an optimum ratio of 2 enzymes per pScaf (Supplementary Figure 12) was used for each reaction. Hydrolysis reactions were carried out at 30 °C and 250 rpm and samples were analyzed after 24 hours to quantify the glucose released. The amount of glucose released was quantified using high-performance liquid chromatography (HPLC) (Shimadzu, Kyoto, Japan) using a refractive index detector and a ROA-organic acid column (300 × 7.8 mm; Phemomenex®, Torrance, CA, USA) with elution in 5 mM sulfuric acid at 0.6 mL/min and 60 °C for 35 minutes.
Fermentation was performed by first washing yeast displaying aScaf3-mSEA twice with YP medium (1% yeast extract, 2% peptone), before resuspending to concentration of 3.75×109 cells/3 mL YP (OD600 = 50) containing 0.001% ergosterol, 0.042% Tween 80, and 1% or 2% PASC under anaerobic conditions. Yeast displaying only aScaf3-pScaf complexes without enzymes loaded was used as a negative control. Fermentation was carried out anaerobically at 30 °C and 250 rpm, and ethanol titer was measured from the fermentation broth at the indicated time points using HPLC protocol described above.
Supplementary Material
Acknowledgements
This work was partly supported by the National Science Foundation (NSF) under grant no. 1511720, 1645229 and CAREER Award 1653611, by the National Institutes of Health (NIH) under grant no. CA191952 and OD020053, and by MCubed at the University of Michigan. H.G. and J.L. were partly supported by Basic Science Research Program through the National Research Foundation of Korea (2017R1A2B3011676, 2013M3A6A8073184) and by WTU joint research grant from Konkuk University. The authors would like to thank Dr. L. Zhang and B. D. Hill for assistance with the confocal microscopy experiments, and C. Jackman for the help with setting up the anaerobic chamber for fermentation.
Footnotes
Data Availability
The data that support the plots within this paper and other findings of this study are available from the last corresponding author upon reasonable request.
Supplementary Information
Supplementary information is available free of charge on the journal website.
Competing Interests
The authors declare no competing interests.
References
- 1.Alper H & Stephanopoulos G Engineering for biofuels: exploiting innate microbial capacity or importing biosynthetic potential? Nat. Rev. Microbiol 7, 715–23 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Wheeldon I et al. Substrate channelling as an approach to cascade reactions. Nat. Chem 8, 299–309 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Avalos JL, Fink GR & Stephanopoulos G Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat. Biotechnol 31, 335–341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dodds DR & Gross RA Chemistry. Chemicals from biomass. Science 318, 1250–1251 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Gupta NK, Fukuoka A & Nakajima K Amorphous Nb2O5 as a Selective and Reusable Catalyst for Furfural Production from Xylose in Biphasic Water and Toluene. ACS Catal. 7, 2430–2436 (2017). [Google Scholar]
- 6.Zinoviev S et al. Next-generation biofuels: Survey of emerging technologies and sustainability issues. ChemSusChem 3, 1106–1133 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Huber GW, Iborra S & Corma A Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering. Chem. Rev 106, 4044–4098 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Isikgor FH & Becer CR Lignocellulosic biomass: a sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem 6, 4497–4559 (2015). [Google Scholar]
- 9.Caes BR, Teixeira RE, Knapp KG & Raines RT Biomass to Furanics: Renewable Routes to Chemicals and Fuels. ACS Sustain. Chem. Eng 3, 2591–2605 (2015). [Google Scholar]
- 10.Himmel ME & Bayer EA Lignocellulose conversion to biofuels: current challenges, global perspectives. Curr. Opin. Biotechnol 20, 316–317 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Yeh TM et al. Hydrothermal catalytic production of fuels and chemicals from aquatic biomass. J. Chem. Technol. Biotechnol 88, 13–24 (2013). [Google Scholar]
- 12.Wen F, Nair NU & Zhao H Protein engineering in designing tailored enzymes and microorganisms for biofuels production. Curr. Opin. Biotechnol 20, 412–419 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horn SJ, Vaaje-Kolstad G, Westereng B & Eijsink VG Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Percival Zhang Y-H, Himmel ME & Mielenz JR Outlook for cellulase improvement: screening and selection strategies. Biotechnol. Adv 24, 452–481 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Schwarz WH The cellulosome and cellulose degradation by anaerobic bacteria. Appl. Microbiol. Biotechnol 56, 634–649 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Lynd LR, Weimer PJ, van Zyl WH & Pretorius IS Microbial Cellulose Utilization: Fundamentals and Biotechnology. Microbiol. Mol. Biol. Rev 66, 739–739 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shang BZ & Chu JW Kinetic modeling at single-molecule resolution elucidates the mechanisms of cellulase synergy. ACS Catal. 4, 2216–2225 (2014). [Google Scholar]
