

Abstract
Males and females differ in body composition and fat distribution. Using a mouse model that segregates gonadal sex (ovaries and testes) from chromosomal sex (XX and XY), we showed that XX chromosome complement in combination with a high-fat diet led to enhanced weight gain in the presence of male or female gonads. We identified the genomic dosage of Kdm5c, an X chromosome gene that escapes X chromosome inactivation, as a determinant of the X chromosome effect on adiposity. Modulating Kdm5c gene dosage in XX female mice to levels that are normally present in males resulted in reduced body weight, fat content, and food intake to a degree similar to that seen with altering the entire X chromosome dosage. In cultured preadipocytes, the levels of KDM5C histone demethylase influenced chromatin accessibility (ATAC-Seq), gene expression (RNA-Seq), and adipocyte differentiation. Both in vitro and in vivo, Kdm5c dosage influenced gene expression involved in extracellular matrix remodeling, which is critical for adipocyte differentiation and adipose tissue expansion. In humans, adipose tissue KDM5C mRNA levels and KDM5C genetic variants were associated with body mass. These studies demonstrate that the sex-dependent dosage of Kdm5c contributes to male/female differences in adipocyte biology and highlight X-escape genes as a critical component of female physiology.
Keywords: Genetics, Metabolism
Keywords: Adipose tissue, Mouse models

Introduction
Adipose tissue plays a prominent role in metabolic regulation, and dysregulation of adipose tissue storage is a risk factor for cardiometabolic disease (1). Sex is a key determinant in the regulation of adipose tissue storage and distribution (2–4). Women tend to store fat subcutaneously in the femorogluteal area, whereas men and postmenopausal women tend to accumulate fat in the abdominal region (3). Estrogen replacement in postmenopausal women reduces central adipose tissue, which has focused attention on gonadal hormones as a key mediator of sex differences in adiposity (5, 6).
In addition to gonadal hormones, there is evidence that genetic factors that differ between males and females influence body fat. For example, male and female mouse and human embryos differ in body composition even before gonadal development (7, 8). The key genetic difference between females and males is the presence of XX versus XY chromosomes. Since individuals with XX chromosomes typically have female gonads and those with XY chromosomes have male gonads, it is difficult to ascertain the contributions of each component to a trait such as adiposity. However, evidence for a role of sex chromosome complement in human adiposity comes from studies of Klinefelter syndrome subjects (XXY), who have increased body fat mass compared with matched XY counterparts, even before puberty (9, 10). However, the mechanisms underlying the apparent effects of X chromosome dosage effects on adiposity in these individuals are not understood.
To better distinguish the contributions of gonads and sex chromosomes to adiposity, we used the four core genotypes (FCG) mouse model, which generates XX and XY mice with ovaries and XX and XY mice with testes (11). Analysis of these 4 genotypes allows a determination of whether a specific trait is influenced by gonadal sex (ovaries, female; or testes, male), chromosomal sex (XX or XY), or an interaction between the two (12). We previously demonstrated that if adult FCG mice are gonadectomized to remove the acute effects of gonadal hormones, XX mice with ovaries or testes develop increased adiposity compared with XY mice with ovaries or testes (13). Furthermore, we determined that the presence of 2 copies of the X chromosome was responsible for increased adiposity in gonadectomized XX mice, regardless of the presence of a Y chromosome.
A likely mechanism by which the XX chromosome complement could influence sex differences is through the expression of genes that escape X chromosome inactivation (14). In mammals, the X chromosome carries more than 800 protein coding genes that are not present on the Y chromosome, such that XX females have a double genomic dose of these genes compared with XY males. To normalize the expressed dosage of X chromosome genes between the sexes, most genes on 1 X chromosome in XX cells are transcriptionally silenced early in development through X chromosome inactivation. However, some X chromosome genes escape inactivation to varying degrees and are expressed from both alleles in XX cells (15, 16). These “X-escape genes” are often conserved between mouse and human, and several have functions that could influence gene or protein expression broadly across the genome. We previously identified 4 X-escape genes that are expressed at more than 50% higher levels in XX compared with XY metabolic tissues and include histone modifying enzymes, an RNA helicase, and a translation initiation factor (13). We hypothesize that the differential genomic dosage of X-escape genes in XX compared with XY cells contributes to sex differences in cell processes that lead to metabolic phenotypes.
In this study, we identify a role for X chromosome dosage in determining diet-induced weight gain in mice with intact ovaries or testes. Subsequently, we identify the X-escape gene Kdm5c as a key contributor to X chromosome dosage effect on adiposity. Kdm5c (also known as Jarid1c) encodes a histone demethylase, which removes tri- and dimethyl marks from histone 3 lysine 4 (H3K4) to regulate chromatin access by transcription factors (17). By modulating Kdm5c levels in the mouse, we demonstrate that Kdm5c gene dosage is a determinant of sex differences in adiposity. We further characterize the role of Kdm5c dosage in chromatin landscape, gene expression, and differentiation of cultured preadipocytes. Finally, we assess the association of KDM5C gene expression levels and genetic variation with human body weight traits.
Results
An XX chromosome complement accelerates diet-induced weight gain in mice with male or female gonads.
In our previous study, we detected effects of sex chromosome complement on adiposity in FCG mice that had been gonadectomized to remove the acute effects of gonadal secretions (13). We assessed whether the influence of sex chromosome complement on body weight is evident in the presence of circulating gonadal hormones in adult C57BL/6 FCG mice. In mice fed a standard chow diet (~6% of calories from fat), those with testes weighed more than those with ovaries from 10–32 weeks of age, regardless of XX or XY chromosome complement (Figure 1A). More in-depth analysis of a separate cohort of chow-fed mice at 14 weeks of age revealed effects of both gonadal and chromosomal components on body weight parameters. Body weight was higher in mice with male compared with female gonads by an average of 6.1 grams, but XX mice also had higher body weight than XY by nearly 2 grams (Figure 1D). Body composition was influenced primarily by gonadal type, with gonadal males having 44% higher absolute lean mass and gonadal females having a greater proportion of body mass as fat (fat mass %) (Figure 1D). Interactions between sex chromosomes and gonadal type were also noted in the percentages of fat and lean mass (Figure 1D).
Figure 1. XX mice gain more weight than XY mice on a high-fat diet.

(A) Body weight curves of gonadally intact FCG mice fed a chow diet. XX and XY refer to chromosome type; F, female gonads (ovaries); M, male gonads (testes). n = 11 XX female; n = 11 XX male; n = 12 XY female; n = 16 XY male. (B) Body weight and (C) percentage weight gained in FCG mice over 10 weeks on high-fat diet, starting at 14 weeks of age. n = 8 mice per genotype (B–G). (D) Body weight, fat mass, lean mass, percentage of fat mass, and percentage of lean mass on chow and (E) after 10 weeks of high-fat feeding. (F) Representative mice after 10 weeks of high-fat diet (individuals with median body weight shown for each genotype). (G) Gonadal and inguinal fat pad mass normalized to kidney mass. (H) Food intake measured at 1 week (left) and at 8 weeks (right) of high-fat diet feeding during the dark (black horizontal bar) and light (white horizontal bar) periods of the circadian cycle (note different y axis scales). n = 4 mice per genotype. Data are represented as mean ± SD. Data in A–C were analyzed by repeated measures 3-way ANOVA (factors of gonadal sex, sex chromosomes, and age). Data in D–H were analyzed by 2-way ANOVA. Statistically significant differences for sex chromosome complement and for gonadal sex are denoted by brackets. Int., significant interactions of sex chromosome complement and gonadal sex. *P < 0.05; **P < 0.01; ***P < 0.001; †P < 0.0001; ‡P < 0.00001. HFD, high-fat diet.
We previously noted that a high-fat diet amplified the effect of sex chromosome complement on adiposity in mice following gonadectomy (13). Here, we assessed the contribution of sex chromosome complement on adiposity in gonadally intact animals. At 14 weeks of age, mice were fed a high-fat diet (60% calories from fat, 26% from carbohydrate) for 10 weeks. All genotypes gained weight (Figure 1B), but XX mice with either gonadal type increased their body weight by a greater proportion than XY mice (Figure 1C). After 10 weeks of a high-fat diet, XX mice had a greater percentage of fat mass than XY mice of both gonadal types (Figure 1, E and F). Gonadal and inguinal fat depots were larger in XX compared with XY mice by 34% and 64%, respectively (Figure 1G). These results indicate that gonadal sex is a primary determinant of body weight and adiposity in mice fed a chow diet and that XX chromosome complement promotes accelerated weight and fat gain in response to a high-fat diet.
