

Abstract
To figure out which diagnosis is more suitable and which antiepileptic drugs are more sensitive to epileptic negative myoclonus (ENM) as the first seizure type in atypical benign epilepsy with centrotemporal spikes.
We reviewed the electroencephalogram (EEG) database of Linyi People's Hospital Affiliated to Shandong University and medical records of patients with ENM onset. The characteristics of epileptic seizures, onset age, treatment process, growth and development history, past disease history, family history, degree of mental deterioration, cranial imaging, and video-EEG were studied retrospectively and followed up.
There were 4 cases with ENM onset and 1 with continuous ENM, 3 males and 1 female. The onset age was from 2 years 3 months to 8 years 7 months. The cranial magnetic resonance imaging (MRI) and developmental quotient, as well as the family, personal, and past disease history, were normal. Frequent falls and drops were the main clinical manifestations. Five months after the onset of ENM, case 1 had focal seizures in sleep. ENM was the first and only manifestation in all the other 3 children. Discharges of interictal EEG were in bilateral rolandic areas, especially in midline areas (Cz, Pz), electrical status epilepticus in sleep was found in 3 cases. One child was sensitive to levetiracetam, the other 3 were sensitive to clonazepam.
ENM can affect the upper or lower extremities. ENM as the first or only symptom was a special phenomenon in benign epilepsy with centrotemporal spikes (BECTS) variants. Ignorance of midline spikes mainly in Cz or Pz in BECTS might lead to missed diagnosis of ENM. Whether benzodiazepines are viable as a choice of BECTS variants with electrical status epilepticus in sleep when ENM is the first symptom still needs a large sample evidence-based observation.
Keywords: benign epilepsy with centrotemporal spikes, benzodiazepines, electrical status epilepticus in sleep, epileptic negative myoclonus, video-EEG
1. Introduction
Epileptic negative myoclonus (ENM) is defined as a transient inhibition of muscle activity. Some studies have shown that ENM is produced by inhibiting the primary sensorimotor cortex of the affected limbs through magnetoencephalography.[1] ENM is not a specific seizure type of a particular epileptic syndrome. It has been reported to occur in West syndrome,[2] partial epilepsy, general epilepsy, or in other diseases or induced by some drugs.[3–8] ENM often occurs in the course of various types of epilepsy. Most ENM occurs after the diagnosis of benign childhood epilepsy with central temporal spikes (BECTS). It is rarely reported that ENM is the first onset and (or) the only symptom of epilepsy.
The clinical treatment and diagnosis and neuroelectrophysiological examinations of 4 epileptic children with negative myoclonus onset and rolandic discharges were retrospectively analyzed in the present study. This can help us to judge the prognosis and outcome of this type of epilepsy.
2. Methods
We reviewed the electroencephalogram (EEG) database of Linyi People's Hospital Affiliated to Shandong University and medical records of patients aged 0–18 years with ENM onset between 2013 and 2018. The children were followed up for seizures, head imaging, video-EEG characteristics, onset age, treatment process, growth, and development history, previous disease history, family history, and degree of intellectual regression. The study protocol was approved by the ethical committee of the Linyi people's hospital (No. 13002).
2.1. Electro-physiological examination of nervous system
All the children completed video-EEG monitoring in our hospital from 1 to several times. Each child was examined the day after sleep deprivation, with 19 EEG recording electrodes being placed in accordance with the international 10 to 20 system. Patients were monitored for about 4 hours, including open-close eye tests, hyperventilation, and at least 1 complete wake-sleep cycle. All the children underwent orthostatic straightening arms tests during video-EEG monitoring to detect ENM. Methods: The children, while in an awake state, were kept in standing positions with their arms repeatedly extended forward. At the same time, electromyography (EMG) monitoring was performed.
2.2. Methods and criteria for intelligence determination
Toddlers and infants under 4 years old were assessed with the Gesell Development Scale; children aged 4 to 6 were assessed with the Wechsler Pre-school Intelligence Scale, and children aged 7 to 14 were assessed with the Wechsler Intelligence Scale.
