

Abstract
Microbes produce low-molecular-weight alcohols from sugar, but these metabolites are difficult to separate from water and possess relatively low heating values. A combination of photo-, organo-, and enzyme catalysis is shown here to convert C4 butanol (BuOH) to C8 2-ethylhexenal (2-EH) using only solar energy to drive the process. First, alcohol dehydrogenase (ADH) catalyzed the oxidation of BuOH to butyraldehyde (BA), using NAD+ as a cofactor. To prevent back reaction, NAD+ was regenerated using a platinum-seeded cadmium sulfide (Pt@CdS) photocatalyst. An amine-based organocatalyst then upgraded BA to 2-EH under mild aqueous conditions rather than harsh basic conditions in order to preserve enzyme and photocatalyst stability. The process also simultaneously increased total BuOH conversion. Thus, three disparate types of catalysts synergistically generated C8 products from C4 alcohols under green chemistry conditions of neutral pH, low temperature, and pressure.
Keywords: enzyme catalysis, photocatalysis, organocatalysis, green chemistry, nonequilibrium processes
Graphical Abstract
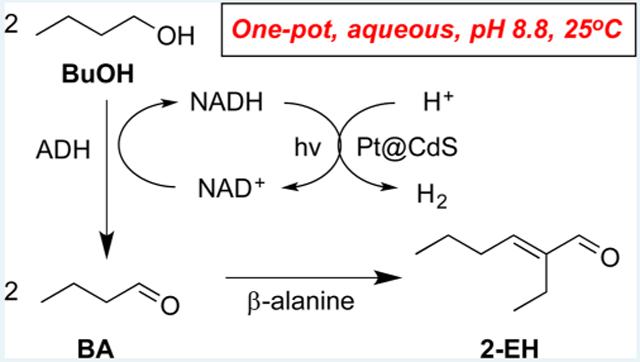
Both environmental pressures and limitations in resources have driven exploration in not only producing carbon-neutral fuels but also the synthesis of commodity chemicals.1-4 Researchers have utilized microbes to convert sugars into carbon-containing products, using either common strains, such as E. coli or Clostridium or by reprogramming cellular machinery to produce biofuels 1-9 and drugs.10-12 However, the separation of such water-soluble chemicals from a complex aqueous medium that contains cells, nutrients, and multiple products is nontrivial and low yielding. For example, distillation of low-molecular-weight alcohols such as ethanol or butanol from water requires fractional, multicolumn distillation, which often approaches or even exceeds the heating value of the BuOH itself.13,14 In situ extraction is of limited utility because common metabolites possess poor partition coefficients in immiscible organic solvents. To help mitigate these challenges and increase the energetic value of cellular fermentation, we report the first example of upgrading a C4 alcohol into the higher-molecular-weight C8 hydrocarbon 2-ethylhexenal (2-EH), using a combination of photo-, organo-, and enzyme catalysis under ambient aqueous conditions with only solar light as an energy source (see Scheme 1).
Scheme 1.

Overall Reaction Scheme for Multicatalytic Conversion of Butanol (BuOH) to n-Butyraldehyde (BA) to 2-Ethylhexenal (2-EH)a
aBuOH oxidation to BA is catalyzed by alcohol dehydrogenase (ADH), converting NAD+ to NADH in the process. The NAD+ cofactor is then regenerated along with H2 by photocatalysis on a Pt@ CdS nanorod. The BA product is then upgraded to 2-EH via aldol condensation catalyzed by β-alanine. Measured turnover frequencies are given next to each catalyst.
This process is inspired by nature’s ability to use multiple catalysts to synthesize higher-order carbonaceous species under completely ambient conditions via the continuous shifting of multiple equilibria. Although each of the three catalysts function efficiently as separate components, when combined, they must operate under the same mild conditions, without mutual interference.
