

Abstract
BACKGROUND
While mitochondria play an important role in innate immunity, the relationship between mitochondrial dysfunction and inflammation in heart failure (HF) is poorly understood. In this study we aimed to investigate the mechanistic link between mitochondrial dysfunction and inflammatory activation in peripheral blood mononuclear cells (PBMCs), and the potential antiinflammatory effect of boosting the NAD level.
METHODS
We compared the PBMC mitochondrial respiration of 19 hospitalized patients with stage D HF with that of 19 healthy participants. We then created an in vitro model of sterile inflammation by treating healthy PBMCs with mitochondrial damage–associated molecular patterns (MitoDAMPs) isolated from human heart tissue. Last, we enrolled patients with stage D HF and sampled their blood before and after taking 5 to 9 days of oral nicotinamide riboside (NR), a NAD precursor.
RESULTS
We demonstrated that HF is associated with both reduced respiratory capacity and elevated proinflammatory cytokine gene expressions. In our in vitro model, MitoDAMP-treated PBMCs secreted IL-6 that impaired mitochondrial respiration by reducing complex I activity. Last, oral NR administration enhanced PBMC respiration and reduced proinflammatory cytokine gene expression in 4 subjects with HF.
CONCLUSION
These findings suggest that systemic inflammation in patients with HF is causally linked to mitochondrial function of the PBMCs. Increasing NAD levels may have the potential to improve mitochondrial respiration and attenuate proinflammatory activation of PBMCs in HF.
TRIAL REGISTRATION
ClinicalTrials.gov NCT03727646.
FUNDING
This study was funded by the NIH, the University of Washington, and the American Heart Association.
Keywords: Cardiology, Inflammation
Keywords: Cardiovascular disease, Drug therapy, Mitochondria
Introduction
Worldwide, heart failure (HF) is among the top causes of morbidity and mortality. In the United States alone, annual direct medical expenditures for HF exceed $30 billion (1–3). It has long been postulated that a chronic sterile inflammatory state in HF may be involved in disease progression and may contribute to worse clinical outcomes (4, 5). While proinflammatory cytokines such as TNF-α, IL-1, IL-6, and IL-18 have been demonstrated to have a negative inotropic effect in various experimental models (6), results of anti–TNF-α clinical trials in HF were discouraging (7, 8). More recently, the CANTOS trial of anti–IL-1B monoclonal antibody therapy in atherosclerosis showed promising results in the HF subgroup analysis (9). These conflicting results likely reflect the complexity and intricacy of the cytokine-mediated mechanisms of HF. A better understanding of the molecular mechanisms by which peripheral monocytes process and respond to proinflammatory signals is imperative to the development of targeted novel therapeutics for HF.
Mitochondrial dysfunction contributes to the development of HF via multiple mechanisms (10). Damage-associated molecular pattern released from mitochondria (MitoDAMP) has been implicated to activate the TLR/NFKB axis of peripheral monocytes in cardiac injury, leading to cytokine production and systemic inflammation (4, 5, 11). Furthermore, production of reactive oxygen species (ROS) by mitochondria has been proposed as a critical step in the activation of NLRP3 inflammasome and secretion of proinflammatory cytokines (12, 13). To date, it has not been determined whether mitochondrial dysfunction of circulating immune cells contributes to the heightened inflammatory state in patients with HF.
Recently, nicotinamide riboside (NR), a precursor in the NAD salvage pathway (14), has been demonstrated to enhance respiratory function, reduce mitochondrial ROS (mtROS) production, and reduce IL-1B production in peripheral blood mononuclear cells (PBMCs) (15). In addition, NR was shown to reduce the production of TNF-α and IL-6 and upregulate antiinflammatory molecule, adiponectin, in mouse hepatocytes (16) and reduce circulating of IL-5 and IL-6 in healthy elderly subjects (17). Last, systemic NAD repletion by NR or NMN (nicotinamide mononucleotide) has been shown to be cardioprotective in multiple murine models of cardiomyopathy (18–20). These observations raise the possibility that targeting NAD metabolism might benefit patients with HF by modulating the HF-associated inflammatory state.
To further explore this possibility we (a) compared PBMC respiratory function and inflammatory cytokine expression of patients with heart failure with reduced ejection fraction (HFrEF) and healthy participants, (b) investigated the mechanistic link between mitochondrial dysfunction and inflammatory activation in PBMCs, and (c) explored the possibility that targeting mitochondrial metabolism by increasing NAD level with NR might attenuate PBMC inflammatory activation in vitro and in patients with HFrEF.
Results
Baseline characteristics of study participants.
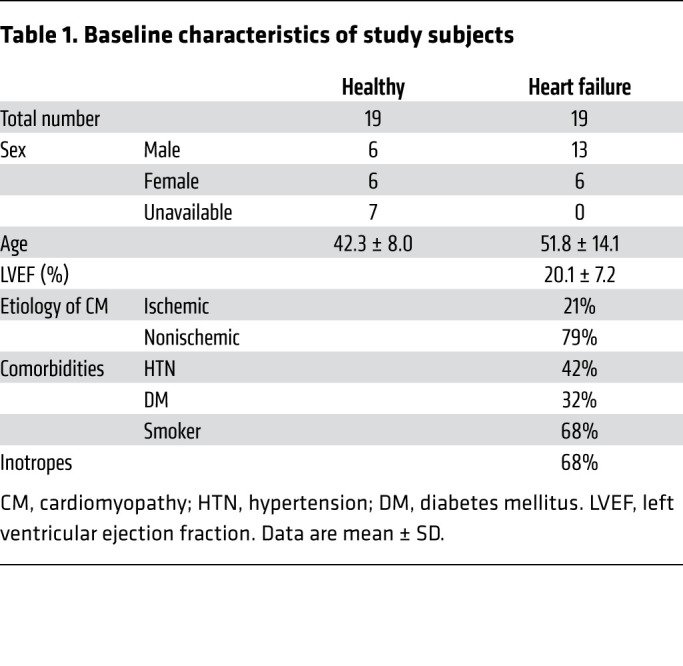
A total of 19 patients with stage D HF and 19 healthy participants were recruited. As shown in Table 1, the subjects with HF were predominantly male with a mean left ventricular ejection fraction (LVEF) of 20% ± 7%, among which approximately 80% had nonischemic etiology of cardiomyopathy, and 68% were on inotropic support when their blood samples were obtained. Mean age comparison between healthy and HF groups by Mann-Whitney test showed a P value of 0.034.
