

Abstract
Lung cancer is the world's most common malignancies and ranks first among all cancer-related deaths. Lung adenocarcinoma (LUAD) is the most frequent histological type in lung cancer. Its pathogenesis has not yet been fully elucidated, so it is of great significance to explore related genes for elucidating the molecular mechanism involved in occurrence and development of LUAD.
To explore the crucial genes associated with LUAD development and progression, microarray datasets GSE7670, GSE10072, and GSE31547 were acquired from the Gene Expression Omnibus (GEO) database. R language Limma package was adopted to screen the differentially expressed genes (DEGs). The clusterProfiler package was used for enrichment analysis and annotation of the Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genome (KEGG) pathways for DEGs. The Search Tool for the Retrieval of Interacting Genes database (STRING) was used to construct the protein interaction network for DEGs, while Cytoscape was adopted to visualize it. The functional module was screened with Cytoscape's MCODE (The Molecular Complex Detection) plugin. The crucial genes associated with LUAD were identified by cytoHubba plugin. Kaplan–Meier plotter online tool was used to perform survival analysis of the hub gene.
Three hundred twenty-one DEGs in total were screened, of which 105 were upregulated and 216 were downregulated. It was found that some GO terms and pathways (e.g., collagen trimer, extracellular structure organization, heparin binding, complement and coagulation cascades, malaria, protein digestion and absorption, and PPAR signaling pathway) were considerably enriched in DEGs. UBE2C, TOP2A, RRM2, CDC20, CCNB2, KIAA0101, BUB1B, TPX2, PRC1, and CDK1 were identified as crucial genes. Survival analysis showed that the overexpression of UBE2C, TOP2A, RRM2, CDC20, CCNB2, KIAA0101, BUB1B, TPX2, and PRC1 significantly reduced the overall survival of LUAD patients. One of the crucial genes: UBE2C was validated by immunohistochemistry to be upregulated in LUAD tissues.
This study screened out potential biomarkers of LUAD, providing a theoretical basis for elucidating the pathogenesis and evaluating the prognosis of LUAD.
Keywords: bioinformatic analysis, differentially expressed genes, lung adenocarcinoma
1. Introduction
Lung cancer, with increasing morbidity and mortality, is the world's most frequently occurred malignant tumor that endanger people's health. On the basis of the findings of the World Health Organization (WHO), lung cancer morbidity and mortality are ranked first.[1] In 2018, 2.09 million people worldwide suffered from lung cancer (11.6% of the world's total cancer), and the number of deaths was 1.76 million (18.4% of the total cancer among the world). Lung cancer is divided into 2 kinds, that is, nonsmall cell lung cancer (NSCLC) and small cell lung cancer, accounting for 85% and 15% of lung cancer, respectively. NSCLC includes 3 histological types, including adenocarcinoma, large cell carcinoma, and squamous cell carcinoma. In many countries, adenocarcinoma has outnumbered squamous cell carcinoma as the most frequently occurred kind of lung cancer tissue. The occurrence and development of lung adenocarcinoma (LUAD) is a complex process involving the abnormal expression and genetic variation of multiple genes. BCL-2 stimulates the growth and progression of LUAD by inhibiting apoptosis.[2] Exon 21 L858R point mutation and exon 19 deletion are the most common type of EGFR mutations, which induce cell proliferation, inhibit apoptosis, and promote angiogenesis and tumor metastasis.[3] High expression of MACC1, KIF2A, and SR-B1 is closely related to progression and adverse prognosis of LUAD patients.[4–6] However, the molecular mechanism of the development of LUAD is still far from being proven, and it is difficult to implement effective early diagnosis and treatment. Therefore, it is of great significance to elucidate the molecular mechanisms involved in the occurrence and progression of LUAD.
Microarray technology has undergone rapid development during the last decades. Gene expression microarrays are powerful tools for studying the differential expression of genes related to the carcinogenesis and progression of LUAD. Many studies of LUAD integrate cross-platform gene expression data.[7,8] As cross-platform gene chips are constructed using different standards, the conclusions drawn from those studies will also reduce the reliability. In this study, differentially expressed genes (DEGs) in LUAD and adjacent tissues were identified using R software package, while LUAD gene expression data from the same platform were taken from the database called Gene Expression Omnibus (GEO). The function of DEGs was analyzed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Searching Tool for the Retrieval of Interacting Genes (STRING; https://string-db.org) was utilized to establish the Protein-Protein interaction (PPI) network. The cytoHubba plugin identified 10 hub genes associated with LUAD. The Kaplan--Meier survival curve reveals the hub genes’ prognostic value for LUAD. The use of a variety of bioinformatics methods provides clues and evidence for further study of the molecular mechanisms of LUAD development.
2. Materials and methods
2.1. Microarray data and data processing
Acquired from the GEO database of the National Center for Biotechnology Information (NCBI), the gene expression profile of GSE7670, GSE10072, and GSE31547 data sets based on the GPL96 platform was utilized. Among them, GSE7670 contains 27 pairs of paired LUAD tissues and adjacent tissues, GSE10072 contains 49 cases of normal human lung tissues and 58 cases of LUAD tissues, and GSE31547 contains 30 cases of LUAD tissues and 20 cases of adjacent tissues. Raw expression data were subjected to background correction, quantile normalization and log2 transformation through the RMA algorithm of the affy software package. The probe ID was then converted to the gene name by the GPL96 platform file.
2.2. Identification of DEGs
According to the inclusion criteria of |log2FC| > 1 and adjusted P < .05, DEGs were screened through the R language limma package.
