

Abstract
BACKGROUND:
The dura mater can be easily biopsied during most cranial neurosurgical operations. We describe a protocol that allows for robust generation of induced pluripotent stem cells (iPSCs) and neural progenitors from acutely harvested dura mater.
OBJECTIVE:
To generate iPSCs and neural progenitor cells from dura mater obtained during ventriculoperitoneal shunt surgery.
METHODS:
Dura was obtained during ventriculoperitoneal shunt surgery for normal pressure hydrocephalus from a 60-year-old patient with severe cognitive impairment. Fibroblasts were isolated from the dural matrix and transduced with nonintegrating Sendai virus for iPSC induction. A subset of successfully generated iPSC clones underwent immunocytochemical analysis, teratoma assay, karyotyping, and targeted neural differentiation.
RESULTS:
Eleven iPSC clones were obtained from the transduction of an estimated 600,000 dural fibroblasts after 3 passages. Three clones underwent immunocytochemical analysis and were shown to express the transcription factors OCT-4, SOX2, and the embryonic cell markers SSEA-4, TRA-1–60, and Nanog. Two clones were tested for pluripotency and formed teratomas at the injection site in immunodeficient mice. Three clones underwent chromosomal analysis and were found to have a normal metaphase spread and karyotype. One clone underwent targeted neural differentiation and formed neural rosettes as well as TuJ1/SOX1-positive neural progenitor cells.
CONCLUSIONS:
IPSCs and neural progenitor cells can be efficiently derived from the dura of patients who need to undergo cranial neurosurgical operations. IPSCs were obtained with a nonintegrating virus and exhibited a normal karyotype, making them candidates for future autotransplantation after targeted differentiation to treat functional deficits.
Keywords: Dura, Fibroblast, Induced pluripotent stem cell, Neural progenitor cell, Regenerative medicine, Shunt surgery
INTRODUCTION
Cell restoration therapy with neural stem and progenitor cells holds great promise for the treatment of various neurological and neurosurgical diseases such as stroke, traumatic brain injury, Alzheimer disease or Parkinson disease (2, 3, 11, 12). Transplantation of human neural stem cells from adult donors has proven inefficient in replacing lost neurons after stroke. In contrast to fetal-derived progenitor cells, adult neural progenitor cells predominantly differentiate down a glial lineage pathway and have poor capacity to generate neurons in vivo (14). Even neural progenitor cells derived from the developing cortex lose their ability to form neurons over time and eventually undergo senescence after expansion in vitro (24).
Induced pluripotent stem cells (iPSCs) provide a better means of generating neurons, as these cells are capable of replicating the period of fetal neuronal production, which is largely complete by midgestation (19). Often, iPSCs are derived from skin fibroblasts (7) or from hematopoietic progenitor cells (26). Neurosurgeons can easily obtain a dural sample during shunt surgery (e.g., hydrocephalus after aneurysmal subarachnoid hemorrhage, hydrocephalus after germinal matrix hemorrhage) or during craniotomies (e.g., hemicraniectomy for traumatic brain injury or stroke). This raises the question whether iPSCs and neural progenitor cells can reliably be generated from dura mater tissue obtained during surgery.
Although the generation of iPSCs from postmortem dura mater has been described before (18), the derivation of iPSCs from a small piece of acutely harvested dura mater for therapeutic purposes has not yet been published. We present the efficient production of iPSCs and neural progenitor cells with normal karyotype from dura mater fibroblasts of a 60-year-old man who underwent ventriculoperitoneal shunt surgery for normal pressure hydrocephalus. Because the patient also suffered from failure to thrive resulting in modified Rankin scale 3 disability, he may be considered as an example of a patient who could benefit in the future from transplantation of his own iPSCs-derived neural progenitor cells using this technique.
METHODS
Dural Harvest
The local institutional review board (#364524–7) and the stem cell research oversight (#1106) committees approved the dural harvest to generate iPSCs. The patient was a 60-year-old man with impaired cognition and a modified Rankin scale of 3 due to failure to thrive in the setting of traumatic brain injury and chronic alcoholism. He cannot read or write and never held a job. He underwent a craniotomy for right frontal hematoma evacuation in 2012 at an outside hospital. He was subsequently diagnosed with normal pressure hydrocephalus because he had ventriculomegaly on magnetic resonance imaging, and his gait improved after a large-volume lumbar puncture. He was scheduled for placement of a left frontal ventriculoperitoneal shunt. The family consented to the dural harvest. During surgery, a left frontal burr hole was placed with a perforator. The edges of the burr hole were undercut with a No. 4 Kerrison punch (Aesculap, Center Valley, Pennsylvania, USA). The dura was not coagulated before the harvest. The dura was harvested as a 1-cm piece with a No. 11 scalpel by cutting the dura in a circular fashion along the edges of the burr hole. The dural specimen was placed in Hank’s buffered solution (GIBCO, Grand Island, New York, USA) and stored overnight at 4°C. The sample was frozen to −90°C with a Gordinier Electronics controlled rate freezer (Roseville, Michigan, USA) in 10% dimethyl sulfoxide (Sigma, St. Louis, Missouri, USA), 45% fetal bovine serum (FBS; JR Scientific, Woodland, California, USA), and 45% dura fibroblast medium consisting of 10% FBS, 2 mM glutamax (GIBCO), 0.1 mM 2-mercaptoethanol (GIBCO), and 100 U/mL–0.1 mg/mL penicillin-streptomycin (HyClone, Logan, Utah, USA) in Dulbecco’s modified eagle medium: nutrient mixture F12 (DMEM/F12; HyClone).
Fibroblast Isolation and Passaging
The sample was thawed for 1 minute at 37°C and washed twice in phosphate-buffered saline (PBS; HyClone), followed by 2 washes in DMEM (HyClone). The sample was cut into pieces measuring 100 μm to 3 mm using a sterile scalpel. One drop of sterile silicon grease (Corning, Tewksbury, Massachusetts, USA) was added to the center of the wells of a 6-well plate (Corning), and 4–5 pieces of the dura were placed around each drop. A coverslip (Fisher Scientific, Waltham, Massachusetts, USA) was placed on top of the silicon and samples and 2 mL of biopsy culture media consisting of 10% FBS, 2 mM glutamax, 0.1 mM 2-mercaptoethanol, 0.1 mM Minimal Essential Medium Non-Essential Amino Acids Solution (GIBCO), 1× antibiotic-antimycotic solution (GIBCO), and 1× nucleosides (Millipore, Temecula, California, USA) in DMEM was added to each well. The sample was incubated for 6 days at 37°C in 5% CO2. After 6 days, the medium was switched to 2 mL/well of fibroblast medium, and medium was changed every other day. After 16 days, fibroblasts were confluent and all coverslips were lifted. Fibroblast medium was removed, and cells were rinsed with PBS. Fibroblasts were incubated for 5 minutes with 500 μL TrypLE (1×; Life Technologies, Carlsbad, California, USA), then diluted with 500 μL fibroblast medium and replated in T25 flasks (Corning). Fibroblasts were passaged 3 times before transduction. Fibroblasts were cryopreserved after centrifugation at 1000 rpm for 5 minutes at room temperature and resuspension in 10% dimethyl sulfoxide/45% FBS/45% dura fibroblast medium.