3. Results
There were 4 (0.86‰, 4/4650) EEG records with ENM as the first seizure type selected. There were 4 children, 3 male, and 1 female, aged from 2 years and 3 months to 8 years and 7 months, with frequent drops and falls as clinical manifestations (Table 1). All the patients had provided informed consent for publication of the cases.
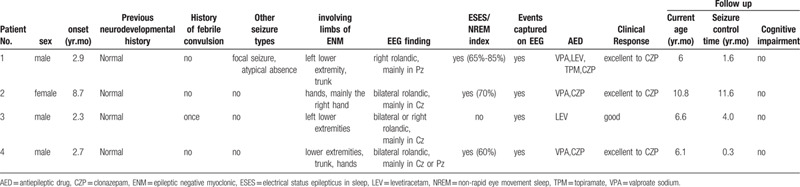
Table 1.
The clinical and follow-up data of 4 patients having atypical benign partial epilepsy with epileptic negative myoclonic.
3.1. Clinical Case
Case 1, male, 6 years old, onset age of 2 years and 9 months with seizure type 1 manifested as sudden weakness of his left leg when walking or standing, followed by falling, after falling the patient could quickly stand up again; this process occurred about 7 to 8 times a day, sometimes up to 20 times a day; and walking was unsteady. These seizures were found as continuous ENM in V-EEG, the left lower extremity flexion corresponds to the high amplitude spike slow wave in the right rolandic area of EEG, especially in Pz, and with the short rest of EMG in the left lower extremity. Interictal EEG: electrical status epilepticus in sleep (ESES), spike-slow-wave index (SWI) in non-rapid eye movement sleep was 65% to 85%. Two months after onset, seizure type 2 was characterized by nodding and tilting of the body, isolation or rhythm, sometimes accompanied by decreased consciousness. During the same period, EEG showed high-amplitude spike slow waves in the bilateral rolandic area, which could spread widely. At the same time, EMG of the upper extremity showed intermittent rest, which was judged to be an atypical absence seizure caused by continuous negative myoclonic seizures. Five months after onset, seizure type 3 appeared as 1 side of the limb flexion and shaking, consciousness is not completely lost, unable to speak, lasting about 1–2 minutes, from half an hour after he went to sleep, three times in 1 week. The patient was insensitive to valproate sodium (VPA), levetiracetam and topiramate. When clonazepam (CZP) was added, the seizures were controlled for 1 and a half years, and video-EEG was restored to normal.
Case 2, female, 10 years and 8 months, with the onset of negative myoclonus from 8 years and 7 months old which was manifested as multiple drops of both hands or right hand during awakening, with the most frequent being 10 to 20 drops per day and the longest interval of no seizures was 2 months. Spike and slow wave in both parietal rolandic areas and mainly in midline areas (Cz), and SWI was 70%. No other seizure types occurred. Seizures were poorly controlled by VPA. After 5 months of onset when CZP was added, the seizures stopped gradually, and the EEG was significantly improved without ESES. The amplitudes of spikes and slow waves were lower than 50uv. Two months later, the parents stopped CZP by themselves. The child had frequent seizures and ESES again. CZP was used again at 8 months after onset. The seizures stopped again. There was no seizure for more than 1 year and 6 months.
Case 3, male, 6 years and 6 months, with the onset of negative myoclonus at 2 years and 3 months old, manifested as the left lower limb collapsing suddenly after bending the knee during the awakening period, and could quickly get up, once every 3–5 days. Spikes and slow waves were mainly in the central midline area (Cz) of the right rolandic areas, with no ESES in the sleep period. Seizures were gradually stopped a week after the addition of levetiracetam. There was no seizure in 4 years, and epileptiform discharges were significantly reduced.
