

Summary:
Ferroptosis is a non-apoptotic mode of regulated cell death that is iron- and lipid peroxidation-dependent. As new mechanistic insight into ferroptotic effectors and how they are regulated in different disease contexts is uncovered, our understanding of the physiological and pathological relevance of this mode of cell death continues to grow. Along these lines, a host of pharmacological modulators of this pathway have been identified, targeting proteins involved in iron homeostasis; the generation and reduction of lipid peroxides; or cystine import and glutathione metabolism. Also, of note, many components of the ferroptosis cascade are target genes of the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2), indicating its critical role in mediating the ferroptotic response. In this review, we discuss the in vitro, in vivo, and clinical evidence of ferroptosis in disease, including a brief discussion of targeting upstream mediators of this cascade, including NRF2, to treat ferroptosis-driven diseases.
Keywords: Ferroptosis, Cell death, Lipid peroxidation, Iron, Ferritinophagy, Cancer, Neurodegeneration, Diabetes, Cardiovascular Disease, NRF2
Graphical Abstract
Introduction:
Cell death is an integral process that occurs throughout all forms of life. Aside from accidental cell death (ACD), which is an unregulated necrotic form of cell death that only occurs after the most extreme of insults, the majority of cell death occurs via some form of regulated cell death (RCD). Different from ACD, RCD pathways involve a precise series of events executed by a designated group of effector molecules. As such, the type of RCD elicited by a particular stressor depends on which subset of effector molecules is activated, which in turn dictates the final mechanism by which the cell dies. The original classification of cell death types, which was based on key differences in the structure and morphology of the dying cell, originated in the early 1970s, and was simply termed type I (apoptotic), type II (autophagic), and type III (necrotic) (Schweichel and Merker, 1973). However, an ever-growing body of evidence has evolved over the past few decades to reveal that while many of the morphological features of cell death fit into these three categories, the stimuli, molecular mechanisms, and key mediators of these cascades vary greatly depending on cell type and context. For this reason, as many as 12 modes of RCD have now been proposed, including intrinsic and extrinsic apoptosis, mitochondrial permeability transition-driven necrosis, necroptosis, pyroptosis, parthanatos, entotic cell death, NETotic cell death, lysosome-dependent and autophagy-dependent cell death, immunogenic cell death, and ferroptosis, each of which is comprised of its own subset of pro-death machinery designed to carry out the initiation, propagation, and execution of a specific cell death response (Galluzzi et al., 2018). Importantly, RCD plays a role throughout the normal development and lifespan of humans, ranging from contributing to organ, digit, and limb development during embryogenesis, to sacrificing overly damaged, defunct, or infected cells during the response to environmental injury/infection, as well as driving the normal aging process (Fuchs and Steller, 2011; Jorgensen et al., 2017; Suzanne and Steller, 2013; Tower, 2015). Accordingly, targeting the different effectors or stimuli that drive the major RCD cascades is a critical area of interest across disease states, including neurodegeneration, diabetes, cardiovascular disease, infection, and cancer, due to the desire to prevent or initiate RCD based on the disease context (Tang et al., 2019).
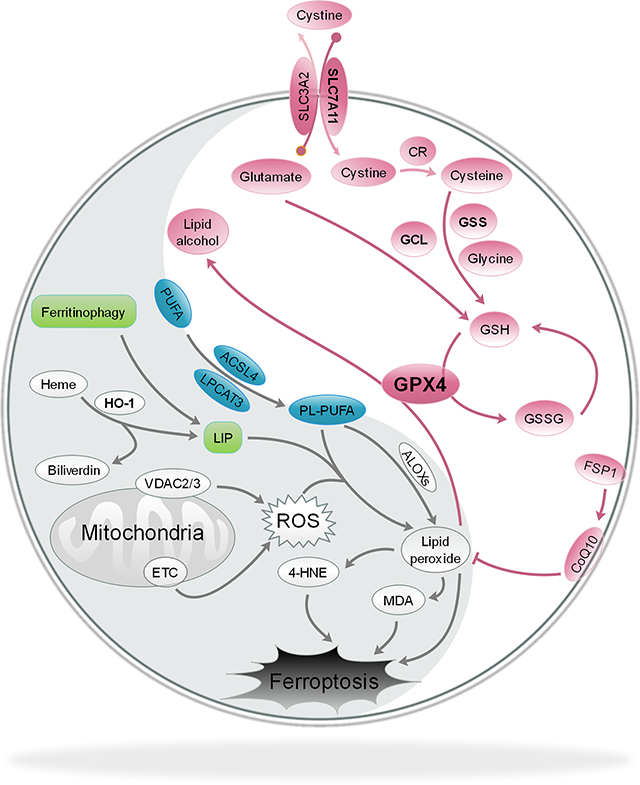
One of the more recently identified modes of RCD is ferroptosis, a lipid peroxidation-dependent form of cell death that often occurs as a result of an accumulation of free iron. The term “ferroptosis” was coined in 2012 by Brent Stockwell’s group following the discovery of two small molecules—eradicator of RAS and ST-expressing cells (Erastin) and RAS selective lethal 3 (RSL3)—that induced a non-apoptotic form of cell death in mutant HRAS-expressing foreskin fibroblasts that could be prevented by iron chelating agents or lipophilic antioxidants (Dixon et al., 2012; Dolma et al., 2003; Yang and Stockwell, 2008). It is important to note, however, that while the term ferroptosis is relatively new, evidence of key components that drive this form of RCD have actually been around since the 1950s (reviewed in (Hirschhorn and Stockwell, 2019)). For example, a study in 1955 by Harry Eagle and colleagues identified that cystine deprivation resulted in a morphologically distinct mode of cell death, which he later went on to show was a result of depleted glutathione (Cohen and Eagle, 1961; Eagle, 1955, 1959). Cystine is imported into the cell via the system xc- cystine/glutamate antiporter, where it is then oxidized to cysteine and utilized for glutathione synthesis. In fact, erastin, mentioned above as one of the recently identified inducers of ferroptosis, is a system xc- inhibitor (Dixon et al., 2012). Further studies in the 1970s confirmed that cystine deprivation and subsequent glutathione depletion were key initiators of cell death both in vitro and in vivo, and also indicated that increased production of lipid-based reactive species played a critical role, as the addition of lipophilic antioxidants (i.e. α-tocopherol) prevented death (Bannai et al., 1977; Mitchell et al., 1973). The production of lipid peroxides occurs as a result of oxidative attack on polyunsaturated fatty acids (PUFAs) by a number of reactive oxygen species (Chen et al.), a process termed lipid peroxidation. These lipid peroxides, which are electrophilic and thus very reactive in nature, have long been known to have numerous cytotoxic effects, and are now well-established drivers of the ferroptotic cascade (Ayala et al., 2014; Conrad et al., 2018).