First, the enzyme alcohol dehydrogenase (ADH) was utilized to convert BuOH to n-butyraldehyde (BA), which in the process converts NAD+ to NADH (Scheme 1). Alcohol dehydrogenases are common enzymes that run reversible alcohol oxidations and aldehyde reductions; therefore, to promote BuOH oxidation, the cofactor NAD+ must be regenerated. Semiconductor photocatalysts have been well-studied for water splitting applications and have sufficiently wide bandgaps for photo-oxidizing NADH.15-23 In this work, we show that, upon photoirradiation, Pt-decorated CdS (Pt@ CdS) nanorods (NRs) oxidize NADH back to NAD+ and reduce protons to H2 in the process, thereby pushing enzymatic equilibria toward BA formation. In the absence of NAD+ regeneration by the Pt@CdS, significantly less BA production was observed due to the rapid back reaction by the enzyme to BuOH. In order to promote aldehyde coupling, the previously identified organocatalyst β-alanine was added to the mixture to help catalyze aldol condensation of BA (C4) to 2-ethylhexenal (C8) directly in aqueous buffer at mild pH.24-29 The 2-EH that forms can then be easily extracted from water by phase separation or partitioning into an immiscible organic phase, such as glycerol tributyrate. These results show the promise of upgrading cell metabolites directly in water to higher-order, higher-energy hydrocarbons that can also be easily extracted from the reaction mixture. The H2 produced by this process can also be further utilized in future work for hydrogenating products further to generate fully saturated carbonaceous materials that can also undergo additional aldol reactions.
The first step in building this catalytic cycle was to design a method to sustain continuous endothermic conversion of alcohols to more reactive aldehydes using only light as an energy source. Alcohol dehydrogenase (ADH) has excellent catalytic activity and will convert alcohols to aldehydes without overoxidation. However, if a continuous source of NAD+ cofactor is not supplied as an electron acceptor, the buildup of NADH will result in back reaction to BuOH.30 Because of the energy levels of CdS, platinum-decorated CdS nanocrystals (Pt@CdS) were chosen as photocatalysts for oxidizing NADH to NAD+, while producing H2 from the Pt sites. As H2 can also easily be measured by sampling the headspace by gas chromatography (GC) analysis, monitoring H2 formation over time allows for indirect measurement of ADH oxidation of BuOH to BA without disturbing the reaction.
First, CdS nanorods (NRs) were synthesized using previously published procedures by Robinson et al.31 The resulting nanorods had dimensions of ~6 nm in diameter and an average length of 80 nm (Figure 1). Next, Pt seeds were nucleated on the CdS NRs (Pt@CdS) by photodeposition using the method developed by Dukovic et al.32 to generate NRs that had, on average, 1–2 Pt seeds per CdS; this nanostructure had a first absorption onset peak at 480 nm, which corresponds to a band gap energy of 2.6 eV. In order to stabilize these nanostructures in water, the ligands on the as synthesized Pt@CdS NRs underwent ligand exchange with 2-mercaptoethanol (ME) to impart water solubility to the particles. In previous studies, we showed that, while negatively charged small molecule ligands such as thioglycolic acid reduced H2 production from Pt@CdS, neutral groups such as ME did not.33 After ligand exchange, excess ME was removed by microcentrifuge filtration.
Figure 1.

(a) Ultraviolet–visible light (UV-vis) spectrum of Pt-decorated CdS nanorods. (b) Transmission electron microscopy (TEM) image of Pt-decorated CdS nanorods. Scale bar = 100 nm. (c) Production of H2 over a reaction time of 3 h from 25 nM Pt@CdS (■) in the presence and (▲) in the absence of 3 mM NADH, as well as (●) in the absence of irradiation.
Next, in order to test NAD+ regeneration from Pt@CdS, 25 nM Pt@CdS was mixed with 3 mM NADH and degassed with Ar for 1 h in 1 M phosphate buffer (PB). Then, the Pt@CdS and NADH solutions were photoirradiated with a solar simulator, and at varying time points, aliquots were removed from the gas phase and analyzed via GC to determine H2 production. Because of the valence-band level of the CdS and the oxidation potential of NADH, 7.0 αwmol of H2 were detected in the vapor phase after 60 min and continued to increase up to 9.4 αwmol after 3 h (see Figure 1, as well as Figure S1 in he Supporting Information). The oxidation of NADH by Pt@CdS was also confirmed using NMR spectroscopy (Figure S2 in the Supporting Information). Without photoirradiation, only 2 nmol of H2 was detected. Finally, photoirradiation of 25 nM Pt@CdS in the absence of NADH produced 764 nmol of H2, which may be attributed to water oxidation or consumption of ligands on the nanoparticles.