We assessed energy balance in the mice represented in Figure 1B at 1 week and 8 weeks of high-fat diet feeding. All 4 genotypes exhibited typical diurnal variations in locomotor activity (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI140223DS1). Mice with female gonads were more active than those with male gonads, as measured by locomotion along the horizontal plane (Supplemental Figure 1B). There were no significant differences among the genotypes in energy expenditure or respiratory quotient (RQ) (Supplemental Figure 1, C and D). After 1 week of a high-fat diet, food intake was increased by both XX chromosome complement and male gonadal sex during both the dark and light phases (Figure 1H). After 8 weeks of a high-fat diet, XX chromosomes and male gonads led to increased food intake specifically during the light phase of the circadian cycle (Figure 1H). Thus, energy balance is influenced by a complex interplay among sex chromosome complement, gonadal type, and the circadian cycle.
We also examined the contributions of gonadal and sex chromosome types to glucose homeostasis at 10 weeks of high-fat feeding. Males had higher fasting glucose and insulin levels as well as higher homeostatic model assessment–estimated insulin resistance (HOMA-IR) compared with females (Supplemental Figure 2A). Insulin levels and HOMA-IR values were also higher in XX compared with XY mice. The presence of female gonads led to more rapid clearance of the glucose bolus in glucose tolerance tests (Supplemental Figure 2B). These results suggest compound effects of gonadal and chromosomal sex on glucose homeostasis in response to a high-fat diet.
Kdm5c gene dosage recapitulates X chromosome dosage effects on adiposity.
We hypothesized that differences between XX and XY mice in diet-induced adiposity are related to higher expression levels of specific X chromosome genes that escape silencing by X chromosome inactivation (15, 16). We previously demonstrated that such X-escape genes exhibit higher mRNA expression levels in tissues of XX compared with XY mice that had been gonadectomized (13). Four genes in particular showed robust differences in expression levels based on X chromosome dosage — Kdm5c, Kdm6a, Ddx3x, and Eif2s3x. These 4 genes are ubiquitously expressed across tissues (18), and we found that they exhibit elevated expression in XX compared with XY mouse subcutaneous and gonadal adipose tissue depots in gonadally intact mice fed a high-fat diet for 10 weeks (Figure 2, A–D). The X-escape genes are also expressed at higher levels in human tissues from females compared with males, as illustrated for human KDM5C (Figure 2E).
Figure 2. Genes that escape X inactivation have elevated expression in XX vs.

XY mice, and female versus male human tissues. mRNA expression levels of Kdm5c (A), Kdm6a (B), Ddx3x (C), and Eif2s3x (D) in gonadal fat and inguinal fat depots of gonadally intact FCG mice fed a high-fat diet for 10 weeks as determined by qPCR. n = 8 per genotype. Expression was normalized to 36B4 mRNA levels. (E) Expression of human KDM5C in metabolic tissues (subcutaneous adipose, visceral adipose, hypothalamus, liver, skeletal muscle, pancreas, and small intestine [Sm intest]). Data are from the GTEx Project (51). For A–D, data are represented as mean ± SD, and data were analyzed by 2-way ANOVA. Significant comparisons for sex chromosome complement and for gonadal sex are denoted by brackets. *P < 0.05; **P < 0.01; ***P < 0.001; †P < 0.0001; ‡P < 0.00001.
We selected Kdm5c as a candidate gene to test for the X chromosome effect on adiposity. Supporting evidence for this candidate includes a previous report showing that members of the KDM5 gene family of histone demethylases influence adipocyte differentiation in cultured cells (19). To test this candidate, we generated WT (Kdm5c+/+) and hemizygous (Kdm5c+/–) mice on a C57BL/6 background. All mice were XX female mice, such that the only difference was the presence of 1 versus 2 functional Kdm5c gene alleles. Male mice were not studied because Kdm5c–/Y male embryos were nonviable on the inbred C57BL/6 background, possibly reflecting a requirement for KDM5C to support fetal development.
The reduced Kdm5c genomic dose in Kdm5c+/– mice reduced the levels of KDM5C protein to approximately half the levels observed in tissues of Kdm5c+/+ littermates (Figure 3A). This was comparable to the differential KDM5C protein levels in XX compared with XY mouse tissues (Figure 3A). On a chow diet, Kdm5c+/– mice had lower body weight throughout life and 20% lower body fat at 26 weeks of age (Figure 3, B and C). On the high-fat diet, Kdm5c+/+ mice gained weight more rapidly than Kdm5c+/– mice, with a body weight increase of 75% and 30%, respectively, over 13 weeks of high-fat diet feeding (Figure 3E). Percentage of fat mass was nearly 30% higher in Kdm5c+/+ compared with Kdm5c+/– mice after 8 weeks on the high-fat diet (Figure 3F). The reduced body weight in Kdm5c+/– mice was associated with reduced food intake during the light phase of the circadian cycle (Figure 3, D and G). Thus, Kdm5c genomic dosage had an impact that mirrored the effect of X chromosome dosage on body fat, weight gain, and food intake reported in our previous studies (13, 20).
Figure 3. Kdm5c dosage affects body weight, fat mass, and food intake.

(A) KDM5C protein levels in whole brain extracts of gonadally intact female mice with 1 or 2 copies of Kdm5c (left) and in FCG mice (right panel). (B) Growth curves of chow-fed female Kdm5c+/+ mice (n = 18–45) and Kdm5c+/– mice (n = 7–18). (C) Percentage fat mass of chow-fed Kdm5c+/+ mice (n = 6) and Kdm5c+/– mice (n = 8) at 26 weeks of age. (D) Food intake of 10-week-old chow-fed Kdm5c+/+ mice (n = 20) and Kdm5c+/– mice (n = 13) during dark (black horizontal bar) and light (white horizontal bar) phases. (E) Growth curves of female Kdm5c+/+ (n = 13) mice and Kdm5c+/– mice fed a high-fat diet (n = 9). (F) Proportional fat mass and (G) food intake of Kdm5c+/+ mice (n = 13) and Kdm5c+/– mice (n = 9) after 8 weeks of high-fat diet. Values are represented as mean ± SD. Data were analyzed by ANOVA (for growth curves) or by t test. **P < 0.01.
Kdm5c gene dosage influences gene expression and chromatin structure during adipogenesis.
Kdm5c gene dosage influenced adipocyte size distribution in vivo. Quantitation of adipocyte size in histological sections from inguinal white adipose tissue of mice fed a high-fat diet for 10 weeks revealed a shift in the distribution toward larger adipocytes in Kdm5c+/– compared with Kdm5c+/+ mice (Figure 4A). We hypothesized that KDM5C may influence the recruitment of preadipocytes to differentiate into adipocytes during diet-induced adipose tissue expansion. To explore this, we investigated the relative levels of KDM5C protein in preadipocytes and mature adipocytes in mouse adipose tissue and in the 3T3-L1 preadipocyte cell line. We fractionated white and brown adipose tissues from C57BL/6J mice into stromal vascular cells and mature adipocytes. KDM5C protein was present almost exclusively in the stromal vascular fraction (Figure 4B), which contains adipocyte stem cells as well as endothelial and immune cell types (21). In 3T3-L1 cells, Kdm5c mRNA was present in preadipocytes and throughout adipocyte maturation, but KDM5C protein was detected primarily in preadipocytes and was extinguished as cells differentiated and markers of mature adipocytes appeared (e.g., fatty acid binding protein 4 [FABP4]) (Figure 4C).
Figure 4. Kdm5c gene dosage influences adipocyte size in vivo and adipocyte differentiation in vitro.

(A) Adipose tissue histology and adipocyte size distribution from inguinal fat pads of Kdm5c+/+ and Kdm5c+/– mice fed a high-fat diet for 10 weeks. Original magnification, ×100. Adipocyte sizes were determined from cumulative scoring of adipocytes in histological sections from each of 6 mice per genotype. Size distribution was different between the 2 genotypes. Mann-Whitney U test, P < 0.0001. (B) Immunoblot of KDM5C protein in the stromal vascular (SV) fraction, mature adipocyte fraction, and whole tissue fractions of brown adipose tissue (BAT), gonadal white adipose tissue (Gon. WAT), and inguinal white adipose tissue (Ing. WAT) from C57BL/6J female mice (30 μg protein loaded per each lane). MW, molecular weight standards. Western blots are representative of 2 experiments. (C) Kdm5c mRNA (upper) and protein levels (lower) in differentiating 3T3-L1 preadipocytes from 2 days before confluence through day 8 after addition of differentiation cocktail (25 μg protein loaded per lane). FABP4 is expressed in mature adipocytes; GAPDH was used as loading control. Western blots are representative of 2 or more experiments. (D) Timeline of Kdm5c knockdown in 3T3-L1 preadipocytes and collection of samples analyzed in E–G. (E) Kdm5c mRNA levels in 3T3-L1 preadipocytes after siRNA knockdown performed at day –3 relative to confluence. Data are representative of 3 experiments. (F) Cell number increases in 3T3-L1 preadipocytes transfected 3 days before confluence with Kdm5c or nonspecific siRNA. n = 4 samples per treatment. Data are representative of 2 experiments. (G) Appearance and quantification of oil red O staining in 3T3-L1 cells after knockdown at day –3 and harvest at day 6 of adipocyte differentiation. Scale bar: 200 μm. n = 6 samples per treatment. Data are representative of 2 experiments. **P < 0.01; ***P < 0.0001.