- 18.Percival Zhang YH et al. A transition from cellulose swelling to cellulose dissolution by o-phosphoric acid: Evidence from enzymatic hydrolysis and supramolecular structure. Biomacromolecules 7, 644–648 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Artzi L, Bayer EA & Moraïs S Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nat. Rev. Microbiol 15, 83–95 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Fierobe H-P et al. Action of designer cellulosomes on homogeneous versus complex substrates: controlled incorporation of three distinct enzymes into a defined trifunctional scaffoldin. J. Biol. Chem 280, 16325–16334 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Moraïs S et al. Cellulase-Xylanase Synergy in Designer Cellulosomes for Enhanced Degradation of a Complex Cellulosic Substrate. MBio 1, e00285–10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vazana Y, Moraïs S, Barak Y, Lamed R & Bayer EA Interplay between Clostridium thermocellum family 48 and family 9 cellulases in cellulosomal versus noncellulosomal states. Appl. Environ. Microbiol 76, 3236–3243 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fierobe HP et al. Degradation of cellulose substrates by cellulosome chimeras: Substrate targeting versus proximity of enzyme components. J. Biol. Chem 277, 49621–49630 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Sun J, Wen F, Si T, Xu JH & Zhao H Direct conversion of xylan to ethanol by recombinant Saccharomyces cerevisiae strains displaying an engineered minihemicellulosome. Appl. Environ. Microbiol 78, 3837–3845 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith MR, Khera E & Wen F Engineering Novel and Improved Biocatalysts by Cell Surface Display. Ind. Eng. Chem. Res 54, 4021–4032 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.You C, Zhang X-Z, Sathitsuksanoh N, Lynd LR & Zhang Y-HP Enhanced microbial utilization of recalcitrant cellulose by an ex vivo cellulosome-microbe complex. Appl. Environ. Microbiol 78, 1437–1444 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wieczorek AS & Martin VJJ Engineering the cell surface display of cohesins for assembly of cellulosome-inspired enzyme complexes on Lactococcus lactis. Microb. Cell Fact 9, 69 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Y, Zhang Y-HP & Lynd LR Enzyme-microbe synergy during cellulose hydrolysis by Clostridium thermocellum. Proc. Natl. Acad. Sci 103, 16165–16169 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen F, Sun J & Zhao H Yeast surface display of trifunctional minicellulosomes for simultaneous saccharification and fermentation of cellulose to ethanol. Appl. Environ. Microbiol 76, 1251–1260 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang Y, Si T, Ang EL & Zhao H An Engineered Pentafunctional Minicellulosome for Simultaneous Saccharification and Ethanol Fermentation in Saccharomyces cerevisiae. Appl. Environ. Microbiol 80, 6677–6684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai S-L, Oh J, Singh S, Chen R & Chen W Functional assembly of minicellulosomes on the Saccharomyces cerevisiae cell surface for cellulose hydrolysis and ethanol production. Appl. Environ. Microbiol 75, 6087–6093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bugada LF, Smith MR & Wen F Engineering Spatially Organized Multienzyme Assemblies for Complex Chemical Transformation. ACS Catal. 8, 7898–7906 (2018). [Google Scholar]
- 33.Tsai S-LL, DaSilva NA & Chen W Functional display of complex cellulosomes on the yeast surface via adaptive assembly. ACS Synth. Biol 2, 14–21 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Fan L-H, Zhang Z-J, Yu X-Y, Xue Y-X & Tan T-W Self-surface assembly of cellulosomes with two miniscaffoldins on Saccharomyces cerevisiae for cellulosic ethanol production. Proc. Natl. Acad. Sci. U. S. A 109, 13260–13265 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matano Y, Hasunuma T & Kondo A Display of cellulases on the cell surface of Saccharomyces cerevisiae for high yield ethanol production from high-solid lignocellulosic biomass. Bioresour. Technol 108, 128–133 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Meyer A et al. Optimization of a whole-cell biocatalyst by employing genetically encoded product sensors inside nanolitre reactors. Nat. Chem 7, 673–678 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Bligaard T et al. Toward Benchmarking in Catalysis Science: Best Practices, Challenges, and Opportunities. ACS Catal. 6, 2590–2602 (2016). [Google Scholar]