The conversion of BuOH to BA by ADH in the presence of NAD+ was next determined by measuring NADH formation over time via UV-vis spectroscopy (Figure S3 in the Supporting Information). Since the known equilibrium constant (Keq = 2.7 × 10–4)34 indicates that ADH from S. cerevisiae favors the reduction of BA to BuOH, NAD+ must be regenerated from NADH to push the reaction equilibrium toward BA formation. To study if irradiation of photocatalysts such as CdS would improve BA formation, the amount of BA formed from a mixture of ADH, Pt@CdS, and NAD+ was determined next. For this, BuOH concentration was held at 50 mM to simulate the typical fermentation yields,35,36 and 1 M PB at pH 8.8 was utilized for optimal ADH activity.30 50 mM BuOH, 25 nM Pt@ CdS, and different concentrations of NAD+ (0, 0.3, and 3.0 mM) were first mixed in buffer and the solutions were then degassed with Ar for 1 h. Next, corresponding concentrations of ADH (0, 10, 100 units, respectively) were injected into the degassed solutions and the enzyme reaction was allowed to run for 15 min before photoirradiation. At varying time points, aliquots were removed from the gas phase to measure H2 formation. As shown in Figure 2, H2 was produced steadily with a linear dependence on time and the amount of H2 formed was dependent on the amount of ADH added. This dependence on ADH indicated that the enzymatically generated NADH was acting as an electron source for Pt@CdS to produce H2. At the end of the 3 h reaction, the aqueous solution was next extracted via the addition of CDCl3 that contained tetramethylsilane (TMS), which was added as an internal standard for 1H NMR spectroscopy. Because the actual amount of BA formed by the enzyme in water is difficult to quantify directly because of the equilibrium with its hydrate form,37 the amount of BA was determined by comparison to standards that adjusted for partitioning of BA between the buffer and CDCl3, which accounted for hydrate formation (Figure S4 in the Supporting Information). The amount of BA produced by this study was found to be 0.2, 0.7, and 2.2 mM from 0, 10, and 100 units of ADH (0, 0.3, and 3 αM), respectively, by integration of the 1H NMR spectra (Figure S5 in the Supporting Information). For comparison, the same experiment was run at 100 units ADH without irradiation, which produced 0.5 mM BA after 3 h (Figure 2). This result shows that, in the absence of NAD+ regeneration, the equilibrium shifts toward BuOH, yielding considerably lower amounts of BA.
Figure 2.

(a) Production of H2 from Pt@CdS and (b) comparison of BA generation from BuOH with different concentrations of ADH and NAD+. Includes control measurement in the absence of irradiation. Error bars indicate one standard deviation (SD) from triplicate measurements.
The C4 BA was then converted to C8 2-EH, using aldol condensation conditions that were compatible with the enzyme. In previous research, we showed that certain amino-acid organocatalysts promoted aldol condensation effectively at mild pH and temperature, as compared to common aldol conditions at high pH (>11), temperatures, and organic solvents.28 We found that amine-based organocatalysts such as lysine and β-alanine converted BA to 2-EH with yields of ~65% when initial BA concentrations were held at 70 mM and β-alanine was held at 550 mM, which still maintained low toxicity to bacteria. Since, in this work, the amount of BA produced from ADH and Pt@CdS was found to be low (on a millimolar scale (mM)) and aldol condensation is a second-order reaction, we tested the reactivity of 550 mM β-alanine with 1–5 mM BA at pH 8.8 and 1 M PB. At the end of 3 h, the aqueous media was extracted with TMS/CDCl3 and both BA and 2-EH concentrations were measured using 1H NMR spectra, compared to a calibration curve, as described above (see Figure S6 in the Supporting Information). As shown in Figure 3, 550 mM β-alanine and 1, 2, and 5 mM BA all showed approximately quantitative conversion of BA to 2-EH. ADH activity was also tested in the presence of β-alanine to confirm that no enzyme inhibition was occurring (see Figure S3).
