

Abstract
Purpose:
Describe the clinical and molecular findings of patients with Kabuki syndrome (KS) who present with hypoglycemia due to congenital hyperinsulinism (HI), and assess the incidence of KS in patients with HI.
Methods:
We documented the clinical features and molecular diagnoses of 10 infants with persistent HI and KS via a combination of sequencing and copy-number profiling methodologies. Subsequently, we retrospectively evaluated 100 infants with HI lacking a genetic diagnosis, for causative variants in KS genes.
Results:
Molecular diagnoses of KS were established by identification of pathogenic variants in KMT2D (n = 5) and KDM6A (n = 5). Among the 100 infants with HI of unknown genetic etiology, a KS diagnosis was uncovered in one patient.
Conclusions:
The incidence of HI among patients with KS may be higher than previously reported, and KS may account for as much as 1% of patients diagnosed with HI. As the recognition of dysmorphic features associated with KS is challenging in the neonatal period, we propose KS should be considered in the differential diagnosis of HI. Since HI in patients with KS is well managed medically, a timely recognition of hyperinsulinemic episodes will improve outcomes, and prevent aggravation of the preexisting mild to moderate intellectual disability in KS.
Keywords: Hypoglycemia, hyperinsulinism, Kabuki syndrome, KDM6A, KMT2D
INTRODUCTION
Transient hypoglycemia is a common condition among neonates, typically resolving within the first few days of life. However, persistent neonatal hypoglycemia is a more significant finding that warrants further investigation.1 One of the leading causes of persistent neonatal hypoglycemia is dysregulated insulin secretion by the pancreatic β-cells, termed congenital hyperinsulinism (HI). Affected neonates with the condition have inappropriately high levels of insulin at birth, causing recurrent episodes of hypoglycemia.2 Timely recognition of this condition is challenging, as the symptoms of hypoglycemia can be subtle in newborns.3 Hyperinsulinemic hypoglycemic episodes in neonates, if not recognized, are a leading cause of developmental delays and permanent neurologic damage.4 Pathogenic variants in 11 genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, UCP2, INSR, HNF1A, HNF4A, SLC16A1, PGM1) have been reported as monogenic causes of nonsyndromic HI.5–14 The most common are inactivating pathogenic variants in the genes that encode for the two subunits of the pancreatic β-cell ATP-sensitive potassium channel, ABCC8 and KCNJ11.5,6 Pathogenic variants in these two genes account for the majority of cases of HI, however approximately 50% of patients have an unknown genetic etiology.15
In addition to monogenic nonsyndromic causes of HI, several multisystemic genetic syndromes are associated with HI. The underlying molecular mechanisms that lead to HI in most of these syndromes are unclear. A significant proportion of patients with Beckwith-Wiedemann syndrome will present with HI in their childhood, whereas it has been noted as an infrequent occurrence in patients with Kabuki, Sotos, Costello, or Turner syndromes respectively.16–19
Kabuki syndrome (KS) is a multiple congenital anomaly syndrome characterized by five cardinal manifestations: characteristic dysmorphic facial features, craniofacial/skeletal anomalies, dermatoglyphic abnormalities, mild to moderate intellectual disability and postnatal growth deficiencies.20,21 Patients with KS typically have characteristic dysmorphic facial features, including long palpebral fissures of the eye, eversion of the lower lateral eyelid, long and dense eyelashes, arched and broad eyebrows, depressed nasal tip, thin upper lip vermilion coupled with a full lower lip, and prominent ears. Skeletal and dermatoglyphic abnormalities are also commonly observed, and postnatal growth deficiencies may present as short stature and/or failure to thrive. Other findings that may be observed at a variable rate in KS include persistent hypoglycemia, congenital cardiovascular defects, congenital hypothyroidism, seizures, hypotonia, gastrointestinal problems, feeding difficulties, and increased susceptibility to infections.22 More than 75% of patients with KS harbor pathogenic variants in KMT2D (previously known as MLL2) located at 12q13.13 that typically occur de novo and in rare cases may be inherited (autosomal dominant Kabuki syndrome 1, KS1),23–26 and 5–8% have pathogenic variants in KDM6A located at Xp.11.3 that are most frequently de novo (X-linked dominant Kabuki syndrome 2, KS2).27 Both male and female patients with KDM6A-associated KS have been reported; affected females may have skewed X-inactivation or have haploinsufficiency of the KDM6A protein.27,28 KMT2D and KDM6A encode for proteins that play an important role in the epigenetic regulation of transcriptionally active chromatin by interacting with each other in the protein complex known as ASCOM.29 Pathogenic variants in these two genes cause abnormal chromatin remodeling, resulting in genome-wide effects impacting a range of organ systems. Fifteen to twenty percent of patients with a clinical diagnosis of KS may not carry variants in either KMT2D or KDM6A genes, respectively. In a more recent publication, other genes and loci that potentially contribute to a Kabuki-like syndrome have been discussed.29
It was initially proposed that patients with KS caused by pathogenic variants in the KMT2D gene were more likely to be affected with classic facial dysmorphic features, in addition to a higher association with neonatal feeding problems, kidney abnormalities, skeletal joint dislocations, and palatal malformations.25 A recent study hypothesized that hypoglycemia due to HI may be a more common manifestation associated with KS2.30 Although neonatal or infantile hypoglycemia seem to manifest in approximately 8–10% of patients with KS, the presentation of hypoglycemia secondary to HI in patients with KS was noted to be rare and found to have occurred in only 1 of 313 (0.3%) patients.31 In addition to hyperinsulinism, growth hormone deficiency and adrenal insufficiency have been proposed as possible contributing factors to persistent hypoglycemia in KS.25,30
In this study, we characterized the presenting features of 10 newly identified individuals who came to medical attention due to neonatal hypoglycemia secondary to hyperinsulinism. Genetic evaluation revealed pathogenic variants in either KDM6A or KMT2D in all 10 cases, confirming a molecular diagnosis of KS. After the identification of these 10 cases, we adopted a retrospective approach and sought to determine the incidence of KS in HI patients who were diagnosed in the neonatal period. Thus, we evaluated 100 infants with a clinical diagnosis of HI and an unknown molecular diagnosis, for possible pathogenic variants in KDM6A and KMT2D.