Case 4, male, 6 years and 1 month, continuous negative myoclonus onset at 2 years and 7 months old, manifested as frequent falls during awake period, sometimes accompanied by trunk shaking, tilting to the right, hands shaking, 2 to 3 times a day, the longest interval was 10 months. The child's seizures aggravated at 5 years and 6 months old and the patient began to see a doctor, with spike and slow waves discharge of the interictal EEG mainly in the central line area (Cz) of bilateral rolandic areas, and SWI was 60%. Frequent negative myoclonic seizures nearly status epilepticus were monitored, seizures and intermittent discharge disappeared after static diazepam as shown in Figure 1. The seizures were controlled within 1 week after VPA was added. Two months after treatment, the EEG did not show any seizure and the discharge during the interval of seizure was significantly reduced, and the amplitude decreased. The seizures occurred again 4 months after treatment, the EEG was aggravated, and the seizures stopped again by adding CZP. At present, there was no seizure in more than 3 months.
Figure 1.
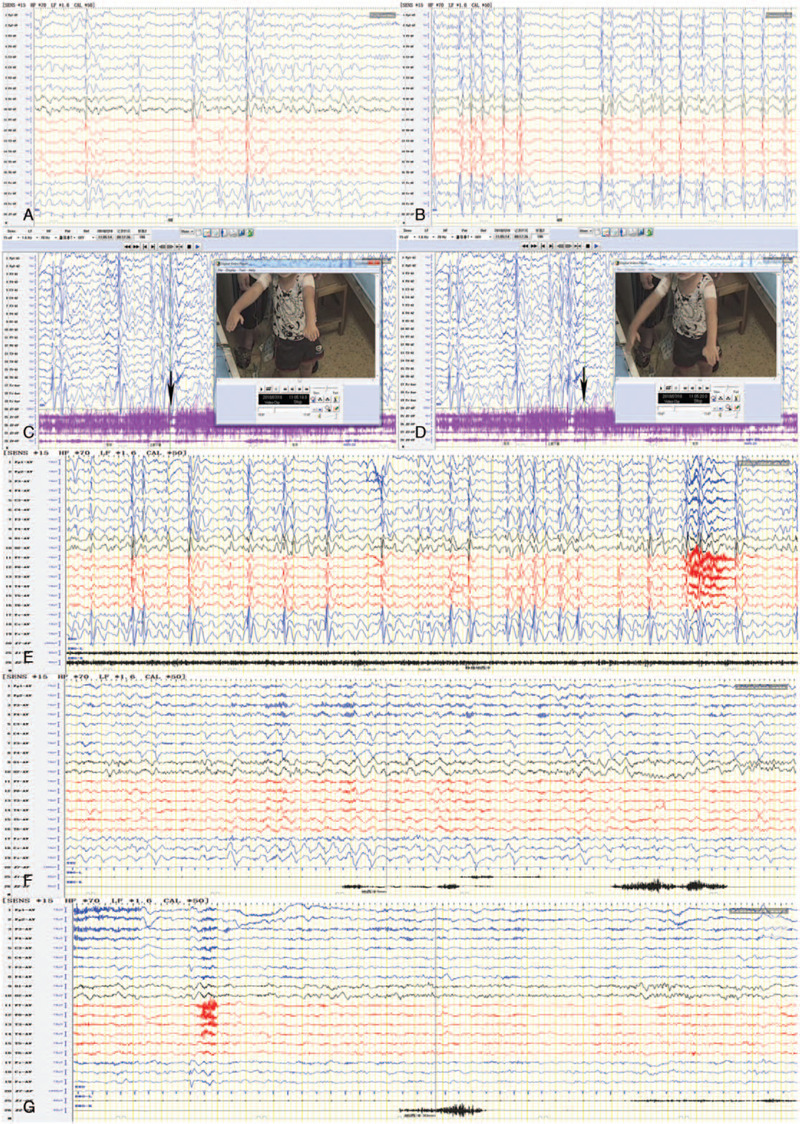
Case 4 A. bilateral spike and slow wave firing in rolandic area, mainly in midline area; B. Sleep period, electrical status epilepticus in sleep (ESES), non-rapid eye movement sleep (NERM) index 60%; C and D. frequent negative myoclonic seizures (red arrows); E. EEG of awake period before diazepam; F. 5 minutes after diazepam; G. 30 minutes after diazepam, intermittent discharge and seizures disappeared.