Critically, the cell has a number of defense systems in place to deal with increased production of lipid peroxides, including glutathione (GSH) and GSH-utilizing enzymes. Among these, the glutathione peroxidases, specifically glutathione peroxidase 4 (GPX4), are phospholipid hydroperoxidases that catalyze the GSH-dependent reduction of membrane lipid peroxides into their less reactive alcohol form. This key enzyme is indispensable during development, as whole-body knockout results in embryonic lethality, and numerous studies have shown that loss of GPX4 expression or function enhances susceptibility to oxidative stress-induced cell death in a variety of contexts (Cardoso et al., 2017; Hangauer et al., 2017; Ingold et al., 2018; Kagan et al., 2017; Yang et al., 2014; Yant et al., 2003). As one might expect, its integral role in mitigating lipid peroxidation makes GPX4 a critical anti-ferroptotic mediator, and a number of ferroptosis inducers, including RSL3, inhibit GPX4 to activate the ferroptotic cascade (Gaschler et al., 2018; Seibt et al., 2019; Yang et al., 2014). Consistently, the loss of GPX4 functionality, along with increased free iron availability and oxidation of PUFAs, were recently proposed to constitute the “Hallmarks of Ferroptosis”, or the three main events associated with the initiation of this pathway (Dixon and Stockwell, 2019). Of note, many of the redox and iron metabolism proteins responsible for preventing these hallmarks from initiating the ferroptotic cascade, i.e. ferritin light and heavy chain (FTL/FTH1), the catalytic and regulatory subunits of glutamate-cysteine ligase (GCLC/GCLM), and even GPX4 itself, are established target genes of nuclear factor erythroid 2-related factor 2 (NRF2), a key transcription factor responsible for maintaining the metabolic, redox, and proteostatic balance of the cell, particularly during stress (Dodson et al., 2019). Under basal conditions, a kelch-like ECH-associated protein 1-cullin3-ring box protein 1 (KEAP1-CUL3-RBX1) E3 ubiquitin ligase complex constantly targets NRF2 for proteasomal degradation. However, during increased oxidative stress, or if KEAP1, CUL3, or NRF2 are mutated, NRF2 can no longer be ubiquitylated and degraded, allowing newly translated NRF2 to translocate to the nucleus and activate transcription of antioxidant response element (ARE)-containing genes, many of which play a key role in preventing the initiation of ferroptosis (Figure 1). Thus, the balance between pro- and anti-ferroptotic effectors plays a critical role in determining whether or not the cell lives or dies under certain stress conditions.
Figure 1: NRF2 regulates genes involved in preventing ferroptosis.

Under homeostatic conditions, NRF2 is ubiquitylated and targeted for proteasomal degradation by a KEAP1-CUL3-RBX1 E3 ubiquitin ligase complex. Under conditions of oxidative/electrophilic stress, or as a result of mutations in KEAP1, CUL3, or NRF2 itself, NRF2 is no longer degraded, allowing for nuclear translocation and activation of antioxidant response element (ARE)-containing genes. NRF2 transcriptional targets are involved in mediating iron/metal metabolism, the catabolism/detoxification of reactive intermediates, and glutathione synthesis and metabolism, all of which play a key role in preventing initiation of ferroptosis.
While ferroptosis was originally discovered in the context of screening for novel therapeutic agents to kill RAS-mutated cancer cells, the relevance of this mode of cell death has expanded to other pathological settings as well. In cancer, initiating ferroptotic cell death is a desirable outcome, particularly in cancer types that have shown resistance to pro-apoptotic chemotherapeutic agents; however, in other pathologies, such as neurodegeneration, type II diabetes, ischemic injury, or cardiovascular disease, ferroptosis is a negative outcome that contributes to injury or enhances disease progression. This duality, much like that of other RCD pathways, has rapidly pushed the field towards understanding not only the pathophysiological, but also the physiological relevance of ferroptosis across an increasing array of disease settings, creating a number of exciting avenues of therapeutic discovery. In this review, we will discuss the key initiators and effectors of the ferroptotic cascade, including relevant models and possible pharmacological approaches for understanding and targeting this mechanism of RCD in disease. Finally, since NRF2 is a key ferroptotic mediator, we will examine if modulating NRF2 activity directly is a viable therapeutic approach for treating ferroptosis-driven diseases.
Key mechanisms of the ferroptotic cascade:
As more and more mechanistic insight into ferroptotic cell death comes to light, three key features continue to be the main pathological pillars underlying this mode of cell death: 1) Iron accumulation, 2) Increased lipid peroxidation, and 3) Inability to efficiently reduce lipid peroxides. Specifically, a breakdown in the systems designed to prevent these maladaptive features from causing excessive damage is what leads to initiation of the ferroptotic cascade. Here, we will discuss these critical features of ferroptosis, including the important pro- and anti-ferroptotic mediators that dictate this pathway.
Excessive iron accumulation
As the name indicates, “ferroptosis” is an iron-dependent form of cell death. Iron, is essential for human existence, playing a role in electron transfer during oxidative phosphorylation, as well as transporting oxygen throughout the blood as part of the heme group in hemoglobin. As shown in Figure 2, iron exists in two forms, ferric (Fe3+) or ferrous (Fe2+), the majority of which is incorporated into proteins or stored by ferritin, with the relatively low remainder of free iron constituting the labile iron pool (LIP). Importantly, the transport, metabolism, and storage of iron is tightly regulated, as increased levels of free, redox-active iron (Fe2+) can increase the production of reactive oxygen species (Chen et al.) via the Fenton reaction, leading to an increased production of lipid peroxides, which will be discussed in more detail below. Extracellular Fe3+ is bound by transferrin (TF) and endocytosed into the cell by transferrin receptor 1 (TFRC). Once internalized, the complex dissociates in the late endosome/lysosome due to the increasingly acidic pH. Newly freed Fe3+ is then reduced to Fe2+ by the metalloreductase STEAP3 and transported out of the endosome/lysosome into the cytosol by the divalent metal transporter (DMT1). In the cytosol, Fe2+ can then be reduced and stored as Fe3+ by ferritin, exported back out of the cell by ferroportin (FPN1/SLC40A1), metabolized and incorporated into iron-containing proteins, or exist as part of the transient LIP (Kakhlon and Cabantchik, 2002)(Figure 2). Proper maintenance of iron homeostasis is integral to cellular survival, which may be part of the reason why excessive iron triggers its own specific form of cell death.
Figure 2: Iron metabolism and ferritinophagy and their regulation by NRF2.

Ferric iron (Fe3+) in the blood is bound and transported by transferrin (Ferretti et al.). Fe3+-TR is bound by transferrin receptor 1 (TFRC) and endocytosed. In the endosome, the acidic pH promotes dissociation of TFRC and Fe3+, which is then reduced to Fe2+ by the metalloreductase STEAP3. Fe2+ is then transported to the cytosol by divalent metal transporter 1 (DMT1), contributing to the labile iron pool (LIP). Fe2+ can be exported out of cells by ferroportin (FPN1), incorporated into iron-containing proteins, or bound and stored by ferritin as Fe3+. Ferritinophagy is the autophagy-dependent degradation of ferritin via nuclear receptor coactivator 4 (NCOA4). Following ferritin degradation, newly freed Fe3+ is reduced to Fe2+ by STEAP3 and exported from the lysosome into the cytosol by DMT1 to again become part of the LIP, where it can subsequently react with ROS via the Fenton reaction. Importantly, a number of key iron metabolism, storage, and transport, as well as key autophagy/ferritinophagy initiation genes are transcriptionally regulated by NRF2.
Sensitivity to ferroptosis is mediated, at least in part, by alterations to key iron processing pathways. For example, the oncogenic RAS cells that were utilized during the initial discovery of ferroptosis inducers have increased iron content due to upregulation of TFRC, and downregulation of FTH1 and FTL, rendering them more sensitive to ferroptosis. Conversely, increased expression of proteins involved in iron-sulfur cluster biogenesis, such as CDGSH iron sulfur domain 1 (CISD1; mitoNEET) and the cysteine desulfurase NFS1, have been shown to desensitize hepatocellular carcinoma (HCC) and non-small cell lung tumor cells to ferroptosis, respectively (Alvarez et al., 2017; Yuan et al., 2016a). Another recently identified contributor to increased intracellular free iron is the autophagic degradation of ferritin by nuclear receptor coactivator 4 (NCOA4), a process termed “ferritinophagy” (Mancias et al., 2014) (Figure 2). While ferritinophagy has been shown to have some physiological roles, for example, in mediating iron release from ferritin for heme synthesis during erythropoiesis (Mancias et al., 2015), it has also been shown to play a critical role in initiating ferroptosis under certain pathological conditions. For example, in HT-1080 fibrosarcoma and PANC1 pancreatic adenocarcinoma cell lines, erastin-induced ferroptosis was inhibited by genetic ablation of NCOA4, whereas overexpression significantly enhanced the ferroptotic phenotype (Hou et al., 2016). Overall, these studies clearly indicate the need for tight regulation of free iron levels, supporting the notion that dysregulation of iron metabolism is a key driver of ferroptosis.