PATIENTS AND METHODS
All patients involved in this study were diagnosed with persistent hyperinsulinism with onset in the neonatal period (Table 1). The patients were diagnosed with HI according to a set of criteria that included examination of the levels of plasma insulin, β-hydroxybutyrate, free fatty acids, and glycemic response to glucagon when plasma glucose was low.32 Of note, patients with HI do not necessarily present with high insulin levels, but they have an inappropriate level of insulin for their glucose measurements. Peripheral blood samples from patients 1–2 and 10 were referred to the molecular diagnostic laboratory at the University of Chicago for genetic evaluation of HI, and patient 3 was diagnosed with HI and referred for evaluation of KS at the University of Chicago. Patient 4 was identified at Boston Children’s Hospital; patients 5–7 were identified at Cook Children’s Medical Center and patients 8 and 9 were identified at the University of Exeter Medical School.
Table 1.
Clinical history of 10 patients who presented with Congenital Hyperinsulinism (CHI)
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Current Age | 8 months | 18 months | 13 months | 14 months | 5 years |
| Gender | Male | Female | Male | Female | Male |
| Ethnicity & Family history | Italian/German/Slovakian, nonconsanguineous | Caucasian parents, maternal depression, history of alcohol abuse/dependence, gastric bypass, epilepsy, tobacco use 1/2 pack per day. Paternal heart condition, hypertrophic cardiomyopathy with defibrillator | Hispanic/Latino, maternal family history of asthma, paternal diabetes age of onset 28 years | Italian/Iranian, nonconsanguineous | Hispanic, nonconsanguineous |
| Age at delivery | 37 5/7 weeks via NSVD to G1P0 28 years-old mother | 38 6/7 weeks via NSVD to G5P0040 36 years-old mother | 39 6/7 weeks via NSVD to G2P2 24-year-old mother | 39 4/7 weeks via NSVD | 39 weeks via NSVD |
| Birth weight | 6 lbs 15 oz | 7 lbs 3 oz | 7 lbs 8 oz | 6 lbs 0.7 oz | 7 lbs 8 oz |
| Perinatal complications | Tachypnea requiring CPAP, hypoglycemia | Increased work of breathing after feeding | Desaturations on DOL3 | IUGR at 32 weeks | – |
| Presentation of hypoglycemia | DOL1 | DOL1 | DOL1 | DOL1 | DOL8 |
| Plasma glucose (mg/dL) Ref: 80-120[Lowest recorded] | 51[29] | 21[21] | 44[12] | 75.6[25.2/34.2] | 47[40] |
| Insulin (uIU/ml = mU/L) | 10.9 | 8.1 | 3.9 | 16.3 | 40 |
| Beta hydroxybutyrate (mmol/L) | 0.04 | NA | 0.15 | NA | 0.04 |
| Free fatty acids (mmol/L) | 0.28 | NA | NA | NA | NA |
| Glycemic response to glucagon (mg/dL) | 70 | 110 (taken during assessment for response to diazoxide) | Response to glucagon noted but no value available | NA | 20 |
| Treatment of hypoglycemia | Diazoxide 4.5 mg/kg/day. Started at 15 mg/kg/day | Diazoxide 5 mg/kg/day | Diazoxide 4 mg/kg/day | Initially with chlorothiazide 0.2 ml twice/day, diazoxide 3 mg/kg three times/ day | Diazoxide until 3 months of age |
| Was normoglycemia achieved? | Yes | Yes | Yes | Yes | Yes |
| Patient 6 | Patient 7 | Patient 8 | Patient 9 | Patient 10 | |
| Current Age | 3 years | 9 months | 18 months | 17 months | 7 months |
| Gender | Female | Male | Female | Female | Male |
| Ethnicity & Family history | Caucasian | Asian | Caucasian, nonconsanguineous | Iranian/Italian, nonconsanguineous | Ecuadorian |
| Age at delivery | 39 weeks via NSVD | 37 weeks via C-section due to nonreassuring fetal heart tones | 39 1/7 weeks via elective C-section for breech presentation | 39 4/7 weeks | 38 weeks via C-section |
| Birth weight | 7 lbs 14 oz | 5 lbs 5 oz | 11 lbs 2 oz | 6 lbs 4 oz | 6 lbs 8 oz |
| Perinatal complications | Polyhydramnios with tobacco use and asthma in the mother | Single umbilical artery, IUGR, and nuchal cord | PPHN requiring sildenafil | None | 15-day NICU stay for respiratory distress colostomy at DOL 1, Surgeries: colostomy, PSARP, g-tube and fundoplication |
| Presentation of hypoglycemia | DOL7 | DOL7 | DOL1 | DOL1 | Likely early DOL but only discovered while coming from Ecuador for ostomy reversal during preoperatory work at 7 months old |
| Plasma glucose (mg/dL) Ref: 80–120[Lowest recorded] | 35[31] | 46[30] | 17 | 25 | 41[33] |
| Insulin (uIU/ml = mU/L) | 8.8 | 1 | 8.5 | 16.3 | 6.9 |
| Beta hydroxybutyrate (mmol/L) | 0.21 | 0.22 | 0.6 | 0.4 | 0.6 |
| Free fatty acids (mmol/L) | 1.82 | NA | 0.3 | 0.6 | 1.3 |
| Glycemic response to glucagon (mg/dL) | 42 | 82 | NA | NA | 80 |