Table 1. Baseline characteristics of study subjects.
PBMCs from patients with HFrEF showed reduced respiratory capacity and elevated proinflammatory cytokine gene expression.
Purified PBMCs from study participants were subjected to the standard Seahorse Mito Stress Test (Figure 1A and ref. 21). Mean PBMC basal respiration (oxygen consumption rate [OCR]) trended lower in subjects with HF than healthy subjects though not reaching statistical significance (Figure 1B). The FCCP-induced (trifluoromethoxy carbonylcyanide phenylhydrazone–induced) maximal OCR was significantly lower in patients with HF as compared with healthy participants (Figure 1C). Together, these findings suggest that PBMC respiratory capacity is impaired in patients with HFrEF. Furthermore, consistent with previous reports that HFrEF is associated with a proinflammatory state, we found that mRNA levels of NLRP3, a key component of the inflammasome in monocytes and macrophages, as well as proinflammatory cytokines (IL-1B, IL-18, TNF-α), were significantly higher in patients with stage D HFrEF as compared with healthy participants. IL-6 showed a similar trend but did not reaching statistical significance (Figure 1D). Although subjects with HF had a slightly higher monocyte-to-lymphocyte ratio in the PBMCs (Supplemental Table 2; supplemental material available online with this article; https://doi.org/10.1172/JCI138538DS1), it is unlikely that it accounted for the reduced maximal respiration or increased cytokine expression. It has been shown that basal and maximal OCR per microgram protein are comparable between healthy monocytes and lymphocytes (22), and monocytes are roughly 2 times larger in volume than lymphocytes (23, 24). Thus, a higher fraction of monocytes in PBMCs would increase rather than decrease OCR. Moreover, the IL-1B, IL-6, and TNF-α productions in response to LPS simulation were shown to be comparable between monocytes and PBMCs (25).
Figure 1. Heart failure is associated with a reduced maximal respiration and elevated proinflammatory cytokine gene expressions in PBMCs.

(A) Representative OCR plot upon various inhibitor injections in a standard Seahorse Mito stress test, comparing PBMCs from healthy subjects and those with stage D HF. Oligomycin A: inhibitor of complex V. FCCP (trifluoromethoxy carbonylcyanide phenylhydrazone): uncoupling agent by permeabilizing inner mitochondrial membrane. Antimycin A: inhibitor of complex III. Rotenone: inhibitor of complex I. (B and C) Basal and FCCP-induced maximal respiration of PBMCs from healthy subjects (n = 19) and subjects with stage D HF (n = 19), respectively. OCR data normalized via log10 transformation were subjected to ordinary unpaired 2-tailed parametric test (Welch’s t test). Normal distribution was assessed by Kolmogorov-Smirnov test. (D) Relative mRNA levels of NLRP3 and proinflammatory cytokines of PBMCs of healthy subjects and those with stage D HF by RT-qPCR. NLRP3 (healthy n = 9, HF n = 9), IL-1B (n = 9, n = 9), IL-6 (n = 12, n = 11), IL-18 (n = 7, n = 9), TNF-α (n = 9, n = 9). Mean mRNA level of healthy subjects normalized to 1. mRNA data analyzed with unpaired nonparametric 2-tailed t test. All data shown in mean ± SEM.
MitoDAMP induces PBMC respiratory impairment and inflammatory cytokine gene expression, which can be attenuated by inhibition of the NLRP3 inflammasome axis.
To explore the potential cellular mechanisms linking mitochondrial respiratory function and cytokine production, we sought to create in vitro models to mimic the proinflammatory state in HF. We first treated PBMCs from healthy participants with lipopolysaccharide (LPS), an endotoxin known to trigger proinflammatory activation in peripheral monocytes. Four hours of LPS treatment resulted in a sharp decline in PBMC maximal OCR (Supplemental Figure 1A) as well as increases in mRNA expression for NLRP3 and proinflammatory cytokines (Supplemental Figure 1B), most notably, IL-6.
Mitochondrial content, by virtue of its evolutionary origin, can elicit an immunogenic response independent of HF status (26). It was recently reported that the plasma level of mitochondrial DNA is elevated in patients with HF (27). To test whether the release of DAMP from damaged mitochondria could elicit a sterile inflammatory state in HF, MitoDAMP was extracted by lysing mitochondria isolated from myocardial tissue of patients with end-stage HF undergoing left ventricular assist device (LVAD) surgeries (Figure 2A). Similar to LPS, MitoDAMP treatment of healthy PBMCs induced a marked elevation of proinflammatory cytokine gene expressions (Figure 2B).
Figure 2. MitoDAMP induces PBMC respiratory impairment and inflammatory cytokine gene expression, and the latter can be partially attenuated by inhibition of the NLRP3 inflammasome axis.

(A) Schematics of the MitoDAMP extracted from mitochondria purified from human myocardial tissue. (B) Relative mRNA levels of NLRP3 and proinflammatory cytokines of healthy PBMCs after 4-hour treatments of vehicle or MitoDAMP. Vehicle normalized to 1. P value was determined by paired 2-tailed t test. n = 4. (C) Mitochondrial ROS levels of healthy PBMCs after MitoDAMP treatment. Zero hour normalized to 100%. P value cut off of 0.05 was determined by paired 2-tailed t test. n = 3. (D) Maximal respiration of healthy PBMCs after 4-hour treatment of vehicle or MitoDAMP with or without 0.5 mM MitoTempo, n = 4. P value determined by 1-way ANOVA with multiple pairwise comparisons. (E) Percent change of NLRP3 and cytokine mRNA levels of healthy PBMCs after 4-hour treatment of MitoDAMP with 0.5 mM MitoTempo (n = 4) or 1 μM MCC950 (n = 5) relative to MitoDAMP alone. P value was determined by paired 2-tailed t test. (F) Maximal respiration of healthy PBMCs after 4-hour treatments of vehicle or MitoDAMP with or without 1 μM MCC950. n = 3. P value determined by 1-way ANOVA with multiple pairwise comparisons. All data shown in mean ± SEM.