As mentioned above, many of the key iron storage, metabolism, and transport proteins (i.e. FTL, FTH1, SLC40A1, and BLVRA/B) are regulated at the transcriptional level by NRF2 (Kerins and Ooi, 2017). While the bulk of the iron-related NRF2 target genes serve anti-ferroptotic functions, there is some evidence in the literature of NRF2 downstream targets that promote the ferroptotic cascade. Perhaps the most notable example is heme-oxygenase 1 (HMOX1), which catalyzes the cleavage of heme to form biliverdin, carbon monoxide, and ferrous iron (Fe2+). Intriguingly, the pro-ferroptotic effects of withaferin A and BAY11–7085 have both been shown to occur in an NRF2-HMOX1-dependent manner in neuroblastoma and glioblastoma cell lines, respectively (Chang et al., 2018; Hassannia et al., 2018). Furthermore, erastin-induced ferroptotic cell death in HT-1080 cells was also shown to be driven by increased HMOX1 expression (Kwon et al., 2015). Thus, NRF2-mediated HMOX1 expression could be a critical driver of the ferroptotic cascade in certain contexts. Significantly, it has also been proposed that a number of key autophagy initiation proteins contain putative antioxidant response elements (AREs) in their promoters (Pajares et al., 2016). As such, NRF2 regulation of not only iron processing, but also autophagy activation proteins (i.e. ATG5, ATG7, ULK1/2) needed for ferritinophagy to occur, could play a key role in mediating ferritinophagy-dependent activation of ferroptosis in a number of pathologies, including neurodegeneration and cancer. Therefore, NRF2 may actually serve either an anti- or pro-ferroptotic role depending on the pathological context.
Increased lipid peroxidation
Oxidative attack on PUFAs in the plasma membrane, as well as organellar membranes, is a significant source of lipid peroxides. While membrane lipid peroxidation can occur non-enzymatically as a result of excessive accumulation of iron and reactive species, enzymatic peroxidation of lipids, i.e. by lipoxygenases (ALOX 5/12/15), can also propagate the formation of lipid peroxides (Dobrian et al., 2011). Accordingly, increased expression of ALOX15 is often correlated with increased ferroptotic cell death (Yang et al., 2016). ALOXs generate reactive lipids via metabolism of membrane PUFA-containing phospholipids, which are generated by acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). Importantly, ACLS4 has been shown to be a critical mediator of the pro-ferroptotic cascade, as pharmacological inhibition or genetic ablation prevents initiation of ferroptosis by a number of ferroptosis inducers (Doll et al., 2017; Yuan et al., 2016b). Regardless of the mechanism, membrane lipid peroxidation can result in significant damage to cellular membrane structure, and its end-products, 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), are also highly reactive and have been shown to form adducts with proteins and DNA resulting in significant cytotoxic effects (Ayala et al., 2014; Barrera et al., 2015). There are a number of potent initiators of lipid peroxidation, including ROS (i.e. hydroxyl and hydroperoxyl radicals), reactive nitrogen species (peroxynitrite), as well as the actual end products of lipid peroxidation themselves (4-HNE and MDA) (Yin et al., 2011). Discussed briefly above, supplementation with lipophilic antioxidants—vitamin E or coenzyme Q10 (CoQ10/ubiquinone) and their derivatives—has been shown to prevent lipid peroxidation and thus mitigate ferroptosis in certain contexts (Angeli et al., 2017; Shimada et al., 2016). However, a number of endogenous defenses are also in place to prevent excessive accumulation of reactive species, including lipid peroxides. For instance, a newly discovered anti-ferroptotic gene, ferroptosis suppressor 1 (AIFM2/FSP1) was just recently identified as a critical regenerator of endogenous CoQ10 in the membrane, with inhibition of FSP1 resulting in a robust activation of the ferroptotic cascade in a GPX4-null setting (Bersuker et al., 2019; Doll et al., 2019). Intriguingly, Chip-Seq analysis to identify NRF2-regulated genes in sulforaphane-treated cells indicated that NRF2 can bind to the promoter of AIFM2, indicating that this regulator of the ferroptotic cascade may also be an NRF2 target gene (Chorley et al., 2012). These findings suggest that there is some built in redundancy to ensure the cell is adequately protected from membrane lipid peroxidation. Thus, while an accumulation of iron and subsequent iron-dependent Fenton chemistry are significant contributors to increased lipid peroxidation in a number of pathogenic settings, deficiencies in the numerous defense systems put in place to prevent excessive lipid peroxidation from occurring also plays a key role. In the following section, we will discuss the key redox-based anti-ferroptotic mediators, and how their loss of function is associated with activation of the ferroptosis pathway.
Inability to effectively reduce lipid peroxides
Perhaps the most widely studied anti-ferroptotic mediator, at least regarding the prevention of membrane lipid peroxidation, is the phospholipid hydroperoxidase GPX4. GPX4 deficiency, as a result of genetic ablation, mutation, or pharmacological inhibition, is a well-established initiator of ferroptosis, and the role of this protein in mediating this cascade across a variety of pathophysiological contexts has been reviewed extensively elsewhere (Angeli et al., 2017; Maiorino et al., 2018; Seibt et al., 2019). Intriguingly, a number of cancer cells have actually been shown to survive and proliferate following genetic deletion of GPX4 (Viswanathan et al., 2017), suggesting that GPX4 inhibition alone may be insufficient to kill certain cancer cell types. An important regulator of not only GPX4 expression, but also other important antioxidant defense system components is NRF2. Mentioned previously, NRF2 target genes regulate not only key aspects of iron metabolism, but also the catabolism of xenobiotics and reactive aldehydes, as well as glutathione synthesis, NADPH regeneration, and GSH-dependent reduction of thioredoxins and peroxiredoxins, making it a multifaceted regulator of the anti-ferroptotic response (Dodson et al., 2019). Accordingly, deficiencies in not only GPX4, but also a host of other NRF2 targets, including SLC7A11, FTH1/FTL, SLC40A1, and PRDX6, among others, can lead to initiation of ferroptosis, or enhanced susceptibility of cancer cells to pro-ferroptotic agents (Hassannia et al., 2019). The important role that NRF2 and its downstream targets play in preventing ferroptosis make this pathway an attractive therapeutic target for treating ferroptosis-driven diseases, which will be discussed in detail below.
Finally, while an increase in iron-dependent lipid peroxidation and a decrease in the defense systems designed to keep excessive iron and lipid peroxide formation at bay are known initiators of the ferroptotic cascade, very little is understood regarding how lipid peroxides are actually involved in the final stages of ferroptotic death. While a “ballooning” phenotype has been demonstrated as the final morphological feature of cells undergoing ferroptosis, this morphology is not observed in all cell types that demonstrate other key features of ferroptotic death. Thus, future studies to determine how lipid peroxides elicit the ferroptotic phenotype, as well as the identification of downstream effectors that mediate the final death phenotype, are still needed. Identification of specific effectors of the ferroptotic cascade is of particular importance, as there is significant overlap between ferroptosis and other modes of RCD. For example, the production of ROS and lipid peroxides is a common feature of apoptosis, necroptosis, pyroptosis, and autophagy-dependent cell death, among others (Tang et al., 2019). Mitochondrial changes, including ROS production, release of pro-death factors, and morphological alterations, are prevalent features of not only ferroptosis, but apoptosis, necroptosis, and pyroptosis as well (Bock and Tait, 2020). The lysosome, another critical regulator of cellular function, also plays a central role in mediating ferroptosis, necroptosis, pyroptosis, autophagy-dependent cell death, and lysosome-dependent cell death following certain stimuli (Ballabio and Bonifacino, 2020). In fact, along with ferritinophagy discussed above, the lysosome, autophagy, and ferroptosis are closely linked on multiple levels, as numerous effectors of macroautophagy (i.e. Beclin 1), lipophagy (i.e. Rab7), and chaperone-mediated autophagy (i.e. LAMP2A) have all been linked to initiation of ferroptotic cell death (Zhou et al., 2019). Finally, many transcriptional regulators of the ferroptotic machinery, such as NRF2 and p53, have also been shown to regulate key components of other RCD pathways, such as apoptosis (Aubrey et al., 2018; Stepkowski and Kruszewski, 2011). As such, a clear delineation of the contribution of ferroptotic cell death, versus that of other related modes of RCD, should be considered when investigating the mechanisms underlying a cell’s demise. Of course, determining the relevance of ferroptosis in specific pathological settings will be facilitated by the continued discovery of specific stimuli, morphological features, and downstream effectors associated with the ferroptotic cascade.
