

Abstract
Importance
Adults with Down syndrome (DS) are at high‐risk of revealing Alzheimer's disease (AD) pathology, in part due to the triplication of chromosome 21 encoding the amyloid precursor protein. Adults with DS are uniformly affected by AD pathology by their 30′s and have a 70% to 80% chance of clinical dementia by their 60′s. Our previous studies have assessed longitudinal changes in amyloid beta (Aβ) accumulation in DS.
Objective
The goal of the present study was to assess the presence of brain tau using [18F]AV‐1451 positron emission tomography (PET) in DS and to assess the relationship of brain tau pathology to Aβ using Pittsburgh Compound B (PiB)‐PET.
Design
Cohort study
Setting
Multi‐center study
Participants
Participants consisted of a sample of individuals with DS and sibling controls recruited from the community; exclusion criteria included contraindications for magnetic resonance imaging (MRI) and/or a medical or psychiatric condition that impaired cognitive functioning.
Exposures
PET brain scans to assess Aβ ([11C]PiB) and tau ([18F]AV‐1451) burden.
Main outcomes and measures
Multiple linear regression models (adjusted for chronological age, sex and performance site) were used to examine associations between regional [18F]AV‐1451 standard uptake value ratio (SUVR) (based on regions associated with Braak stages 1‐6) and global [11C]PiB SUVR (as both a continuous and dichotomous variable).
Results
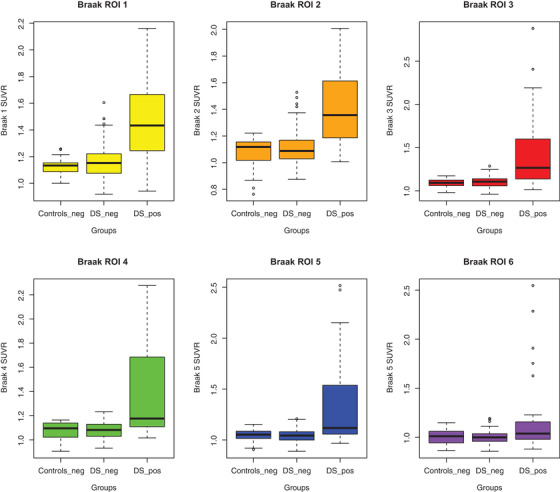
A cohort of 156 participants (mean age = 39.05, SD(8.4)) were examined. These results revealed a significant relationship between in vivo Aβ and tau pathology in DS. As a dichotomous variable, [18F]AV‐1451 retention was higher in each Braak region in PiB(+) participants. We also found, based on our statistical models, starting with the Braak 3 region of interest (ROI), an acceleration of [18F]AV‐1451 SUVR deposition with [11C]PiB SUVR increases.
Keywords: Down syndrome, PET amyloid, TAU, Alzheimer's disease
1. INTRODUCTION
Definitive diagnosis of Alzheimer's disease (AD) relies on the demonstration of amyloid beta (Aβ) plaques and tau‐containing neurofibrillary tangles at autopsy. 1 , 2 More recently, the availability of positron emission tomography (PET) ligands for Aβ, such as [11C]PiB 3 and for tau, such as [18F]AV‐1451 4 have allowed in vivo examination of the relationship of cognition to AD pathophysiology. These studies have demonstrated that tau pathology is more closely related to cognitive decline than Aβ2 and is associated with AD diagnosis, 5 amyloid positivity, 6 and cognitive function. 7
Adults with Down syndrome (DS) are uniformly affected by AD‐related pathology, characterized by the presence of Aβ plaques by their fourth decade (Wisniewski 1985; Hyman 1992; Lemere 1996), and they demonstrate an increased prevalence of AD. From a large body of data, it is clear that overproduction of amyloid precursor protein (APP), in part due to the triplication of chromosome 21 encoding APP, is associated with a high risk of AD in DS and the appearance of clinical AD at an earlier age. 8 , 9
Aβ PET studies in DS have identified a distinct pattern amyloid deposition, beginning predictably in mid‐life, 10 , 11 which has been confirmed in an autopsy study of DS. 12 , 13 Tau PET studies in DS are relatively new, with only one small study (n = 12) demonstrating increasing tau burden with age and amyloid positivity and a correlation between tau burden and cognitive impairment, 14 mimicking the findings observed in late‐onset AD using [18F] AV‐1451. 5 Based on these data, we hypothesize that higher regional Aβ measured by PiB‐PET will be associated with increased neurofibrillary tangles (NFTs) measured using AV‐1451 PET. This cross‐sectional PET study aims to assess in DS the relationship of global Aβ to regional neurofibrillary tangles (NFTs) characterized by Braak staging.