2.3. Pathway and DEGs functional enrichment analysis
The enrichment analysis of GO and KEGG is widely used in the study of gene chip data. ClusterProfiler is a powerful R package that supports not only GO and KEGG enrichment analysis, but also excellent visualization of analysis results.[9] In this study, the clusterProfiler package was used to annotate and enrich the GO and KEGG pathways for DEGs. Function annotation of DEGs is helpful to find out the biological functions and pathways of these genes involved in the carcinogenesis of LUAD. P < .05 was determined as a cut-off criterion for significant enrichment.
2.4. PPI network establishment and module identification
STRING is an online program for predicting protein interactions. The DEGs’ protein interaction network was constructed by STRING with the smallest required interaction score ≥0.4. Cytoscape was used to visualize the network, and the functional module was screened with Cytoscape's MCODE (The Molecular Complex Detection) plugin. The parameters for screening function module were: degree cut-off = 2, MCODE scores > 5, Max depth = 100, node score cut-off = 0.2, k-score = 2. GO and KEGG pathway analysis was carried out on the genes in the functional modules using clusterProfiler package.
2.5. Identifying and analyzing hub genes
CytoHubba ranks nodes based on their attributes in the network. It provides a variety of topological analysis methods, of which MCC is the newly proposed method. In this paper, the top 10 genes ranked by the MCC method are defined as the hub genes. Survival analysis of the hub gene was carried out using the Kaplan--Meier plotter online tool (http://kmplot.com/analysis/). The relationship between gene expression patterns and tumor grading and staging was demonstrated using the oncomine database (https://www.oncomine.org).
2.6. Immunohistochemistry of UBE2C
Ten pairs of LUAD and adjacent normal tissues with complete clinicopathological information were collected from Huai’an First People's Hospital. The experiment was approved by the Medical Ethics Committee of Huai’an First People's Hospital, and each participant signed a written informed consent. The operation procedures of immunohistochemistry were reported in previous study.[10] All sections were incubated overnight at 4°C with anti-UBE2C (abcam, 1:300 dilution). Each immunostained section was independently read by 2 pathologists who did not know the clinicopathological data. The criteria for judging the results of immunohistochemistry has been described in previous study.[10] Wilcoxon signed-rank test was used to analyze the expression of UBE2C in LUAD and its adjacent tissues.
3. Results
3.1. Identification of DEGs
Seven hundred fifty, 801, and 709 DEGs were screened from the 3 sets of gene chips GSE7670, GSE10072, and GSE31547, of which 144, 508, and 338 genes were upregulated, and 606, 293, and 371 genes were downregulated, respectively. The false-positive rate of DEGs screening results was reduced by a Venn diagram. Finally, 321 overlapped DEGs were identified (Fig. 1A), including 105 upregulated genes and 216 downregulated genes.
Figure 1.

Venn diagram, PPI network and the most significant module of DEGs. (A) DEGs were screened from the expression data sets, and the overlapped parts were used as DEGs for subsequent analysis. The screening criteria were fold change >2 and P < .01. (B) The PPI network for DEGs was constructed by string and visualized with Cytoscape. (C) The most significant module consisting of 19 expressed genes was obtained from the PPI network using the Cytoscape plugin MCODE. High expression and low expression genes are labeled with red and blue, respectively.
3.2. Pathway and functional enrichment analysis of DEGs
GO enrichment analysis of DEGs revealed that DEGs were considerably enriched in 31 cellular components (CCs), 441 biological processes (BPs), and 35 molecule functions (MFs). The top 10 BPs included extracellular structure organization, extracellular matrix organization, leukocyte migration, regulation of body fluid levels, cell-substrate adhesion, regulation of angiogenesis, negative regulation of immune system process, regulation of vasculature development, regulation of response to wounding, and positive regulation of response to external stimulus (Table 1; Fig. 2A). The top 10 CCs included extracellular matrix, collagen-containing extracellular matrix, collagen trimer, platelet alpha granule, fibrillar collagen trimer, banded collagen fibril, apical part of cell, membrane raft, membrane microdomain, and membrane region (Table 1; Fig. 2A). The top 10 MFs included heparin binding, glycosaminoglycan binding, extracellular matrix structural constituent, sulfur compound binding, growth factor binding, cytokine binding, integrin binding, amide binding, amyloid-beta binding, and protease binding (Table 1; Fig. 2A). Furthermore, the significant enrichment KEGG pathway of DEGs included complement and coagulation cascades, malaria, protein digestion and absorption, and PPAR signaling pathway (Table 1; Fig. 2B).
Table 1.
Pathway and functional enrichment analysis of DEGs in LUAD samples.

Figure 2.
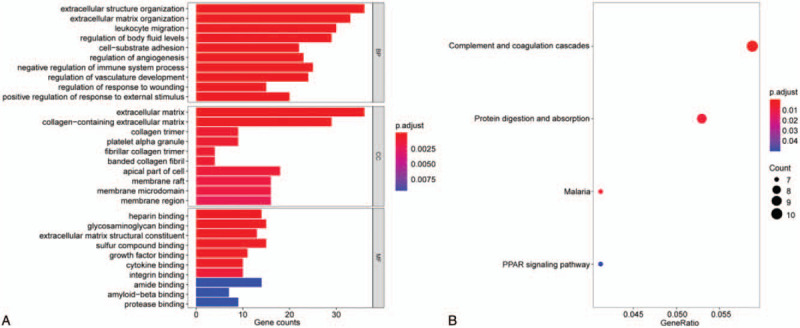
GO term and KEGG pathway enrichment analysis for DEGs were performed by clusterProfiler package. (A) GO enrichment analysis. (B) KEGG pathway enrichment analysis.