The relevance of ferroptosis in human health and disease:
As mentioned above, ferroptotic cell death has been demonstrated to play a role in a variety of diseases, serving a beneficial or detrimental role depending on the pathophysiological context. On the one hand, ferroptosis, much like other forms of RCD, is thought to play an integral role in removing overly damaged or dysfunctional cells to preserve the functionality of the stressed cell or organ system. As such, this cascade may play an essential role in tumor suppression by preventing pro-tumorigenic maladaptive changes; and, targeting chemoresistant tumor types with ferroptosis inducers remains an active and important area of interest. On the other hand, in pathological settings where cell death exacerbates disease onset and progression, such as cardiovascular disease, diabetes, and neurodegeneration, inhibition of ferroptosis is desired in order to mitigate excessive cell loss and prevent irreparable damage (Figure 3). In this section, we will review the role of ferroptosis in different disease states and discuss the key models and pharmacological modulators of ferroptosis that are being utilized to better understand and target this mode of RCD in disease.
Figure 3: Ferroptosis and its role in disease.

Ferroptosis has been shown to play a role in neurodegeneration, cardiovascular disease, diabetes, and cancer, among others. In cancer, the therapeutic goal is to activate ferroptosis to kill tumor cells that are resistant to other modes of cell death. In neurodegeneration, type II diabetes, and cardiovascular disease, the therapeutic goal is to prevent ferroptosis-induced cell loss to slow onset or mitigate disease progression. Inducers and inhibitors of ferroptosis that have been shown to affect experimental models of each disease in vitro and in vivo, or patients in clinic, are also included.
Ferroptosis in cancer
Induction of ferroptosis to kill resistant cancer cell types is a promising strategy for both neoadjuvant and adjuvant chemotherapy. The majority of ferroptosis-inducing compounds discovered to date target key caretakers of redox homeostasis (i.e. system xc- and GPX4), discussed in detail above, to collapse the existing redox balance and initiate the ferroptotic cascade. This is of particular importance when considering chemoresistance, as many cancer cell types are known to be resistant to ROS-inducing chemotherapeutic agents, in part through increased expression of key antioxidant proteins, making compounds that modulate these proteins attractive therapeutically. For example, knockdown of GPX4 results in increased lipid peroxidation and subsequent ferroptotic cell death in renal cell carcinoma cells, with deferoxamine (DFO; iron chelator) or vitamin E (lipophilic antioxidant) treatment preventing development of the ferroptotic phenotype (Yang et al., 2014). Similarly, genetic or pharmacological inhibition of SLC7A11 has also been shown to induce ferroptosis in cisplatin-resistant head and neck cancer cell lines, indicating that loss of function of these key anti-ferroptotic mediators enhances cancer cell death (Roh et al., 2016). As mentioned previously, both GPX4 and SLC7A11 are established NRF2 target genes, and increased expression of these genes is associated with a poorer prognosis in endometrial, ovarian, pancreatic, liver, lung, and renal cancers, among others (Daher et al., 2019; Kerins et al., 2018; Lim et al., 2019; Romero et al., 2017; Sui et al., 2019). Aberrant expression of NRF2 itself is also associated with poorer prognosis in a number of cancer types (Kitamura and Motohashi, 2018), and a correlation between high NRF2 and system xc- levels has been associated with decreased ferroptosis and increased malignancy in the progression of advance stage glioblastomas (Singer et al., 2015). It is already well-established that NRF2, as well as many of its downstream target genes, plays a key role in dictating cancer initiation and progression (Rojo de la Vega et al., 2018). These recent studies also suggest that the NRF2 pathway can modulate cancer cell sensitivity to ferroptosis inducers, as many of the currently identified pharmacological agents and adjuvant therapies that induce ferroptosis target proteins in the NRF2 signaling pathway (i.e. system xc-, GPX4, and GCL). It has also been shown that p53, a critical cell cycle regulator which when mutated enhances tumor progression, may actually carry out its tumor suppressive functions by enhancing susceptibility to ferroptosis via reducing the expression of SLC7A11 (Jiang et al., 2015). Similarly, the tumor suppressor BRCA-1-associated protein 1 (BAP1), a nuclear deubiquitinase that mediates histone deubiquitination, has also been shown to suppress SLC7A11 expression and inhibit tumor development via induction of ferroptosis (Zhang et al., 2019a; Zhang et al., 2018; Zhang et al., 2019b). Thus, targeting key oncogenic transcription factors, such as NRF2, BAP1, and mutant p53, as well as their anti-ferroptotic downstream targets, to initiate ferroptosis in chemoresistant cell types remains a viable therapeutic approach.
There are several pharmacological modulators that can induce ferroptotic cell death both in vitro and in vivo, including RSL3, erastin, sorafenib, artemisinin (Ferretti et al.), dihydroartemisinin (DHA), sulfasalazine (SAS), buthionine sulfoximine (BSO), and ferroptosis inducing 56 (FIN56) (Dixon et al., 2012; Dixon et al., 2014; Eling et al., 2015; Lin et al., 2016; Shin et al., 2018; Stockwell et al., 2017; Sun et al., 2016b; Yang et al., 2014). As discussed earlier, RSL3 and erastin were identified as being selectively lethal against genetically engineered oncogenic mutant HRAS fibroblasts (Yang and Stockwell, 2008). Mechanistically, RSL3 binds to and inactivates GPX4, resulting in an accumulation of reactive lipid species and activation of the ferroptotic cascade (Weiwer et al., 2012; Yang et al., 2014). These studies suggest that there is a significant relationship between GPX4 function, ferroptosis, and oncogenic RAS activity, however, the mechanisms behind this relationship needs further elucidation. In acute lymphoblastic leukemia cells, RSL3 cooperates with the second mitochondrial-derived activator of caspases (Smac) mimetic BV6 to induce ROS-dependent cell death in an apoptosis-independent manner. Furthermore, genetic or pharmacological inhibition of lipid peroxidation by GPX4 overexpression, or treatment with ferrostatin (Fer-1) or DFO, respectively, significantly decreased RSL3/BV6-induced cell death (Dachert et al., 2016). Together, these data suggest that RSL3-induced ferroptosis could be a viable chemotherapeutic option for certain cancers, particularly those with high GPX4 or iron storage/export (i.e. FTL/FTH1, TF, and FPN1) protein expression, although combination with other chemotherapies may still be necessary.
Erastin, a piperazinyl-quinazolinone compound, depletes glutathione by inhibiting cystine import via system xc- (Dixon et al., 2014; Yang et al., 2014). Much of the early work characterizing erastin-dependent pro-ferroptotic mechanisms was done in HT-1080 fibrosarcoma cells, with ferroptosis induction by this compound later being confirmed in a number of different cancer cell lines (Dixon et al., 2012; Dixon et al., 2014). Cystine import is critical for maintaining the intracellular cysteine pool, and many cancer cells are considered to be “cystine-addicted” (Ma et al., 2015; Sweiry et al., 1995), as glutathione depletion resulting from cystine/cysteine deprivation can eventually lead to excessive lipid peroxidation and ferroptotic cell death. Similar to RSL3, erastin/BV6 cotreatment induced ferroptotic cell death in acute lymphoblastic leukemia cells. However, unlike the RSL3/BV6 combination, neither upregulation of GPX4 nor treatment with Fer-1 were able to rescue cells from erastin/BV6-induced death, despite reducing the levels of lipid peroxides (Dachert et al., 2016). Treatment with erastin also sensitized normal and cisplatin-resistant A2780 ovarian carcinoma cells to cisplatin treatment (Sato et al., 2018). Potent anti-tumor effects have also been observed with sulfasalazine (SAS), another established inhibitor of system xc-, which was developed 80 years ago as an anti-inflammatory drug (Guan et al., 2009). In vivo, SAS inhibits tumor growth by inducing cystine depletion and subsequent ferroptotic death in a prostate cancer xenograft model (Doxsee et al., 2007). Additionally, SAS alleviated glioma-induced seizures in mice, while in human glioma cell lines, SAS potentiated the chemotherapeutic effect of the well-established drug temozolomide (TMZ) (Buckingham et al., 2011; Robert et al., 2015). SAS has also been shown to sensitize gliomas to radiotherapy (Sleire et al., 2015). These studies indicate that an adjuvant therapeutic approach utilizing erastin or SAS in combination with chemo- or radiotherapy could be a successful approach for treating ovarian carcinomas, leukemias, gliomas, and possibly other cancers as well.