| Treatment of hypoglycemia | Diazoxide (partially responsive), octreotide, pancreatectomy due to diffuse disease, at 3 years old managed with Somatuline, g-button feeds, overnight feeds | Diazoxide, responsive and well controlled. At 4 months of age stopped due to pulmonary hypertension. Solcarb to help maintain blood glucose. Now managed with feeds | Partial pancreatectomy and diazoxide 10 mg/kg/day | Diazoxide 3 mg/kg/ day | Diazoxide 10 mg/kg/ dose three times a day |
| Was normoglycemia achieved? | No | Yes | Yes | Yes | Yes |
Laboratory values for plasma glucose, insulin, beta-hydroxybutyrate, and free fatty acids were recorded during a critical sample collection
NSVD normal spontaneous vaginal delivery, CPAP continuous positive airway pressure, IUGR intrauterine growth retardation, DOL day of life, PPHN persistent newborn pulmonary hypertension, PSARP posterior sagittal anorectoplasty, NICU neonatal intensive care unit, CHI congenital hyperinsulinism
Of the 10 probands reported in this study, pathogenic variants in the KS genes, KMT2D and KDM6A, were identified by either array-comparative genomic hybridization (array-CGH) (n = 2, patients 1 and 2), Sanger sequencing of the KS genes (n = 3, patients 3, 8, and 9), targeted next-generation sequencing (NGS) of a HI gene panel (n = 1, patient 10), or clinical whole-exome sequencing (WES) (n = 4, patients 4–7). Patients 1 and 2 received array-CGH copy-number analysis of the associated HI genes (ABCC8, KCNJ11, HADH, HNF1A, HNF4A, INSR, GCK, GLUD1, SLC16A1, UCP2), and of the KS genes (KDM6A, KMT2D) using a high-resolution, custom-designed, exon-targeted 4×180K array-CGH platform (Agilent Technologies, Santa Clara, CA). Patient 10 was tested on a targeted NGS panel that included 10 nonsyndromic HI genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, UCP2, INSR, HNF1A, HNF4A, SLC16A1) and the KS genes (KDM6A and KDM2D). Written informed consent was obtained to allow for the use of the medical history, genetic testing report, and photographs (if available) as approved by the institutional review board at the different institutions.
Retrospective analysis of HI infants for pathogenic variants in the KS genes
For the purpose of this study, we focused our retrospective analysis on patients who received negative testing results on a panel of 10 genes associated with nonsyndromic HI at the University of Chicago, and were less than 2 years old at the time of testing. Previous genetic analysis consisted of a combined targeted next-generation sequencing (NGS) panel33 and copy-number analysis by array-CGH of 10 HI genes: ABCC8, KCNJ11, HADH, HNF1A, HNF4A, INSR, GCK, GLUD1, SLC16A1, and UCP2. We have recently updated this panel to include genes that are associated with syndromic forms of HI including the KS genes, KDM6A and KMT2D. From our patient database, we selected 100 patients that fit the above criteria and analyzed them on the updated HI panel to specifically identify mutations in the KDM6A and KMT2D genes.
RESULTS
Clinical details and genetic findings
Patient 1 presented with a biochemical profile consistent with HI and tachypnea on his first day of life (DOL1) (Table 1). He was able to achieve normoglycemia through diazoxide therapy. The patient was referred for testing of the original nonsyndromic HI gene panel by NGS and targeted array-CGH at 1 week of age, the results of which were negative. However, a hemizygous deletion of exons 3 through 24 of the KDM6A gene was identified by array-CGH due to the inclusion of these two genes on the array library design. The deletion was found to be in an apparent mosaic state (Fig. 1a). Duplex polymerase chain reaction (PCR) of exons 10 and 19 of the KDM6A gene was performed to confirm the presence of the KDM6A deletion in this patient. This analysis revealed the presence of a faint band corresponding to the KDM6A exons 10 and 19 in the patient’s DNA (Fig. 1b). Sequence analysis performed on the PCR products obtained from the duplex PCR reactions using sequencing primers for KDM6A exons 10 and 19 confirmed the specificity of these products. These collective results provide confirmation of the KDM6A deletion in this patient and also suggest that the deletion likely occurred postfertilization and is thus present in a mosaic state in this individual. The detection of a pathogenic variant in KDM6A provided the patient with a molecular diagnosis of KS. His facial features were unremarkable during the neonatal period, however starting at age 4 months facial features become more characteristic for KS (Figure 1c and Table 2).
Fig. 1. Clinical features and/or molecular characterization of Patients 1-4.