3.3. PPI network of DEGs and module identification
The STRING online tool was adopted to construct a protein--protein interaction network (PPI) for 321 DEGs (Fig. 1B). Through the Cytoscape plugin MCODE, the most important module containing 19 expressed genes was acquired from the PPI network (Fig. 1C). GO and KEGG analysis of the core module indicated the BPs involved were principally nuclear division, organelle fission, mitotic sister chromatid segregation, and DNA strand elongation (Table 2; Fig. 3A). The CCs involved were mainly spindle, anaphase-promoting complex, and nuclear ubiquitin ligase complex (Table 2; Fig. 3A). The MFs involved were kinesin binding and DNA-dependent ATPase activity (Table 2; Fig. 3A). The main signaling pathways included P53 signaling pathway, cell cycle, and oocyte meiosis (Table 2; Fig. 3B).
Table 2.
Pathway and functional enrichment analysis of DEGs in the most significant module.

Figure 3.
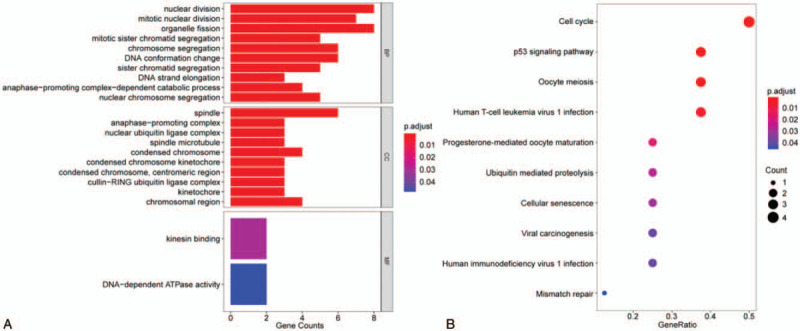
GO term and KEGG pathway enrichment analysis of DEGs in the most significant module were performed by clusterProfiler package. (A) GO enrichment analysis. (B) KEGG pathway enrichment analysis.
3.4. Identification and analysis of hub genes
Ten hub genes in total were extracted from cytoHubba. Table 3 summarizes the abbreviations, full names, and functions of these hub genes. Kaplan--Meier plotter database was used to carry out a survival analysis of 10 hub genes. As indicated by the result, LUAD patients with high expression of UBE2C, TOP2A, RRM2, CDC20, CCNB2, KIAA0101, BUB1B, TPX2, PRC1 were substantially related to poor overall survival (Fig. 4). There were no CDK1 survival curve data in the Kaplan--Meier plotter database, so it was not displayed. Among these genes, UBE2C, TOP2A, and KIAA0101 have node degrees exceeding 20, suggesting that they are likely to play an essential role in the occurrence and progression of LUAD. As indicated by the Oncomine database analysis, the expression of UBE2C, TOP2A, and KIAA0101 was increased (Fig. 5)[11–14] and positively correlated with pathological grade and TNM stage of LUAD (Fig. 6).
Table 3.
Function list of 10 hub genes.

Figure 4.
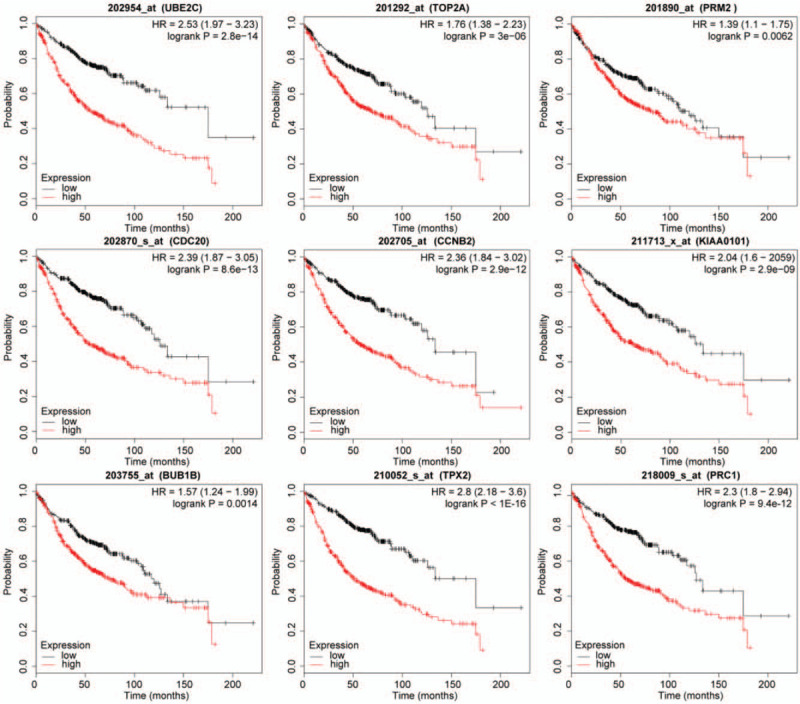
Overall survival analysis of hub genes was performed by Kaplan--Meier plotter database (CDK1 survival curve data is not available in the database and is not displayed).
Figure 5.