Case 1 presented atypical absence seizures at the second month and focal seizures at the 5th month after onset. Negative myoclonic seizures were the first and only manifestations in the other 3 cases, continuous ENM presented in 3 cases (1, 2, 4). There were 3 cases of ESES in rolandic regions, and the highest amplitude areas were Cz/Pz. All the patients had normal cranial MRI and developmental quotient, no special family history or personal history, no regression of development after onset. There was a history of simple febrile convulsion in case 3. Three Cases were sensitive to benzodiazepines. Although the EEG still had epileptiform discharge, the discharge index and amplitude were significantly reduced. Case 1 was detected by whole-exome sequencing, and no definite pathogenic mutation was found.
4. Discussion
Negative myoclonus epilepticus is a nonspecific motor disorder occurring in different epileptic syndromes.[2,9–11] This dyskinesia is caused by dysfunction of different anatomical sites, including cortical lesions in the premotor and motor areas. Negative myoclonic can involve 1 or both limbs clinically, resulting in temporary loss of muscle strength, thereby interfering with the coordination of movement, resulting in instability. When the upper limbs are involved in the performance of shaking hands, there is droping something suddenly when holding; when the trunk is involved in the performance of nodding, there is body tilt; when the lower limbs are involved in the performance of standing or walking, there is instability and falling. And lower limbs were less commonly involved than upper extremities. Three of the children in this group complained mainly of falls, which were easily missed or misdiagnosed as atonic, tonic, or myoclonic-atonic seizures. We usually need to use video-EEG to further clarify the type of seizures. EMG resting is time-locked to epileptiform discharge in the contralateral cortex, and there is no previous EMG burst.[12,13] Negative myoclonic seizures were monitored by video-EEG in 4 patients. When the EEG showed high amplitude spikes and slow waves in the high rolandic area in awake periods for 1 to several seconds, the contralateral limb EMG rested. Epileptiform discharges in interictal and ictal period EEG were mainly located in the midline region in this group. And the main clinical involvement was lower extremity and trunk. And 3/4 patients had continuous ENM. We inferred that continuous ENM as the first symptoms of rolandic epilepsy with ESES may be related to the discharge location which is mainly in the midline area (Cz or Pz). A similar situation had not been mentioned in previous reports of ENM, maybe because of the absence of midline area in early EEG monitoring, which needs to be observed with large sample evidence-based observation.
In addition to ENM, the other major feature of this group of children was ESES. ESES is a special EEG phenomenon as the sustained spike and slow wave which were induced by sleep. Recent studies on ESES genetics have found that SLC9A6, ATN1, SRPX2, OPA3, KCNQ2, KCNA2, GRIN2A,[14]CNKSr2, SLC6A1, KCNB1, SCN2A, and some CNVs play a role in the genesis of ESES lineage.[14,15] In this study, case 1 was detected by whole-exome sequencing, and no definite pathogenic gene mutation was found. We know the genetic basis of BECTs is polygenic and complex.[16] In the next step, we will expand the samples of this special phenotype in BECT for GWAS analysis to understand the mechanisms, it will also help us to know why the current patients have a lower chance of BECTS type epileptic syndrome. Adamantadine,[17] steroid hormone,[18] and surgery[19] have been reported to have therapeutic effects on ESES, whereas benzodiazepines may be an appropriate alternative.[19,20] Sánchez Fernández and Inutsuka[21,22] found diazepam could significantly reduce the spikes and slows wave index of the non-rapid eye movement sleep sleep period, in the long-term follow-up, a few cases recurred, and the spikes and slows wave index returned to the level before treatment. Repeating the diazepam treatment still has a certain effect, and with fewer side effects and short medication time. With the treatment of ESES, the decline of the SWI is usually accompanied by a cognitive improvement.[23] ESES can occur in a series of epileptic syndromes characterized by epileptic seizures, ESES, and cognitive impairment. It is commonly called ESES-related syndromes, which include epilepsy with continued slow spike and slow wave in slow wave sleep, acquired epileptic aphasia syndrome, BECTS and its variants. BECTS is the most common epileptic syndrome in childhood,[24,25] accounting for 15%-24% of children with epilepsy; onset age is 2 to 10 years old, and the typical form of seizures is focal seizures in sleep. Antiepileptic drugs have a high curative effect in most cases. Recently studies had shown that children with BECTS may suffer from impairment of speech, oropharyngeal motor function, cognition, and memory.[26–28] 4.6% to 5.8% of the children with BECTS had atypical changes during the course of the disease, which were characterized by increased frequency of habitual epileptic seizures, new types of seizures, and ESES. Studies have shown that the BECTS variant can be distinguished by monitoring the high and middle frequency waves of EEG,[29] but we didn’t find it in our study.