As shown in Figure 2C, MitoDAMP treatment resulted in a surge of mtROS production within 2 hours, which persisted for 6 hours. Mitochondrial maximal OCR was significantly impaired by MitoDAMP (Figure 2D). mtROS generation has been reported to be one of the key activators of the NLRP3 inflammasome (28), thus the role of NLRP3 inflammasome in connecting respiratory impairment and cytokine production was tested. Treating the PBMCs with MitoTempo, a nitroxide-based mitochondrial-specific ROS scavenger, in the presence of MitoDAMP, did not change maximal OCR (Figure 2D) but markedly attenuated cytokine gene expressions (Figure 2E). Moreover, treatment with MCC950, a specific inhibitor of the NLRP3 inflammasome, resulted in a similar attenuation of the MitoDAMP-induced surge on IL-6 gene expression (Figures 2E), while maximal OCR was marginally improved (Figure 2F). These findings suggest that MitoDAMP-induced mtROS production and NLRP3 activation contributes to proinflammatory cytokine gene expression in PBMCs. However, inhibition of either mechanism alone appears to minimally affect maximal OCR, suggesting that mtROS generation is downstream of respiratory impairment.
Secreted IL-6 impairs mitochondrial respiration by reducing complex I activity.
Among the cytokines studied in the in vitro experiments, increases in gene expression levels in response to proinflammatory stimuli were consistently highest for IL-6. Further, MitoDAMP stimulation of PBMCs resulted in a surge of secreted IL-6 protein within 4 hours (Figure 3A). We therefore hypothesized that the MitoDAMP-induced IL-6 secretion mediated the observed mitochondrial respiratory impairment in an autocrine fashion. Concurrent treatment with LMT28, an inhibitor of IL-6 receptor B (GP130), prevented the MitoDAMP-induced decline in maximal OCR (Figure 3B). Conversely, treating healthy PBMCs with recombinant IL-6 resulted in a dose-dependent decline of maximal OCR (Figure 3C). A significant reduction of maximal OCR by IL-6 was observed at concentrations as low as 0.1 ng/mL, which is comparable to the plasma concentrations of IL-6 during acute cardiac decompensation (29), as well as with the level of secreted IL-6 achieved at 4 hours following MitoDAMP stimulation in our in vitro experiments (Figure 3A). Consistently, treatment of IL-6 decreased the maximal OCR of monocytes isolated from healthy subjects (Supplemental Figure 3). In contrast, recombinant IL-1B and IL-18 treatments did not result in a significant decrease in PBMC maximal OCR (Supplemental Figure 4). Together, these results suggest that autocrine activation of IL-6 is a key mediator of MitoDAMP-induced respiratory impairment.
Figure 3. Secreted IL-6 from MitoDAMP stimulation impairs mitochondrial respiration by reducing complex I activity.

(A) Secreted IL-6 protein level by ELISA of healthy PBMCs after 2-hour (n = 3) or 4-hour (n = 6) treatment of vehicle or MitoDAMP. P value determined by paired 2-tailed t test. (B) Maximal respiration of healthy PBMCs after 4-hour treatments of vehicle or MitoDAMP with or without 100 μM LMT28, a specific inhibitor of the IL-6 receptor b (GP 130). n = 4. (C) Maximal respiration of healthy PBMCs after 4-hour treatments of vehicle or increasing concentrations of human recombinant IL-6. n = 4. B and C analyzed with 1-way ANOVA with multiple pairwise comparisons. (D) Representative Seahorse plot of baseline and post-drug treatment OCR of healthy PBMCs after 4-hour treatments of vehicle or IL-6 (1 ng/mL). FCCP; uncoupling agent by permeabilizing inner mitochondrial membrane; Rotenone (Rot): complex I inhibitor; Antimycin A (AA): complex III inhibitor; TMPD/Ascorbate: exogenous electron donor for complex IV. n = 3. (E) Quantitation of D. (F) Complex I activity of healthy PBMCs after 4-hour treatments of vehicle or IL-6 (1 ng/mL). n = 5. Vehicle normalized to 1. E and F analyzed by paired 2-tailed t test. All data shown in mean ± SEM.
Next, we searched for the end-effector of IL-6–induced reduction of maximal OCR. To this end, we compared changes in OCR in response to various electron transport chain (ETC) complex inhibitors in sequence in order to identify those complex(es) whose activity was affected by IL-6 treatment. As shown in Figure 3D, complex I inhibition by rotenone virtually eliminated all FCCP-induced OCR such that subsequent inhibition of complex III by Antimycin A only resulted in minimal OCR reduction, suggesting complex II was a minor contributor to the overall ETC activity in our system. Moreover, when TMPD (N,N,N′,N′-Tetramethyl-p-Phenylenediamine) and ascorbate were added to the system as electron donors for cytochrome C, which drives OCR through complex IV, we observed no difference in OCR between vehicle- and IL-6–treated PBMCs. These results suggest that IL-6 affected function of complex I function, but not of complexes II or IV. The contributions of each complex to the OCR in our assay system are summarized in Figure 3E. Consistent with these findings, direct measurement of complex I activity showed that IL-6 treatment reduced complex I enzymatic activity (Figure 3F). Together, these results suggest that IL-6 reduces mitochondrial respiration via reduction of complex I activity.
Increasing NAD+ levels with NR suppresses proinflammatory response in PBMCs in vitro.
Based on the recent observations that NAD augmentation by NR enhances respiratory function and reduces mtROS and IL-1B productions in PBMCs (15), we hypothesized that NR may ameliorate the reductions in maximal OCR and cytokine production seen with MitoDAMP stimulation. As shown in Figure 4, A and B, concurrent NR treatment of healthy PBMCs in the presence of MitoDAMP resulted in a reduction of IL-6 secretion and proinflammatory cytokine gene expressions, respectively. Importantly, NR treatment partially prevented the MitoDAMP-induced decline of maximal OCR (Figure 4C). A similar effect of NR in enhancing maximal OCR and reducing proinflammatory cytokine gene expressions is also observed in LPS-treated PBMCs (Supplemental Figure 1C).