Our findings focused attention on the potential role of KDM5C in preadipocytes. We assessed the effects of manipulating Kdm5c levels on preadipocyte replication and differentiation (Figure 4D). Kdm5c knockdown by 70% to 80% in subconfluent 3T3-L1 preadipocytes led to a greater than 20% reduction in preadipocyte proliferation (Figure 4, E and F). Furthermore, Kdm5c knockdown during the preadipocyte period blunted lipid accumulation (as assessed by oil red O staining and quantitation) by more than 50% in mature adipocytes that were assessed 6 days after addition of differentiation cocktail (Figure 4G).
We hypothesized that Kdm5c dosage influences adipogenesis through its role in histone modification to regulate genome access to transcription factors. We assessed the effects of Kdm5c knockdown in 3T3-L1 preadipocytes on chromatin accessibility (ATAC-Seq) and gene expression (RNA-Seq) in 3T3-L1 preadipocytes. Kdm5c siRNA was administered 1 day before preadipocyte confluence (day –3), and RNA-Seq and ATAC-Seq analyses were performed at confluence (day –2) and for 2 subsequent days (day –1 and day 0) (Figure 5A). Comparison of the transcriptional profile between Kdm5c knockdown and control cells (treated with a nonspecific siRNA) revealed differential gene expression as early as 1 day after knockdown, with increasing numbers of differentially expressed genes on each subsequent day (Figure 5A). Analysis of differentially expressed genes revealed several pathways that were significantly enriched (Figure 5A). The top pathway (adjusted P < 10–5) contained genes implicated in extracellular matrix (ECM) remodeling (Figure 5A), a process that is critical during the early stages of adipocyte differentiation as well as in the maintenance and expansion of adipose tissue mass (22). Differentially expressed genes involved in ECM organization included matrix metalloproteinases (Mmp3, Mmp9, Mmp13), metallopeptidases (Adamts1, Adamts2, Adamts5, Adamts14), and integrins (Itga2, Itga3, Itga7, Itgb7, Itgb8) (Supplemental Figure 3). Additional enriched pathways (adjusted P < 0.035 to 10–3) included immune signaling and interleukin pathways (Figure 5A; see also Supplemental Figure 3).
Figure 5. Kdm5c knockdown in preadipocytes alters gene expression and chromatin accessibility at promoters enriched in adipogenic transcription factor motifs.

(A) Differential gene expression induced by Kdm5c knockdown in 3T3-L1 preadipocytes. Top: timeline of sample generation (n = 5 samples for each treatment at each time point). Volcano plots show genes that are differentially expressed (EdgeR) in response to Kdm5c knockdown (light blue dots). Differentially expressed genes with greater than 1.25-fold difference were analyzed for pathway enrichment, and genes belonging to 3 most significant pathways are highlighted with colored dots: red dots, genes involved in ECM organization (ECM); dark blue dots, genes in immune signaling pathways; purple dots, genes in interleukin pathway. (B) ATAC-Seq analysis of the same samples in A showed altered chromatin accessibility after Kdm5c knockdown at genomic locations indicated (n = 4 samples for each treatment at each time point). Alterations at proximal gene promoters (within 1.5 kb of transcription start sites) progressively increased each day after knockdown. (C) Differentially expressed genes from day 0 were overlaid with differential ATAC-Seq promoter peaks from day 0, and 48% of the corresponding genes showed overlap in the 2 data sets. (D) Genes that showed differential mRNA expression and ATAC-Seq promoter peaks showed enrichment for specific transcription factor motifs in the promoter region.
Histone demethylases act to establish or maintain open or closed chromatin states. We performed ATAC-Seq (23) to identify regions of open chromatin that are influenced by Kdm5c levels in 3T3-L1 cells. Kdm5c knockdown in 3T3-L1 preadipocytes resulted in significant differential accessibility across the genome (Figure 5B). A focus on differential ATAC-Seq peaks that occurred within promoter regions (1 kb upstream and 0.5 kb downstream from the transcription start site) identified 441 gene promoters with differential peaks in control compared with Kdm5c knockdown cells (based on DESeq2 analysis, P < 0.05). At day 0, nearly half of the genes that were differentially expressed after Kdm5c knockdown also showed an enrichment in ATAC-Seq peaks in the proximal promoter (Figure 5C and Supplemental Figure 4), suggesting that these may be genomic sites at which KDM5C levels influence gene expression through alterations in chromatin accessibility. Among these sites, there was an enrichment for targets of transcription factors such as C/EBPβ, CREB1, and KLF (Figure 5D), which have known roles in adipogenesis (24–26).
The studies of Kdm5c knockdown in 3T3-L1 cells revealed pathways that are proximal targets of KDM5C action in cultured preadipocytes. To verify these results in vivo, we compared gene expression in subcutaneous adipose tissue from Kdm5c+/+ and Kdm5c+/– mice. Adipose tissue was fractionated into stromal vascular and mature adipocytes for RNA-Seq analysis. Kdm5c haploinsufficiency caused altered gene expression in both fractions (Figure 6, A and B), and many of these differentially expressed genes were also significantly altered by Kdm5c knockdown in 3T3-L1 cells (Figure 6, A and B). Since KDM5C protein levels are low in mature adipocytes (Figure 4B), the altered gene expression that we observed in mature adipocytes of Kdm5c+/– mice may reflect events during early stages of adipocyte differentiation. Pathway enrichment analysis showed strong similarities between in vivo gene expression alterations due to Kdm5c haploinsufficiency and the effects of Kdm5c knockdown in 3T3-L1 preadipocytes. In particular, reduced Kdm5c dosage both in vivo and in vitro altered gene expression related to ECM remodeling and immune and interferon signaling pathways (Figure 6C). These results illustrate that modulation of Kdm5c genomic dose from levels normally present in females to levels normally present in males elicits substantial differences in adipose tissue gene expression and affects processes that are implicated in adipocyte differentiation and adipose tissue expansion.
Figure 6. Modulation of Kdm5c gene dosage in vivo alters adipose tissue gene expression in ECM organization and immune signaling pathways.

RNA-Seq was performed in (A) stromal vascular (SV) and (B) mature adipocyte (MAT) fractions of inguinal fat from Kdm5c+/+ and Kdm5c+/– mice (n = 3 mice per genotype). Volcano plots show genes that are differentially expressed (EdgeR) between the 2 genotypes (light blue) and genes that also belong to the pathways that were enriched after Kdm5c knockdown in 3T3-L1 preadipocytes from Figure 5A (gold dots). (C) Pathways that are enriched for genes differentially expressed between both Kdm5c+/+ and Kdm5c+/– adipose tissue fractions (>1.25-fold) and in Kdm5c knockdown in 3T3-L1 cells.
KDM5C mRNA correlation with BMI and genetic variants associated with human adiposity.
Our studies in the mouse and in cultured cells establish a positive relationship between Kdm5c levels and adiposity. To determine whether this relationship is conserved in humans, we assessed whether KDM5C expression levels are correlated with BMI and whether genetic variation at the KDM5C locus is associated with body weight traits. An analysis of KDM5C gene expression in adipose tissue of 776 female twins from the TwinsUK cohort (27) revealed a strong positive correlation between KDM5C expression and BMI (P = 5 × 10–10) (Supplemental Figure 5).