Another promising pro-ferroptotic chemotherapeutic drug candidate is caspase 3/7-independent lethal 56 (CIL56), which was shown to induce cell death in the absence of activation of caspases 3 and 7. CIL56, FIN56 (ferroptosis inducing 56), and many of their analogs were found to selectively target oncogenic RAS cancer cells and induce ferroptosis via promoting the degradation of GPX4 and reducing the abundance of intracellular CoQ10 (antioxidant) (Shimada et al., 2016). This mechanism differs from RSL3, which inhibits the catalytic activity of GPX4. Although the exact mechanism of degradation of GPX4 is unknown, the enzymatic activity of acetyl-CoA carboxylase (Ferretti et al.) is required, as inhibition of ACC via 5-(tetradecyloxy)-2-furoic acid (TOFA) prevented GPX4 degradation (Shimada et al., 2016). This same study also demonstrated that FIN56 can bind to and activate squalene synthase, an enzyme involved in isoprenoid biosynthesis, that potently induced ferroptosis independently of GPX4 degradation. Overall, these findings indicate that FIN56, much like its predecessors, could be a useful drug in targeting cancers, including those with oncogenic RAS mutations.
Targeting ferroptosis in neurodegeneration and neuronal injury
Although the ferroptotic phenotype was originally identified in cancer cells, recent efforts have also expanded into detecting and measuring the role of ferroptosis in other diseases, including neuronal injury and neurodegeneration (Morris et al., 2018). The association between oxidative stress, lipid peroxidation, mitochondrial dysfunction, iron dysregulation, and neurodegeneration has long been appreciated (Guo et al., 2013; Lee et al., 2006). Elevated levels of iron and increased lipid peroxidation are consistently detected in the post-mortem brain tissue and cerebral spinal fluid of Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, motor neuron disease, and multiple sclerosis patients (Adibhatla and Hatcher, 2010; Bradley-Whitman and Lovell, 2015; Shichiri, 2014; Sugiyama and Sun, 2014). Iron is an essential element for normal brain development and cognitive function, and as mentioned above, iron transport, metabolism, and storage are tightly regulated, as a prolonged increase in the pool of redox-active iron can have numerous deleterious effects (Agrawal et al., 2017). During normal aging, iron progressively accumulates in specific neuronal and glial cell subtypes that constitute certain regions of the brain (Belaidi and Bush, 2016). Not surprisingly, brain regions with the highest levels of iron accumulation are also the ones associated with the onset and progression of a number of different neurological diseases. Accordingly, an accumulation of free iron has been shown to cause neuronal cell death via a number of different RCD pathways, including apoptosis, necroptosis, and ferroptosis (Dixon and Stockwell, 2014). Thus, a great deal of effort has been made towards preventing or reducing iron accumulation to treat neurodegenerative diseases. Along these lines, treatment with the iron chelators DFO and deferiprone showed some efficacy in clinical trials for both Alzheimer’s and Parkinson’s disease, mainly resulting in improvements to motor function, indicating that chelation of iron may improve certain symptoms associated with these diseases (Crapper McLachlan et al., 1991; Devos et al., 2014).
Aside from excessive accumulation of free iron contributing to age-related neurodegenerative disorders, other neuronal initiators of the ferroptotic cascade have also been determined. For example, neuronal cells were demonstrated to be sensitive to both RSL3 and erastin, which fits neuronal reliance on a heightened reductive state to prevent ROS-induced damage. Additionally, the ferroptosis inhibitors DFO, Fer-1, and liproxstatin-1 were able to prevent mitochondrial damage in neurons associated with RSL3-dependent inhibition of GPX4 and generation of damaging free radicals (Jelinek et al., 2018). Neuron-specific deletion of GPX4 resulted in selective loss of CA3 hippocampal interneurons and a neurodegenerative phenotype in the hippocampus (Seiler et al., 2008). Several other neurodegenerative disorders including dementia have clinicopathological features that are consistent with ferroptosis; however, the mechanisms underlying ferroptosis in these disease settings, as well as the relevance in enhancing disease progression, still need further clarification (Belaidi and Bush, 2016; Brettschneider et al., 2015; Chiang et al., 2017; Williams et al., 2006). Outside the realm of neurodegeneration, ferroptotic cell death has also been shown to play a role in other neurological disorders and neuronal injury as well. Signs of ferroptosis have been identified in neuronal cell death during cerebral ischemic injury (Speer et al., 2013) and glutamate-induced neurotoxicity in organotypic hippocampal slice cultures (Dixon et al., 2012). Similarly, two classes of ferroptosis inhibitors, ferrostatins and liproxstatins, were shown to ameliorate the damage associated with traumatic and hemorrhagic brain injury (Chen et al., 2015; Gascon et al., 2016; Zille et al., 2017). Overall, these studies indicate that ferroptosis drives the tissue damage and neuronal loss associated with a number of neuronal pathologies, and that targeting relevant ferroptotic effectors for therapeutic intervention is a promising approach for the treatment of neuronal injury and neurodegenerative disease.
Ferroptosis as a driver of cardiovascular disease
Similar to its detrimental role in neurodegeneration, ferroptosis has also been linked to the exacerbation of cardiovascular disease. Iron overload (IO), which typically occurs as a result of excess iron in the body due to an inherited genetic disorder (i.e. hemochromatosis) or receiving multiple blood transfusions, plays a pathogenic role in cardiotoxicity and has been identified in several cardiovascular conditions in both animal models and human patients (Gujja et al., 2010). IO cardiomyopathy is the result of excessive iron accumulation in the myocardium, which leads to damage and cardiac dysfunction (Kremastinos and Farmakis, 2011). Additionally, it has been suggested that abnormal myocardial iron status plays a role in diabetic patients with heart failure, and that chelation therapy could improve the prognosis of coronary microvascular adaptation (Nitenberg et al., 2002). Iron chelators DFO, deferiprone, and deferasirox, have also been shown to negate the pathogenic effects of IO due to transfusions and pressure overload, suggesting a possible therapeutic approach for managing IO-driven heart failure (Brittenham, 2011; Liu et al., 2018). While these studies indicate that IO-linked myocardial damage could result in ferroptosis, causing a subsequent loss of cardiac function, further clinical evidence that ferroptosis actually occurs in these conditions is still needed. However, there is a great deal of experimental evidence to support iron-induced myocardial ferroptosis. When treated with the anthracycline drug doxorubicin (DOX), cardiomyocytes exhibited key features of ferroptotic cell death, including iron accumulation and lipid peroxidation, which could be reversed by Fer-1 (Fang et al., 2019). Similarly, treatment with erastin, RSL3, or ferric iron led to an increase in both intracellular free iron levels and iron-mediated oxidative stress in cardiomyocytes, which again could be reversed by cotreatment with Fer-1, indicating that iron-induced oxidative stress is a key trigger of ferroptotic death in the heart (Lapenna et al., 2018). Due to the well-established relationship between iron and ferroptosis, these findings suggest that ferroptosis may be a key driver of several cardiovascular pathologies, and that proper management of iron levels, or chelation during situations where excess iron is present, could be an effective preventative measure and treatment strategy for cardiovascular diseases.