Oncomine analysis showed that (A) UBE2C, (B) TOP2A, and (C) KIAA0101 were highly expressed in lung cancer tissues compared with normal tissues.[11–14]
Figure 6.
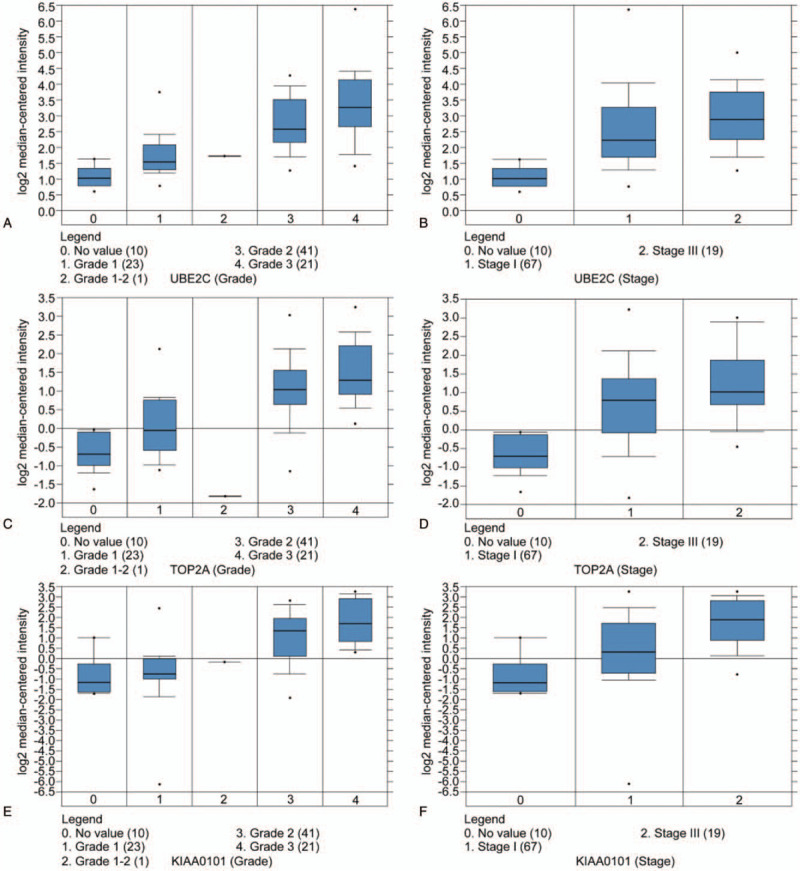
Association between the expression of UBE2C, TOP2A, KIAA0101, and tumor grade and stage in the Beer Lung Adenocarcinoma dataset. (A,B) UBE2C mRNA expression in LUAD compared with normal tissues. (C,D) TOP2A mRNA expression in LUAD compared to normal tissues. (E,F) KIAA0101 mRNA expression in LUAD compared to normal tissues.
3.5. Immunohistochemistry of UBE2C
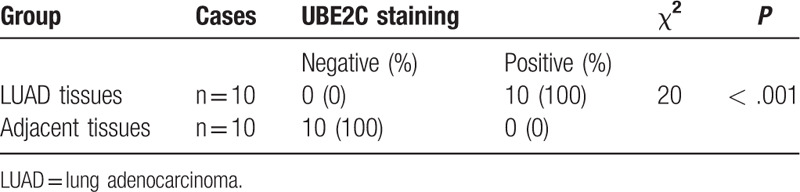
The representative immunostaining images of UBE2C in 10 pairs of LUAD and paracancerous tissues are shown in Figure 7. UBE2C was mainly expressed in the nucleus. The positive expression of UBE2C in LUAD and adjacent normal tissues were 10 (100%) and 0 (0%) respectively (Table 4, P < .001), which proved that the expression of UBE2C in LUAD was higher than that in paracancerous tissues.
Figure 7.
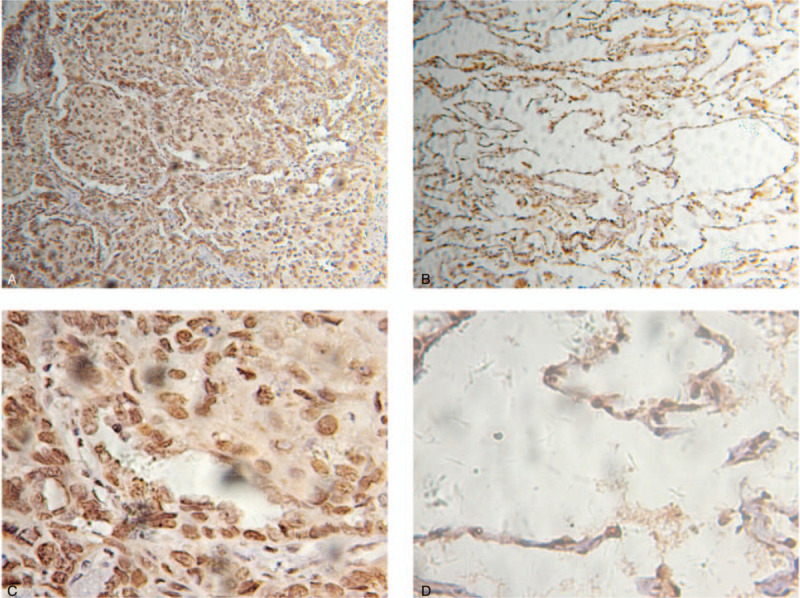
The expression of UBE2C in LUAD and adjacent normal tissues was analyzed by immunohistochemical staining. (A) Immunostaining of LUAD tissues (×100). (B) Immunostaining of normal tissues (×100). (C) Immunostaining of LUAD tissues (×400). (D) Immunostaining of normal tissues (×400).