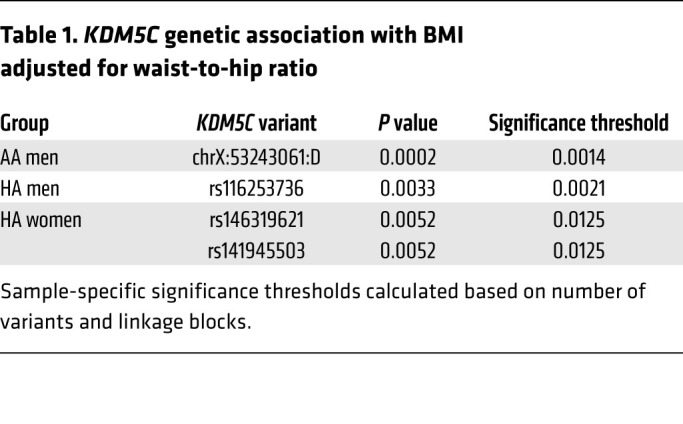
To assess whether genetic variation at the KDM5C gene locus might influence body weight characteristics, we tested variants spanning the KDM5C gene for association with BMI adjusted for waist-to-hip ratio in the Multi-Ethnic Study of Atherosclerosis (28). We assessed association in 3 ethnic groups that each had 700 or more samples for each sex: African American (AA), Hispanic American (HA), and European American (EA). We performed association analyses in men and women separately within each ethnic group, and the variants that met requirements for association analysis in each group are shown in Supplemental Figure 6. To correct for multiple testing, we adjusted for linkage disequilibrium (LD) among the SNPs analyzed and calculated significant P values for each group, as delineated in Methods. Due to differential LD structure by ethnic group and sex (Supplemental Figure 7), distinct P values were calculated for each group. Applying these criteria, we found significant associations between adiposity traits and KDM5C noncoding variants in AA men, HA men, and HA women (Table 1 and Supplemental Figure 6). We did not detect associations in EA samples.
Table 1. KDM5C genetic association with BMI adjusted for waist-to-hip ratio.
Model for the role of sex differences in KDM5C dosage and adiposity.
Taken together, our results demonstrate that inherent differences between males and females in the genomic dosage of the X chromosome — and specifically, the Kdm5c gene — influence adipose tissue accrual and diet-induced expansion in the mouse (Figure 7). The increased genomic dose of Kdm5c in XX compared with XY cells leads to elevated expression due to escape from X chromosome inactivation. Presumably related to the KDM5C histone demethylase activity, the higher KDM5C levels in XX cells influence the chromatin landscape and gene expression in preadipocytes to modulate genes involved in ECM remodeling and other processes. Consistent with the results in our model systems, individual differences in KDM5C expression levels and genetic variants in the KDM5C gene are also associated with adiposity traits in humans.
Figure 7. Proposed mechanism for X chromosome dosage effects on adiposity.

The Kdm5c genomic dose present in XX cells (upper panel) leads to higher levels of KDM5C histone demethylase than in cells with a single Kdm5c gene due to XY or Kdm5c+/– genotype (lower panel). KDM5C protein levels modulate histone methylation and influence gene expression and positively correlate with adiposity traits in the mouse and in humans. Xi, transcriptionally inactivated X chromosome; Xa, active X chromosome.
Discussion
Sex has a strong influence on fat accumulation and distribution. Traditionally, sex differences in adiposity have been attributed to gonadal hormones. Using mice in which gonadal type segregates independently from sex chromosome complement, we found that gonads are the key determinant of adiposity in mice fed a chow diet, but that XX chromosome complement promotes more rapid weight and fat accumulation than XY in response to a high-fat diet. We hypothesized that the higher genome dose and expression levels of specific genes that escape X chromosome inactivation contribute to the differences between XX and XY mice. We tested Kdm5c as a candidate for X chromosome dosage effects on adiposity and demonstrated that modulation of Kdm5c gene dosage closely replicated several features of modulating X chromosome dosage, including effects on body fat composition, weight gain on a high-fat diet, and increased food intake during the inactive part of the circadian cycle. We also demonstrated a role for Kdm5c dosage on adipocyte proliferation and differentiation, the regulation of chromatin accessibility and gene expression, and body weight traits in humans.
It is likely that Kdm5c dosage affects very early stages of adipocyte recruitment and differentiation, since we detected KDM5C protein in preadipocytes (or the stromal vascular fraction in vivo) and not in mature adipocytes. Accordingly, Kdm5c knockdown in 3T3-L1 preadipocytes led to reduced cell proliferation and reduced lipid accumulation after cells were differentiated. The reduced lipid accumulation in mature adipocytes following Kdm5c knockdown in vitro or Kdm5c+/– gene dosage in vivo is likely a reflection of fewer cells recruited to differentiate to mature adipocytes, rather than a direct effect of KDM5C on lipid synthesis. As a result, mature adipocytes in Kdm5c+/– fat pads may be fewer, but tend toward larger sizes, to accommodate greater fat storage per cell in mice that are challenged with excess dietary fat. Our results are in agreement with the previous demonstration that members of the KDM5 family of histone demethylases (Kdm5a, Kdm5b, and Kdm5c) influence 3T3-L1 adipogenesis (19). However, Kdm5c is unique among the KDM5 genes in its location on the X chromosome and its differential expression levels between males and females. Kdm5c mRNA is detected in many tissues (although protein levels have not been assessed), and KDM5C mutations have been associated with neurodevelopmental disorders and cancer (29, 30). Thus, it is possible that differential Kdm5c levels in nonadipose tissues (such as hypothalamus) may also contribute to sex differences in energy balance.
The modulation of Kdm5c levels was associated with alterations in expression of genes involved in ECM remodeling and interferon signaling both in vivo and in vitro. ECM reorganization is critical for adipose tissue expansion and occurs through the breakdown and resynthesis of proteins, proteoglycans, and polysaccharides to allow the enlargement of existing adipocytes, adipogenesis from precursor cells, and angiogenesis (31–33). Among the ECM genes that were influenced by Kdm5c dosage is Mmp3, which encodes a metalloproteinase that is differentially expressed in male and female C57BL/6J mouse adipose tissue and is associated with sex differences in diet-induced adipose tissue expansion (34). Genes associated with interferon signaling were also influenced by Kdm5c dosage. Interferons stimulate several transduction pathways that have been associated with adipocyte differentiation, including JAK/STAT, MAP kinase, and PI3K/AKT pathways (35, 36). Importantly, nearly half of the changes in gene expression that resulted from Kdm5c knockdown in 3T3-L1 cells correlated with alterations in chromatin accessibility of gene promoters as assessed by ATAC-Seq. This suggests that many of the observed alterations in gene expression may result from KDM5C-mediated histone modifications at relevant gene promoters, but further studies to directly map KDM5C binding sites in preadipocytes await the availability of an antibody that exhibits adequate specificity for genome-wide chromatin immunoprecipitation studies.
In mouse models of obesity, a commonly held generalization is that a high-fat diet induces greater weight gain in males compared with females (37–39). This is an oversimplification, as several factors influence the outcome of a high-fat diet, including genetic background and age. In many mouse strains, male mice gain more absolute weight and fat mass than females; however, there are numerous mouse strains in which females gain more absolute fat mass (40). Furthermore, given the typical differences in body size between males and females, it is most relevant to compare percentage of body weight or fat gained during a high-fat diet, as we have done in the analysis of our FCG mouse phenotypes. Age is also a determinant of sex-dependent response to a high-fat diet. In C57BL/6 mice (the genetic background of our FCG and Kdm5c haploinsufficient mice), a high-fat diet fed to juveniles leads to greater absolute and proportional weight gain in males compared with females, but in adult or middle-aged mice (similar to ages used in our diet studies), female C57BL/6 mice gain a greater proportion of their body weight than males (41). Our studies with FCG and Kdm5c “genomic-dose” models vary specific components of naturally occurring sex differences to demonstrate that expression of an individual X-escape gene influences adipose tissue accrual. The same effects are expected to be active in standard XX female versus XY male mice.
The relationship that we observed between KDM5C levels and adiposity traits in the mouse are further supported by our findings in humans. Specifically, we detected a strong positive correlation between adipose tissue KDM5C expression levels and BMI and identified genetic variants in human KDM5C that are associated with BMI normalized to waist-to-hip ratio. The latter trait is notable because waist-to-hip ratio reflects differences in fat distribution that commonly occur between men and women, with men having a propensity to store visceral fat around the waist and women typically having more subcutaneous fat in the hip region. The genetic associations were most prominent in AA and HA men and in HA women. The associations in men may be more readily identified because males carry either 0 or 1 copy of the minor allele (since they have a single X chromosome), whereas women carry 0, 1, or 2 copies, leading to 3 genotypic groups for each variant. We therefore require a larger sample to detect an association with the same effect size in women compared with men (i.e., at a minor and major allele frequency of p and q in a total sample of n, we require sample sizes for women of n × p2, n × 2 × p × q, and n × q2 for the 3 genotypic groups, but sample sizes of n × p and n × q for the 2 genotypic groups in men). Our results in humans support our findings that KDM5C levels influence adiposity and also raise the possibility that variations in KDM5C-mediated histone modifications may contribute to individual differences in obesity.