The possible role of ferroptosis in diabetes
It is well known that increased oxidative stress in β-islet cells of the pancreas can lead to cell death and loss of pancreatic function during type II diabetes (Gerber and Rutter, 2017). However, very few studies have investigated whether or not increases in iron and the production of lipid peroxides can lead to ferroptotic death of β-islet cells. Recently, it was shown that human β-islet cells are susceptible to erastin and RSL3-induced ferroptosis, which could be reversed by Fer-1 (Bruni et al., 2018). Interestingly, this same study found that treatment of β-islet cells with erastin, Fer-1, or both compounds in combination, prior to transplantation into an immunodeficient recipient mouse did not diminish their functionality, although the authors state this could be due to limitations in their model (Bruni et al., 2018). It is worthwhile to note that despite the limitations of this study, inhibiting ferroptosis may prove to be a successful strategy for improving β-islet cell transplantation for type I diabetic patients, although further evidence indicating that ferroptosis is affecting transplantation and that anti-ferroptotic agents such as DFO and Fer-1 have a protective effect, are needed prior to employing this strategy. Notably, a close relationship between iron, β-cell function, and insulin secretion also exists, and IO, a known inducer of the ferroptotic cascade discussed earlier, has been linked to increased diabetic complications (i.e. chronic inflammation and insulin resistance), suggesting that ferroptotic death could be playing a role. Much like β-cell transplantation, direct evidence of the importance of ferroptosis, as compared to the well-established role of apoptosis in driving pancreatic dysfunction during type II diabetes, is still lacking. Nevertheless, this study indicates that the connection between ferroptosis and β-islet dysfunction, as well as its possible role in other relevant diabetic tissues during type II diabetes, warrants further investigation.
Targeting NRF2 to modulate ferroptosis in disease:
Due to its role in combating oxidative stress, as well as mediating numerous aspects of metabolic and protein homeostasis, NRF2 continues to be a promising therapeutic target for the treatment of neurodegeneration, cancer, diabetes, cardiovascular disease, and kidney disease (de Haan, 2011; Jeong et al., 2006; Schmidlin et al., 2019a; Schmidlin et al., 2019b). As mentioned briefly above, a number of proteins whose function are closely tied to the ferroptosis cascade are NRF2 target genes. Consequently, pharmacological modulation of NRF2 itself to induce or inhibit ferroptosis is a viable area of interest (Figure 4). However, very few studies have shown that activation of NRF2 can directly prevent initiation of ferroptotic cell death. It is also important to note that while pharmacological activation of NRF2 could play a beneficial role in heart disease and preventing tumor initiation, activation of NRF2 following malignant transformation may facilitate cancer cell survival in certain cancer types through increasing resistance to ferroptosis. Thus, pharmacological activation of NRF2 could be a useful preventive measure in certain disease contexts, but an inhibitor of NRF2 may prove more useful in treating certain pathologies, such as ferroptosis-resistant cancers. Along these lines, there has been a bit more experimental evidence supporting the notion that NRF2 inhibition, either as a primary or adjuvant chemotherapy, can sensitize resistant cancer cells to cell death, including via ferroptosis (Dodson et al., 2019; Panieri and Saso, 2019). For example, hepatocellular carcinoma cells treated with brusatol, a known NRF2 inhibitor, were sensitized to sorafenib treatment due to inhibition of metallothionein-1G (MT-1G) (Sun et al., 2016a). As MT-1G plays a vital role in the regulation of lipid peroxidation, loss of MT-1G induced ferroptosis and increased the anticancer activity of sorafenib. Inhibition of NRF2 with trigonelline also sensitized head and neck cancer cells to RSL3- and artesunate-induced ferroptosis both in vitro and in vivo (Roh et al., 2017; Shin et al., 2018). While more studies are needed to elucidate the relationship between NRF2 inhibition, cancer treatment, and ferroptosis, these results provide key evidence that NRF2 inhibitors may be a promising chemotherapeutic approach for treating cancer.
Figure 4: Modulation of NRF2 to target ferroptosis-driven diseases.

NRF2 regulates the expression of enzymes responsible for glutathione synthesis (GLC and GSS), as well as preventing lipid peroxidation and reducing oxidized CoQ10, a key membrane antioxidant (GPX4 and FSP1). Lipid peroxides are key initiators of ferroptosis, which has been shown to be involved in numerous diseases, including cancer, type II diabetes, cardiovascular disease, and neurodegeneration.
In addition to the NRF2 pathway, modulation of other key metabolic/signaling pathways that mediate the ferroptotic cascade have also been shown to have possible therapeutic benefit. For example, inhibition of glutaminolysis, which is critical for serum deprivation-induced ferroptosis, prevented initiation of ferroptosis and reduced ischemia-reperfusion-induced heart injury (Gao et al., 2015). As discussed in detail above, numerous studies have shown that iron chelation to prevent lipid peroxidation remains a viable option for preventing ferroptosis in the context of cardiovascular disease, neurodegeneration, and diabetes. Similarly, in addition to containing AREs in their promotor regions, iron response elements (IREs) have also been identified in the untranslated region of mRNA encoding FTL, FTH, and FPN1 (Bogdan et al., 2016). Translation of IRE-containing transcripts is governed by iron responsive element binding proteins 1 and 2 (IREB1/2), which bind to the IRE in iron replete conditions to inhibit translation of their targets. Importantly, IREB2 was shown to be a critical pro-ferroptotic driver of erastin-induced ferroptosis (Dixon et al., 2012). Thus, modulation of iron levels can affect ferroptosis at multiple levels and continues to be a viable strategy for enhancing or preventing ferroptotic death depending on the disease context. Finally, other critical signaling pathways, such as p53, protein kinase C (PKC), and the RAS/MAPK pathway, have also been linked to regulating ferroptosis, particularly in a cancer setting (Dixon and Stockwell, 2019; Jiang et al., 2015; Xie et al., 2016); however, whether or not targeting these pathways can result in ferroptosis-specific effects, or cause death via a different mechanism, still needs to be elucidated. Thus, targeting the ferroptotic cascade, as well as its upstream mediators, remains an active and ongoing area of interest in modulating this pathway in disease.
Conclusions and Future Perspectives:
Our understanding of ferroptosis, much like other forms of RCD following their initial discovery, continues to grow as an ever-increasing number of studies begin to clarify the relevant mechanisms and disease contexts related to this pathway. Perhaps one of the most intriguing aspects of ferroptotic cell death is the unique subset of activators, inhibitors, and their respective targets that differentiate it from other prominent RCD cascades. While its unique nature has generated a number of chemotherapeutic possibilities for treating resistant cancers, the actual (patho)physiological relevance of this pathway, including viable translational approaches to target it, is still evolving. This is particularly true for neurodegeneration, cardiovascular disease, and diabetes, where ferroptosis might underlie the neuronal, cardiomyocyte, and β-cell loss associated with these diseases. Another emerging aspect of ferroptosis-related research is the prominent role that the transcription factor NRF2 and its myriad of transcriptional targets play in preventing, or in certain contexts possibly promoting, activation of the ferroptotic cascade. This infers that careful consideration should be given regarding what proteins, particularly in the NRF2 pathway, represent viable targets to initiate ferroptosis in tumor cell types, without harming their normal counterparts. Another interesting therapeutic possibility is utilizing pharmacological activation of NRF2 or other important anti-ferroptotic mediators to treat diseases where preventing ferroptosis is more favorable. The answers to these questions will of course be context-dependent and rely upon the continued discovery of the experimental and clinical relevance of this pathway, as its stimuli and molecular mechanisms continue to be unraveled.
Acknowledgements:
The authors are funded by the following grants from the National Institutes of Health: ES026845 (DDZ), DK109555 (DDZ), ES004940 (DDZ), ES007091 (CJS), and ES006694 (a center grant).