Table 4.
UBE2C expression between LUAD and adjacent tissues.
4. Discussion
LUAD as a nonsmall cell lung cancer is the world's most frequently occurred cancers. In some areas, LUAD has the highest incidence of lung cancer, with extremely high morbidity and mortality.[1] Traditional screening methods have poor sensitivity to early diagnosis of LUAD. Most patients are diagnosed with LUAD in an advanced stage, missing the optimal opportunity for treatment. Some therapeutic regimens are not significant for the improvement of patient survival, so it is particularly necessary to study the mechanism of LUAD. It is currently believed that the carcinogenesis of lung cancer is a multifactor, multistep process, and its specific pathogenesis is still unclear. Gene chip technology enables rapid, high-throughput research on gene expression, which greatly accelerates the process of studying the pathogenesis, clinical types, diagnosis, treatment, and prognosis of lung cancer at the gene level.
In this study, 3 gene chip data were downloaded from the GEO database. Through a comparative analysis of the DEGs in LUAD tissues and normal tissues or adjacent tissues, 321 DEGs were screened, of which 216 were downregulated genes and 105 were upregulated genes. KEGG pathway enrichment analysis and GO enrichment analysis were carried out on DEGs. The DEGs were mainly enriched in functions related to extracellular matrix, cell adhesion, and angiogenesis, such as extracellular structure organization, extracellular matrix organization, leukocyte migration, cell-substrate adhesion, regulation of angiogenesis, regulation of vasculature development. Recently, numerous researches have shown that the degradation of extracellular matrix and basement membrane plays a decisive role in the invasion and metastasis of cancer cells.[15–17] Cell adhesion is the key to invasion and metastasis of cancer.[18,19] Increasing number of researches and experiments have found that cell adhesion molecules are closely linked to lung cancer.[20,21] Angiogenesis exists in the whole development courses and progression of malignant tumors and is closely related to tumor development, invasion and metastasis.[22] Moreover, it has been found that membrane rafts are strongly associated with various cancers such as LUAD,[23] breast cancer,[24] colorectal cancer,[25] and pancreatic cancer.[26] In short, all these theoretical results are consistent with our findings. GO enrichment analysis demonstrated that the variations in the most remarkable module were enriched primarily in nuclear division, organelle fission, mitotic sister chromatid segregation, and DNA strand elongation, whereas changes in KEGG were chiefly enriched in P53 signaling pathway, cell cycle and Oocyte meiosis.
CytoHubba, a network analysis plugin, was adopted to screen the top 10 genes as hub genes. Among these genes, UBE2C, TOP2A, and KIAA0101 have node degrees exceeding 20. In recent years, it was found that UBE2C was lowly expressed in normal tissues while highly expressed in a variety of tumors such as lung cancer,[27] breast cancer,[28] ovarian cancer,[29] esophageal cancer,[30] and gastric cancer.[31] Overexpression of UBE2C positively correlates to poor prognosis in tumors.[27,31] Upregulation of UBE2C promotes tumor cell proliferation, invasion, and drug resistance,[32] and downregulation of UBE2C can reverse the resistance of NSCLC cells to cisplatin and can be used as a potential target for tumor therapy.[33] TOP2A is an ATP-dependent hydrolase and synthetase that plays a vital role in cells. It catalyzes the cleavage and religation of double-stranded DNA duplexes and is involved in cell division, mitotic chromosome pairing, chromosome segregation, gene recombination and transcription, and DNA damage and repair. More and more studies have shown that TOP2A is closely related to cellular proliferation, metastasis, prognosis, and drug resistance in lung cancer.[34–36] In addition, TOP2A is highly expressed in breast cancer,[37] ovarian cancer,[38] prostate cancer,[39] and colon cancer,[40] and its expression level can be used as an indicator for tumor diagnosis, treatment, and prognosis evaluation. KIAA0101, also known as PCLAF (PCNA-associated factor), is receiving more and more attention due to its role in tumor progression and metastasis. Increased expression of KIAA0101 was detected in several human tumors including lung cancer,[41] breast cancer,[42] and liver cancer.[43] The high expression of KIAA0101 can significantly affect the regulation of cell cycle.[44] Downregulation of KIAA0101 expression by miR-197–5p can significantly reduce sarcoma cell proliferation and induce cell senescence.[45] The ribonucleotide reductase subunit M2 (RRM2) is not only closely related to DNA synthesis, but also has an effect on the biological behavior, metastatic potential, and anti-cancer drug resistance. Inhibition of RRM2 expression can reduce the activity of ribonucleotide reductase, hinder the growth of cancer cells, increase tumor cell apoptosis, inhibit tumor metastasis, and reverse drug resistance. According to various studies recently, RRM2 can be used as a target for anti-lung cancer drugs.[46] RRM2 is highly expressed in lung cancer, negatively correlated with overall survival, and positively correlated with lymph node metastasis.[47] Cell division cycle 20 (CDC20) is an important regulator of cell cycle checkpoints by directly binding and activating anaphase-promoting complex (APC) in the process of regulating cell mitosis. CDC20 is highly expressed in lung cancer,[48] gastric cancer,[49] and other malignant tumor tissues. Meanwhile, CDC20 is also a poor prognostic factor for colorectal cancer[50] and lung cancer.[51] CCNB2 is an important part of the class B cyclin family. CCNB2 is highly expressed in lung cancer and liver cancer, and its expression level is significantly related to clinical pathology and poor prognosis.[52,53] In colorectal cancer, the NF-Y subunit binds to the DNA fragment on the CNNB2 promoter and regulates the activity of the CCNB2 promoter.[54] Recent studies have shown that the mitotic checkpoint serine/threonine kinase B (BUB1B) is important in tumorigenesis and development. This protein is an important functional protein in mitosis and it is critical in the growth of various cancers.[55,56] Overexpression of BUB1B may be involved in the occurrence of chromosomal instability and promote tumorigenesis and progression.[57] TPX2 is involved in the mitotic spindle microtubule function by activating the cell cycle kinase Aurora A, which is highly expressed in NSCLC and shows poor overall survival.[58] PRC1 is upregulated in LUAD. Deregulating PRC1 expression can hinder the invasion and proliferation of LUAD cells by inhibiting cell proliferation, stimulating apoptosis and cell cycle arrest.[59] Cyclin-dependent kinase 1 (CDK1) is an essential cell cycle regulator, and its increased expression can lead to a high risk of cancer recurrence and low survival rate for patients with LUAD.[48] Downregulation of CDK1 can inhibit cell proliferation and is likely to become a potential target for tumor therapy.[60]
5. Conclusion
This study identified 321 DEGs and screened 10 hub genes, providing a rationale for future study of the molecular mechanism of LUAD development. However, gene expression profiling only detects mRNA expression of DEGs. Additional researches and experiments are necessary to confirm the genes expression at the protein level.