2. METHODS
2.1. Study design and participants
All participants, n = 156 (135 DS and 21 controls), mean age = 39.05 (SD = 8.4) underwent [11C]PiB and [18F]AV‐1451 scans as well as clinical and neuropsychological examination. All participants had a clinical diagnosis based on consensus case conference. The consensus for our 135 DS participants was as follows: 108 were non‐demented, non–mild cognitive impaired (MCI), 8 had a clinical diagnosis of MCI, 11 had a diagnosis of dementia, and for 8 participants the clinical consensus was unable to determine a diagnosis. 15 Based on our published PiB thresholds, 16 all of our participants were categorized as DS‐PiB(−) or DS‐PiB(+); briefly, any DS participant with PiB regional values exceeding the threshold in at least one region of the six regions measured (anterior cingulate, frontal cortex, striatum, precuneus, lateral temporal, and parietal cortices) is defined as DS‐PiB(+). In addition, we also included a sibling‐control sample; all were classified as PiB(−).
Image Acquisition Imaging data were acquired at four sites: The University of Pittsburgh using a Siemens Prisma (magnetic resonance imaging; MRI) and a Siemens mCT Biograph (PET), the University of Wisconsin‐Madison using a General Electric Discovery MR750 (MRI) and a Siemens HR+ (PET), the University of Cambridge using a General Electric SIGNA PET/MR (MRI and PET), and Banner Health using a General Electric Discovery MR750 (MRI) and a General Electric Discovery 710 (PET). T1‐weighted MR images were acquired for each subject for anatomical reference.
PET tracers (15 mCi [11C]PiB or 10 mCi [18F]AV‐1451, nominal) were administered as bolus injections, over approximately 30 seconds, followed by a saline flush. Subjects were imaged over time ranges that included 50‐70 minutes post‐injection for the case of [11C]PiB and 80‐100 minutes post‐injection for the case of [18F]AV‐1451. For each subject, [18F]AV‐1451 scans were obtained in time proximity and, most frequently, on the same day following the [11C]PiB scans.
PET images were reconstructed into 45‐minute time frames spanning the range 50‐70 minutes post‐injection ([11C]PiB) or 80‐100 minutes post‐injection ([18F]AV‐1451). Image reconstruction was performed using the manufacturer's software and included corrections for scatter, deadtime, random coincidences, and radioactive decay.
2.2. Image processing
The multiframe PET images were visually inspected for frame‐to‐frame motion. If necessary, motion correction was performed using a set of stable frames (averaged) as a reference. Frames requiring correction were registered to the reference using PMOD. Single‐frame PET images were formed by averaging over the 50‐70 minute post‐injection frames for PiB and over 80‐100 minutes for AV‐1451. Each subject's MRI was manually aligned to anterior‐commissure/posterior commissure (ACPC) orientation. The single‐frame PET images (PiB and AV‐1451) were registered to this using the registration tool of PMOD (https://www.pmod.com/web/) via maximization of normalized mutual information.