Transduction of Fibroblasts into iPSCs
Fibroblasts were plated into 2 wells of a 6-well plate at a concentration of 300,000 cells per well with fibroblast medium. Cells were transduced 1 day later with the CytoTune 2.0 Sendai virus (Life Technologies) at a multiple of infection of 5:5:3 (KOS:c-Myc:Klf4) and incubated at 37°C overnight. Day 1 after transduction, the medium was replaced with fresh fibroblast medium to remove the CytoTune 2.0 Sendai reprogramming virus (Life Technologies). On days 3 and 5 after transduction, the medium was again replaced with fresh fibroblast medium. On day 6, mouse embryonic fibroblast (MEF) dishes were prepared. Twelve 100-mm dishes were coated for 1 hour at room temperature with 0.1% gelatin (Life Technologies). The gelatin was then aspirated and 10 mL of MEF medium containing 10% FBS (JR Scientific), 2 mM glutamax in DMEM High Glucose (HyClone) with 1.6 × 106 mouse embryonic fibroblasts (GlobalStem, Gaithersburg, Maryland, USA) was added to each dish. On day 7 after transduction, the human fibroblast cells were washed once with Dulbecco’s PBS (DPBS) and subsequently lifted from the 6-well plates with 0.5 mL TrypLE Select reagent (Life Technologies) at 37°C. After 2 minutes, 2 mL of fibroblast medium was added, and cells were centrifuged for 4 minutes at 1000 rpm in 15 mL conical tubes (Corning). Cells were resuspended in fibroblast medium and plated at a concentration of 217,500 fibroblasts per 100-mm MEF dish in a total of 12 dishes. One day later, the medium was changed to iPSC medium (20% KnockOut Serum Replacement, 100 nM Minimal Essential Medium Non-Essential Amino Acids Solution, 1% glutamax, 55 nM β-mercaptoethanol, and 30 ng/mL basic fibroblast growth factor (bFGF) in Knockout DMEM/F12; Life Technologies). The iPSC medium was changed every other day for 10 days. Twelve days after plating the human fibroblasts on MEF dishes, 32 iPSC-like colonies were collected manually with a 10-μL pipette tip. They were plated as individual clones in their own wells in 12-well plates. Eleven of the original 32 iPSC-like clones were passaged 6 days later into 1 well each of 6-well MEF plates. Each colony was manually dissected out using a 10-μL pipette tip. The colonies were then washed out of each well, broken up gently with a 1000-μL pipette tip and placed in fresh MEF wells for expansion. All 11 iPSC clones could be passaged a third and fourth time. After passage 5, all iPSC clones were frozen with the Gordinier Electronics freezing system.
Immunocytochemistry of iPSCs
Three of the 11 iPSC clones were randomly chosen for immunocytochemistry to verify pluripotency. Clones were analyzed with the Pluripotent Stem Cell 4-Marker Immunocytochemistry Kit (Life Technologies). Clones were fixed with 4% paraformaldehyde in 0.1 M DPBS for 15 minutes at room temperature in 6-well plates (Corning). Cells were stored for 6 days in 1× wash buffer (0.1 M DPBS; Life Technologies). They were then incubated at room temperature for 15 minutes in permeabilization solution (1% saponin in DPBS; Life Technologies), which was followed by incubation in blocking solution (3% bovine serum albumin in DPBS; Life Technologies) for 30 minutes. Clones in one 6-well plate were incubated with a combination of mouse immunoglobulin (Ig)G3 anti-SSEA4 (1:100; Life Technologies) and rabbit anti-OCT4 (1:100, Life Technologies) overnight at 4°C. After 3 washes in 1× wash buffer, clones were incubated with the secondary antibodies Alexa Fluor 488 goat anti-mouse IgG3 (1:250; Life Technologies) and Alexa Fluor 594 donkey anti-rabbit (1:250; Life Technologies) for 1 hour at room temperature in blocking solution. Clones in another 6-well plate were incubated with a combination of rat anti-SOX2 (1:100; Life Technologies) and mouse IgM anti-TRA-1–60 (1:100; Life Technologies) overnight at 4°C. After 3 washes in 1× wash buffer, clones were incubated with the secondary antibodies Alexa Fluor 488 Donkey anti-rat (1:250; Life Technologies) and Alexa Fluor 594 goat anti-mouse IgM (1:250; Life Technologies) for 1 hour at room temperature in blocking solution. A third 6-well plate was stained with polyclonal rabbit anti-Nanog antibody (1:200; PeproTech, Rocky Hill, New Jersey, USA) overnight at 4°C, washed 3 times with 1× wash buffer and incubated with Alexa Fluor 594 donkey anti-rabbit antibody (1:250; Life Technologies) for 1 hour at room temperature in blocking solution. All 6-well plates were subsequently washed 3 times with wash buffer and counterstained with 2 drops of NucBlue Fixed Cell Stain (4′,6-diamidino-2-phenylindole; Life Technologies). Cells were directly imaged after staining with a Nikon (Tokyo, Japan) Ti-U inverted microscope using a Photometrics (Tucson, Arizona, USA) CoolSnap camera.
Transcriptome Profiling of Fibroblasts Before and After Transduction
Transcriptome analysis was performed using Applied Biosystems’ (Foster City, California, USA) TaqMan hPSC Scorecard Kit (A15871) per the manufacturer’s instructions. Briefly, fibroblasts and iPSC were grown, passaged, and lifted as previously described. Ribonucleic acid (RNA) was isolated using Direct-zol RNA MiniPrep (Zymo Research, Irvine, California, USA), and the RNA was immediately quantified using a NanoDrop. Single-stranded complementary deoxyribonucleic acid (cDNA) was then generated from 1 μg fresh total RNA by reverse transcription using the high-capacity cDNA reverse transcription kit with RNase inhibitor (Applied Biosystems). The reverse transcriptase reaction was run in a BioRad S1000 Thermal Cycler (Hercules, California, USA). The cDNA was immediately used for a quantitative reverse transcriptase–polymerase chain reaction using the TaqMan hPSC Scorecard Panel (Applied Biosystems) using one 96-well FAST plate per sample. The cDNA was loaded to the plate along with 2× TaqMan Fast Advanced Master Mix in equal parts, and the reaction was run in the Applied Biosystems StepOnePlus using Standard Curve method. The gene expression data generated by the quantitative reverse transcriptase–polymerase chain reaction experiment were analyzed using the web-based hPSC Scorecard Analysis Software provided by Applied Biosystems at www.lifetechnologies.com/scorecarddata.
Teratoma Assay
Two of the 11 iPSC clones were chosen for a teratoma assay to establish pluripotency. Clones were passaged 11 times on MEFs using 30 ng/mL bFGF iPSC medium until a total volume of four 6-well plates was obtained for each clone. Subsequently, each 6-well plate was treated with Collagenase IV (210 U/mL; Worthington, Lakewood, New Jersey, USA) for 10 minutes at 37°C. Cells were scraped from each well and centrifuged at 850 rpm for 3 minutes. Cells from each 6-well plate were resuspended separately in 125 μL of 30% Matrigel/PBS (BD, San Jose, California, USA) to a concentration of 1 × 106 cells/125 μL and loaded into a 1-mL insulin syringe (BD). Eight adult male nonobese diabetic/severe combined immunodeficiency common gamma chain −/− mice (stock number 005557; Jackson Laboratories, Sacramento, California, USA) were injected subcutaneously into the left hind limb with 1 of the 8 samples derived from the 2 iPSC clones. Mice were housed in ventilated Tecniplast (West Chester, Pennsylvania, USA) cages and fed Harlan (Indianapolis, Indiana, USA) 2918 irradiated chow ad libitum. After 2.5 months, mice were submitted to the Comparative Pathology Laboratory for complete necropsy evaluation. Mice were euthanized by carbon dioxide narcosis, evaluated for any gross evidence of tumor formation, and representative sets of tissues, including the liver, kidneys, spleen, pancreas, adrenal gland, gallbladder, lungs, heart, brain, gastrointestinal tract, urogenital tract, and left hind limb, were collected for histology. Collected tissues were fixed in 10% neutral-buffered formalin, paraffin-embedded, routinely processed, and stained with hematoxylin and eosin. A board-certified veterinary pathologist evaluated tissue sections for any evidence of tumor and/or teratoma formation.