Author contributions
Data curation: Wu-Bi Zhou.
Methodology: Jing-Jing Dai.
Validation: Bing Wang.
Writing – original draft: Jing-Jing Dai, Bing Wang.
Writing – review & editing: Bing Wang.
Footnotes
Abbreviations: BPs = biologic processes, BUB1B = Serine/Threonine Kinase B, CCNB2 = Cyclin B2, CCs = cellular components, CDC20 = Cell Division Cycle 20, CDK1 = Cyclin Dependent Kinase 1, DEGs = differentially expressed genes, GEO = gene expression omnibus, GO = gene ontology, KEGG = Kyoto Encyclopedia of Genes and Genome, KIAA0101 = proliferating cell nuclear antigen (PCNA)-associated factor, LUAD = lung adenocarcinoma, MCODE = The Molecular Complex Detection, MFs = molecule functions, PPI = protein--protein interaction, PRC1 = Protein Regulator Of Cytokinesis 1, RRM2 = Ribonucleotide Reductase Regulatory Subunit M2, STRING = Search Tool for the Retrieval of Interacting Genes, TOP2A = DNA Topoisomerase II Alpha, TPX2 = targeting protein for Xklp2, UBE2C = Ubiquitin Conjugating Enzyme E2 C.
How to cite this article: Dai JJ, Zhou WB, Wang B. Identification of crucial genes associated with lung adenocarcinoma by bioinformatic analysis. Medicine. 2020;99:44(e23052).
The authors report no conflicts of interest.
The authors did not receive financial support for this study.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- [1].Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- [2].Nalluri S, Ghoshal-Gupta S, Kutiyanawalla A, et al. TIMP-1 inhibits apoptosis in lung adenocarcinoma cells via interaction with Bcl-2. PLoS One 2015;10:e0137673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J 2010;277:301–8. [DOI] [PubMed] [Google Scholar]
- [4].Guo T, Zhao S, Li Z, et al. Elevated MACC1 expression predicts poor prognosis in small invasive lung adenocarcinoma. Cancer Biomark 2018;22:301–10. [DOI] [PubMed] [Google Scholar]
- [5].Xie T, Li X, Ye F, et al. High KIF2A expression promotes proliferation, migration and predicts poor prognosis in lung adenocarcinoma. Biochem Biophys Res Commun 2018;497:65–72. [DOI] [PubMed] [Google Scholar]
- [6].Feng H, Wang M, Wu C, et al. High scavenger receptor class B type I expression is related to tumor aggressiveness and poor prognosis in lung adenocarcinoma: a STROBE compliant article. Medicine (Baltim) 2018;97:e0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang X, Gao C, Liu L, et al. DNA methylation-based diagnostic and prognostic biomarkers of nonsmoking lung adenocarcinoma patients. J Cell Biochem 2019;120:13520–30. [DOI] [PubMed] [Google Scholar]
- [8].Zhang MY, Liu XX, Li H, et al. Elevated mRNA Levels of AURKA, CDC20 and TPX2 are associated with poor prognosis of smoking related lung adenocarcinoma using bioinformatics analysis. Int J Med Sci 2018;15:1676–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang W, Wang Z, Zhao J, et al. A novel molecular and clinical staging model to predict survival for patients with esophageal squamous cell carcinoma. Oncotarget 2016;7:63526–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 2002;8:816–24. [DOI] [PubMed] [Google Scholar]
- [12].Garber ME, Troyanskaya OG, Schluens K, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A 2001;98:13784–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stearman RS, Dwyer-Nield L, Zerbe L, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am J Pathol 2005;167:1763–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Su LJ, Chang CW, Wu YC, et al. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 2007;8:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Y, Zhang H, Gong H, et al. miR-182 suppresses invadopodia formation and metastasis in non-small cell lung cancer by targeting cortactin gene. J Exp Clin Cancer Res 2018;37:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Singh SS, Bhatt MLB, Kushwaha VS, et al. Role of matrix metalloproteinase 13 gene expression in the evaluation of radiation response in oral squamous cell carcinoma. J Carcinog 2017;16:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Walter C, Davis JT, Mathur J, et al. Physical defects in basement membrane-mimicking collagen-IV matrices trigger cellular EMT and invasion. Integr Biol 2018;10:342–55. [DOI] [PubMed] [Google Scholar]
- [18].Chen X, Hu X, Li Y, et al. Resveratrol inhibits Erk1/2-mediated adhesion of cancer cells via activating PP2A-PTEN signaling network. J Cell Physiol 2019;234:2822–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Martinucci B, Minatel BC, Cucielo MS, et al. Basement membrane extract attenuates the more malignant gene expression profile accentuated by fibronectin in prostate cancer cells. Mol Cell Biochem 2019;451:131–8. [DOI] [PubMed] [Google Scholar]