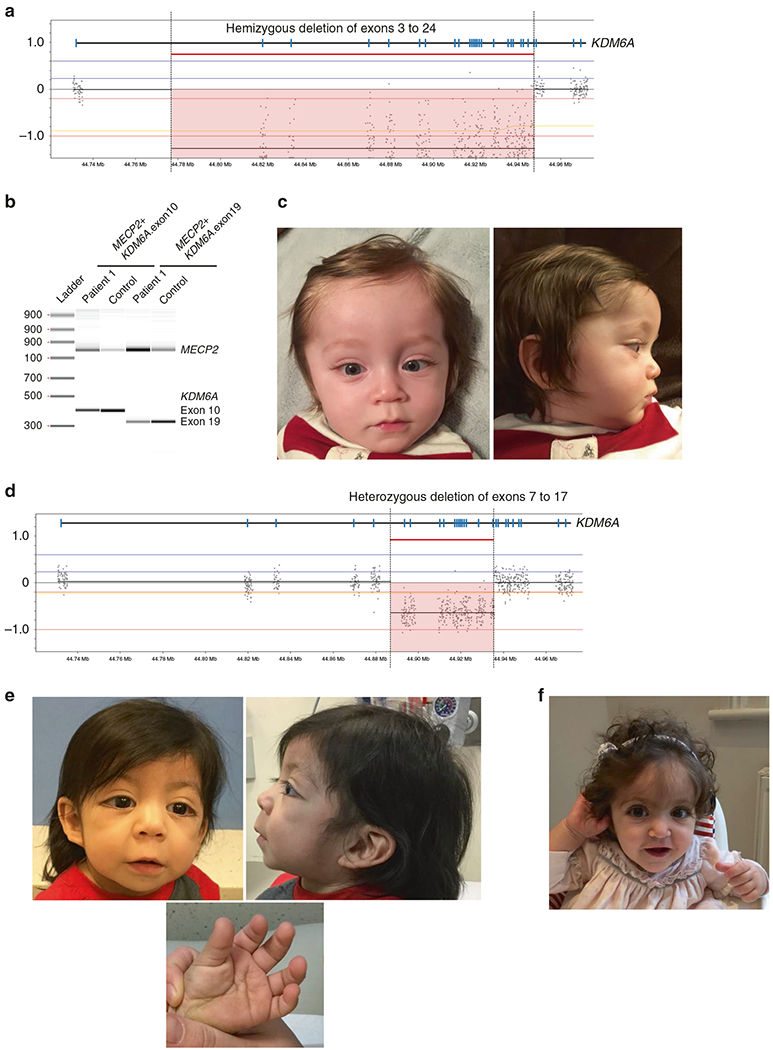
a Array-CGH results for patient 1 showing a hemizygous deletion involving exons 3–24 of KDM6A (arr[hg19] Xp11.3(44818602_44947539)×0). The reference sequence used for KDM6A is NM_021140.3. b Confirmation analysis of the deletion by duplex-PCR of exons 10 and 19 of KDM6A using a control amplicon in the MECP2 gene revealed the presence of a faint band corresponding to the KDM6A exons 10 and 19 in the patient’s DNA, suggesting the likely mosaic nature of the deletion. c Facial features of patient 1 at 9 months of age, showing long palpebral fissures, long eyelashes, and thin vermilion of the upper lip. d Array-CGH results for patient 2 providing evidence of a heterozygous deletion involving exons 7–17 of KDM6A (arr[hg19] Xp11.3(44892185_ 44935785)×1). e Patient 3 at 13 months old with the typical facial appearance and presence of persistent fingertip pads. f Patient 4 at 14 months old with the typical facial appearance of KS
Table 2.
Kabuki syndrome features of the 10 patients
| Features | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|
| Age of KS diagnosis | 2 months | 18 months | 6 months | 6 months | 1 year |
| Genotype | Mosaic hemizygous deletion of KDM6A exons 3–24 arr[hg19]Xp11.3(44818602_ 44947539)×0 | De novo deletion of KDM6A exons 7–17 arr[hg19] Xp11.3 (44892185_ 44935785)×1 | De novo KMT2D c.603_604dup (p. Gly202Alafs*7) | De novo KDM6A c.357C>G (p.Tyr119*) | De novo KMT2D c.11149C>T (p.Gln3717*) |
| Facial | Long palpebral fissures | Long eyelashes, low-set ears, protruding ear lobes | Long palpebral fissures with lateral eversion of eyelid | Long palpebral fissures, eversion of lateral third of lower eyelid, arched/sparse eyebrows, depressed/flat nasal tip, large/dysmorphic ears | Long palpebral fissures, short nasal septum, arching eyebrows |
| Eye | Left homonymous hemianopsia visual field defect, exotropia (since resolved), and bilateral hyperopia | Left-side strabismus | – | – | – |
| Oral | Thin upper lip vermilion | – | Thin upper lip vermilion | – | – |
| Skeletal | Hip dysplasia | – | Sacral dimple with hair tuft | Hip dysplasia | – |
| Hand | – | Fleshy finger pads | 5th finger clinodactyly, tapering fingers, abnormal palmar creases, fleshy finger pads | Fetal fingertip pads | – |
| Neurological | Congenital partial agenesis of the corpus callosum with right paramedian posterior interhemispheric cyst, developmental delay, seizures | Decreased symmetric muscle tone and strength | Developmental delay | Neonatal hypotonia | Hearing loss, developmental delay |
| Cardiovascular | Small atrial septal defect and resolving biventricular hypertrophy | – | – | – | Atrial septal defect |
| Gastrointestinal | Feeding difficulties and gastroesophageal reflux | – | Feeding difficulties | Feeding difficulties and gastroesophageal reflux | Feeding difficulties |
| Urogenital | – | – | Dysplastic left kidney, renal cysts in right | – | – |
| Endocrine | Short stature: height <5th percentile | – | Short stature: 5th percentile in height | Short stature | – |
| Features | Patient 6 | Patient 7 | Patient 8 | Patient 9 | Patient 10 |
| Age of KS diagnosis | 15–18 months | 9 months | 11 months | 7 months | 7 months |
| Genotype | De novo KMT2D c.709del (p. Glu237Serfs*24) | De novo KMT2D c.8366G>A (p.Arg2789Gln) | De novo KDM6A c.2074_2075del (p. Gln692Glyfs*37) | De novo KDM6A c.357C>G, (p.Tyr119*) | De novo KMT2D c.6613delinsAA, (p.Ala2205Asnfs*38) |
| Facial | Long palpebral fissures, short nasal septum, low set ear | Long palpebral fissures | Depressed nasal bridge | Long eyelashes | Arched eyebrows |
| Eye | – | – | Prominent eyes | – | Long eyelashes, blue sclerae |
| Oral | – | – | Small jaw | High arch palate, small jaw | |
| Skeletal | – | Sacral dimple | Left DDH with Pavlik harness | – | |
| Hand | – | – | – | Reduced subcutaneous fat | |
| Neurological | Hearing loss, developmental delay | Developmental delay | Sacral dimple | Truncal hypotonia, mild developmental delay | Developmentally delayed |
| Cardiovascular | – | One umbilical artery | PDA and small PFO with mild concentric LVH | – | Heart murmur, ventricular septal defect |
| Gastrointestinal | Feeding difficulties | Feeding difficulties | Feeding difficulties, cows milk protein intolerance | Feeding difficulties | Imperforate anus, GERD |
| Urogenital | – | – | – | – | Undescended testes |
| Endocrine | – | – | Premature thelarche | Poor weight gain and short stature | Low weight and height |
DDH developmental dysplasia of the hip, PDA patent ductus arteriosus, PFO patent foramen ovale, GERD gastroesophageal reflux disease