Karyotyping
Three iPSC clones were chosen for karyotyping. IPSC lines were plated in a 6-well plate. Each well was treated with 20 μL colcemid (GIBCO) for 2 hours in a 37°C incubator to arrest the mitotic cells in metaphase. Colonies were lifted with Accutase (Stem Cell Technologies, Vancouver, British Columbia, Canada) and subsequently centrifuged at 850 rpm for 3 minutes. The cell pellet was resuspended in 0.067 M KCl (Sigma) hypotonic solution and incubated for 20 minutes at room temperature. A 3:1 methanol (Fisher Scientific):acetic acid (Sigma-Aldrich) fixative solution was added to the hypotonic solution and incubated for 5 minutes at room temperature. This was followed by 3 treatments in a 3:1 methanol (Fisher Scientific):acetic acid (Sigma-Aldrich) fixative solution, with each treatment incubated for 1 hour at room temperature. Samples were dropped onto clean wet slides and were aged in an oven at 90°F for 1–2 hours. Afterward, slides were immersed in Trypsin (HyClone) for 30–40 seconds and rinsed in FBS and saline. After an additional rinse in saline, slides were stained in 12.5% Giemsa (GIBCO) in Gurrs buffer (GIBCO) for 2–3 minutes. Slides were rinsed in distilled water and air-dried. Microscopic analysis included scanning of all slides, the count of a minimum of 20 metaphases, analysis of a minimum of 7 metaphases, and karyotyping a minimum of 2 metaphases.
Neural Differentiation and Staining
Colonies were grown on MEF plates in media containing 30 ng/mL bFGF iPSC medium. When clones had grown to large colonies, medium was removed and replaced with 1 mL PBS. Marked colonies were manually dissected out with a P10 pipette tip. Colonies were pooled in a 15-mL conical tube (Corning) and centrifuged for 3 minutes at 850 rpm. The pellet was incubated for 3 minutes at 37°C with Accutase and cells were triturated afterward with a P1000 pipette tip. Subsequently, 2 mL of mTeSR1 medium (Stem Cell Technologies) was added to the cells. Cells were centrifuged again at 850 rpm and resuspended in 1 mL mTeSR1 medium. Cells were plated at a concentration of 25,000 cells per well on poly-l-ornithine (100 μg/mL; Sigma) and laminin (3 μg/mL; Sigma) coated 48-well plates (Corning) in mTeSR1 and 10 μM ROCKi (Calbiochem, San Diego, California, USA). Cells were incubated for 2 days at 37°C at 5% CO2. On day 3, cells were incubated in STEMdiff APEL medium (Stem Cell Technologies) with 10 μM SB421542 (Stemgent, Cambridge, Massachusetts, USA) and 250 nM LDN193189 (Stemgent). LDN193189 and SB421542 were used for dual SMAD inhibition to rapidly differentiate iPSCs into early neuroectoderm (4). Medium was changed every other day. On day 11, half of the media was replaced with neural differentiation medium (Neurobasal media (GIBCO), B27 without retinoic acid (1×; GIBCO), 2 mM glutamax and penicillin-streptomycin (100 U/mL/0.1 mg/mL). On day 13, another half of the media was replaced with neuronal differentiation medium. After day 15, the medium was completely exchanged for neuronal differentiation medium every other day. On day 22, cells were fixed with 4% paraformaldehyde for 12 minutes. Cells were washed 3 times in PBS and incubated with permeabilizing/blocking solution (2% Triton X-100 and 1% goat serum [GIBCO] in PBS) for 1 hour at room temperature. Differentiated cells were then incubated with mouse monoclonal TuJ-1 antibody (1:1000; Abcam, Cambridge, Massachusetta, USA) and rabbit polyclonal anti-SOX1 antibody (1:250; Abcam) overnight at 4°C. Cells were washed 3 times with PBS and incubated with secondary antibodies Alexa Fluor 488 goat anti-mouse IgG (Invitrogen, Carlsbad, California, USA) and Alexa Fluor 594 goat anti-rabbit IgG (Invitrogen) at 1:1000 at room temperature for 1 hour. Cells were counterstained and mounted with 2.5 drops of Vectashield Mounting Medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, California, USA). Images were taken immediately with a Nikon Ti-U microscope (Tokyo, Japan) using a Photometrics (Tucson, Arizona, USA) CoolSnap camera.
RESULTS
A 1-cm piece of dura could easily be harvested during shunt surgery and was cut into smaller pieces ranging from 100 μm to 3 mm (Figure 1A). Migration of fibroblasts out of the dural matrix was first observed after 6 days (Figure 1B), increased after 8 days (Figure 1C), and was robust after 16 days (Figure 1D). After 16 days, enough fibroblasts had migrated out of the dural matrix to perform the first passage.
Figure 1.