- [20].Yang Y, Jiang Y, Xie D, et al. Inhibition of cell-adhesion protein DPYSL3 promotes metastasis of lung cancer. Respir Res 2018;19:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chae YK, Choi WM, Bae WH, et al. Overexpression of adhesion molecules and barrier molecules is associated with differential infiltration of immune cells in non-small cell lung cancer. Sci Rep 2018;8:1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen E, Tang C, Peng K, et al. ANGPTL6-mediated angiogenesis promotes alpha fetoprotein-producing gastric cancer progression. Pathol Res Pract 2019;215:152454. [DOI] [PubMed] [Google Scholar]
- [23].Zeng J, Zhang H, Tan Y, et al. Aggregation of lipid rafts activates c-met and c-Src in non-small cell lung cancer cells. BMC Cancer 2018;18:611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ewaschuk JB, Newell M, Field CJ. Docosahexanoic acid improves chemotherapy efficacy by inducing CD95 translocation to lipid rafts in ER(–) breast cancer cells. Lipids 2012;47:1019–30. [DOI] [PubMed] [Google Scholar]
- [25].Ye DM, Ye SC, Yu SQ, et al. Drug-resistance reversal in colorectal cancer cells by destruction of flotillins, the key lipid rafts proteins. Neoplasma 2019;66:576–83. [DOI] [PubMed] [Google Scholar]
- [26].Gupta VK, Sharma NS, Kesh K, et al. Metastasis and chemoresistance in CD133 expressing pancreatic cancer cells are dependent on their lipid raft integrity. Cancer Lett 2018;439:101–12. [DOI] [PubMed] [Google Scholar]
- [27].Ma R, Kang X, Zhang G, et al. High expression of UBE2C is associated with the aggressive progression and poor outcome of malignant glioma. Oncol Lett 2016;11:2300–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mo CH, Gao L, Zhu XF, et al. The clinicopathological significance of UBE2C in breast cancer: a study based on immunohistochemistry, microarray and RNA-sequencing data. Cancer Cell Int 2017;17:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Martinez-Canales S, Lopez de Rodas M, Nuncia-Cantarero M, et al. Functional transcriptomic annotation and protein-protein interaction analysis identify EZH2 and UBE2C as key upregulated proteins in ovarian cancer. Cancer Med 2018;7:1896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Palumbo A, Jr, Da Costa NM, De Martino M, et al. UBE2C is overexpressed in ESCC tissues and its abrogation attenuates the malignant phenotype of ESCC cell lines. Oncotarget 2016;7:65876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang HQ, Zhao G, Ke B, et al. Overexpression of UBE2C correlates with poor prognosis in gastric cancer patients. Eur Rev Med Pharmacol Sci 2018;22:1665–71. [DOI] [PubMed] [Google Scholar]
- [32].Xiong Y, Lu J, Fang Q, et al. UBE2C functions as a potential oncogene by enhancing cell proliferation, migration, invasion, and drug resistance in hepatocellular carcinoma cells. Biosci Rep 2019;39:BSR20182384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guo J, Jin D, Wu Y, et al. The miR 495-UBE2C-ABCG2/ERCC1 axis reverses cisplatin resistance by downregulating drug resistance genes in cisplatin-resistant non-small cell lung cancer cells. EBioMedicine 2018;35:204–21. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [34].Yan S, Shun-Chang J, Li C, et al. Topoisomerase II alpha expression and the benefit of adjuvant chemotherapy for postoperative patients with non-small cell lung cancer. BMC Cancer 2010;10:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Syahruddin E, Oguri T, Takahashi T, et al. Differential expression of DNA topoisomerase II alpha and II beta genes between small cell and non-small cell lung cancer. Jpn J Cancer Res 1998;89:855–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wessel I, Jensen LH, Renodon-Corniere A, et al. Human small cell lung cancer NYH cells resistant to the bisdioxopiperazine ICRF-187 exhibit a functional dominant Tyr165Ser mutation in the Walker A ATP binding site of topoisomerase II alpha. FEBS Lett 2002;520:161–6. [DOI] [PubMed] [Google Scholar]
- [37].Zheng H, Li X, Chen C, et al. Quantum dot-based immunofluorescent imaging and quantitative detection of TOP2A and prognostic value in triple-negative breast cancer. Int J Nanomedicine 2016;11:5519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Faggad A, Darb-Esfahani S, Wirtz R, et al. Topoisomerase IIalpha mRNA and protein expression in ovarian carcinoma: correlation with clinicopathological factors and prognosis. Mod Pathol 2009;22:579–88. [DOI] [PubMed] [Google Scholar]