Figure 4. NR attenuates MitoDAMP-induced PBMC respiratory impairment and proinflammatory cytokine production in vitro.

(A) Secreted IL-6 protein by ELISA of healthy PBMCs after 4-hour treatment of vehicle or 1 mM NR in the presence of MitoDAMP. P value determined by paired 2-tailed t test. (B) Percent change of NLRP3 and cytokine mRNA levels of healthy PBMCs after 4-hour treatment of MitoDAMP with 1 mM NR relative to MitoDAMP only. P value was determined by paired 2-tailed t test. n = 4. (C) Maximal respiration of healthy PMBCs after 4-hour treatment of vehicle, MitoDAMP, or MitoDAMP with 1 mM NR. n = 4. P value determined by ordinary 1-way ANOVA with multiple pairwise comparisons. B and C shown in mean ± SEM.
NR enhances mitochondrial respiration and reduces proinflammatory cytokine production in human HF.
We subsequently tested the effect of NR on the PBMCs of patients with HF in the absence of MitoDAMP. Incubation with NR in vitro increased the basal and maximal OCR in the PBMCs of both healthy subjects and subjects with HF (Figure 5, A and B, respectively). However, reductions of NLRP3 and proinflammatory cytokine gene expressions in HF PBMCs were minimal (Supplemental Figure 2), indicating that a 4-hour exposure to NR may be insufficient to revert the inflammatory state. To investigate this more closely, we enrolled patients with stage D HF to take NR orally for 5 to 9 days (Figure 6). Blood samples were obtained before and after NR administration. As shown in Figure 5C, oral NR administration resulted in increases of whole blood NAD+ levels, as previously seen in healthy subjects (30). Furthermore, NR treatment resulted in a consistent enhancement of the basal and maximal OCR in healthy and HF PBMCs (Figure 5, D and E), and an up to 30-fold reduction of proinflammatory cytokine gene expressions (Figure 5F). Background information of the study subjects can be found in Supplemental Table 1. No adverse effects associated with NR were observed among the 5 participants.
Figure 5. NR enhances mitochondrial respiration and reduces proinflammatory cytokine production in HF.

(A and B) Ratios of basal and maximal respiration of healthy or HF PBMCs after 4-hour 1 mM NR treatments relative to vehicle, respectively. n = 8 and n = 10, respectively. (C) Whole blood NAD+ level of subjects with stage D HF before or after 5 to 9 days of oral NR administration. (D and E) Basal and maximal respiration of PBMCs from subjects with stage D HF before and after oral NR administration, respectively. (F) Relative mRNA levels of NLRP3 and inflammatory cytokines of PBMCs from subjects with stage D HF before and after NR administration. Post-NR mRNA level normalized to 1. n = 4. (A and B) P values determined by unpaired 2-tailed t test, and data shown in mean ± SEM. (C–F) P values determined by paired 2-tailed t test.
Figure 6. Study design of oral NR administration in patients with HF.

Discussion
This study demonstrates a critical role of mitochondrial function in the inflammatory activation of peripheral immune cells in patients with HFrEF. We found that proinflammatory molecules (e.g., damage-associated molecular patterns released from dysfunctional mitochondria, likely via the Toll-like receptor cascade) trigger increased expressions of proinflammatory cytokines, particularly IL-6. Secreted IL-6 appears to feed back in an autocrine manner to impair mitochondrial respiration by inhibiting complex I activity and inducing mitochondrial ROS production, resulting in assembly of the NLRP3 inflammasome and increased secretion of proinflammatory cytokines such as IL-1B and IL-18 (13, 28, 31, 32). Secreted IL-1B can feed back to further potentiate the NFKB axis, resulting in a vicious cycle (ref. 33 and Figure 7).
Figure 7. Model of DAMP-induced monocyte activation.

The priming signal involves interaction of MitoDAMP with TLRs to stimulate the expression of inflammasome components and proinflammatory cytokines via activation of transcription factor NFKB. The secreted IL-6 in the priming step feeds back in an autocrine manner to impair mitochondrial respiration by inhibiting complex I activity and inducing mitochondrial ROS production, which leads to the assembly of the NLRP3 inflammasome to active caspase 1. Caspase-1, in turn, cleaves pro–IL-1B to IL-1B. Secreted IL-1B feeds back to further potentiate the NFKB axis.
Based on the results of our in vitro experiments, we speculate that the effect of NR represents an upstream mechanism which preserves mitochondrial respiration and reduces ROS production in the presence of an inflammatory trigger. On the other hand, mtROS and NLRP3 are downstream of the mitochondrial dysfunction; therefore, their inhibitions do not result in improvement of maximal OCR. Although the IL-6 mRNA level was reduced by mitochondrial ROS and NLRP3 inflammasome inhibition, the treatment may not be sufficient to normalize the secreted IL-6 level and remove mitochondrial impairment in our experimental setting. The molecular mediators by which NR treatment improves maximal OCR and reduces cytokine production in PBMCs warrant further investigation.
It has been demonstrated that circulating IL-6 is upregulated in both acute myocardial infarction and chronic HF; the former was thought to be cardioprotective, but if left unchecked, chronic IL-6 elevation can result in maladaptive hypertrophy and reduced LVEF (34, 35), potentially perpetuating HF. To date, there has not been a randomized clinical trial antagonizing IL-6 during post–myocardial infarction remodeling or in chronic HF. The present study suggests that IL-6 assumes an important signaling role connecting mitochondrial function and inflammation in peripheral immune cells. In cultured adipocytes, in vitro IL-6 treatment resulted in a decrease of mitochondrial membrane potential, cellular ATP production, an increase in intracellular ROS, and a reduction of respiratory reserve capacity (36). On the contrary, IL-6 was shown to hyperpolarize mitochondrial inner membrane in CD4+ cells (37). While the exact mechanism remains elusive, there has been evidence suggesting that a small fraction of STAT3, a downstream effector of the IL-6R axis, localizes in the mitochondria to regulate the electron transport chain and ATP production (38). Whether mitochondrial STAT3 is responsible for inhibiting complex I activity during chronic sterile inflammatory states, such as HF, is currently underexplored.