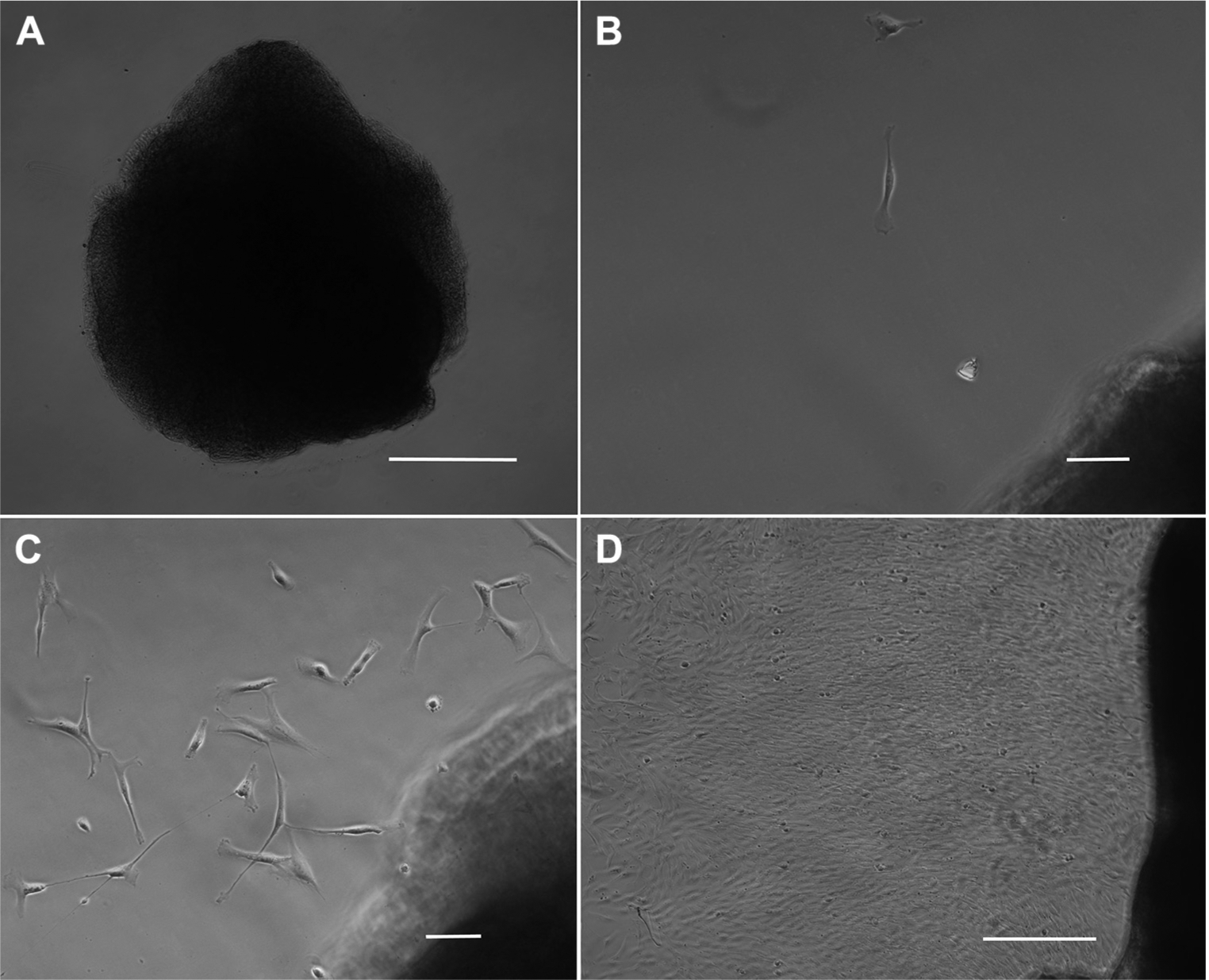
Isolation of dural fibroblasts. (A) An approximately 150-μm piece of dura was placed into a well of a 6-well plate adjacent to a drop of silicon grease underneath a coverslip and incubated in fibroblast media. Scale bar, 50 μm. (B) After 6 days, single fibroblasts have migrated out of the dural matrix. Scale bar, 10 μm. (C) After 8 days, more fibroblasts have left the dural matrix. Scale bar, 10 μm. (D) After 16 days, there is robust migration of fibroblasts out of the dural matrix, and cells are ready for the first passage. Scale bar, 50 μm.
After 3 passages, fibroblasts could be plated (Figure 2A) and transduced with the Sendai virus carrying the reprogramming factors described by Takahashi and Yamanaka (21). The CytoTune 2.0 Kit (Life Technologies) was chosen for reprogramming because it offers higher reprogramming efficiency, faster clearance of vectors, and lower cytotoxicity compared with the previous kit. At 19 days after transduction, iPSC-like colonies could be visualized (Figure 2B). A total of 32 colonies were picked for passage, and 11 of these colonies continued to form iPSC-like colonies after 2 passages (Figure 2C).
Figure 2.

Transduction of dural fibroblasts. (A) After 3 passages, fibroblasts were plated into 6-well plates and transduced with the Sendai virus. (B) 19 days after transduction, single induced pluripotent stem cell (iPSC)-like colonies could be easily visualized. (C) 11 of the 32 clones picked continued to form iPSC-like colonies after 2 passages. Scale bars, 10 μm.
Three of the 11 clones (clones 1, 8, and 9) were chosen for immunocytochemical analysis for markers of pluripotency. Clone 9 expressed Oct-4 (Figure 3A), SSEA-4 (Figure 3B), SOX2 (Figure 3D), TRA-1–60 (Figure 3E), and Nanog (Figure 3G). The other 2 clones were also found to express markers or pluripotency (Supplemental Figure 1).
Figure 3.
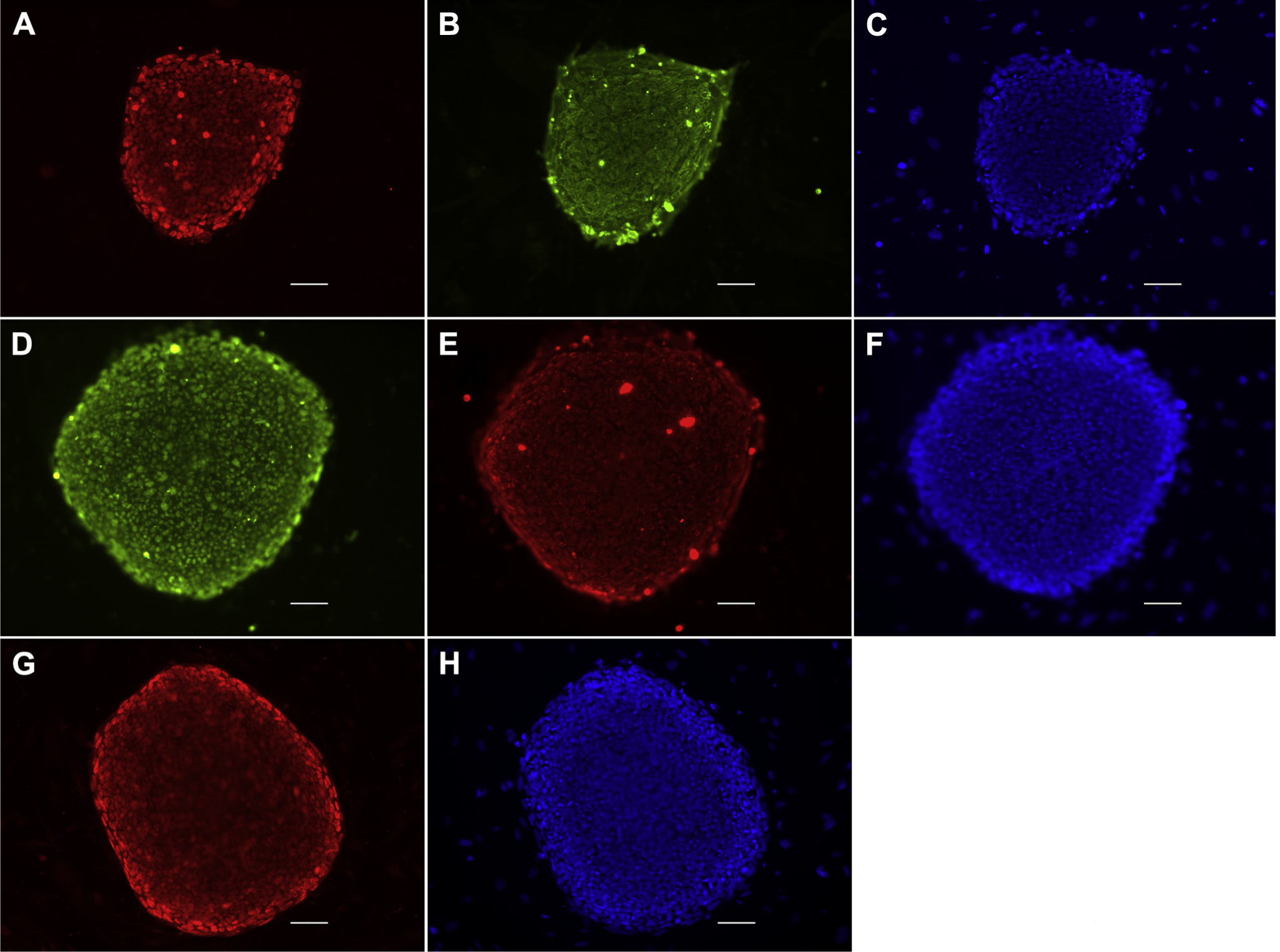
Immunocytochemical analysis. Three clones were analyzed with immunocytochemistry for pluripotency. Clone 9 shows that it expresses the pluripotency markers OCT4 (A) and SSEA-4 (B), 4′,6-diamidino-2-phenylindole (DAPI) (C). The clone also expresses SOX2 (D) and TRA-1–60 (E), DAPI (F). The pluripotency transcription factor Nanog is expressed as well (G), DAPI (H). Scale bars, 10 μm. OCT4, octamer-binding transcription factor 4; SOX2, (sex-determining region Y)-box 2; SSEA-4, stage-specific embryonic antigen 4; TRA-1–60, tumor rejection antigen 1–60.