We propose that genes that escape X inactivation have a role in determining several sex differences in normo- and pathophysiology. Recently, the X-escape gene Kdm6a was shown to modulate sex differences in T cell biology in a mouse model of experimental autoimmune encephalomyelitis (42). Both Kdm5c and Kdm6a belong to a special subgroup of X-escape genes that have paralogous genes on the Y chromosome (43). Since these X and Y paralogs do not recombine with one another, they have diverged over evolutionary time such that the gene pairs may no longer retain the same molecular functions. For example, in the case of Kdm6a (also known as Utx), in vivo complementation studies suggest that Uty (the Y chromosome paralog of Utx) cannot fully substitute for critical roles of Utx during embryonic development (44). The Y chromosome paralog of Kdm5c, known as Kdm5d, has been implicated in the regulation of gene expression in fibroblasts, in cardiomyocyte differentiation, and in renal cell carcinomas (45–47). KDM5C and KDM5D proteins exhibit conserved protein domains for histone demethylase activity, but experimentally determined H3K4 demethylase activity appeared substantially lower for KDM5D than KDM5C (48). Thus, it is possible that the physiological roles of Kdm5c and Kdm5d genes are not equivalent due to differences in protein activity, gene regulation, and/or other factors. Further studies to determine the physiological roles of such X-Y gene pairs should shed additional light on the regulation of sexually dimorphic traits.
In conclusion, our findings highlight the effects of sex chromosome complement — and particularly, the special class of X chromosome genes that escape inactivation — on sex differences in metabolism. Our studies demonstrate that sexual imbalance in Kdm5c expression levels influence diet-induced adipose tissue accrual. The experimental reduction of Kdm5c genomic dosage in XX females to that which is normally present in XY males led to reduced diet-induced adipose tissue expansion, likely regulated by changes in gene expression in processes such as ECM reorganization. Further studies will assess the potential contribution of KDM5C levels to processes that rely upon successful adipose tissue expansion, such as pregnancy, as well as the role of sex differences in Kdm5c gene dosage in tissues beyond adipose tissue.
Methods
Animals.
FCG C57BL/6J mice were from a colony maintained at UCLA. XX female mice were mated with XY–(Sry+) male mice to generate XX, XX(Sry+), XY–, and XY–(Sry+) offspring. Genotyping was performed by PCR to detect the presence of the Sry transgene (forward: AGCCCTACAGCCACATGATA; reverse: GTCTTGCCTGTATGTGATGG) and Y chromosome–specific sequence (forward: CTGGAGCTCTACAGTGATGA; reverse: CAGTTACCAATCAACACATCAC) (14).
Floxed Kdm5c mice have been described previously (49). At UCLA, floxed Kdm5c mice were backcrossed to C57BL/6J to generation N11 and then crossed with C57BL/6J Ella:Cre mice (Jackson Laboratory) to generate heterozygous knockouts of the floxed allele through Cre-lox recombination. Subsequent generations were crossed with C57BL/6J mice to eliminate Cre and the floxed allele. Because male Kdm5/–/Y mice were not viable on a C57BL/6 inbred background, we studied female XX Kdm5c+/+ and Kdm5c+/– mice produced by mating Kdm5c+/– females with WT males.
FCG mice were housed 2 per cage with identical genotypes, and mice of the Kdm5c gene dosage model were housed 2–4 per cage. All mice were maintained under a 12-hour light/12-hour dark cycle. Mice were initially fed a Purina Rodent Laboratory Chow diet containing 5%–6% fat from calories (Purina 5001). Where indicated, adult FCG and Kdm5c mice were fed a high-fat diet (S3282, Bio-Serve; 60% calories from fat, 26% from carbohydrate, 15% from protein).
Body composition and energy balance.
Mice were weighed at weekly intervals. Body composition was measured on conscious mice using a Mouse Minispec apparatus with Echo Medical Systems software. Food intake was measured per cage over three 24-hour periods at 0800 and 1800 hours. Mice were housed individually in calibrated Oxymax metabolic chambers (Columbus Instruments) to detect O2 consumption, CO2 production, locomotor activity, and heat production over 72 hours at weeks 1 and 10 of the high-fat diet. Mice had ad libitum access to food and water. Average RQ, cumulative heat production and cumulative locomotor activity were calculated from the second dark and second light intervals of the 72-hour measurement period. Heat production was determined as follows: ([3.815 + 1.232 × (observed respiratory exchange ratio [RER])] × [observed oxygen consumption (VO2)]). This parameter does not contain a body weight term and thus avoids complications that could occur due to differences in body weight.
Glucose homeostasis.
Blood samples were obtained by retroorbital bleeding at time of dissection after fasting (0800–1300 hours). Plasma was prepared by centrifugation of whole blood at 3,400g for 10 minutes at 4°C for glucose and insulin measurements (50). HOMA-IR was calculated using the following equation: ([glucose mmol/L × insulin microunits/mL]/22.5).
Glucose tolerance tests were performed after 10 weeks of a high-fat diet. After fasting (0800–1300 hours), mice were administered an intraperitoneal glucose bolus (2 mg per gram body weight). Glucose measurements were performed at 0, 15, 30, 60, 120, and 180 minutes with an AlphaTrak Glucometer by tail nick.
Quantitative RT-PCR.
Gonadal and inguinal fat tissues were flash-frozen in liquid nitrogen and stored at –80°C. RNA was isolated from tissue using TRIzol (Life Technologies). First-strand cDNA was generated with iScript (Bio-Rad). Quantitative reverse-transcription PCR (RT-PCR) was performed with a Bio-Rad CFX Connect Real-Time PCR Detection System using SsoAdvanced SYBR Green Supermix and 36B4 mRNA as a normalization control. Primers were as follows: Kdm5c (ACCCACCTGGCAAAAACATTGG, ACTGTCGAAGGGGGATGCTGTG); Ddx3x (GGATCACGGGGTGATTCAAGAGG, CTATCTCCACGGCCACCAATGC); Kdm6a (CCAATCCCCGCAGAGCTTACCT, TTGCTCGGAGCTGTTCCAAGTG); Eif2s3x (TTGTGCCGAGCTGACAGAATGG, CGACAGGGAGCCTATGTTGACCA); and 36B4 (AGATGCAGCAGATCCGCAT, GTTCTTGCCCATCAGCACC).
GTEx human gene expression data.
Gene expression data for KDM5C was obtained from the Genotype-Tissue Expression (GTEx) Consortium, a large-scale RNA-Seq project that characterized the human transcriptome across numerous tissues obtained from hundreds of human donors as postmortem samples (51, 52).
Adipose tissue histology.
Adipose tissues were fixed overnight in 10% formalin. Sections of 7 μm were obtained and stained with H&E at the UCLA Translational Pathology Core Laboratory. For adipocyte area measurements, sections were digitalized (Zeiss Observer D1 with ×10 objective) and the cell surface quantified with the Adiposoft plugin in FIJI in arbitrary units (53). Frequency counts were performed using bins at increments of 250.
Adipose tissue fractionation and Western blot analysis.
Adipose tissue from 2 mice was pooled and minced in digestion buffer (100 mM HEPES, 120 mM NaCl, 50 mM KCl, 5 mM glucose, 1 mM CaCl2, 1.5% BSA) containing 20 mg/mL collagenase I (MilliporeSigma). After 1 hour digestion at 37°C, the sample was filtered through a 70 μm mesh and debris was pelleted at 50g. The supernatant and top fat layer were spun at 200g to separate the stromal vascular (pellet) and mature adipocyte (top layer) fractions. Each fraction was resuspended in 100 μl lysis buffer (10 mM Tris pH 7.5, 10 mM NaCl, 1 mM EDTA, 0.5% Triton X-100) containing protease and phosphatase inhibitors. Protein levels were analyzed by Western blot as described (54). Primary antibodies used were as follows: KDM5C (Active Motif, catalog 39229), FABP4 (Abcam, catalog ab92501), and GAPDH (GeneTex, catalog GTX100118).
Kdm5c knockdown in 3T3-L1 preadipocytes.
3T3-L1 preadipocytes (Zen-Bio; karyotype XY, 2n = 40, but diploid chromosome number may be unstable) were reverse-transfected with 50 nM nontargeting (control) or Kdm5c siRNA (Dharmacon) with Lipofectamine RNAiMAX (Thermo Fisher Scientific) at differentiation day –3 (subconfluent) or –2 (confluent). For RNA isolation (transcriptome analysis) and nucleus isolation (ATAC-Seq), cells were collected at 24 hours (day –2), 48 hours (day –1), and 72 hours (day 0) after transfection. For proliferation analysis, cells were replated at a low density 24 hours after transfection and cell counts were performed every 24 hours. For analysis of differentiation, differentiation medium was added on day 0 and maintenance medium (all adipocyte media obtained from Zen-Bio) was added on day 2. At day 6, cells were formalin fixed, washed with 60% isopropanol, and stained with ORO (0.20% w/v). After imaging stained cells, ORO was solubilized with 100% isopropanol and quantified by spectrophotometry at an OD of 500 nm.
RNA-Seq analysis.