Figure 3.

(a) 2-EH production from β-alanine catalyzed condensation of BA at varying concentrations (b) Partial NMR spectra in CDCl3 of BA and 2-EH standards and the extraction product of a 5 mM BA condensation reaction. Error bars indicate one SD from triplicate measurements.
Based on the individual results of the enzymatic, photocatalysis, and organocatalysis, we next combined all three catalysts to upgrade BuOH to 2-EH in a single reaction. For this, 50 mM BuOH was mixed with 25 nM Pt@CdS, 3 mM NAD+, and 550 mM β-alanine. The solutions were degassed with Ar for 1 h, followed by injecting 100 units ADH into the reaction. The ADH was allowed to react with BuOH for 15 min, followed by photoirradiation with a solar simulator for 3 h. At various time intervals, the headspace was sampled to measure H2 generation. As shown in Figure 4, H2 was continuously generated, peaking at an amount of 6.4 ywmol after 3 h. Next, the reactions were extracted with CDCl3/TMS. The BA and 2-EH peaks were easily distinguished by 1H NMR spectroscopy by observing the aldehyde peaks at 9.77 and 9.38 ppm, respectively, as well as the alkene doublet of 2-EH at 6.45 ppm (Figure S7 in the Supporting Information). As shown in Figures 4a and 4b, 1.5 mM BA and 1.8 mM 2-EH (5.2 mM equiv of BA) were detected in the solutions with β-alanine, while no 2-EH was detected in solutions without β-alanine. The combined BA and 2-EH data indicate that ~71% of the BA formed was converted to 2-EH in the β-alanine added solution. Because BuOH oxidation provides the only source of BA in this reaction, this study showed that the presence of β-alanine improved BuOH conversion by at least 2-fold, because of biasing of the equilibrium through BA depletion.
Figure 4.

(a) H2 production from Pt@CdS over 3 h of irradiation of a mixture of BuOH, ADH, NAD+, and β-alanine. (b) BA and 2-EH yields with and without β-alanine, as measured by 1H NMR. (c) H2 production after 3 h of reaction with simulated ABE feedstock, instead of pure butanol. (d) Product distribution from this mixture. All error bars indicate one SD from triplicate measurements.
To more closely mimic microbial fermentation mixtures, we next formulated a synthetic acetone–butanol–ethanol (ABE) solution in PB and measured product formation in the presence of all three catalysts under solar irradiation. The typical 3:6:1 molar ratio of naturally produced ABE was simulated with 15 mM acetone, 30 mM BuOH, and 5 mM ethanol for a total metabolite concentration of 50 mM. This mixture was combined with ADH and added to degassed solutions of Pt@CdS, NAD+, and β-alanine. During photoirradiation, the vapor phase was sampled continuously to measure H2 production, and after 3 h, the solutions were extracted with CDCl3 and the products were characterized by 1H NMR spectroscopy. The aldehyde peak at 9.81 ppm was used for determination of acetaldehyde concentration (Figure S8 in the Supporting Information). As shown in Figures 4c and 4d, a mixture of products composed of 1.0 mM acetaldehyde (AA), 1.0 mM BA, and 1.5 mM 2-EH was measured. These results show the successful formation of 2-EH from a complex ABE mixture and the possibility of converting ethanol to higher-order products as well.