Each subject's T1 MRI was parcellated into regions of interest (ROIs) using FreeSurfer v5.3. The standard FreeSurfer pipeline was used for this procedure and modified to obtain a more finely detailed parcellation of the striatum than that produced by FreeSurfer with the built‐in Desikan‐Killiany (DK) atlas. We incorporate the Clinical Imaging Center (CIC) atlas, 17 developed for dopamine imaging, into our analysis. The T1 MRI associated with CIC atlas was processed through FreeSurfer using the standard pipeline, providing a transformation that can be used to put the CIC atlas into internal FreeSurfer space. The net result of the procedure is that any scan processed through FreeSurfer can be labeled with the CIC regions as well as the FreeSurfer/DK regions. With this capability, subject scans were parcellated into FreeSurfer/DK regions, except that the striatum was parcellated using CIC atlas regions. Specifically, the set of FreeSurfer DK regions (right and left) caudate, putamen, and pallidum were replaced by the set of CIC atlas regions (right and left) ventral striatum, dorsal caudate, posterior caudate, anterior putamen, posterior putamen, and pallidum. All FreeSurfer results were inspected and, if necessary, edited for proper anatomical alignment with the MRI.
For some subjects, however, the raw FreeSurfer (FS) results were inadequate, even for editing. Based on work reported by Svarer et al., 18 some of these cases were salvaged using an approach in which scans that were successfully parcellated by FreeSurfer were used as templates for the problematical scans. Briefly, a template ensemble was assembled using 12 subjects (10 DS and 2 controls) from the Neurodegeneration in Aging Down Syndrome (NiAD) population with existing high‐quality FS‐based parcellations. All of the T1 template images were skull stripped using the tissue segmentation function of SPM12 (Statistical Parametric Mapping, www.fil.ion.ucl.ac.uk/spm/software/spm12). Voxels with a combined probability of being white matter, gray matter, or cerebrospinal fluid of >0.0001 were classified as brain tissue and retained in the image. Next, the T1 MR image from each subject to be processed was skull stripped using the procedure described for the templates. For a given subject, each of the 12 template T1 MR images was warped to the skull‐stripped subject image using the “Normalize” function of SPM8, and the existing FS parcellations were carried along using the templates’ warping parameters. Using this method, for each FS region, 12 ROI images warped to the subject's MRI were produced. A “probability image” was then generated by averaging the 12 individual template ROI images. The final probability‐template ROI for the region was constructed by selecting voxels with the highest probability until a volume was achieved that was equal to the volume of that region averaged over the 12 warped templates. This process was repeated for every FS region resulting in a full set of FS‐ROIs for each subject. Final results were inspected and either accepted or rejected, but no ROI editing was performed. Of 156 participants this method was utilized only on 20 participants, all in the DS group.
HIGHLIGHTS
Question: The goal of this study was examine the relationship between amyloid and tau burden in Down syndrome (DS).
Findings: In this multi‐center population‐based cohort study of DS we observed a significant relationship between in vivo amyloid beta (Aβ) and tau pathology in DS, similar to that seen in the typical aging population. We also found, based on our statistical models, an acceleration of [18F]AV‐1451 SUVR deposition with [11C]PiB SUVR increases.
Meaning: These data add to the existing body of literature in DS to contribute to providing the necessary framework to identify appropriate participants for clinical trials, track efficacy of interventions, and track dementia progression in the DS population.
RESEARCH IN CONTEXT
Systematic review: Tau positron emission tomography (PET) studies in Down syndrome (DS) are relatively new, with only one small study (n = 12) demonstrating increasing tau burden with age and amyloid positivity and a correlation between tau burden and cognitive impairment; however, this study was preliminary in nature. No studies have been undertaken to examine the relationship between amyloid and tau burden in DS. Our findings demonstrate a significant relationship between in vivo amyloid beta (Aβ) and tau pathology in DS, similar to those seen in the typical aging population. As a dichotomous variable, [18F]AV‐1451 retention was higher in each Braak region in PiB(+) participants. We also found, based on our statistical models, starting with the Braak 3 region of interest (ROI) an acceleration of [18F]AV‐1451 SUVR deposition with [11C]PiB SUVR increases.