Total RNA was isolated from 3T3-L1 cells or fractionated adipose tissue (mature adipocytes or stromal vascular cells) with miRNeasy Micro Kit (QIAGEN), and integrity was assessed with the 4200 TapeStation System (Agilent). Strand-specific RNA-Seq libraries were constructed with the Universal Plus mRNA-Seq Kit (NuGEN Technologies). The workflow consisted of poly(A) RNA selection, RNA fragmentation, and double-stranded cDNA synthesis with a mixture of random and oligo(dT) priming, followed by end repair to generate blunt ends, adaptor ligation, strand selection, and PCR amplification to produce the final libraries. Amplified libraries were quantified by Qubit dsDNA High Sensitivity Assay Kit and quality checked with the 4200 TapeStation System (Agilent). Different index adaptors were used for multiplexing samples, and sequencing was performed with Illumina NovaSeq 6000 sequencer on a NovaSeq S2 flow cell to produce 50 bp paired-end reads (2 × 50 bp). RNA-Seq alignment was performed with STAR, version 2.7.2a, to the mouse genome GRCm38.97, and the read count per gene was quantified with STAR and the Ensembl Mus_musculus.GRCm38.97 GTF file (55). The average read depth was 36 × 106 reads/sample, and an average of 86% of reads was uniquely aligned to the genome.
Differential gene expression was identified with EdgeR, version 3.26.8, with trimmed mean of M values (TMM) normalization and significance denoted as adjusted P < 0.05. Heatmaps were generated with gplots heatmap.2, version 3.0.1.1, and volcano plots with customized R function. RNA-Seq of 3T3-L1 cells was performed with n = 4 control and n = 4 Kdm5c knockdown samples; RNA-Seq of fractionated mouse tissues was performed with stromal vascular and mature adipocyte samples from 3 Kdm5c+/+ and 3 Kdm5c+/– mice.
ATAC-Seq analysis.
For ATAC-Seq, 50,000 cells per sample were lysed to collect nuclei and treated with Tn5 transposase (Illumina) for 30 minutes at 37°C with gentle agitation, as previously described (56). The DNA was isolated with DNA Clean & Concentrator Kit (Zymo) and PCR amplified and barcoded with NEBNext High-Fidelity PCR Mix (New England Biolabs) and unique dual indexes (Illumina). The ATAC-Seq library amplification was confirmed by real-time PCR, and additional barcoding PCR cycles were added as necessary while avoiding overamplification. Amplified ATAC-Seq libraries were purified with DNA Clean & Concentrator Kit (Zymo). The purified libraries were quantified with Kapa Library Quant Kit (KAPA Biosystems) and quality assessed on 4200 TapeStation System (Agilent). The libraries were pooled based on molar concentrations and sequenced on an Illumina NovaSeq 6000 S2 at an average read depth of 40 × 106 reads/sample at the UCLA Technology Center for Genomics and Bioinformatics.
ATAC-Seq fastq files were trimmed to remove low-quality reads and adapters using Cutadapt, version 2.3 (code.google.com/p/cutadapt/). The reads were aligned to the reference mouse GRCm38/mm10 genome with bowtie2, version 2.2.9 (57); typical alignment was greater than 95%. Peak calling was performed with model-based analysis of ChIP-Seq (MACS2, version 2.1.1) with a q value of 0.05 (58). The peaks for each experimental group were merged into a single bed file, and HTseq, version 0.9.1, was used to count the number of reads that overlap each peak per sample (59). The peak counts were analyzed with DESeq2, version 1.24.0, to identify differentially accessible genomic regions, with significance counted as P < 0.05 (60). The peak counts were visualized with IGV, version 2.6.2 (61). The genomic locations of the peaks were determined and visualized with ChIPseeker, version 1.20.0. (62). ATAC-Seq of 3T3-L1 cells was performed with n = 4 control and n = 4 Kdm5c knockdown samples.
Pathway enrichment analysis.
Pathway analysis for RNA-Seq data was performed with Enrichr (63, 64) for the differentially expressed genes with adjusted P < 0.05 and fold change greater than 1.25. Pathway analysis for ATAC-Seq was performed using peaks with greater than 1.5-fold change upon Kdm5c knockdown and that corresponded to genes that were differentially expressed (adjusted P < 0.05) in the day 0 RNA-Seq analysis.
Human gene expression association analyses.
RNA-Seq data from subcutaneous adipose tissue punch biopsies were available for 766 twins from the TwinsUK study (27). RNA-Seq reads were aligned to the hg19 reference genome using STAR, version 2.4.0.1 (55), as previously described (65). Gene and exon-level quantification was conducted using the Quan tool in the QTLtools package (66). TMM-value normalization (67) was applied to exon counts, and TMM-normalized counts per million (CPMs) were then inverse normalized. Subjects who had not fasted on the day of biopsy were excluded from all further analyses. Inverse normal transformation was applied to all quantitative traits.
For gene expression association analysis, a mixed effects model was then fitted using the lmer function from the lme4 package (68) in R, version 3.3.2. Inverse-normalized exon counts for each exon were treated as a continuous dependent variable and BMI as an independent variable. Covariates added to the model included age, time since last meal, mean GC content, median insert size, and median transcript integrity number (69) as fixed effects; and morning versus afternoon appointment time, date of RNA-Seq, RNA extraction batch, primer index, family,and zygosity as random effects. Significance was calculated by comparing the full model, including BMI, to a null model in which BMI was omitted, using a 1 degree of freedom ANOVA.
Human genetic association.
We tested SNPs in Multi-Ethnic Study of Atherosclerosis (MESA) 1000G phase 1 imputed data at the KDM5C gene locus (Chr:53220000–53254604, genome release GRCh37/hg19) if the sample size in the minor allele carrier group was 10 or greater. MESA has 4 population samples: AA, HA, Han Chinese, and EA, with a sample size of 385–1400 in each ethnic/sex group. The number of SNPs in the KDM5C gene that met the minimum sample requirement for association analysis was different for each population and between men and women. Power analyses performed using QUANTO (https://bio.tools/QUANTO) showed that in samples that contained at least 700 individuals/ethnic group/sex, we had 87% power to detect association if a SNP explained 3% of the phenotypic variance. We therefore carried out association analysis separately for men and women within the AA, HA, and EA groups, which have 700 or more samples in each analysis group.
We assumed an additive model using linear regression, accounting for genotype imputation uncertainty. To adjust for potential confounding effect, we included age, age2, study site, and top 2 principle components (PC1, PC2) in the testing model. The plink.multivariate package in the PLINK program was used in the multivariate analysis for BMI-WHR (70). Given that Bonferroni’s correction is not appropriate here owing to LD structure, we used the Haploview program to investigate the LD structure within each population to define independent blocks/SNPs to adjust for the number of independent tests. Ultimately, significant associations were identified for the AA and HA groups at α = 0.05, determined as follows: in AA men, there were 5 LD blocks and 2 independent SNPs; in AA women, there were 4 LD blocks and 27 independent SNPs; in HA men, there were 2 LD blocks and 4 independent SNPs; and in HA women, there was 1 LD block (Supplemental Figure 6). We used P value cutoffs for significant association as follows: 0.05/(no. of independent blocks + SNPs)/4 study populations, i.e., 0.0014 ( = 0.05/(5 +2 )/4) for AA men; 0.0004 (P = 0.05/(4 + 27)/4) for AA women; 0.0021 (P = 0.05/(2 + 4)/4) for HA men; and 0.0125 (P = 0.05/1/4) for HA women.
Availability of data, code, and biological materials.
Data sets have been deposited in relevant databases. RNA-Seq and ATAC-Seq data associated with Figures 5 and 6 are available in NCBI’s Gene Expression Omnibus database (GEO GSE147994, GSE147997, GSE147998). The human subjects data associated with Table 1 are available in dgGAP (phs000209.v13.p3).
Code used for analysis of RNA-Seq and ATAC-Seq is all freely available from developers, as cited in the Methods section. Data analyses were performed with the following: RNA-Seq alignment with STAR, version 2.4.0.1 or version 2.7.2a; differential gene expression with EdgeR, version 3.26.8; heatmap generation with gplots heatmap.2, version 3.0.1.1; volcano plots with customized R function; Cutadapt, version 2.3 for ATAC-Seq fastq file trimming; read alignment with bowtie2, version 2.2.9; ATAC-Seq peak calling with MACS2, version 2.1.1; peak merging with HTseq, version 0.9.1; differential peak counts with DESeq2, version 1.24.0; peak visualization with IGV, version 2.6.2; peak genomic location with ChIPseeker, version 1.20.0; pathway enrichment analysis with Enrichr; human gene and exon-level quantitation with Quan tool in the QTLtools package; gene expression association analysis using Ime4; power analysis for human SNPs performed using QUANTO; multivariate analysis with PLINK; and LD structure with Haploview.