Generating high-value products from photosynthetically captured carbon is an appealing strategy for sustainable chemistry, but the energy required to recover these products must not negate the carbon neutrality of the products themselves. In this paper, we have demonstrated a one-pot, three-catalyst approach to produce high-value products capable of more-efficient liquid–liquid extraction using only light as an energy source and without sacrificial electron donors. Not only was the C4 butanol, which is a common Clostridium fermentation product, upgraded to C8 2-ethylhexenal, but, in the process, H2 was generated that may be recovered or utilized to further hydrogenate the product for continuous upgrading. This process was run by three types of catalysts—photo-catalysts, organocatalysts, and enzymatic catalysts—running simultaneously under conditions that were favorable for all, reducing the time and cost of product separation between steps. Future work will focus on extending the reaction sequence to both upgrade even smaller metabolites and adding a fourth catalyst for in situ hydrogenation.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Prof. J. Will Medlin for helpful discussions, Dr. Hans Funke for help with gas chromatography, Dr. Liangcan He for help with TEM, and Prof. John Falconer for use of his solar simulator and photocatalysis-GC setup. Research was supported by NIH (No. DP2EB020401) and by the U.S. Dept of Energy (DOE), Office of Science, Basic Energy Sciences (BES) under Award No. DE-SC0006398.
ABBREVIATIONS
- BuOH
n-butanol
- 2-EH
2-ethylhexenal
- ADH
alcohol dehydrogenase
- BA
butyraldehyde
- Pt@CdS
platinum-seeded cadmium sulfide nanorods
- NAD+
β-nicotinamide adenine dinucleotide
- NADH
β-nicotinamide adenine dinucleotide (reduced)
- NRs
nanorods
- ME
2-mercaptoethanol
- PB
phosphate buffer
- TMS
tetramethylsilane
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.6b03213.
Detailed description of experimental procedures, gas chromatograph calibration curve, NMR spectra of extracted products, extraction efficiency data, and enzyme inhibition testing results (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Wargacki AJ; Leonard E; Win MN; Regitsky DD; Santos CNS; Kim PB; Cooper SR; Raisner RM; Herman A; Sivitz AB; Lakshmanaswamy A; Kashiyama Y; Baker D; Yoshikuni Y Science 2012, 335, 308–313. [DOI] [PubMed] [Google Scholar]
- (2).Latif H; Zeidan AA; Nielsen AT; Zengler K Curr. Opin. Biotechnol. 2014, 27, 79–87. [DOI] [PubMed] [Google Scholar]
- (3).Peralta-Yahya PP; Zhang F; del Cardayre SB; Keasling JD Nature 2012, 488, 320–328. [DOI] [PubMed] [Google Scholar]
- (4).Lennen RM; Pfleger BF Curr. Opin. Biotechnol. 2013, 24, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Stephanopoulos G Science 2007, 315, 801–804. [DOI] [PubMed] [Google Scholar]
- (6).Bond-Watts BB; Bellerose RJ; Chang MCY Nat. Chem. Biol. 2011, 7, 222–227. [DOI] [PubMed] [Google Scholar]
- (7).Wu L; Moteki T; Gokhale AA; Flaherty DW; Toste FD Chem. 2016, 1, 32–58. [Google Scholar]
- (8).Deneyer A; Renders T; Van Aelst J; Van den Bosch S; Gabriels D; Sels B Curr. Opin. Chem. Biol. 2015, 29, 40–48. [DOI] [PubMed] [Google Scholar]
- (9).Schwartz TJ; O’Neill BJ; Shanks BH; Dumesic JA ACS Catal. 2014, 4, 2060–2069. [Google Scholar]