All the patients we observed presented mainly or only with ENM. The spike and slow waves were emitted in unilateral or bilateral rolandic regions, and the highest amplitude areas were Cz/Pz, and with ESES. The patients could be diagnosed as BECTS variant type I by literature review, although they do not have the classic onset like other BECTS variants. Three of the 4 patients were boys, and the onset age was less than 3 years old. The onset age was generally much younger. Continuous ENM mainly restricted to lower limbs leading to gait instability or even falls. This is unlike the classical BECTS variant type I which mainly involves upper limbs and mostly appears in isolation. When discharges in the midline region had the highest amplitude, then ENM was mainly restricted to lower limbs, which might because the area that controls the lower limb is closer to the central sulcus in the anatomical position. In our study, a small dose of CZP was taken orally before sleep to avoid the influence (such as drowsiness, unresponsiveness, memory decline) of large dose or daytime CZP on children's daily activities. We found ENM could be well controlled when CZP was added, even if ESES did not improve significantly. Three cases, especially case 4, had frequent, near persistent ENM due to intermittent or near persistent high-amplitude spikes and slow waves in the rolandic area during the awake period of EEG monitoring. And epileptiform discharge and seizures were clearly controlled by intravenous diazepam. In consideration of side effects, VPA was chosen as the first choice, a good curative effect was obtained at the beginning. CZP was preferred to be added after repeated illness because we knew the therapeutic process of case 1 and case 2. The addition of CZP proved effective. The continuous ENM onset in 3 children with ESES was susceptible to benzodiazepine. But because of the small number of samples in our study, whether benzodiazepines are a viable choice of treatment for BECTS variants with ESES when ENM is the first symptom still needs a large sample evidence-based observation.
When ENM appears as the only or main clinical manifestation and is accompanied by ESES, attention should be paid to BECTS variants. For Rolandic epilepsy with ENM onset, the lower limbs and trunk are mainly involved, and epileptiform discharge is mainly in the midline area. Ignorance of midline spikes mainly in Cz or Pz in BECTS might lead to a missed diagnosis of ENM. Whether benzodiazepines as choice for treatment of BECTS variants with ESES when ENM is the first symptom needs a large sample evidence-based observation.
The sample size of this study is small, and not all of them have been tested for genes. In the future, expanding the samples of this special phenotype in BECTS for GWAS analysis may help us understand the mechanisms.
Acknowledgment
Thanks to Rowan Coates for his guidance on English grammar.
Author contributions
Conceptualization: Baomin Li.
Data curation: Li Yang, Na Xu, LiYun Xu, Juan Zhao, Yufen Li.
Investigation: Baomin Li.
Writing – original draft: Li Yang.
Writing – review & editing: Li Yang, Baomin Li.