The reference sequence used for KDM6A is NM_021140.3. The reference sequence used for KMT2D is NM_003482.3
Patient 2 presented with persistent hypoglycemia due to HI on DOL1. The prenatal care was complicated by maternal alcohol exposure. She had an uncomplicated delivery, but was noted to have muscle hypotonia and labored breathing following feedings. With diazoxide therapy she was able to achieve normoglycemia (Table 1). Similar to patient 1, genetic evaluation for HI was negative for 10 nonsyndromic HI genes analyzed by NGS and array-CGH, however a heterozygous deletion of exons 7–17 in KDM6A was detected through array-CGH (Fig. 1d). The patient was evaluated by an endocrinologist after the diagnosis of KS, noting dysmorphic features consisting of long eyelashes, low-set ears, and persistent finger pads (Table 2).
Patient 3 presented with hypoglycemia on DOL1 (Table 1) that was responsive to diazoxide treatment. At age 6 months, an evaluation by a geneticist noted additional findings including long palpebral fissures with lateral eversion of the lower eyelids, hirsutism, fifth finger clinodactyly, tapering fingers, and abnormal palmar creases (Fig. 1e and Table 2). In addition, renal hypoplasia, an additional renal cyst, and a sacral dimple were present in the patient. Chromosomal microarray analysis was normal. Sanger sequencing of KMT2D was performed due to clinical suspicion of KS and demonstrated a pathogenic two base pair duplication in KMT2D, c.603_604dup (p.Gly202Alafs*7).
Patient 4 presented with hypoglycemia on DOL1 and was treated adequately with diazoxide (Table 1). Her perinatal birth history was complicated by intrauterine growth retardation (IUGR) detected perinatally at 32 weeks gestation. Whole-exome sequencing (WES) performed at 1 year of age revealed a de novo heterozygous truncating variant in KDM6A, c.357C>G (p.Tyr119*). She manifested typical Kabuki facies (Fig. 1f), persistent finger pads, feeding difficulties, and short stature (Table 2).
Patient 5 experienced hypoglycemia shortly after birth on DOL1. He required intraveneous fluids with a high glucose infusion rate of 20 mg/kg/min. He was diagnosed with HI on DOL 8, which was well controlled using diazoxide until 3 months of age (Table 1). WES performed at 1 year of age revealed a de novo truncating variant in KMT2D, c.11149C>T (p.Gln3717*). He manifested typical KS facies with long palpebral fissures, short nasal columella, arching eyebrows, in addition to developmental delay, feeding difficulty, hearing loss and atrial septal defect (Table 2).
Patient 6 had a perinatal history complicated by polyhydramnios and maternal history of asthma and tobacco use. She presented with hypoglycemia on DOL1 and was diagnosed with HI on DOL7. She was partially responsive to diazoxide and adequately managed with continuous overnight feeds. At 4 months of age there was evidence of diffuse disease so she received octreotide and achieved partial response. At 1 year of age she received a 98% pancreatectomy (Table 1). Genetic testing for the original nonsyndromic HI panel was negative. At 15–18 months of age she was evaluated by WES and found to harbor a truncating KMT2D variant, c.709del, (p.Glu237Serfs*24). She has typical KS facies with long palpebral fissures, short nasal columella, low-set ears, in addition to developmental delay, feeding difficulty, and hearing loss. At 2 years and 11 months of age, her hypoglycemia was managed medically with lantreotide, g-button feeds, and overnight feeds. She continued to have recurrent episodes of hypoglycemia (Tables 1 and 2).
Patient 7 had a perinatal history complicated by a single umbilical artery, IUGR and nuchal cord. During the neonatal period he was diagnosed with HI at DOL7, and was responsive to diazoxide therapy. Genetic evaluation for the original nonsyndromic HI panel was negative. At 4 months of age he developed pulmonary hypertension, which was resolved by discontinuing diazoxide therapy. His blood glucose was then maintained by Solcarb and at 9 months of age his glucose levels were well managed by feeds (Table 1). WES was performed at 9 months of age and identified a de novo KMT2D variant, c.8366G>A (p.Arg2789Gln). This sequence change alters the last nucleotide of exon 33 of KMT2D. In silico splicing prediction algorithms (MaxEntScan, Human Splicing Finder, and NNSplice) indicate that this sequence change is predicted to weaken the canonical acceptor splice site, and it is thus likely to result in aberrant splicing of KMT2D mRNA. The patient at 9 months of age was noted to have typical KS facies with long palpebral fissures, in addition to developmental delay, feeding difficulty, and a sacral dimple (Table 2).