Interpretation: These findings are among the first to report tau pathology using PET in DS and are, to our knowledge, the largest DS cohort using tau‐PET. Our findings suggest that tau pathology in DS does not differ from that of late‐onset AD, with tau pathology increasing in regions associated with Braak regions in those with significant amyloid pathology. These data track with pathological studies in DS that have not identified clear differences in neurofibrillary tau deposition from late‐onset AD, but unlike these neuropathological studies are not restricted to those with late‐stage disease in all groups. In addition, these data extend our previous findings, identifying a distinct pattern of predominant striatal Aβ deposition that distinguished the Aβ deposition seen in late‐onset AD and DS into tau pathology in DS.
Future directions: These data adding to the existing body of literature in DS contribute to providing the necessary framework to identify appropriate participants for clinical trials, track efficacy of interventions, and track dementia progression in the DS population.
For each subject, PET activity images (registered with the MRI) were sampled using the ROIs determined as described above either using standard FS or our probability template method. Regional values were normalized by dividing by cerebellar gray matter activity, determined using a volume‐weighted average of activity obtained using the FreeSurfer left and right cerebellar cortex ROIs. The result is a set of cerebellum gray matter normalized tissue ratios (standardized uptake value ratios, SUVRs) for each ROI and tracer.
A global region for amyloid status (positive or negative) was composed from the region's anterior cingulate, superior frontal, orbitofrontal, insula, lateral temporal, parietal posterior cingulate, precuneus, putamen, and striatum. The FreeSurfer and CIC atlas regions from which these are composed are listed in Supplemental Table S1. Parenthetically, because of the observation that the striatum of DS subjects shows different patterns of amyloid deposition than in non‐DS sporadic AD, a larger striatal region was used in DS studies than in non‐DS. See Supplemental Table S1.1 and S1.2 for details. [11C]PiB SUVRs were determined for each of these super regions and ultimately for the global region via a volume‐weighted average of the SUVRs of the component regions. Scans were defined to be amyloid positive, PiB (+), or negative PiB (−), based on a global SUVR threshold of 1.36.
Six Braak super regions (Braak regions) corresponding to the six Braak stages 19 were used for the quantitation of [18F]AV‐1451. The Braak region SUVRs were determined from a volume‐weighted average of SUVRs in sets of FreeSurfer regions described in Schöll et al., 20 except that the striatum was not included in Braak region 5. The Braak regions are exclusive in that, for example, Braak region 2 does not include Braak region 1, and so on. As in the case of [11C]PiB, [18F]AV‐1451 SUVRs are normalized by cerebellar gray matter.
2.3. Statistical methods
Descriptive statistics (means and standard deviations) are presented for age and for each Braak Region by DS‐PiB (−), DS‐PiB (+), and Controls‐PiB (−) in Table 1. Frequency percentages are shown for sex and consensus diagnosis (Table 1).
TABLE 1.