Recently, there has been tremendous interest in agents that increase cellular NAD as potential therapeutics for HF. Multiple studies demonstrated that augmenting NAD level is cardioprotective in animal models of cardiomyopathy induced by pressure overload (18–20), iron overload (39), or genetic mutations (19, 40). Most of those studies focus on the effect of NAD in the myocardium, such that boosting myocardial NAD+ leads to activation of NAD-dependent deacetylases (i.e., Sirtuins) or polyADP-ribosylases. Increasing cardiac NAD levels by pharmacological or genetic approaches has been shown to improve myocardial mitochondrial function and energy metabolism partly via the activity and downstream targets of mitochondrial-specific Sirtuins (18, 19, 41, 42).
Results from this study suggest that NAD augmentation may be protective against cardiomyopathy. The reduction of systemic inflammation through inhibition of proinflammatory activation of circulating immune cells allows the system to break from the vicious cycle that perpetuates the disease. Mechanistically, NAD+ repletion by NR has been shown to reduce mtROS production across tissue types (15, 43–46), which suppresses NLRP3 inflammasome/caspase I axis and secretions of active IL-1B and IL-18 (Figure 5 and ref. 28). In addition, NAD+-dependent activities of SIRT1 and SIRT2 in the cytosol have been found to inhibit transcription factor NFKB, resulting in blunted proinflammatory cytokine gene expression (47–49). On the other hand, there is evidence that the NAD+-consuming enzyme CD38 is upregulated in human monocyte-derived macrophages during inflammation, and that CD38 promotes cytokine secretion (50).
To our knowledge, this study is the first to examine whether NAD augmentation has antiinflammatory effects in the context of human HF. Although the human efficacy data are currently lacking, the collective actions of NAD repletion on preserving cardiac function and reducing systemic inflammation may provide a molecular basis for a novel HF therapy.
Limitations of the study.
For our in vitro experiments, whether a 4-hour culture truly reflects the cellular changes in a chronic inflammatory state needs to be substantiated. In addition, in this study, we modeled sterile inflammation in HF using mitochondrial extracts to mimic MitoDAMP. This mixture contains mitochondrial DNA, RNA, proteins, and phospholipids, many of which have been shown to be proinflammatory (11). The specific component(s) in MitoDAMP responsible for triggering PBMC proinflammatory activation are yet to be identified, and quantitative changes of MitoDAMP in peripheral blood of patients with HF warrant further investigation. Moreover, while the small number (n = 4) and short treatment period (5–9 days) of the experiments involving NR administration to patients with stage D HF (Figures 5 and 6) limit our ability to draw conclusions regarding the efficacy or safety of NR, the results pose promising opportunities for future clinical trials to investigate NR’s potential antiinflammatory effect. Last, since the average age of healthy participants in the study was younger than that of patients with HF, a potential influence of age on our findings could not be excluded with the small sample size.
Methods
Study participants.
For baseline Seahorse standard mito stress test, 19 healthy (i.e., with no history of acute or chronic disease) participants and 19 hospitalized patients with stage D HFrEF were enrolled into the study. In the HF group, those requiring temporary mechanical support, such as Impella, intraaortic balloon pump (IABP), or extracorporeal membrane oxygenation (EMCO), were excluded.
For experiments associated with oral NR administration (Figure 5, C–F), 5 hospitalized patients with stage D HF undergoing advanced HF therapy evaluations were put on escalating doses of NR (250 mg twice a day for day 1, 500 mg twice a day for day 2, and 1000 mg twice a day from day 3 on) for 5 to 9 days. Fasting blood samples were obtained for PBMC isolation at baseline and after NR administration. Subject 5 was withdrawn due to a change in clinical course deemed to be independent from NR administration.
PBMC and monocyte isolation.
A quantity of 30–60 mL fasting blood was collected into EDTA-containing Vacutainers (Becton Dickinson; catalog 364606). Blood then was diluted (1:1) with RPMI medium (Corning; catalog 17-105-CV) and applied to Histopaque gradient medium (MilliporeSigma; catalog 10771) using SepMate-50 (STEMCELL Technologies), and centrifuged at 1200g for 10 minutes. The top layer contained the enriched PBMCs, which were collected followed by centrifugation at 300g for 10 minutes. The pellet was resuspended with ACK lysis buffer (Gibco; catalog A1049201) and incubated at room temperature for 5 minutes to remove residual red blood cells. Next, the enriched PBMCs were washed twice with RPMI medium. All PBMC samples were subjected to the baseline Seahorse mito stress test. When available, the remaining cells were used for cytokine mRNA quantitative PCR and in vitro assays. For monocyte isolation, a magnetic bead–based negative-selection monocyte isolation kit (Miltenyi Biotec, 130-096-537) was used per manufacturer’s instructions. Briefly, antibody-conjugated magnetic bead solution was added to isolated PBMCs resuspended in RPMI containing 0.5% BSA and incubated for 20 minutes at 4°C, followed by magnetic column binding and elution. The procedure was repeated once to improve the purity of monocytes. The monocytes were washed once with RPMI before subsequent experiments.
Seahorse mito stress test.
OCR of freshly isolated PBMCs was assessed using a Seahorse XFe24 analyzer (Agilent Technologies). Briefly, purified PBMCs were resuspended in Seahorse base medium (Agilent Technologies; supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose, pH 7.4), and added 106 cells/well at equal volume of 500 μL in Seahorse 24-well plate. The cells were maintained in a non-CO2 incubator and allowed to settle for 30 minutes at 37°C. Next, the plate was centrifuged at 40g for 10 minutes at room temperature without no application of the brake to help cells attach to the bottom of the plate. Drugs used in the assay: 5 μM oligomycin A (MilliporeSigma; catalog 75351), 3 μM trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) (MilliporeSigma; catalog C2920), 1 μM rotenone (MilliporeSigma; catalog A8674), 1 μM antimycin A (MilliporeSigma; catalog R8875), 0.5 mM TMPD (MilliporeSigma; catalog T7394) and 10 mM ascorbate (MilliporeSigma; catalog 95209).