Footnotes
Abbreviations: BECTS = benign childhood epilepsy with central temporal spikes, CZP = clonazepam, EEG = electroencephalogram, EMG = electromyography, ENM = epileptic negative myoclonus, ESES = electrical status epilepticus in sleep, SWI = spike-slow-wave index, TPM = topiramate, VPA = valproate sodium.
How to cite this article: Yang L, Su Q, Xu N, Xu L, Zhao J, Fan C, Li Y, Li B. Continuous epileptic negative myoclonus as the first seizure type in atypical benign epilepsy with centrotemporal spikes. Medicine. 2020;99:44(e22965).
This study was supported by the foundation of Science and Technology Development Plan of Shandong Province (NO.2018WS399), Science and Technology Development Plan of Linyi (NO.201818009), National Key Research and Development Program of China (NO.2016YFC1306202), Key Research and Development Plan in Shandong Province (NO.2016GSF201073), General program of Qilu Hospital of Shandong University (NO.2015QLMS08), Medical and Health Technology Development Program in Shandong Province (No. 2017WS499).
The authors have no conflicts of interest to disclose.
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
- [1].Yoshimura M, Zhang S, Ueda Y, et al. An analysis of epileptic negative myoclonus by magnetoencephalography. Epilepsy Res 2015;110:139–45. [DOI] [PubMed] [Google Scholar]
- [2].Shibata T, Yoshinaga H, Oka M, et al. [West syndrome associated with epileptic negative myoclonus]. No To Hattatsu 2014;46:354–8. [PubMed] [Google Scholar]
- [3].Correia P, Ribeiro JA, Bento C, et al. Negative myoclonus secondary to paroxetine intake. BMJ Case Rep 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim JB, Jung JM, Park MH, et al. Negative myoclonus induced by gabapentin and pregabalin: a case series and systematic literature review. J Neurol Sci 2017;382:36–9. [DOI] [PubMed] [Google Scholar]
- [5].van Samkar A, De Kleermaeker F, Te Riele MGE, et al. Negative myoclonus induced by ciprofloxacin. Tremor Other Hyperkinet Mov (N Y) 2017;7:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Praharaj SK, Vemanna N, Sharma PS. Knee buckling (negative myoclonus) associated with clozapine: is there a dose threshold? Clin Toxicol (Phila) 2015;53:918–9. [DOI] [PubMed] [Google Scholar]
- [7].Gelisse P, Genton P, Velizarova R, et al. Worsening of negative myoclonus by lamotrigine in a case of idiopathic focal epilepsy of children with long-term follow-up. Brain Dev 2012;34:248–50. [DOI] [PubMed] [Google Scholar]
- [8].Kim YB, Park HE, Ryu DW, et al. Chronic negative myoclonus with hypercapnic encephalopathy. Acta Neurol Belg 2017;117:355–7. [DOI] [PubMed] [Google Scholar]
- [9].Guerrini R, Takahashi T. Myoclonus and epilepsy. Handb Clin Neurol 2013;111:667–79. [DOI] [PubMed] [Google Scholar]
- [10].Li X, Wu D, Fernandez IS, et al. Negative myoclonus in a child with anti-NMDA receptor encephalitis. J Neurol Sci 2015;358:532–4. [DOI] [PubMed] [Google Scholar]
- [11].Gunduz A, Kiziltan ME, Coskun T, et al. Electrophysiological findings in Rasmussen's syndrome. Epileptic Disord 2016;18:73–6. [DOI] [PubMed] [Google Scholar]
- [12].Apartis E, Vercueil L. To jerk or not to jerk: a clinical pathophysiology of myoclonus. Rev Neurol (Paris) 2016;172:465–76. [DOI] [PubMed] [Google Scholar]
- [13].Qian P, Li H, Xue J, et al. Scalp-recorded high-frequency oscillations in atypical benign partial epilepsy. Clin Neurophysiol 2016;127:3306–13. [DOI] [PubMed] [Google Scholar]
- [14].Qian P, Yang X, Xu X, et al. Study of GRIN2A mutation in epilepsy-aphasia spectrum disorders. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2018;35:314–8. [DOI] [PubMed] [Google Scholar]