References:
- Adibhatla RM, and Hatcher JF (2010). Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 12, 125–169. [DOI] [PubMed] [Google Scholar]
- Agrawal S, Berggren KL, Marks E, and Fox JH (2017). Impact of high iron intake on cognition and neurodegeneration in humans and in animal models: a systematic review. Nutr Rev 75, 456–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, Sabatini DM, Birsoy K, and Possemato R (2017). NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli JPF, Shah R, Pratt DA, and Conrad M (2017). Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol Sci 38, 489–498. [DOI] [PubMed] [Google Scholar]
- Aubrey BJ, Kelly GL, Janic A, Herold MJ, and Strasser A (2018). How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 25, 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala A, Munoz MF, and Arguelles S (2014). Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014, 360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio A, and Bonifacino JS (2020). Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21, 101–118. [DOI] [PubMed] [Google Scholar]
- Bannai S, Tsukeda H, and Okumura H (1977). Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem Biophys Res Commun 74, 1582–1588. [DOI] [PubMed] [Google Scholar]
- Barrera G, Pizzimenti S, Ciamporcero ES, Daga M, Ullio C, Arcaro A, Cetrangolo GP, Ferretti C, Dianzani C, Lepore A, et al. (2015). Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid Redox Signal 22, 1681–1702. [DOI] [PubMed] [Google Scholar]
- Belaidi AA, and Bush AI (2016). Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem 139 Suppl 1, 179–197. [DOI] [PubMed] [Google Scholar]
- Bersuker K, Hendricks J, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock FJ, and Tait SWG (2020). Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21, 85–100. [DOI] [PubMed] [Google Scholar]
- Bogdan AR, Miyazawa M, Hashimoto K, and Tsuji Y (2016). Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem Sci 41, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley-Whitman MA, and Lovell MA (2015). Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch Toxicol 89, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Lee VM, and Trojanowski JQ (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittenham GM (2011). Iron-chelating therapy for transfusional iron overload. N Engl J Med 364, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni A, Pepper AR, Pawlick RL, Gala-Lopez B, Gamble AF, Kin T, Seeberger K, Korbutt GS, Bornstein SR, Linkermann A, et al. (2018). Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis 9, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, and Sontheimer H (2011). Glutamate release by primary brain tumors induces epileptic activity. Nat Med 17, 1269–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso BR, Hare DJ, Bush AI, and Roberts BR (2017). Glutathione peroxidase 4: a new player in neurodegeneration? Mol Psychiatry 22, 328–335. [DOI] [PubMed] [Google Scholar]
- Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, and Chang WC (2018). Heme oxygenase-1 mediates BAY 11–7085 induced ferroptosis. Cancer Lett 416, 124–137. [DOI] [PubMed] [Google Scholar]
- Chen J, Schenker S, Frosto TA, and Henderson GI (1998). Inhibition of cytochrome c oxidase activity by 4-hydroxynonenal (HNE). Role of HNE adduct formation with the enzyme subunits. Biochim Biophys Acta 1380, 336–344. [DOI] [PubMed] [Google Scholar]
- Chen L, Hambright WS, Na R, and Ran Q (2015). Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J Biol Chem 290, 28097–28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang GC, Mao X, Kang G, Chang E, Pandya S, Vallabhajosula S, Isaacson R, Ravdin LD, Alzheimer’s Disease Neuroimaging, I., and Shungu DC (2017). Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with (1)H-MRS and Pittsburgh Compound-B PET. AJNR Am J Neuroradiol 38, 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, Xue P, Pi J, Kleeberger SR, and Bell DA (2012). Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res 40, 7416–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen EP, and Eagle H (1961). A simplified chemostat for the growth of mammalian cells: characteristics of cell growth in continuous culture. J Exp Med 113, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, and Stockwell BR (2018). Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev 32, 602–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, and Andrews DF (1991). Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337, 1304–1308. [DOI] [PubMed] [Google Scholar]
- Dachert J, Schoeneberger H, Rohde K, and Fulda S (2016). RSL3 and Erastin differentially regulate redox signaling to promote Smac mimetic-induced cell death. Oncotarget 7, 63779–63792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher B, Parks SK, Durivault J, Cormerais Y, Baidarjad H, Tambutte E, Pouyssegur J, and Vucetic M (2019). Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res 79, 3877–3890. [DOI] [PubMed] [Google Scholar]
- de Haan JB (2011). Nrf2 activators as attractive therapeutics for diabetic nephropathy. Diabetes 60, 2683–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, Jonneaux A, Ryckewaert G, Garcon G, Rouaix N, et al. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid Redox Signal 21, 195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, and Stockwell BR (2014). The role of iron and reactive oxygen species in cell death. Nat Chem Biol 10, 9–17. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, and Stockwell BR (2019). The Hallmarks of Ferroptosis. Annu Rev Canc Biol 3, 35–54. [Google Scholar]
- Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, and Nadler JL (2011). Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res 50, 115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M, Castro-Portuguez R, and Zhang DD (2019). NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol, 101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel C, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma S, Lessnick SL, Hahn WC, and Stockwell BR (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. [DOI] [PubMed] [Google Scholar]
- Doxsee DW, Gout PW, Kurita T, Lo M, Buckley AR, Wang Y, Xue H, Karp CM, Cutz JC, Cunha GR, et al. (2007). Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate 67, 162–171. [DOI] [PubMed] [Google Scholar]
- Eagle H (1955). The minimum vitamin requirements of the L and HeLa cells in tissue culture, the production of specific vitamin deficiencies, and their cure. J Exp Med 102, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle H (1959). Amino acid metabolism in mammalian cell cultures. Science 130, 432–437. [DOI] [PubMed] [Google Scholar]
- Eling N, Reuter L, Hazin J, Hamacher-Brady A, and Brady NR (2015). Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2, 517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 116, 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti G, Bacchetti T, Masciangelo S, Nanetti L, Mazzanti L, Silvestrini M, Bartolini M, and Provinciali L (2008). Lipid peroxidation in stroke patients. Clin Chem Lab Med 46, 113–117. [DOI] [PubMed] [Google Scholar]
- Fuchs Y, and Steller H (2011). Programmed cell death in animal development and disease. Cell 147, 742–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Quadri N, Ramasamy R, and Jiang X (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 59, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, et al. (2018). FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol 14, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascon S, Murenu E, Masserdotti G, Ortega F, Russo GL, Petrik D, Deshpande A, Heinrich C, Karow M, Robertson SP, et al. (2016). Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell 18, 396–409. [DOI] [PubMed] [Google Scholar]
- Gerber PA, and Rutter GA (2017). The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid Redox Signal 26, 501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Lo M, Dockery P, Mahon S, Karp CM, Buckley AR, Lam S, Gout PW, and Wang YZ (2009). The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer Chemother Pharmacol 64, 463–472. [DOI] [PubMed] [Google Scholar]
- Gujja P, Rosing DR, Tripodi DJ, and Shizukuda Y (2010). Iron overload cardiomyopathy: better understanding of an increasing disorder. J Am Coll Cardiol 56, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Sun L, Chen X, and Zhang D (2013). Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res 8, 2003–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassannia B, Vandenabeele P, and Vanden Berghe T (2019). Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 35, 830–849. [DOI] [PubMed] [Google Scholar]
- Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, Bayir H, Abhari BA, Angeli JPF, Choi SM, et al. (2018). Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest 128, 3341–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn T, and Stockwell BR (2019). The development of the concept of ferroptosis. Free Radic Biol Med 133, 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, and Tang D (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422 e421. [DOI] [PubMed] [Google Scholar]
- Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, Diederich WE, and Culmsee C (2018). Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic Biol Med 117, 45–57. [DOI] [PubMed] [Google Scholar]