- 38.Boder ET & Wittrup KD Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol 15, 553–557 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Bayer EA, Shimon LJW, Shoham Y & Lamed R Cellulosomes-structure and ultrastructure. J. Struct. Biol 124, 221–234 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Kataeva I, Li XL, Chen H, Choi SK & Ljungdahl LG Cloning and sequence analysis of a new cellulase gene encoding CelK, a major cellulosome component of Clostridium thermocellum: evidence for gene duplication and recombination. J. Bacteriol 181, 5288–5295 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Béguin P, Cornet P & Aubert JP Sequence of a cellulase gene of the thermophilic bacterium Clostridium thermocellum. J. Bacteriol 162, 102–105 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reverbel-Leroy C, Pages S, Belaich A, Belaich JP & Tardif C The processive endocellulase CelF, a major component of the Clostridium cellulolyticum cellulosome: purification and characterization of the recombinant form. J. Bacteriol 179, 46–52 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeng W-Y et al. Structural and functional analysis of three β-glucosidases from bacterium Clostridium cellulovorans, fungus Trichoderma reesei and termite Neotermes koshunensis. J. Struct. Biol 173, 46–56 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Bayer EA, Belaich J-P, Shoham Y & Lamed R The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu. Rev. Microbiol 58, 521–554 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Oliveira C, Carvalho V, Domingues L & Gama FM Recombinant CBM-fusion technology - Applications overview. Biotechnol. Adv 33, 358–369 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Davis KA, Abrams B, Iyer SB, Hoffman RA & Bishop JE Determination of CD4 antigen density on cells: role of antibody valency, avidity, clones, and conjugation. Cytometry 33, 197–205 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Gratama JW et al. Flow cytometric quantitation of immunofluorescence intensity: problems and perspectives. European Working Group on Clinical Cell Analysis. Cytometry 33, 166–78 (1998). [DOI] [PubMed] [Google Scholar]
- 48.Smith MR, Tolbert SV & Wen F Protein-Scaffold Directed Nanoscale Assembly of T Cell Ligands: Artificial Antigen Presentation with Defined Valency, Density, and Ratio. ACS Synth. Biol 7, 1629–1639 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Ripley BD Modelling Spatial Patterns. J. R. Stat. Soc. Ser. B 39, 172–92 (1977). [Google Scholar]
- 50.Wang XX & Shusta EV The use of scFv-displaying yeast in mammalian cell surface selections. J. Immunol. Methods 304, 30–42 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Jindou S et al. Cohesin-dockerin interactions within and between Clostridium josui and Clostridium thermocellum: binding selectivity between cognate dockerin and cohesin domains and species specificity. J. Biol. Chem 279, 9867–9874 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Bae J, Kuroda K & Ueda M Proximity effect among cellulose-degrading enzymes displayed on the Saccharomyces cerevisiae cell surface. Appl. Environ. Microbiol 81, 59–66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erickson HP Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 11, 32–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen L, Mulchandani A & Ge X Spore-displayed enzyme cascade with tunable stoichiometry. Biotechnol. Prog 33, 383–389 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Chen L, Holmes M, Schaefer E, Mulchandani A & Ge X Highly active spore biocatalyst by self-assembly of co-expressed anchoring scaffoldin and multimeric enzyme. Biotechnol. Bioeng 115, 557–564 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Lin JL, Zhu J & Wheeldon I Synthetic Protein Scaffolds for Biosynthetic Pathway Colocalization on Lipid Droplet Membranes. ACS Synth. Biol 6, 1534–1544 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Fu J et al. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol 9, 531–536 (2014). [DOI] [PubMed] [Google Scholar]
- 58.García-Alvarez B et al. Molecular architecture and structural transitions of a Clostridium thermocellum mini-cellulosome. J. Mol. Biol 407, 571–580 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Wood TM & Bhat KM Methods for measuring cellulase activities. Methods Enzymol. 160, 87–112 (1988). [Google Scholar]
- 60.Liu Z et al. Engineering of a novel cellulose-adherent cellulolytic Saccharomyces cerevisiae for cellulosic biofuel production. Sci. Rep 6, 24550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.