- (10).Chang MCY; Keasling JD Nat. Chem. Biol. 2006, 2, 674–681. [DOI] [PubMed] [Google Scholar]
- (11).Ro DK; Paradise EM; Ouellet M; Fisher KJ; Newman KL; Ndungu JM; Ho KA; Eachus RA; Ham TS; Kirby J; Chang MC; Withers ST; Shiba Y; Sarpong R; Keasling JD Nature 2006, 440, 940–943. [DOI] [PubMed] [Google Scholar]
- (12).Ajikumar PK; Tyo K; Carlsen S; Mucha O; Phon TH; Stephanopoulos G Mol. Pharmaceutics 2008, 5, 167–190. [DOI] [PubMed] [Google Scholar]
- (13).Green EM Curr. Opin. Biotechnol. 2011, 22, 337–343. [DOI] [PubMed] [Google Scholar]
- (14).Friedl A FEMS Microbiol. Lett. 2016, 363, DOI: 10.1093/femsle/fnw073. [DOI] [PubMed] [Google Scholar]
- (15).Kalyanasundaram K; Borgarello E; Duonghong D; Grätzel M Angew. Chem., Int. Ed. Engl. 1981, 20, 987–988. [Google Scholar]
- (16).Berr M; Vaneski A; Susha AS; Rodríguez-Fernández J; Döblinger M; Jäckel F; Rogach AL; Feldmann J Appl. Phys. Lett. 2010, 97, 093108. [Google Scholar]
- (17).Simon T; Bouchonville N; Berr MJ; Vaneski A; Adrovic A; Volbers D; Wyrwich R; Döblinger M; Susha AS; Rogach A; Jäckel F; Stolarczyk JK; Feldmann J Nat. Mater. 2014, 13, 1013–1018. [DOI] [PubMed] [Google Scholar]
- (18).Yehezkeli O; de Oliveira DRB; Cha JN Small 2015, 11, 668–674. [DOI] [PubMed] [Google Scholar]
- (19).Walter MG; Warren EL; McKone JR; Boettcher SW; Mi Q; Santori EA; Lewis NS Chem. Rev. 2010, 110, 6446–6473. [DOI] [PubMed] [Google Scholar]
- (20).Liu J; Antonietti M Energy Environ. Sci 2013, 6, 1486–1493. [Google Scholar]
- (21).Amirav L; Alivisatos AP J. Phys. Chem. Lett. 2010, 1, 1051–1054. [Google Scholar]
- (22).Kalisman P; Kauffmann Y; Amirav LJ Mater. Chem. A 2015, 3, 3261–3265. [Google Scholar]
- (23).Kalisman P; Nakibli Y; Amirav L Nano Lett. 2016, 16, 1776–1781. [DOI] [PubMed] [Google Scholar]
- (24).Sirasani G; Tong L; Balskus EP Angew. Chem. Int. Ed. 2014, 53, 7785–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Matthiesen JE; Carraher JM; Vasiliu M; Dixon DA; Tessonnier J-P ACS Sustainable Chem. Eng. 2016, 4, 3575–3585. [Google Scholar]
- (26).Lipshutz BH; Ghorai S Org. Lett. 2012, 14, 422–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).List B; Lerner RA; Barbas CF J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar]
- (28).Domaille DW; Hafenstine GR; Greer MA; Goodwin AP; Cha JN ACS Sustainable Chem. Eng. 2016, 4, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Watanabe Y; Sawada K; Hayashi M Green Chem. 2010, 12, 384. [Google Scholar]
- (30).Racker E J. Biol. Chem. 1950, 184, 313–320. [PubMed] [Google Scholar]
- (31).Robinson RD; Sadtler B; Demchenko DO; Erdonmez CK; Wang L-W; Alivisatos AP Science 2007, 317, 355–358. [DOI] [PubMed] [Google Scholar]
- (32).Dukovic G; Merkle MG; Nelson JH; Hughes SM; Alivisatos AP Adv. Mater. 2008, 20, 4306–4311. [Google Scholar]
- (33).Ma K; Yehezkeli O; Domaille DW; Funke HH; Cha JN Angew. Chem., Int. Ed. 2015, 54, 11490–11494. [DOI] [PubMed] [Google Scholar]
- (34).Leskovac V; Trivic S; Pericin D FEMS Yeast Res. 2002, 2, 481–494. [DOI] [PubMed] [Google Scholar]
- (35).Evans PJ; Wang HY Appl. Environ. Microbiol. 1988, 54, 1662–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Qureshi N; Blaschek HP Biotechnol Prog. 1999, 15, 594–602. [DOI] [PubMed] [Google Scholar]
- (37).Wiberg KB; Morgan KM; Maltz H J. Am. Chem. Soc. 1994, 116, 11067–11077. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.