Transcriptome analysis was performed on one iPSC line (clone 8) and compared with the transcriptional profile of the fibroblasts before transduction. After transduction, genes involved in self-renewal were up-regulated (Figure 4A). Fibroblasts expressed a high amount of mesodermal genes, whereas transduced cells down-regulated mesodermal genes and did not show preferential gene expression of any germ cell layer (Figure 4B, C). Thus, the transcriptome analysis supports the generation of iPSCs.
Figure 4.
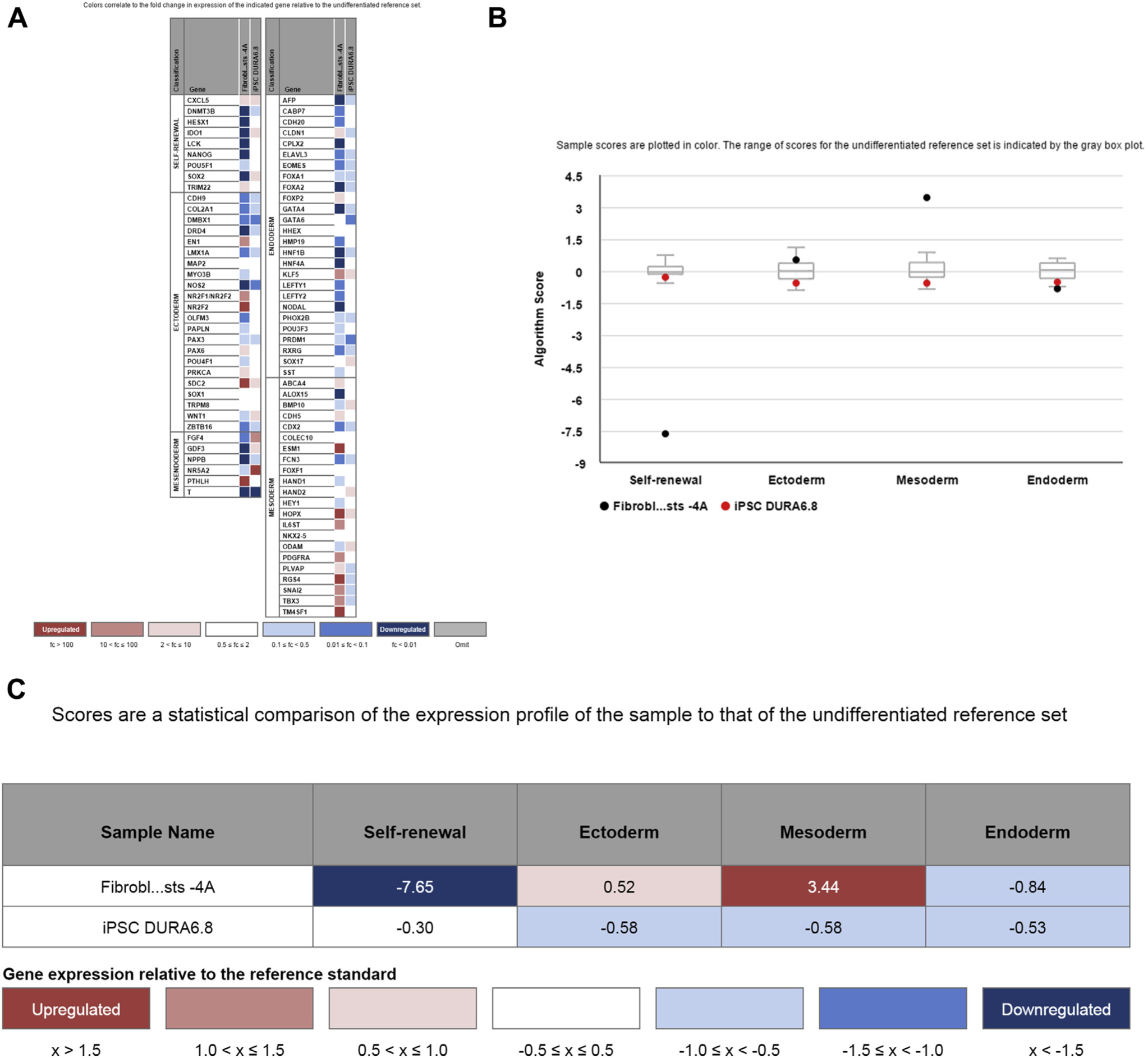
Transcriptome analysis of induced pluripotent stem cells (iPSCs). (A) Transcriptome analysis shows down-regulation of genes involved in self-renewal in fibroblasts compared with an undifferentiated reference set, whereas some of these genes are up-regulated in iPSCs. Only fibroblasts show a mesodermal gene profile. (B, C) Summary analysis of the transcriptome profile confirms down-regulation of mesodermal genes and up-regulation of self-renewal genes after transduction of fibroblasts into iPSCs.
Two clones (clones 8 and 9) were analyzed for pluripotency in a teratoma assay with 4 replicates. Three of 4 mice exhibited teratoma growth in their left hind limb for clones 8 and 9. All 3 germ layers were observed confirming teratoma formation (Figure 5). No metastases or other cancers were observed on histopathologic evaluation.
Figure 5.