PBMC and monocyte culture.
Freshly isolated PBMCs or monocytes from healthy subjects or subjects with HF were resuspended with RPMI medium plus 10% heat inactivated FBS. PBMCs (106 cells/well) were seeded in Seahorse 24-well plates and incubated at 37°C in CO2 incubator with various proinflammatory triggers (vehicle, MitoDAMP, LPS) and compounds (NR, MCC 950, MitoTempo, or LMT-28). After 4 hours incubation, the plate was centrifuged at 40g for 10 minutes at room temperature without no application of the brake to help cells attach to the bottom of the plate. The cells were washed twice and then brought up to 500 μL using Seahorse base medium (Agilent Technologies, supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose, pH 7.4).
MitoDAMP isolation.
Human heart tissue samples (100–200 mg) from the left ventricular apex were collected at the time of left ventricular assist device (LVAD) implantation when tissue from the LVAD core became available. Fresh tissue was stored in ice-cold sterile PBS before mitochondria isolation per prior protocol (19). The purified mitochondria was resuspended in 200 μL MSE buffer (19), and subsequently stored at –80°C. Mitochondria from 6 donors were pooled and lysed by freeze (in liquid nitrogen) and thaw for 3 times, followed by sonication at 30% intensity with 3 seconds on and 3 seconds off for 3 cycles. The lysed mitochondria were then spun down at 10,621g for 10 minutes. Protein concentration of the supernatant (MitoDAMP) was determined by BCA kit. The supernatant was aliquoted and stored at –80°C for future use. For all MitoDAMP stimulation assays, the final protein concentration of MitoDAMP was 0.5 mg/mL (Figure 2A).
IL-6 ELISA.
Isolated PBMCs (4 × 106 cells per sample) from healthy subjects or participants with HF were resuspended in RPMI medium containing 10% heat-inactivated FBS and treated with various proinflammatory triggers (vehicle, MitoDAMP, LPS) and compounds (NR, MCC 950, MitoTempo, or LMT-28) to a final PBMC concentration of 10 × 106 cells per mL. See below for detailed descriptions of the reagents. The samples were incubated in Eppendorf tubes at 37°C in 5% CO2 incubator with caps open for time designated in figure legends. The cultured PBMC samples were centrifuged at 6797g at room temperature. The supernatant was used for ELISA per the manufacturer’s protocol using R&D Systems human IL-6 kit (DY206-05). The cell pellets were stored at –80°C.
RNA isolation and quantitative PCR analysis.
Total RNA was isolated from frozen PBMC pellets using Trizol (Invitrogen) per the manufacturer’s instructions, and cDNA was synthesized using iScript Reverse Transcription Supermix (Bio-Rad) per the manufacturer’s instructions. Real-time PCR was performed in the Corbett rotor gene 6000 real-time PCR machine using iQ SYBR Green Supermix (Bio-Rad) and expression levels of the indicated genes were calculated using the ΔΔCt method. Primer sequences are as follows. NLRP3 forward: GTGTTTCGAATCCCACTGTG; reverse: TCTGCTTCTCACGTACTTTCTG; IL-1B forward: ATGCACCTGTACGATCACTG; reverse: ACAAAGGACATGGAGAACACC; IL-6 forward: CCACTCACCTCTTCAGAACG; reverse: CATCTTTGGAAGGTTCAGGTTG; TNF-α forward: ACTTTGGAGTGATCGGCC; reverse: GCTTGAGGGTTTGCTACAAC; IL-18 forward: CATTGACCAAGGAAATCGGC; reverse: CACAGAGATAGTTACAGCCATACC; 18S rRNA forward: GTAACCCGTTGAACCCCATT; reverse: CCATCCAATCGGTAGTAGCG.
Mitochondrial ROS detection in cultured PBMCs.
To detect mitochondrial ROS production, the treated PBMCs were incubated with 5 μM MitoSOX Red (Life Technologies) and MitoTracker Green (200 nM, Life Technologies) at 37°C for 30 minutes in RPMI medium supplemented with 10% heat-inactivated FBS, and then washed twice with cold PBS before cells were analyzed by flow cytometry on a Cytek Aurora (Cytek Biosciences).
Complex I in vitro assay.
This assay was performed using Abcam Complex I Enzyme Activity Microplate Assay Kit (ab109721), per the manufacturer’s protocol. Briefly, following 4-hour treatment with vehicle or IL-6, 5 × 106 PBMCs were washed with PBS and lysed by detergent for 30 minutes on ice, followed by centrifugation at 16,000g for 20 minutes at 4°C. Supernatant was taken for protein concentration determination. Total protein (12.5 μg) in a final volume of 200 μL was incubated in microplates precoated with anti–complex I antibody for 3 hours at room temperature. After the washing step, NADH and dye were added and optical density (OD) was serially measured at 450 nm over 1 hour. The slope of the curve within the linear range was recorded as complex I activity.
Whole blood NAD+ assays.
These assays were performed as previously described (30). Briefly, 50 μL thawed blood samples were added to tubes containing 1 μg BMP as the internal standard and immediately followed by 300 μL of 4% trichloroacetic acid to precipitate proteins. Calibration standards were similarly prepared except that the matrix was 30 mg/mL BSA spiked with NAD+ and NMN that had been dissolved in methanol. After protein precipitation and high-speed centrifugation, the supernatants were loaded onto Costar Spin-X 0.22 μm spin filters and subject to high speed centrifugation again. The eluates were injected onto an Agilent 1100 series high-performance liquid chromatograph coupled to an Agilent G1956B single-quadrupole mass spectrometer. The mass spectrometer was operated in electrospray ionization mode with positive polarity.
Reagents.