- [15].Kessi M, Peng J, Yang L, et al. Genetic etiologies of the electrical status epilepticus during slow wave sleep: systematic review. BMC Genet 2018;19:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xiong W, Zhou D. Progress in unraveling the genetic etiology of rolandic epilepsy. Seizure 2017;47:99–104. [DOI] [PubMed] [Google Scholar]
- [17].Wilson RB, Eliyan Y, Sankar R, et al. Amantadine: a new treatment for refractory electrical status epilepticus in sleep. Epilepsy Behav 2018;84:74–8. [DOI] [PubMed] [Google Scholar]
- [18].Altunel A, Altunel EO, Sever A. Response to adrenocorticotropic in attention deficit hyperactivity disorder-like symptoms in electrical status epilepticus in sleep syndrome is related to electroencephalographic improvement: a retrospective study. Epilepsy Behav 2017;74:161–6. [DOI] [PubMed] [Google Scholar]
- [19].van den Munckhof B, van Dee V, Sagi L, et al. Treatment of electrical status epilepticus in sleep: a pooled analysis of 575 cases. Epilepsia 2015;56:1738–46. [DOI] [PubMed] [Google Scholar]
- [20].Su TF, Xu SQ, Chen L. Efficacy of levetiracetam combined with short-term clonazepam in treatment of electrical status epilepticus during sleep in children with benign childhood epilepsy with centrotemporal spikes. Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatrics 2014;16:829–33. [PubMed] [Google Scholar]
- [21].Sanchez Fernandez I, Hadjiloizou S, Eksioglu Y, et al. Short-term response of sleep-potentiated spiking to high-dose diazepam in electric status epilepticus during sleep. Pediatr Neurol 2012;46:312–8. [DOI] [PubMed] [Google Scholar]
- [22].Inutsuka M, Kobayashi K, Oka M, et al. Treatment of epilepsy with electrical status epilepticus during slow sleep and its related disorders. Brain Dev 2006;28:281–6. [DOI] [PubMed] [Google Scholar]
- [23].van den Munckhof B, Alderweireld C, Davelaar S, et al. Treatment of electrical status epilepticus in sleep: clinical and EEG characteristics and response to 147 treatments in 47 patients. Eur J Paediatr Neurol 2018;22:64–71. [DOI] [PubMed] [Google Scholar]
- [24].Li R, Wang L, Chen H, et al. Abnormal dynamics of functional connectivity density in children with benign epilepsy with centrotemporal spikes. Brain Imaging Behav 2018;13:985–94. [DOI] [PubMed] [Google Scholar]
- [25].Tan G, Xiao F, Chen S, et al. Frequency-specific alterations in the amplitude and synchronization of resting-state spontaneous low-frequency oscillations in benign childhood epilepsy with centrotemporal spikes. Epilepsy Res 2018;145:178–84. [DOI] [PubMed] [Google Scholar]
- [26].Tristano I, Nicita F, Garone G, et al. Could Rolandic spikes be a prognostic factor of the neurocognitive outcome of children with BECTS? Epilepsy Behav 2018;86:157–62. [DOI] [PubMed] [Google Scholar]
- [27].Wickens S, Bowden SC, D'Souza W. Cognitive functioning in children with self-limited epilepsy with centrotemporal spikes: a systematic review and meta-analysis. Epilepsia 2017;58:1673–85. [DOI] [PubMed] [Google Scholar]
- [28].Fujiwara H, Tenney J, Kadis DS, et al. Cortical morphology, epileptiform discharges, and neuropsychological performance in BECTS. Acta Neurol Scand 2018;138:432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ikemoto S, Hamano SI, Yokota S, et al. Enhancement and bilateral synchronization of ripples in atypical benign epilepsy of childhood with centrotemporal spikes. Clin Neurophysiol 2018;129:1920–5. [DOI] [PubMed] [Google Scholar]