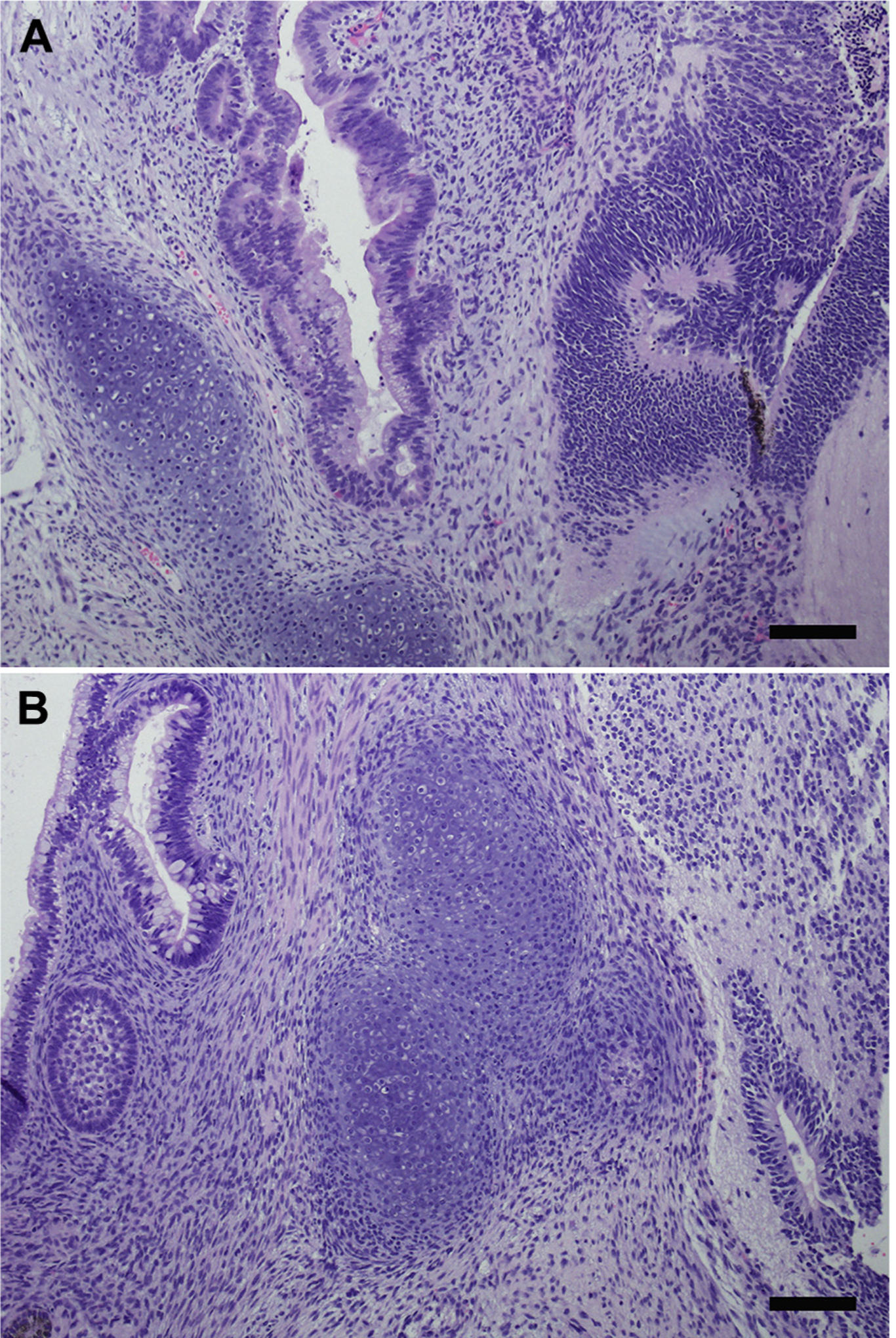
Teratoma assay. (A) Teratoma formation in mice inoculated with clone 8. Tissue types representing all 3 germ layers are present in this neoplasm, including mesoderm (lower left, cartilage), endoderm (center, mucosal epithelium), and ectoderm (right, nervous tissue). (B) Teratoma formation in mice inoculated with clone 9. Tissue types representing all 3 germ layers are present in this neoplasm, including endoderm (upper left, mucosal epithelium), mesoderm (center, cartilage), and ectoderm (middle left, dental stellate reticulum; lower right, nervous tissue). Hematoxylin & eosin stain. Scale bars, 100 μm.
Three clones (clones 1, 8, and 9) underwent chromosomal analysis, and all 3 clones were found to have a normal metaphase spread (Figure 6A) and karyotype (Figure 6B).
Figure 6.
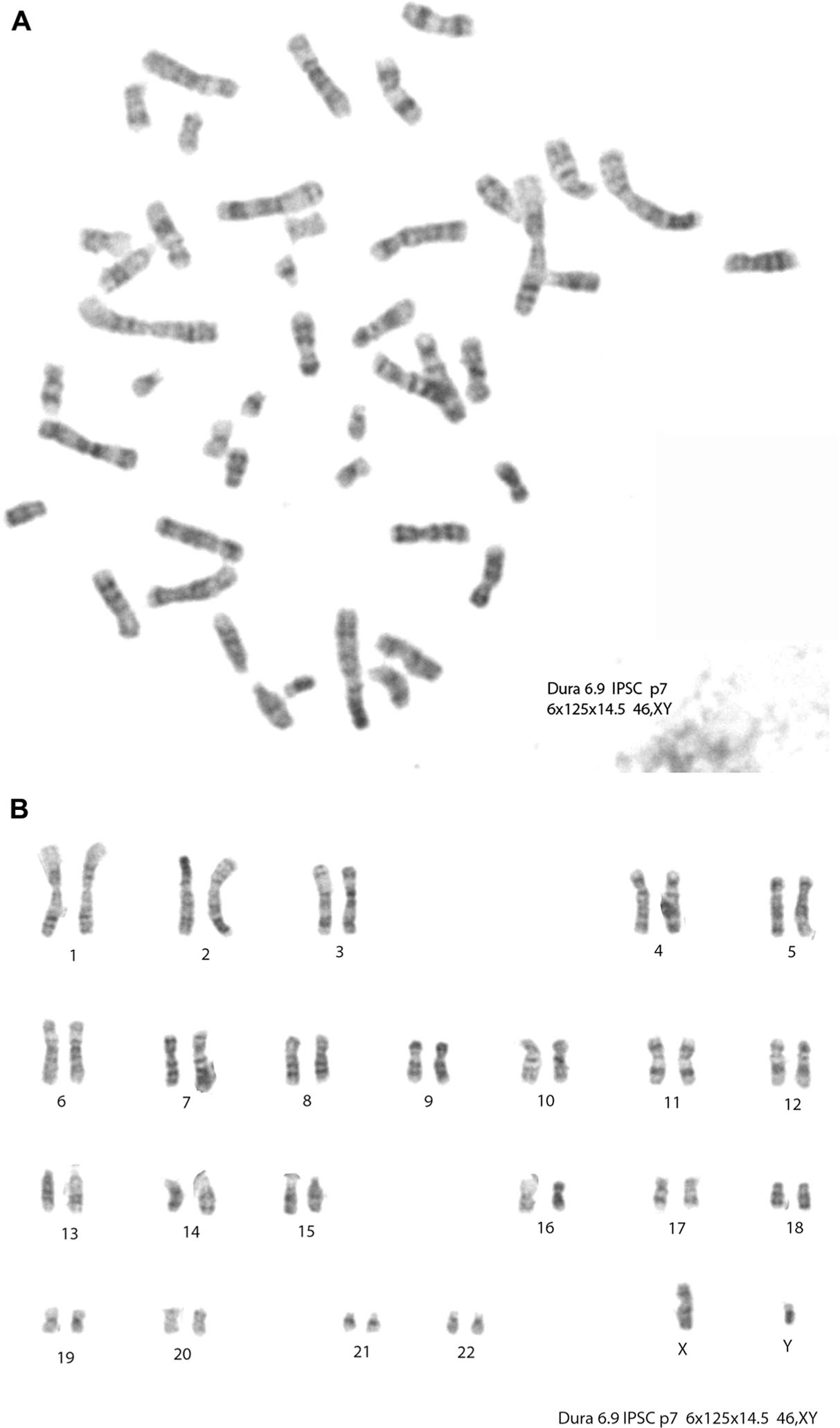
Chromosomal analysis. Three clones underwent chromosomal analysis and were found to have normal karyotypes. Clone 9 shows a normal metaphase spread (A) and karyotype (B).
IPSCs formed neural rosettes after incubation in neural differentiation medium (Figure 7A–C). After 19 days, cells expressed the neuronal marker TuJ-1 (Figure 7D). Most cells coexpressed SOX1, indicating that they were at a neuronal precursor cell stage (Figure 7E–G).
Figure 7.