Lipopolysaccharide (LPS) was purified from Salmonella Minnesota R595 (Enzo Life Sciences, catalog ALX-581-008-L001). MCC 950 stock (MilliporeSigma, catalog PZ0280-5MG) was dissolved in RPMI to 100 μM and stored at –80°C. MitoTempo (MilliporeSigma, catalog SML07375MG) stock solution was dissolved in RPMI to 10 mM and stored at –80°C. LMT-28 (MilliporeSigma, catalog SML1628-5MG)stock solution was dissolved in DMSO to 10 mM and stored at –80°C. Human recombinant IL-6 (Gibco, catalog PHC0064) stock solution was dissolved in RPMI to 1 μg/mL and stored at –80°C. Human recombinant IL-1B and IL-18 (R&D Systems, catalog 201-LB and 9124-IL, respectively) stock solutions were dissolved in RPMI to 1 μg/mL and stored at –80°C.
Nicotinamide riboside.
For in vitro assays, NR was supplied as powder by the manufacturer (Niagen, ChromaDex). Stock solutions (10 mM or 100 mM) dissolved in RPMI with 10% heat inactivated FBS were made on the day of the experiment. For oral administration, NR was supplied by the manufacturer as 250 mg capsules, and manufactured in a GMP-compliant facility according to ISO/IEC 18025:2005 standards.
Statistics.
Statistical analyses were performed using GraphPad Prism 8 (version 8.3.0). For analyses of PBMC respiratory function, the primary outcome was defined as the mean difference in basal OCR and maximal OCR (after FCCP injection), determined by Seahorse mito stress test in PBMCs of healthy subjects versus participants with HFrEF. Mean age comparisons between groups were subjected to an unpaired 2-tailed nonparametric test (Mann-Whitney test). For mRNA expression comparisons between healthy subjects and participants with HF, P value was determined by unpaired parametric 2-tailed t test. In vitro and in vivo before versus after treatment analyses were performed by paired 2-tailed parametric t test or 1-way ANOVA followed by post hoc pairwise multiple comparisons, as appropriate. Error bars in all figures represent SEM.
Study approval.
All studies were approved by the Human Subject Division of University of Washington and the investigation conformed with the principles outlined in the Declaration of Helsinki. For baseline PBMC measurements, blood samples from healthy adult participants were collected under IRB-approved protocol (STUDY00005599), and blood and myocardial tissue samples from patients with HFrEF were collected under a separate IRB-approved protocol (STUDY00002544). For the experiments pertaining to oral NR administration in patients with HFrEF, the study was performed under the IRB-approved protocol STUDY0000543, ClinicalTrials.gov NCT03727646.
Author contributions
BZ and DDHW designed and conducted experiments, and acquired and analyzed data. DDHW wrote the manuscript. YQ, YL, and SA acquired data and edited the manuscript. ASO edited the manuscript and provided reagent. KDO and RT supervised, designed, and conceptualized the project, and edited the manuscript.
Supplementary Material
Acknowledgments
The authors of the manuscript gratefully acknowledge Laura Shireman and Danny Shen for their expertise in measuring whole blood NAD+ levels of patients with HF and Mingyue Zhao for her assistance with cytokine release assays. This study was funded by NIH grant R21 HL126209 (to RT and KDO), NIH grant R01 HL144937 (to KDO and RT), and University of Washington ITHS Catalyst Award (to DDHW). Both BZ (18POST33990352) and DDHW (18POST34030098) are funded by American Heart Association Postdoctoral Fellowships.
Version 1. 08/13/2020
In-Press Preview
Version 2. 10/12/2020
Electronic publication
Version 3. 11/02/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(11):6054–6063.https://doi.org/10.1172/JCI138538.
Contributor Information
Bo Zhou, Email: zb817@u.washington.edu.
Yanhua Qiu, Email: qiuyanhua926@126.com.
Sophia Airhart, Email: sophia.airhart@providence.org.
Yaxin Liu, Email: liuyaxin@uw.edu.
April Stempien-Otero, Email: april@u.washington.edu.
Kevin D. O’Brien, Email: cardiac@uw.edu.
References
- 1.Dickstein K, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM) Eur J Heart Fail. 2008;10(10):933–989. doi: 10.1016/j.ejheart.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Lesyuk W, Kriza C, Kolominsky-Rabas P. Cost-of-illness studies in heart failure: a systematic review 2004-2016. BMC Cardiovasc Disord. 2018;18(1):74. doi: 10.1186/s12872-018-0815-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redfield MM, Jacobsen SJ, Burnett JC, Mahoney DW, Bailey KR, Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA. 2003;289(2):194–202. doi: 10.1001/jama.289.2.194. [DOI] [PubMed] [Google Scholar]
- 4.Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116(7):1254–1268. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakayama H, Otsu K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem J. 2018;475(5):839–852. doi: 10.1042/BCJ20170714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91(11):988–998. doi: 10.1161/01.RES.0000043825.01705.1B. [DOI] [PubMed] [Google Scholar]
- 7.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Anti-TNF Therapy Against Congestive Heart Failure Investigators Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107(25):3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 8.Mann DL, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004;109(13):1594–1602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 9.Everett BM, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139(10):1289–1299. doi: 10.1161/CIRCULATIONAHA.118.038010. [DOI] [PubMed] [Google Scholar]
- 10.Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128(9):3716–3726. doi: 10.1172/JCI120849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sack MN. Mitochondrial fidelity and metabolic agility control immune cell fate and function. J Clin Invest. 2018;128(9):3651–3661. doi: 10.1172/JCI120845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 13.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 14.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31(2):194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Traba J, et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J Clin Invest. 2015;125(12):4592–4600. doi: 10.1172/JCI83260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee HJ, Yang SJ. Nicotinamide riboside regulates inflammation and mitochondrial markers in AML12 hepatocytes. Nutr Res Pract. 2019;13(1):3–10. doi: 10.4162/nrp.2019.13.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elhassan YS, et al. Nicotinamide riboside augments the aged human skeletal muscle NAD+ metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 2019;28(7):1717–1728. doi: 10.1016/j.celrep.2019.