We will provide mouse strains and cell lines described in this study upon request to noncommercial entities for research purposes in agreement with University of California regulations.
Statistics.
In mouse studies, groups were compared using 2-factor ANOVA (NCSS) with main factors of sex (gonadal male vs. gonadal female) and sex chromosome complement (XX vs. XY), and pairs were compared using Student’s t test (2 tailed). For growth curves and the glucose tolerance test, repeated measures ANOVA was performed using time as a variable. For in vitro studies, pairs were assessed with Mann-Whitney nonparametric U tests, and groups were compared using 2-factor ANOVA. Statistically significant comparisons or interactions are presented, with significance defined as P < 0.05. All error bars represent 1 SD.
Study approval.
Mouse studies were conducted in accordance with and were approved by the Institutional Animal Research Committee of UCLA. The TwinsUK project was approved by the Ethics Committee at St Thomas’ Hospital London, where all the biopsies were carried out. Volunteers gave informed consent and signed an approved consent form before biopsy procedures. Volunteers were supplied with an appropriate detailed information sheet regarding the research project and biopsy procedure by mail before biopsies were taken. All MESA participants provided informed consent, and the study was approved by the Institutional Review Board at the Lundquist Institute (formerly Los Angeles BioMedical Research Institute) at Harbor-UCLA Medical Center (IRB protocol 21030-01) or by the Institutional Review Board at the University of Washington (IRB protocol STUDY00009029).
Author contributions
JCL, CBW, APA, and KR designed studies and wrote the manuscript. JCL, XC, and ER performed mouse studies. CBW and LV performed cell culture studies. CBW, RA, FM, MP, and KR analyzed RNA-Seq and ATAC-Seq data. XG, JY, MA, YDIC, and JIR performed human genetic association studies. JSESM and KSS analyzed the TwinsUK gene expression data. SI provided Kdm5c floxed mice. The designation of co–first authors was based on the critical complementary contributions to in vivo studies (JCL) and in vitro studies (CBW), and the order was determined by initial intellectual contributions by JCL.
Supplementary Material
Acknowledgments
The authors thank Maria Palafox, Kristin Harrington, Nikko Gonzales, Ryan Mackie, Haley Hrncir, and Christian O’Donnell for technical assistance. We thank Yang Shi for making Kdm5c-floxed mice freely available. The graphical abstract and model figure were created with BioRender.com. The authors gratefully acknowledge support from the National Institute of Diabetes and Digestive and Kidney Diseases (National Public Health Service U54 DK120324 to KR; R01 DK83561 to APA, XC, and KR), the Ruth L. Kirschstein National Research Service Award (GM007185 to JCL), and the American Heart Association (20POST35100000 to CBW). The GTEx Project was supported by the Common Fund of the Office of the Director of the NIH and by the National Cancer Institute, the National Human Genome Research Institute, the National Heart, Lung, and Blood Institute (NHLBI), the National Institute on Drug Abuse, the National Institute of Mental Health, and the National Institute of Neurological Disorders and Stroke. KSS and JSESM acknowledge the Medical Research Council (MR/M004422/1). MESA and the MESA SHARe project are conducted and supported by the NHLBI in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420, UL1-TR-001881, and DK063491. Generation of TwinsUK gene expression data was funded by European Union Framework Programme 7 grant EuroBATS (no. 259749). TwinsUK is funded by the Wellcome Trust, the Medical Research Council, the European Union, the Chronic Disease Research Foundation (CDRF), Zoe Global Ltd., and the National Institute for Health Research–funded (NIHR-funded) BioResource, Clinical Research Facility, and Biomedical Research Centre based at Guy’s and St. Thomas’ National Health Service (NHS) Foundation Trust in partnership with King’s College London. The authors thank the twins for their participation. The authors also acknowledge use of the research computing facility at King’s College London, Rosalind (https://rosalind.kcl.ac.uk), which is delivered in partnership with the NIHR Biomedical Research Centres at South London and Maudsley and Guy’s and St. Thomas’ NHS Foundation Trusts and partially funded by capital equipment grants from the Maudsley Charity (award 980) and Guy’s and Thomas’ Charity (TR130505). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, King’s College London, or the Department of Health and Social Care.
Version 1. 07/23/2020
In-Press Preview
Version 2. 09/28/2020
Electronic publication
Version 3. 11/02/2020
Print issue publication
Footnotes
JCL’s present address is: Cell and Molecular Biology, Whittier College, Whittier, California, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(11):5688–5702.https://doi.org/10.1172/JCI140223.
Contributor Information
Jenny C. Link, Email: jennychen@ucla.edu.
Carrie B. Wiese, Email: CWiese@mednet.ucla.edu.
Xuqi Chen, Email: xuqichen@physci.ucla.edu.
Rozeta Avetisyan, Email: rozeta@g.ucla.edu.
Emilio Ronquillo, Email: eronquillo@ucla.edu.
Feiyang Ma, Email: mafeiyang321@gmail.com.
Xiuqing Guo, Email: xguo@labiomed.org.
Jie Yao, Email: jyao@lundquist.org.
Matthew Allison, Email: mallison@ucsd.edu.
Jerome I. Rotter, Email: jrotter@lundquist.org.
Kerrin S. Small, Email: kerrin.small@kcl.ac.uk.
Shigeki Iwase, Email: siwase@umich.edu.
Matteo Pellegrini, Email: matteop@mcdb.ucla.edu.
Laurent Vergnes, Email: lvergnes@ucla.edu.
Arthur P. Arnold, Email: arnold@ucla.edu.
Karen Reue, Email: reuek@ucla.edu.
References
- 1.Neeland IJ, et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: a position statement. Lancet Diabetes Endocrinol. 2019;7(9):715–725. doi: 10.1016/S2213-8587(19)30084-1. [DOI] [PubMed] [Google Scholar]
- 2.Link JC, Reue K. Genetic basis for sex differences in obesity and lipid metabolism. Annu Rev Nutr. 2017;37:225–245. doi: 10.1146/annurev-nutr-071816-064827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fried SK, Lee MJ, Karastergiou K. Shaping fat distribution: New insights into the molecular determinants of depot- and sex-dependent adipose biology. Obesity (Silver Spring) 2015;23(7):1345–1352. doi: 10.1002/oby.21133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zore T, Palafox M, Reue K. Sex differences in obesity, lipid metabolism, and inflammation — A role for the sex chromosomes? Mol Metab. 2018;15:35–44. doi: 10.1016/j.molmet.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gambacciani M, et al. Body weight, body fat distribution, and hormonal replacement therapy in early postmenopausal women. J Clin Endocrinol Metab. 1997;82(2):414–417. doi: 10.1210/jcem.82.2.3735. [DOI] [PubMed] [Google Scholar]
- 6.Haarbo J, Marslew U, Gotfredsen A, Christiansen C. Postmenopausal hormone replacement therapy prevents central distribution of body fat after menopause. Metab Clin Exp. 1991;40(12):1323–1326. doi: 10.1016/0026-0495(91)90037-W. [DOI] [PubMed] [Google Scholar]
- 7.Burgoyne PS, Thornhill AR, Boudrean SK, Darling SM, Bishop CE, Evans EP. The genetic basis of XX-XY differences present before gonadal sex differentiation in the mouse. Philos Trans R Soc Lond, B, Biol Sci. 1995;350(1333):253–60 discussion 260. doi: 10.1098/rstb.1995.0159. [DOI] [PubMed] [Google Scholar]
- 8.Bukowski R, et al. Human sexual size dimorphism in early pregnancy. Am J Epidemiol. 2007;165(10):1216–1218. doi: 10.1093/aje/kwm024. [DOI] [PubMed] [Google Scholar]
- 9.Aksglaede L, Molgaard C, Skakkebaek NE, Juul A. Normal bone mineral content but unfavourable muscle/fat ratio in Klinefelter syndrome. Arch Dis Child. 2008;93(1):30–34. doi: 10.1136/adc.2007.120675. [DOI] [PubMed] [Google Scholar]