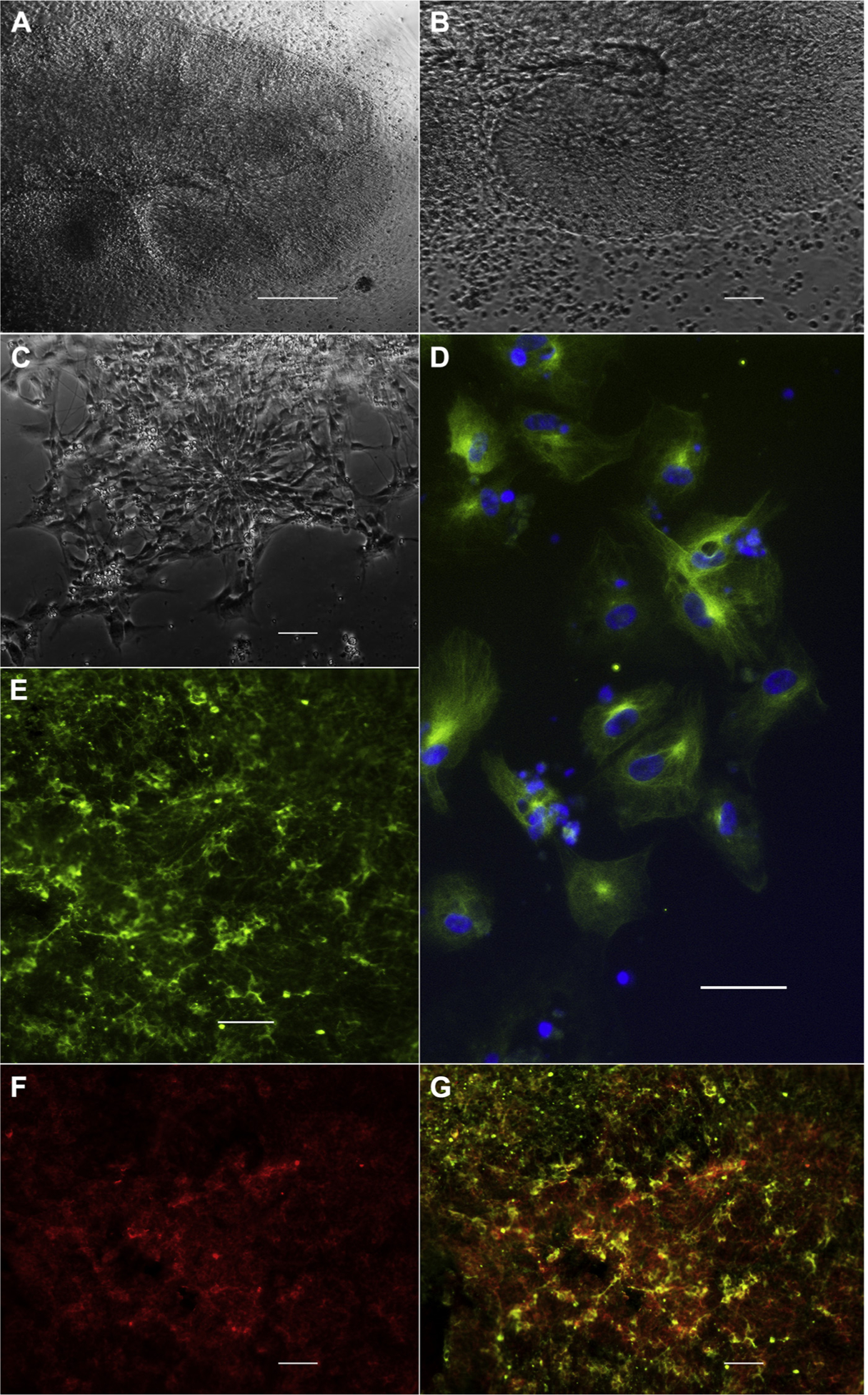
Targeted neural differentiation. After 17 days in neuronal differentiation medium, induced pluripotent stem cell clone 9 forms large (A; scale bar, 50 μm; B, magnification of A; scale bar, 10 μm) neural rosettes. Panel C (scale bar: 10 μm) shows a radial arrangement of elongated columnar cells, which is the morphologic characteristic of a neural rosette. After 19 days, differentiated cells express the neuronal marker TuJ-1 (D; green: TuJ-1; blue: 4′,6-diamidino-2-phenylindole; scale bar, 10 μm). Targeted differentiation leads to the formation of neuronal precursors, which co-express TuJ-1 (E, green) and Sox1 (F, red); TuJ-1/SOX1 (G; scale bar, 10 μm).
DISCUSSION
We show the generation of iPSCs and neural progenitor cells from a small piece of dura obtained from a 60-year-old man with modified Rankin scale 3 disability and normal pressure hydrocephalus. Our patient suffered from cognitive impairment beyond what is expected with normal pressure hydrocephalus. Autologous neural progenitor cells may provide a means in the future to augment or restore damaged circuits and cognition in neurodegenerative disease.
The possibility of iPSC and neural progenitor cell generation from dura mater extends beyond shunt surgery for normal pressure hydrocephalus. Dural harvesting during shunt surgery may also be performed after aneurysmal subarachnoid hemorrhage to generate iPSCs. Approximately 20% of patients develop shunt-dependent hydrocephalus after subarachnoid hemorrhage (13). Some of these patients may develop a stroke in the setting of vasospasm. Neural progenitor cells generated from dural harvests during shunt surgery could potentially be used in the future for autotransplantation to replace or regenerate lost brain tissue after stroke. Another possible application is Parkinson disease where burr holes are also performed during deep brain stimulator placement. Therefore, the protocol may be used to derive, from harvested tissue, a stock of iPSCs to undergo targeted differentiation into dopaminergic neurons for the treatment of Parkinson disease (6). Finally, because the dura mater is readily accessible to neurosurgeons during craniotomies, the protocol may also be used to generate and store iPSCs and neural progenitor cells after craniotomies for treatment of intracranial lesions or after hemicraniectomy for traumatic brain injury or stroke.
IPSCs have been found to carry an epigenetic memory, which is a remnant epigenetic signature of the donor tissue (22). The epigenetic signature has been shown to be an important aspect of further cell differentiation potential (8). Future research needs to answer the question whether dural fibroblasts have a different efficiency than skin fibroblasts in generating neural progenitor cells for autotransplantation in vivo. We do not suggest to replace skin harvests for fibroblasts with the harvest of dural fibroblasts. However, the dura is easily accessible to neurosurgeons during a variety of operations and can easily be banked (e.g., during a ventriculoperitoneal shunt placement or a hemicraniectomy for stroke). Thus, iPSCs can later be derived from such harvested dura obviating the need for skin biopsy.
Reprogramming efficiency to produce iPSCs has been shown to decline with the age of the donor. Although human centenarian cells and postmortem dural fibroblasts have been successfully reprogrammed to form iPSCs (9, 18, 25), the efficiency of iPSC generation has been found to be inversely correlated with age in an in vivo model (5, 23). Our patient was 60 years of age, but this did not seem to prevent efficient iPSC production as 11 iPSC clones could be generated on first attempt. It is controversial whether donor age affects iPSC telomere length and longevity of reprogrammed cells (16). There is, however, evidence that telomeres of iPSCs generated from older patients can obtain the same length as embryonic stem cells after several passages (10, 20). Thus, the age of our patient may not be a major factor influencing the longevity of derived neural progenitor grafts.