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diguet N, et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation. 2018;137(21):2256–2273. doi: 10.1161/CIRCULATIONAHA.116.026099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee CF, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. 2016;134(12):883–894. doi: 10.1161/CIRCULATIONAHA.116.022495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smyrnias I, et al. Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J Am Coll Cardiol. 2019;73(14):1795–1806. doi: 10.1016/j.jacc.2018.12.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholls DG, Darley-Usmar VM, Wu M, Jensen PB, Rogers GW, Ferrick DA. Bioenergetic profile experiment using C2C12 myoblast cells. J Vis Exp. 2010;(46):2511. doi: 10.3791/2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chacko BK, et al. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest. 2013;93(6):690–700. doi: 10.1038/labinvest.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapman EH, Kurec AS, Davey FR. Cell volumes of normal and malignant mononuclear cells. J Clin Pathol. 1981;34(10):1083–1090. doi: 10.1136/jcp.34.10.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zipursky A, Bow E, Seshadri RS, Brown EJ. Leukocyte density and volume in normal subjects and in patients with acute lymphoblastic leukemia. Blood. 1976;48(3):361–371. [PubMed] [Google Scholar]
- 25.Schildberger A, Rossmanith E, Eichhorn T, Strassl K, Weber V. Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflamm. 2013;2013:697972. doi: 10.1155/2013/697972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manfredi AA, Rovere-Querini P. The mitochondrion--a Trojan horse that kicks off inflammation? N Engl J Med. 2010;362(22):2132–2134. doi: 10.1056/NEJMcibr1003521. [DOI] [PubMed] [Google Scholar]
- 27.Dhondup Y, et al. Low circulating levels of mitochondrial and high levels of nuclear DNA predict mortality in chronic heart failure. J Card Fail. 2016;22(10):823–828. doi: 10.1016/j.cardfail.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 28.Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):E3328. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki H, et al. Time-course of changes in the levels of interleukin 6 in acutely decompensated heart failure. Int J Cardiol. 2005;100(3):415–420. doi: 10.1016/j.ijcard.2004.08.041. [DOI] [PubMed] [Google Scholar]
- 30.Airhart SE, et al. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One. 2017;12(12):e0186459. doi: 10.1371/journal.pone.0186459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol. 2013;191(10):5230–5238. doi: 10.4049/jimmunol.1301490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sorbara MT, Girardin SE. Mitochondrial ROS fuel the inflammasome. Cell Res. 2011;21(4):558–560. doi: 10.1038/cr.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281(1):8–27. doi: 10.1111/imr.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terrell AM, Crisostomo PR, Wairiuko GM, Wang M, Morrell ED, Meldrum DR. Jak/STAT/SOCS signaling circuits and associated cytokine-mediated inflammation and hypertrophy in the heart. Shock. 2006;26(3):226–234. doi: 10.1097/01.shk.0000226341.32786.b9. [DOI] [PubMed] [Google Scholar]
- 35.Wollert KC, et al. Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series VIA gp130/leukemia inhibitory factor receptor-dependent pathways. J Biol Chem. 1996;271(16):9535–9545. doi: 10.1074/jbc.271.16.9535. [DOI] [PubMed] [Google Scholar]
- 36.Ji C, et al. IL-6 induces lipolysis and mitochondrial dysfunction, but does not affect insulin-mediated glucose transport in 3T3-L1 adipocytes. J Bioenerg Biomembr. 2011;43(4):367–375. doi: 10.1007/s10863-011-9361-8. [DOI] [PubMed] [Google Scholar]
- 37.Yang R, et al. Mitochondrial Ca2+ and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. Elife. 2015;4:e06376. doi: 10.7554/eLife.06376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang R, Rincon M. Mitochondrial Stat3, the need for design thinking. Int J Biol Sci. 2016;12(5):532–544. doi: 10.7150/ijbs.15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu W, Barrientos T, Mao L, Rockman HA, Sauve AA, Andrews NC. Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Rep. 2015;13(3):533–545. doi: 10.1016/j.celrep.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vignier N, et al. Rescue of biosynthesis of nicotinamide adenine dinucleotide protects the heart in cardiomyopathy caused by lamin A/C gene mutation. Hum Mol Genet. 2018;27(22):3870–3880. doi: 10.1093/hmg/ddy278. [DOI] [PubMed] [Google Scholar]
- 41.Tang X, Chen XF, Chen HZ, Liu DP. Mitochondrial sirtuins in cardiometabolic diseases. Clin Sci (Lond) 2017;131(16):2063–2078. doi: 10.1042/CS20160685. [DOI] [PubMed] [Google Scholar]
- 42.Walker MA, Tian R. Raising NAD in heart failure: time to translate? Circulation. 2018;137(21):2274–2277. doi: 10.1161/CIRCULATIONAHA.117.032626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong G, et al. Administration of nicotinamide riboside prevents oxidative stress and organ injury in sepsis. Free Radic Biol Med. 2018;123:125–137. doi: 10.1016/j.freeradbiomed.2018.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massudi H, Grant R, Guillemin GJ, Braidy N. NAD+ metabolism and oxidative stress: the golden nucleotide on a crown of thorns. Redox Rep. 2012;17(1):28–46. doi: 10.1179/1351000212Y.0000000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schondorf DC, et al. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep. 2018;23(10):2976–2988. doi: 10.1016/j.celrep.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Q, Liu X, Li N, Zhang J, Yang J, Bu P. Sirtuin 3 deficiency aggravates contrast-induced acute kidney injury. J Transl Med. 2018;16(1):313. doi: 10.1186/s12967-018-1690-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rothgiesser KM, Erener S, Waibel S, Lüscher B, Hottiger MO. SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci. 2010;123(Pt 24):4251–4258. doi: 10.1242/jcs.073783. [DOI] [PubMed] [Google Scholar]
- 48.Yang H, et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS One. 2012;7(9):e46364. doi: 10.1371/journal.pone.0046364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeung F, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23(12):2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amici SA, et al. CD38 is robustly induced in human macrophages and monocytes in inflammatory conditions. Front Immunol. 2018;9:1593. doi: 10.3389/fimmu.2018.01593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.