Patient 8 had a birth complicated by persistent pulmonary hypertension of the newborn (PPHN), which required the use of sildenafil. During the neonatal period she had feeding difficulties and presented with HI at DOL1. She received a partial pancreatectomy and her condition was well controlled with the use of diazoxide (Table 1). Genetic evaluation for the original nonsyndromic HI panel at University of Exeter (KCNJ11, ABCC8, GLUD1, HADH, GCK, HNF4A, INSR, SLC16A1, TRMT10A, and HNF1A) was negative. At 11 months of age due to the observation of Kabuki-like features, she received genetic testing for KS that revealed a de novo truncating variant in KDM6A, c.2074_2075del (p. Gln692Glyfs*37). She was noted to have dysmorphic facial features that include a depressed nasal bridge, prominent eyes, and a small jaw, and exhibited premature thelarche. She also had skeletal abnormalities, specifically a left developmental dislocation of the hip (DDH) that was treated with a Pavlik harness, in addition to a sacral dimple. Her cardiovascular abnormalities include a patent ductus arteriosus (PDA), and a small patent foramen ovale (PFO) with mild concentric left ventricular hypertrophy (LVH) of the heart (Table 2).
Patient 9 was born with no known perinatal complications. She presented with hypoglycemia due to HI at DOL1 that was well controlled with diazoxide therapy (Table 1). At 7 months of age she was noted to have dysmorphic features consistent with KS. Genetic testing for KDM6A and KMT2D identified a de novo truncating variant in KDM6A, c.357C>G (p. Tyr119*). She was noted to have long eyelashes, high arch palate and a small jaw, with reduced subcutaneous fat on her hands and truncal hypotonia with mild developmental delay. She also exhibited feeding difficulties with poor weight gain and is of short stature (Table 2).
Patient 10 was born with an imperforate anus, undescended testes, and affected with congenital torticollis and a ventricular septal defect (VSD). He was hospitalized for 15 days after birth for respiratory distress and received a colostomy procedure at DOL1. At 7 months of age he was scheduled to receive an ostomy reversal procedure, and was found during preoperative laboratory work-ups to have severe hypoglycemia. He was responsive to diazoxide treatment and endocrinological evaluation indicated a diagnosis of HI (Table 1). Genetic evaluation for 12 HI genes including the 2 additional KS genes identified a heterozygous pathogenic KMT2D variant, c.6613delinsAA (p.Ala2205Asnfs*38). He was noted to have high arched eyebrows, long eyelashes, and blue sclerae. He also exhibited developmental delay, and is of low weight and height (Table 2).
Retrospective analysis of HI infants for pathogenic variants in the KS genes
To determine the frequency of KS in patients who presented with HI, we reevaluated 100 patients who received a diagnosis of HI under 2 years of age and also tested negative in our laboratory using the original 10-gene nonsyndromic HI panel, which did not include the KS genes at the time of testing. We identified one patient out of a hundred, who harbored a pathogenic variant in KMT2D, c.5135del (p.Lys1712Argfs*10), leading to a molecular diagnosis of KS in this patient (95% confidence interval [CI]: 0.0018–0.0545). This child presented at birth with severe and persistent hypoglycemia and was diagnosed with HI shortly after birth. He was responsive to diazoxide and at age 3 has continued successfully on this treatment. He has classical clinical features of KS including long eyelashes, epicanthus, long palpebral fissures with upslant, large slightly low set ears, and fingertip pads. In addition to the HI, he has developmental delays and feeding difficulties.
DISCUSSION
Genetic defects in genes regulating insulin secretion are responsible for monogenic, non-syndromic forms of persistent HI, although metabolic conditions in the newborn or birth complications in a neonate can also cause presentation of HI.16 In addition, a variety of multisystemic syndromes have also been associated with HI. Kabuki syndrome is associated with the presentation of HI (HI incidence 1:50,000),15 but overall it is a rare condition, with an estimated incidence of 1:32,000 in the Japanese population and 1:86,000 in Australia and New Zealand.20,34 A typical findings have been documented and persistent hypoglycemia due to HI has been described as an uncommon occurrence in KS that has been noted in a limited number of patients, although transient neonatal or infantile hypoglycemia had been observed in as much as 8–10% of patients with KS.31 In addition, hypoglycemia in patients with KS has also been attributed to combined pituitary hormone deficiency,24 growth hormone deficiency,25 and adrenal insufficiency, respectively.35,36
In this study we describe 10 unrelated patients with molecularly confirmed KS who presented with persistent HI during the neonatal period. To assess the incidence of KS in a cohort of unselected HI patients, we retrospectively analyzed 100 individuals of less than 2 years of age who received negative tests results on a HI NGS panel of 10 nonsyndromic genes, for the presence of pathogenic variants in KMT2D and KDM6A. This analysis identified an additional patient who harbored a pathogenic variant in KMT2D, leading to a diagnosis of KS. Of the 11 pathogenic variants identified, 5 were truncating variants in KMT2D, 1 was a splice variant in KMT2D, 3 were truncating variants in KDM6A, and 2 were intragenic deletions in KDM6A, with 1 being a mosaic deletion. To the best of our knowledge, all variants with the exception of c.11149C>T (p.Gln37 1 7*)24 are novel to this study. Our results strongly suggests that genetic evaluation for KS genes, including sequencing and deletion duplication analysis should be carried out during testing for genetic etiologies of HI in neonates, as this approach may inadvertently uncover early diagnoses of KS.