The iPSCs derived from our donor patient were obtained with a nonintegrating virus and exhibited normal karyotypes. Such a protocol may qualify better for generating neural progenitor cells for human clinical trials compared with integrating vector approaches. In our study, the iPSCs were produced under good laboratory practices and not good manufacturing practices (GMP), which are a requirement for human clinical trials. However, our institution has experience in the generation of iPSCs under GMP conditions (1, 15, 17), and additional experience in the GMP manufacturing of neural progenitor cells from other pluripotent stem cell sources. Therefore, transitioning the protocol into GMP manufacturing should not pose a problem.
CONCLUSION
We present the efficient production of iPSCs and neural progenitor cells from a small piece of dura mater obtained from a 60-year-old patient with severe cognitive disability. Further research is needed to address the question whether derived neural progenitor cells can repair damaged circuits and improve outcomes in vivo.
Supplementary Material
{kind=link}
Abbreviations and Acronyms
- bFGF
Basic fibroblast growth factor
- cDNA
Complementary deoxyribonucleic acid
- DMEM
Dulbecco’s modified eagle medium
- DPBS
Dulbecco’s PBS
- FBS
Fetal bovine serum
- GMP
Good manufacturing practices
- Ig
Immunoglobulin
- iPSC
Induced pluripotent stem cell
- MEF
Mouse embryonic fibroblasts
- PBS
Phosphate-buffered saline
- RNA
Ribonucleic acid
Footnotes
Conflict of interest statement: This work was supported by an intramural Clinical and Translational Science Center training grant funded by the California Institute for Regenerative Medicine (CIRM).
REFERENCES
- 1.Abou-El-Enein M, Romhild A, Kaiser D, Beier C, Bauer G, Volk HD, Reinke P: Good Manufacturing Practices (GMP) manufacturing of advanced therapy medicinal products: a novel tailored model for optimizing performance and estimating costs. Cytotherapy 15:362–383, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Ager RR, Davis JL, Agazaryan A, Benavente F, Poon WW, LaFerla FM, Blurton-Jones M: Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 25:813–826, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andres RH, Horie N, Slikker W, Keren-Gill H, Zhan K, Sun G, Manley NC, Pereira MP, Sheikh LA, McMillan EL, Schaar BT, Svendsen CN, Bliss TM, Steinberg GK: Human neural stem cells enhance structural plasticity and axonal transport in the ischaemic brain. Brain 134: 1777–1789, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L: Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng Z, Ito S, Nishio N, Xiao H, Zhang R, Suzuki H, Okawa Y, Murohara T, Isobe K: Establishment of induced pluripotent stem cells from aged mice using bone marrow-derived myeloid cells. J Mol Cell Biol 3:91–98, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Doi D, Samata B, Katsukawa M, Kikuchi T, Morizane A, Ono Y, Sekiguchi K, Nakagawa M, Parmar M, Takahashi J: Isolation of human induced pluripotent stem cell-derived dopaminergic progenitors by cell sorting for successful transplantation. Stem Cell Reports 2:337–350, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karumbayaram S, Lee P, Azghadi SF, Cooper AR, Patterson M, Kohn DB, Pyle A, Clark A, Byrne J, Zack JA, Plath K, Lowry WE: From skin biopsy to neurons through a pluripotent intermediate under Good Manufacturing Practice protocols. Stem Cells Transl Med 1:36–43, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, Cahan P, Huo H, Loh YH, Aryee MJ, Lensch MW, Li H, Collins JJ, Feinberg AP, Daley GQ: Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol 29:1117–1119, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Ait-Hamou N, Leschik J, Pellestor F, Ramirez JM, De Vos J, Lehmann S, Lemaitre JM: Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev 25:2248–2253, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marion RM, Strati K, Li H, Tejera A, Schoeftner S, Ortega S, Serrano M, Blasco MA: Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4: 141–154, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Muller J, Ossig C, Greiner JF, Hauser S, Fauser M, Widera D, Kaltschmidt C, Storch A, Kaltschmidt B: Intrastriatal transplantation of adult human neural crest-derived stem cells improves functional outcome in parkinsonian rats. Stem Cells Transl Med 4:31–43, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nudi ET, Jacqmain J, Dubbs K, Geeck K, Salois G, Searles MA, Smith JS: Combining enriched environment, progesterone, and embryonic neural stem cell therapy improves recovery following brain injury. J Neurotrauma 32:1117–1129, 2015. [DOI] [PubMed] [Google Scholar]
- 13.O’Kelly CJ, Kulkarni AV, Austin PC, Urbach D, Wallace MC: Shunt-dependent hydrocephalus after aneurysmal subarachnoid hemorrhage: incidence, predictors, and revision rates. Clinical article. J Neurosurg 111:1029–1035, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Olstorn H, Moe MC, Roste GK, Bueters T, Langmoen IA: Transplantation of stem cells from the adult human brain to the adult rat brain. Neurosurgery 60:1089–1098; discussion 1098–1099, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Park SS, Caballero S, Bauer G, Shibata B, Roth A, Fitzgerald PG, Forward KI, Zhou P, McGee J, Telander DG, Grant MB, Nolta JA: Long-term effects of intravitreal injection of GMP-grade bone-marrow-derived CD34+ cells in NOD-SCID mice with acute ischemia-reperfusion injury. Invest Ophthalmol Vis Sci 53:986–994, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rohani L, Johnson AA, Arnold A, Stolzing A: The aging signature: a hallmark of induced pluripotent stem cells? Aging Cell 13:2–7, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheu J, Klassen H, Bauer G: Cellular manufacturing for clinical applications. Dev Ophthalmol 53:178–188, 2014. [DOI] [PubMed] [Google Scholar]
- 18.Sproul AA, Vensand LB, Dusenberry CR, Jacob S, Vonsattel JP, Paull DJ, Shelanski ML, Crary JF, Noggle SA: Generation of iPSC lines from archived non-cryoprotected biobanked dura mater. Acta Neuropathol Commun 2:4, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stiles J, Jernigan TL: The basics of brain development. Neuropsychol Rev 20:327–348, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suhr ST, Chang EA, Rodriguez RM, Wang K, Ross PJ, Beyhan Z, Murthy S, Cibelli JB: Telomere dynamics in human cells reprogrammed to pluripotency. PLoS One 4:e8124, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Vaskova EA, Stekleneva AE, Medvedev SP, Zakian SM: “Epigenetic memory” phenomenon in induced pluripotent stem cells. Acta Naturae 5: 15–21, 2013. [PMC free article] [PubMed] [Google Scholar]
- 23.Wang B, Miyagoe-Suzuki Y, Yada E, Ito N, Nishiyama T, Nakamura M, Ono Y, Motohashi N, Segawa M, Masuda S, Takeda S: Reprogramming efficiency and quality of induced Pluripotent Stem Cells (iPSCs) generated from muscle-derived fibroblasts of mdx mice at different ages. PLoS Curr 3:RRN1274, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright LS, Prowse KR, Wallace K, Linskens MH, Svendsen CN: Human progenitor cells isolated from the developing cortex undergo decreased neurogenesis and eventual senescence following expansion in vitro. Exp Cell Res 312:2107–2120, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Yagi T, Kosakai A, Ito D, Okada Y, Akamatsu W, Nihei Y, Nabetani A, Ishikawa F, Arai Y, Hirose N, Okano H, Suzuki N: Establishment of induced pluripotent stem cells from centenarians for neurodegenerative disease research. PLoS One 7: e41572, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye L, Muench MO, Fusaki N, Beyer AI, Wang J, Qi Z, Yu J, Kan YW: Blood cell-derived induced pluripotent stem cells free of reprogramming factors generated by Sendai viral vectors. Stem Cells Transl Med 2:558–566, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.