- Jeong WS, Jun M, and Kong AN (2006). Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxid Redox Signal 8, 99–106. [DOI] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, and Gu W (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Rayamajhi M, and Miao EA (2017). Programmed cell death as a defence against infection. Nat Rev Immunol 17, 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakhlon O, and Cabantchik ZI (2002). The labile iron pool: characterization, measurement, and participation in cellular processes(1). Free Radic Biol Med 33, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Kerins MJ, Milligan J, Wohlschlegel JA, and Ooi A (2018). Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci 109, 2757–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerins MJ, and Ooi A (2017). The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid Redox Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura H, and Motohashi H (2018). NRF2 addiction in cancer cells. Cancer Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremastinos DT, and Farmakis D (2011). Iron overload cardiomyopathy in clinical practice. Circulation 124, 2253–2263. [DOI] [PubMed] [Google Scholar]
- Kwon MY, Park E, Lee SJ, and Chung SW (2015). Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6, 24393–24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapenna D, Ciofani G, Pierdomenico SD, Giamberardino MA, and Porreca E (2018). Iron status and oxidative stress in the aged rabbit heart. J Mol Cell Cardiol 114, 328–333. [DOI] [PubMed] [Google Scholar]
- Lee DW, Andersen JK, and Kaur D (2006). Iron dysregulation and neurodegeneration: the molecular connection. Mol Interv 6, 89–97. [DOI] [PubMed] [Google Scholar]
- Lim JKM, Delaidelli A, Minaker SW, Zhang HF, Colovic M, Yang H, Negri GL, von Karstedt S, Lockwood WW, Schaffer P, et al. (2019). Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci U S A 116, 9433–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, Wang L, and Liang G (2016). Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett 381, 165–175. [DOI] [PubMed] [Google Scholar]
- Liu B, Zhao C, Li H, Chen X, Ding Y, and Xu S (2018). Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem Biophys Res Commun 497, 233–240. [DOI] [PubMed] [Google Scholar]
- Ma MZ, Chen G, Wang P, Lu WH, Zhu CF, Song M, Yang J, Wen S, Xu RH, Hu Y, et al. (2015). Xc- inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett 368, 88–96. [DOI] [PubMed] [Google Scholar]
- Maiorino M, Conrad M, and Ursini F (2018). GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid Redox Signal 29, 61–74. [DOI] [PubMed] [Google Scholar]
- Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC, and Harper JW (2015). Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, and Kimmelman AC (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, and Brodie BB (1973). Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther 187, 185–194. [PubMed] [Google Scholar]
- Morris G, Berk M, Carvalho AF, Maes M, Walker AJ, and Puri BK (2018). Why should neuroscientists worry about iron? The emerging role of ferroptosis in the pathophysiology of neuroprogressive diseases. Behav Brain Res 341, 154–175. [DOI] [PubMed] [Google Scholar]
- Nitenberg A, Ledoux S, Valensi P, Sachs R, and Antony I (2002). Coronary microvascular adaptation to myocardial metabolic demand can be restored by inhibition of iron-catalyzed formation of oxygen free radicals in type 2 diabetic patients. Diabetes 51, 813–818. [DOI] [PubMed] [Google Scholar]
- Pajares M, Jimenez-Moreno N, Garcia-Yague AJ, Escoll M, de Ceballos ML, Van Leuven F, Rabano A, Yamamoto M, Rojo AI, and Cuadrado A (2016). Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12, 1902–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panieri E, and Saso L (2019). Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid Med Cell Longev 2019, 8592348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert SM, Buckingham SC, Campbell SL, Robel S, Holt KT, Ogunrinu-Babarinde T, Warren PP, White DM, Reid MA, Eschbacher JM, et al. (2015). SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med 7, 289ra286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JL, Kim EH, Jang H, and Shin D (2017). Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol 11, 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JL, Kim EH, Jang HJ, Park JY, and Shin D (2016). Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett 381, 96–103. [DOI] [PubMed] [Google Scholar]
- Rojo de la Vega M, Chapman E, and Zhang DD (2018). NRF2 and the Hallmarks of Cancer. Cancer Cell 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez-Rivera FJ, et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Kusumi R, Hamashima S, Kobayashi S, Sasaki S, Komiyama Y, Izumikawa T, Conrad M, Bannai S, and Sato H (2018). The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci Rep 8, 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidlin CJ, Dodson MB, Madhavan L, and Zhang DD (2019a). Redox regulation by NRF2 in aging and disease. Free Radic Biol Med 134, 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidlin CJ, Dodson MB, and Zhang DD (2019b). Filtering through the role of NRF2 in kidney disease. Arch Pharm Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweichel JU, and Merker HJ (1973). The morphology of various types of cell death in prenatal tissues. Teratology 7, 253–266. [DOI] [PubMed] [Google Scholar]
- Seibt TM, Proneth B, and Conrad M (2019). Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med 133, 144–152. [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Shichiri M (2014). The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr 54, 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, and Stockwell BR (2016). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 12, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Kim EH, Lee J, and Roh JL (2018). Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med 129, 454–462. [DOI] [PubMed] [Google Scholar]
- Singer E, Judkins J, Salomonis N, Matlaf L, Soteropoulos P, McAllister S, and Soroceanu L (2015). Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis 6, e1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleire L, Skeie BS, Netland IA, Forde HE, Dodoo E, Selheim F, Leiss L, Heggdal JI, Pedersen PH, Wang J, et al. (2015). Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene 34, 5951–5959. [DOI] [PubMed] [Google Scholar]
- Speer RE, Karuppagounder SS, Basso M, Sleiman SF, Kumar A, Brand D, Smirnova N, Gazaryan I, Khim SJ, and Ratan RR (2013). Hypoxia-inducible factor prolyl hydroxylases as targets for neuroprotection by “antioxidant” metal chelators: From ferroptosis to stroke. Free Radic Biol Med 62, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepkowski TM, and Kruszewski MK (2011). Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic Biol Med 50, 1186–1195. [DOI] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama A, and Sun J (2014). Immunochemical detection of lipid hydroperoxide- and aldehyde-modified proteins in diseases. Subcell Biochem 77, 115–125. [DOI] [PubMed] [Google Scholar]
- Sui S, Zhang J, Xu S, Wang Q, Wang P, and Pang D (2019). Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis 10, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Niu X, Chen R, He W, Chen D, Kang R, and Tang D (2016a). Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 64, 488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, and Tang D (2016b). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzanne M, and Steller H (2013). Shaping organisms with apoptosis. Cell Death Differ 20, 669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweiry JH, Sastre J, Vina J, Elsasser HP, and Mann GE (1995). A role for gamma-glutamyl transpeptidase and the amino acid transport system xc- in cystine transport by a human pancreatic duct cell line. J Physiol 485 (Pt 1), 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Berghe TV, Vandenabeele P, and Kroemer G (2019). The molecular machinery of regulated cell death. Cell Res 29, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower J (2015). Programmed cell death in aging. Ageing Res Rev 23, 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, MacPherson L, Dandapani S, Palmer M, Stockwell BR, Schreiber SL, et al. (2012). Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett 22, 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TI, Lynn BC, Markesbery WR, and Lovell MA (2006). Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol Aging 27, 1094–1099. [DOI] [PubMed] [Google Scholar]
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, and Tang D (2016). Ferroptosis: process and function. Cell Death Differ 23, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, and Stockwell BR (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113, E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, and Stockwell BR (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, and Prolla TA (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med 34, 496–502. [DOI] [PubMed] [Google Scholar]
- Yin H, Xu L, and Porter NA (2011). Free radical lipid peroxidation: mechanisms and analysis. Chem Rev 111, 5944–5972. [DOI] [PubMed] [Google Scholar]
- Yuan H, Li X, Zhang X, Kang R, and Tang D (2016a). CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun 478, 838–844. [DOI] [PubMed] [Google Scholar]
- Yuan H, Li X, Zhang X, Kang R, and Tang D (2016b). Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478, 1338–1343. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Koppula P, and Gan B (2019a). Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle 18, 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, Sirohi K, Li X, Wei Y, Lee H, et al. (2018). BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol 20, 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhuang L, and Gan B (2019b). BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol Cell Oncol 6, 1536845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, and Tang D (2019). Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. [DOI] [PubMed] [Google Scholar]
- Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, Milner TA, Jonas EA, and Ratan RR (2017). Neuronal Death After Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 48, 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]