The majority of patients with KS harbor pathogenic variants in KMT2D and only 2–8% of cases are due to pathogenic variants in the KDM6A gene.37 In our cohort (n = 11), we observed 5 pathogenic variants (45.5%) in KDM6A, which is evident enrichment over the general frequency of KDM6A pathogenic variants (2–8%) in KS, suggesting that patients with KS harboring pathogenic KDM6A variants (KS2) may have an increased likelihood of presenting with HI as compared with patients with KS who harbor pathogenic variants in KMT2D (KS1). This is an intriguing finding that warrants further investigations in a larger cohort of KS patients, but emphasizes the need for clinicians to more carefully evaluate KS2 patients for hypoglycemic episodes.
Overall, persistent hypoglycemia secondary to HI has been described as an atypical finding in patients with KS.31 In a study by Genevieve et al., the frequency of neonatal hypoglycemia among patients with KS was 21/313 (6.7%); however, HI was suspected in only one patient (0.3%), although it is unclear if a proper assessment of HI was conducted for each of these patients in this study.31 The presentation of HI in patients with KS has also only been documented in single case reports..4,38,39 Our data indicate that the rate of HI among patients with KS may be higher than previously suggested as more patients undergo genetic evaluation. It is possible that only the most severe and persistent HI cases may be documented, as milder or transient forms of HI cases may remain undetected. In our retrospective study on the cohort of 100 HI patients with negative molecular testing at our institution, we identified a single patient with a molecular diagnosis of KS. This indicates that the incidence of KS in neonates with HI without an identifiable genetic cause may be around 1% (95% CI: 0.0018–0.0545), although the number of studied cases is too small to draw any final conclusions.
The 10 patients on whom clinical information was available were documented to have dysmorphic features typical of KS and exhibited developmental delays and feeding difficulties. This is in agreement with previous reports documenting typical phenotypic findings in patients with KS and HI. Patient 1 received a relatively early diagnosis of KS as he was undergoing genetic evaluation for HI. The other patients were diagnosed in the range of 6 months to 18 months in age. This agrees with previous observations that the early diagnosis of KS in the neonatal period is challenging as the typical facial features of KS may not be not clearly evident in young children.37 For a patient with an early diagnosis of KS (especially for KS2), it is important to monitor for possible hypoglycemic episodes that could aggravate their neurological development. Conversely, for a neonate who presents with HI, it is important to consider KS as one of the differential diagnoses. An early diagnosis of KS in the neonatal period can prompt further clinical evaluation for other underlying conditions that may be present in patients with KS, such as congenital heart defects and hip dysplasias that require prompt medical attention. Early diagnosis of KS after an initial presentation of HI also allows the family to receive appropriate prognostic counseling, and could potentially prompt earlier referral to special education or early intervention services to allow maximum developmental achievement for these patients, who are at risk for delays and intellectual disability.
Although the etiology of HI in patients with KS is still unclear, we observe that in the majority of our patients (9/10), the HI was adequately managed with the use of diazoxide, as similarly described in a previous publication.4 Because hypoglycemia in HI of patients with KS can be managed using diazoxide, timely diagnosis of the condition is crucial to improving outcomes for these patients.
In conclusion, the results of this study indicate that patients with KS caused by pathogenic variants in KDM6A (KS2) may be at higher risk to develop HI during the neonatal period, than those with pathogenic variants in KMT2D (KS1). Patients with KS with a history of HI do not appear to have additional specific phenotypic features that would differentiate them from patients without HI. In patients with a diagnosis of KS, awareness of the risk of HI and early recognition of hypoglycemic episodes is important to mitigate the potential neurologically damaging effects of untreated hypoglycemia. The typical features of KS, including the characteristic dysmorphic facial features, may be challenging to recognize in the neonatal period. Thus, we recommend considering KS in the differential diagnosis of persistent HI and also including the comprehensive evaluation (sequencing and deletion/ duplication analysis) of the KS-associated genes KMT2D and KDM6A during genetic testing for HI. Early diagnosis may aid in appropriate follow-up and treatment of the hypoglycemic events in patients with KS, since these patients are likely to respond to the use of diazoxide. With increased genetic testing for KS in patients with HI, we may find that the presentation of HI in patients with KS, though an infrequent occurrence, could be more common than previously postulated.
Supplementary Material
ACKNOWLEDGEMENTS
S.E. is a Wellcome Trust Senior Investigator. We would like to acknowledge the valuable participation of the patients and their families in our study.
Footnotes
DISCLOSURE
The authors declare no conflicts of interest.