- 10.Bardsley MZ, Falkner B, Kowal K, Ross JL. Insulin resistance and metabolic syndrome in prepubertal boys with Klinefelter syndrome. Acta Paediatr. 2011;100(6):866–870. doi: 10.1111/j.1651-2227.2011.02161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnold AP, Chen X. What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front Neuroendocrinol. 2009;30(1):1–9. doi: 10.1016/j.yfrne.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mauvais-Jarvis F, Arnold AP, Reue K. A guide for the design of pre-clinical studies on sex differences in metabolism. Cell Metab. 2017;25(6):1216–1230. doi: 10.1016/j.cmet.2017.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, et al. The number of x chromosomes causes sex differences in adiposity in mice. PLoS Genet. 2012;8(5):e1002709. doi: 10.1371/journal.pgen.1002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burgoyne PS, Arnold AP. A primer on the use of mouse models for identifying direct sex chromosome effects that cause sex differences in non-gonadal tissues. Biol Sex Differ. 2016;7:68. doi: 10.1186/s13293-016-0115-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berletch JB, Yang F, Disteche CM. Escape from X inactivation in mice and humans. Genome Biol. 2010;11(6):213. doi: 10.1186/gb-2010-11-6-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tukiainen T, et al. Landscape of X chromosome inactivation across human tissues. Nature. 2017;550(7675):244–248. doi: 10.1038/nature24265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harmeyer KM, Facompre ND, Herlyn M, Basu D. JARID1 histone demethylases: emerging targets in cancer. Trends Cancer. 2017;3(10):713–725. doi: 10.1016/j.trecan.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu C, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10(11):R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brier AB, et al. The KDM5 family is required for activation of pro-proliferative cell cycle genes during adipocyte differentiation. Nucleic Acids Res. 2017;45(4):1743–1759. doi: 10.1093/nar/gkw1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, et al. Sex differences in diurnal rhythms of food intake in mice caused by gonadal hormones and complement of sex chromosomes. Horm Behav. 2015;75:55–63. doi: 10.1016/j.yhbeh.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bora P, Majumdar AS. Adipose tissue-derived stromal vascular fraction in regenerative medicine: a brief review on biology and translation. Stem Cell Res Ther. 2017;8(1):145. doi: 10.1186/s13287-017-0598-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Croissandeau G, Chrétien M, Mbikay M. Involvement of matrix metalloproteinases in the adipose conversion of 3T3-L1 preadipocytes. Biochem J. 2002;364(Pt 3):739–746. doi: 10.1042/BJ20011158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang JW, Klemm DJ, Vinson C, Lane MD. Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein beta gene during adipogenesis. J Biol Chem. 2004;279(6):4471–4478. doi: 10.1074/jbc.M311327200. [DOI] [PubMed] [Google Scholar]
- 25.Wu Z, Wang S. Role of kruppel-like transcription factors in adipogenesis. Dev Biol. 2013;373(2):235–243. doi: 10.1016/j.ydbio.2012.10.031. [DOI] [PubMed] [Google Scholar]
- 26.Takata Y, Kitami Y, Yang ZH, Nakamura M, Okura T, Hiwada K. Vascular inflammation is negatively autoregulated by interaction between CCAAT/enhancer-binding protein-delta and peroxisome proliferator-activated receptor-gamma. Circ Res. 2002;91(5):427–433. doi: 10.1161/01.RES.0000031271.20771.4F. [DOI] [PubMed] [Google Scholar]
- 27.Buil A, et al. Gene-gene and gene-environment interactions detected by transcriptome sequence analysis in twins. Nat Genet. 2015;47(1):88–91. doi: 10.1038/ng.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bild DE, et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol. 2002;156(9):871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 29.Vallianatos CN, Iwase S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics. 2015;7(3):503–519. doi: 10.2217/epi.15.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Cubas AA, Rathmell WK. Epigenetic modifiers: activities in renal cell carcinoma. Nat Rev Urol. 2018;15(10):599–614. doi: 10.1038/s41585-018-0052-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crewe C, An YA, Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest. 2017;127(1):74–82. doi: 10.1172/JCI88883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruiz-Ojeda FJ, Méndez-Gutiérrez A, Aguilera CM, Plaza-Díaz J. Extracellular matrix remodeling of adipose tissue in obesity and metabolic diseases. Int J Mol Sci. 2019;20(19):E4888. doi: 10.3390/ijms20194888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin D, Chun TH, Kang L. Adipose extracellular matrix remodelling in obesity and insulin resistance. Biochem Pharmacol. 2016;119:8–16. doi: 10.1016/j.bcp.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Y, Lee MJ, Ido Y, Fried SK. High-fat diet-induced obesity regulates MMP3 to modulate depot- and sex-dependent adipose expansion in C57BL/6J mice. Am J Physiol Endocrinol Metab. 2017;312(1):E58–E71. doi: 10.1152/ajpendo.00128.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richard AJ, Stephens JM. The role of JAK-STAT signaling in adipose tissue function. Biochim Biophys Acta. 2014;1842(3):431–439. doi: 10.1016/j.bbadis.2013.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dean JT, et al. Resistance to diet-induced obesity in mice with synthetic glyoxylate shunt. Cell Metab. 2009;9(6):525–536. doi: 10.1016/j.cmet.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Smith DL, Keating KD, Allison DB, Nagy TR. Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice. Obesity (Silver Spring) 2014;22(10):2147–2155. doi: 10.1002/oby.20811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang LL, et al. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity (Silver Spring) 2010;18(3):463–469. doi: 10.1038/oby.2009.273. [DOI] [PubMed] [Google Scholar]
- 39.Dakin RS, Walker BR, Seckl JR, Hadoke PW, Drake AJ. Estrogens protect male mice from obesity complications and influence glucocorticoid metabolism. Int J Obes (Lond) 2015;39(10):1539–1547. doi: 10.1038/ijo.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parks BW, et al. Genetic architecture of insulin resistance in the mouse. Cell Metab. 2015;21(2):334–347. doi: 10.1016/j.cmet.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salinero AE, Anderson BM, Zuloaga KL. Sex differences in the metabolic effects of diet-induced obesity vary by age of onset. Int J Obes (Lond) 2018;42(5):1088–1091. doi: 10.1038/s41366-018-0023-3. [DOI] [PubMed] [Google Scholar]
- 42.Itoh Y, et al. The X-linked histone demethylase Kdm6a in CD4+ T lymphocytes modulates autoimmunity. J Clin Invest. 2019;129(9):3852–3863. doi: 10.1172/JCI126250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jegalian K, Page DC. A proposed path by which genes common to mammalian X and Y chromosomes evolve to become X inactivated. Nature. 1998;394(6695):776–780. doi: 10.1038/29522. [DOI] [PubMed] [Google Scholar]
- 44.Shpargel KB, Sengoku T, Yokoyama S, Magnuson T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012;8(9):e1002964. doi: 10.1371/journal.pgen.1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mizukami H, et al. KDM5D-mediated H3K4 demethylation is required for sexually dimorphic gene expression in mouse embryonic fibroblasts. J Biochem. 2019;165(4):335–342. doi: 10.1093/jb/mvy106. [DOI] [PubMed] [Google Scholar]
- 46.Meyfour A, Pahlavan S, Ansari H, Baharvand H, Salekdeh GH. Down-regulation of a male-specific H3K4 demethylase, KDM5D, impairs cardiomyocyte differentiation. J Proteome Res. 2019;18(12):4277–4282. doi: 10.1021/acs.jproteome.9b00395. [DOI] [PubMed] [Google Scholar]
- 47.Arseneault M, et al. Loss of chromosome Y leads to down regulation of KDM5D and KDM6C epigenetic modifiers in clear cell renal cell carcinoma. Sci Rep. 2017;7:44876. doi: 10.1038/srep44876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwase S, et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128(6):1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 49.Iwase S, et al. A mouse model of X-linked intellectual disability associated with impaired removal of histone methylation. Cell Rep. 2016;14(5):1000–1009. doi: 10.1016/j.celrep.2015.12.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Link JC, et al. Increased high-density lipoprotein cholesterol levels in mice with XX versus XY sex chromosomes. Arterioscler Thromb Vasc Biol. 2015;35(8):1778–1786. doi: 10.1161/ATVBAHA.115.305460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carithers LJ, et al. A novel approach to high-quality postmortem tissue procurement: the GTEx project. Biopreserv Biobank. 2015;13(5):311–319. doi: 10.1089/bio.2015.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang P, et al. Lipin 2/3 phosphatidic acid phosphatases maintain phospholipid homeostasis to regulate chylomicron synthesis. J Clin Invest. 2019;129(1):281–295. doi: 10.1172/JCI122595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31(14):2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 63.Chen EY, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glastonbury CA, Couto Alves A, El-Sayed Moustafa JS, Small KS. Cell-type heterogeneity in adipose tissue is associated with complex traits and reveals disease-relevant cell-specific eQTLs. Am J Hum Genet. 2019;104(6):1013–1024. doi: 10.1016/j.ajhg.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Delaneau O, Ongen H, Brown AA, Fort A, Panousis NI, Dermitzakis ET. A complete tool set for molecular QTL discovery and analysis. Nat Commun. 2017;8:15452. doi: 10.1038/ncomms15452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using Ime4. Journal of Statistical Software. 2015;67(1):51 [Google Scholar]
- 69.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28(16):2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 70.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.