- [39].Labbe DP, Sweeney CJ, Brown M, et al. TOP2A and EZH2 provide early detection of an aggressive prostate cancer subgroup. Clin Cancer Res 2017;23:7072–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang R, Xu J, Zhao J, et al. Proliferation and invasion of colon cancer cells are suppressed by knockdown of TOP2A. J Cell Biochem 2018;119:7256–63. [DOI] [PubMed] [Google Scholar]
- [41].Kato T, Daigo Y, Aragaki M, et al. Overexpression of KIAA0101 predicts poor prognosis in primary lung cancer patients. Lung Cancer 2012;75:110–8. [DOI] [PubMed] [Google Scholar]
- [42].Lv W, Su B, Li Y, et al. KIAA0101 inhibition suppresses cell proliferation and cell cycle progression by promoting the interaction between p53 and Sp1 in breast cancer. Biochem Biophys Res Commun 2018;503:600–6. [DOI] [PubMed] [Google Scholar]
- [43].Abdelgawad IA, Radwan NH, Hassanein HR. KIAA0101 mRNA expression in the peripheral blood of hepatocellular carcinoma patients: association with some clinicopathological features. Clin Biochem 2016;49:787–91. [DOI] [PubMed] [Google Scholar]
- [44].Cheng Y, Li K, Diao D, et al. Expression of KIAA0101 protein is associated with poor survival of esophageal cancer patients and resistance to cisplatin treatment in vitro. Lab Invest 2013;93:1276–87. [DOI] [PubMed] [Google Scholar]
- [45].Jain N, Roy J, Das B, et al. miR-197-5p inhibits sarcomagenesis and induces cellular senescence via repression of KIAA0101. Mol Carcinog 2019;58:1376–88. [DOI] [PubMed] [Google Scholar]
- [46].Rahman MA, Amin AR, Wang D, et al. RRM2 regulates Bcl-2 in head and neck and lung cancers: a potential target for cancer therapy. Clin Cancer Res 2013;19:3416–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tryfonidis K, Papadaki C, Assele S, et al. Association of BRCA1, ERCC1, RAP80, PKM2, RRM1, RRM2, TS, TSP1, and TXR1 mRNA expression levels between primary tumors and infiltrated regional lymph nodes in patients with resectable non-small cell lung cancer. Pharmacogenomics J 2019;19:15–24. [DOI] [PubMed] [Google Scholar]
- [48].Liu WT, Wang Y, Zhang J, et al. A novel strategy of integrated microarray analysis identifies CENPA, CDK1 and CDC20 as a cluster of diagnostic biomarkers in lung adenocarcinoma. Cancer Lett 2018;425:43–53. [DOI] [PubMed] [Google Scholar]
- [49].Kim Y, Choi JW, Lee JH, et al. Spindle assembly checkpoint MAD2 and CDC20 overexpressions and cell-in-cell formation in gastric cancer and its precursor lesions. Hum Pathol 2019;85:174–83. [DOI] [PubMed] [Google Scholar]
- [50].Wu WJ, Hu KS, Wang DS, et al. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J Transl Med 2013;11:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kato T, Daigo Y, Aragaki M, et al. Overexpression of CDC20 predicts poor prognosis in primary non-small cell lung cancer patients. J Surg Oncol 2012;106:423–30. [DOI] [PubMed] [Google Scholar]
- [52].Qian X, Song X, He Y, et al. CCNB2 overexpression is a poor prognostic biomarker in Chinese NSCLC patients. Biomed Pharmacother 2015;74:222–7. [DOI] [PubMed] [Google Scholar]
- [53].Li R, Jiang X, Zhang Y, et al. Cyclin B2 overexpression in human hepatocellular carcinoma is associated with poor prognosis. Arch Med Res 2019;50:10–7. [DOI] [PubMed] [Google Scholar]
- [54].Park SH, Yu GR, Kim WH, et al. NF-Y-dependent cyclin B2 expression in colorectal adenocarcinoma. Clin Cancer Res 2007;13:858–67. [DOI] [PubMed] [Google Scholar]
- [55].Dong S, Huang F, Zhang H, et al. Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues predicts poor survival in pancreatic ductal adenocarcinoma. Biosci Rep 2019;39:BSR20182306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhuang L, Yang Z, Meng Z. Upregulation of BUB1B, CCNB1, CDC7, CDC20, and MCM3 in tumor tissues predicted worse overall survival and disease-free survival in hepatocellular carcinoma patients. Biomed Res Int 2018;2018:7897346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rio Frio T, Lavoie J, Hamel N, et al. Homozygous BUB1B mutation and susceptibility to gastrointestinal neoplasia. N Engl J Med 2010;363:2628–37. [DOI] [PubMed] [Google Scholar]
- [58].Schneider MA, Christopoulos P, Muley T, et al. AURKA, DLGAP5, TPX2, KIF11 and CKAP5: five specific mitosis-associated genes correlate with poor prognosis for non-small cell lung cancer patients. Int J Oncol 2017;50:365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhan P, Zhang B, Xi GM, et al. PRC1 contributes to tumorigenesis of lung adenocarcinoma in association with the Wnt/beta-catenin signaling pathway. Mol Cancer 2017;16:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shi Q, Zhou Z, Ye N, et al. MiR-181a inhibits non-small cell lung cancer cell proliferation by targeting CDK1. Cancer Biomark 2017;20:539–46. [DOI] [PubMed] [Google Scholar]