REFERENCES
- 1.Sweet CB, Grayson S, Polak M. Management strategies for neonatal hypoglycemia. J Pediatr Pharmacol Ther. 2013;18:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanley CA. Hyperinsulinism in infants and children. Pediatr Clin North Am. 1997;44:363–74. [DOI] [PubMed] [Google Scholar]
- 3.Stanley CA. Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. J Clin Endocrinol Metab. 2016;101:815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Subbarayan A, Hussain K. Hypoglycemia in Kabuki syndrome. Am J Med Genet A. 2014;164A:467–71. [DOI] [PubMed] [Google Scholar]
- 5.Thomas PM, Cote GJ, Wohllk N, et al. The molecular basis for familial persistent hyperinsulinemic hypoglycemia of infancy. Proc Assoc Am Physicians. 1996;108:14–9. [PubMed] [Google Scholar]
- 6.Thomas PM, Cote GJ, Wohllk N, et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. 1995;268:426–9. [DOI] [PubMed] [Google Scholar]
- 7.Stanley CA, Lieu YK, Hsu BY, et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. 1998;338:1352–7. [DOI] [PubMed] [Google Scholar]
- 8.Glaser B, Kesavan P, Heyman M, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338:226–30. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Chen P, Palladino A, et al. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem. 2010;285:31806–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.González-Barroso MM, Giurgea I, Bouillaud F, et al. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS ONE. 2008;3:e3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Højlund K, Hansen T, Lajer M, et al. A novel syndrome of autosomal-dominant hyperinsulinemic hypoglycemia linked to a mutation in the human insulin receptor gene. Diabetes. 2004;53:1592–8. [DOI] [PubMed] [Google Scholar]
- 12.Stanescu DE, Hughes N, Kaplan B, et al. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J Clin Endocrinol Metab. 2012;97:E2026–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otonkoski T, Jiao H, Kaminen-Ahola N, et al. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am J Hum Genet. 2007;81:467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tegtmeyer LC, Rust S, van Scherpenzeel M, et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med. 2014;370:533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnoux JB, Verkarre V, Saint-Martin C, et al. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis. 2011. ;6:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapoor RR, James C, Hussain K. Hyperinsulinism in developmental syndromes. Endocr Dev. 2009;14:95–113. [DOI] [PubMed] [Google Scholar]
- 17.Alexander S, Ramadan D, Alkhayyat H, et al. Costello syndrome and hyperinsulinemic hypoglycemia. Am J Med Genet A. 2005;139:227–30. [DOI] [PubMed] [Google Scholar]
- 18.Cappella M, Graziani V, Pragliola A, et al. Hyperinsulinemic hypoglycaemia in a Turner syndrome with ring (X). Case Rep Pediatr. 2015;2015:561974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toda N, Ihara K, Kojima-Ishii K, et al. Hyperinsulinemic hypoglycemia in Beckwith-Wiedemann, Sotos, and Kabuki syndromes: a nationwide survey in Japan. Am J Med Genet A. 2017;173:360–7. [DOI] [PubMed] [Google Scholar]
- 20.Niikawa N, Kuroki Y, Kajii T, et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. 1988;31:565–89. [DOI] [PubMed] [Google Scholar]
- 21.Adam MP, Hudgins L. Kabuki syndrome: a review. Clin Genet. 2005;67:209–19. [DOI] [PubMed] [Google Scholar]
- 22.Bögershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet. 2013;83:201–11. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Bögershausen N, Alanay Y, et al. A mutation screen in patients with Kabuki syndrome. Hum Genet. 2011;130:715–24. [DOI] [PubMed] [Google Scholar]
- 24.Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banka S, Veeramachaneni R, Reardon W, et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20:381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paulussen AD, Stegmann AP, Blok MJ, et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat. 2011;32:E2018–25. [DOI] [PubMed] [Google Scholar]
- 27.Lederer D, Grisart B, Digilio MC, et al. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 2012;90:119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyake N, Mizuno S, Okamoto N, et al. KDM6A point mutations cause Kabuki syndrome. Hum Mutat. 2013;34:108–10. [DOI] [PubMed] [Google Scholar]
- 29.Lintas C, Persico AM. Unraveling molecular pathways shared by Kabuki and Kabuki-like syndromes. Clin Genet 2017. January 31. [DOI] [PubMed] [Google Scholar]
- 30.Banka S, Lederer D, Benoit V, et al. Novel KDM6A (UTX) mutations and a clinical and molecular review of the X-linked Kabuki syndrome (KS2). Clin Genet. 2015;87:252–8. [DOI] [PubMed] [Google Scholar]
- 31.Geneviève D, Amiel J, Viot G, et al. Atypical findings in Kabuki syndrome: report of 8 patients in a series of 20 and review of the literature. Am J Med Genet A. 2004;129A:64–8. [DOI] [PubMed] [Google Scholar]
- 32.Ferrara C, Patel P, Becker S, et al. Biomarkers of insulin for the diagnosis of hyperinsulinemic hypoglycemia in infants and children. J Pediatr. 2016;168:212–9. [DOI] [PubMed] [Google Scholar]
- 33.Alkorta-Aranburu G, Carmody D, Cheng YW, et al. Phenotypic heterogeneity in monogenic diabetes: the clinical and diagnostic utility of a gene panel-based next-generation sequencing approach. Mol Genet Metab. 2014;113:315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White SM, Thompson EM, Kidd A, et al. Growth, behavior, and clinical findings in 27 patients with Kabuki (Niikawa-Kuroki) syndrome. Am J Med Genet A. 2004;127A:118–27. [DOI] [PubMed] [Google Scholar]
- 35.Ma KH, Chow SN, Yau FT. Isolated adrenocorticotropin deficiency in a child with Kabuki syndrome. J Pediatr Endocrinol Metab. 2005;18:607–9. [DOI] [PubMed] [Google Scholar]
- 36.Takagi M, Ishii T, Torii C, et al. A novel mutation in SOX3 polyalanine tract: a case of Kabuki syndrome with combined pituitary hormone deficiency harboring double mutations in MLL2 and SOX3. Pituitary. 2014;17:569–74. [DOI] [PubMed] [Google Scholar]
- 37.Bögershausen N, Gatinois V, Riehmer V, et al. Mutation update for Kabuki syndrome genes KMT2D and KDM6A and further delineation of X-linked Kabuki syndrome subtype 2. Hum Mutat. 2016;37:847–64. [DOI] [PubMed] [Google Scholar]
- 38.Gohda Y, Oka S, Matsunaga T, et al. Neonatal case of novel KMT2D mutation in Kabuki syndrome with severe hypoglycemia. Pediatr Int. 2015;57:726–8. [DOI] [PubMed] [Google Scholar]
- 39.Gole H, Chuk R, Coman D. Persistent hyperinsulinism in Kabuki syndrome 2: case report and literature review. Clin Pract. 2016; 6:848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.