Descriptive statistics for groups for each Braak ROI
| Variable | All N = 156 | Controls‐PiB (−)n = 21 | DS‐PiB (−)n = 73 | DS‐PiB (+)n = 62 |
|---|---|---|---|---|
| Consensus Dx 0 = no MCI/no dementia | 0 (108) | NA | 0 (71) | 0 (37) |
| 1 = MCI | 1(8) | 1 (0) | 1 (8) | |
| 2 = dementia | 2 (11) | 2 (0) | 2 (11) | |
| 3 = unable to determine | 3 (8) | 3 (2) | 3 (6) | |
| Sex | M (76, 48%) | M (5, 24%) | M (36, 38%) | M (35, 56%) |
| Age at AV‐1451 scan | 39.04 (9.00) | 39.00 (12.46) | 33.61(4.80) | 45.45 (7.15) |
| Braak Region 1 | 1.23 (0.22) | 1.12 (0.07) | 1.14 (0.10) | 1.38 (0.27) |
| Braak Region 2 | 1.17 (0.21) | 1.06 (0.13) | 1.09 (0.10) | 1.31 (0.26) |
| Braak Region 3 | 1.18 (0.25) | 1.09(0.05) | 1.10 (0.06) | 1.30 (0.36) |
| Braak Region 4 | 1.14 (0.21) | 1.08(0.07) | 1.08 (0.07) | 1.25 (0.29) |
| Braak Region 5 | 1.12 (0.25) | 1.04 (0.06) | 1.04 (0.06) | 1.23 (0.36) |
| Braak Region 6 | 1.05 (0.21) | 1.00 (0.08) | 1.00 (0.06) | 1.10 (0.30) |
To assess differences among DS‐PiB(−), DS‐PiB(+), and Controls‐PiB(−), multiple linear regression models were used with dummy coding (0/1) for the groups and performance sites (group 1 is the DS‐PiB (+), group 2 is the DS‐PiB (−), and Controls‐PiB (−) are the reference category) for each Braak ROI. The linear model used for each Braak ROI is presented below:
| (1) |
where:
-
i)
represents the intercept (average Braak ROI SUVR value over the whole sample, for each ROI respectively);
-
ii)
is the dummy variable for group factor for subject i, and coefficient represents the difference in the mean of tau SUVR between the DS‐PiB (+) subjects compared to the controls, whereas represents the difference in the mean of Tau SUVR between the DS‐PiB (−) subjects compared to the controls, taking into the account the effect of site.
-
iii)
is the dummy variable for site factor for subject i, and the coefficients for site represent the differences in the mean of tau SUVR between that specific site and the reference site when groups are fixed.
Controls are considered to be the reference category for groups and the site variable is included to account for variability due to data being collected at multiple sites (with UK as the reference site). The same model was rerun including age at scan and all the results are presented in Table 2 in the form of mean differences between the groups along with 95% confidence interval (CI), unadjusted (model 1) as well as adjusted for age at scan (model 1+age). Based on the examination of scatter and LOESS (locally weighted scatterplot smoothing) plots of tau SUVR as a function of Global PiB SUVR and guided by F‐tests, we considered either linear or quadratic models for Braak ROIs 1 through 6. The F‐tests were performed to test if the addition of the quadratic term significantly added to explaining the variability in the model. Based on these evaluations, we used only a linear term for Braak ROIs 1 and 2 (model 2) and added a quadratic term for Braak ROIs 3, 4, 5, and 6 (model 3) with model terms described above.
| (2) |
and
| (3) |
The regression coefficient estimates along with 95% CI unadjusted (model 2 and 3) and adjusted for age (model 2 and 3 including age) are presented in Table 3. These estimates can be considered as effect sizes, since they represent the amount of change in tau SUVR for each one‐unit change in PiB SUVR when site and age are fixed.
TABLE 2.
Estimated mean difference between groups for each Braak ROI
| Region | Groups compared | Estimated mean differences and 95% CI | Estimated mean differences and 95% CI (age adjusted) |
|---|---|---|---|
| Braak 1 | DS‐PiB (+) vs Controls‐PiB (−) | 0.23 (0.13, 0.32) | 0.19 (0.10, 0.30) |
| DS‐PiB (−) vs Controls‐PiB (−) | −0.01(−0.10, 0.08) | 0.01 (−0.08, 0.11) | |
| DS‐PiB (+) vs DS‐PiB (−) | 0.24 (0.18, 0.30) | 0.18 (0.11, 0.26) | |
| Braak 2 | DS‐PiB (+) vs Controls‐PiB (−) | 0.25 (0.15, 0.34) | 0.20 (0.10, 0.29) |
| DS‐PiB (−) vs Controls‐PiB (−) | 0.02 (−0.07, 0.11) | 0.06 (−0.03, 0.15) | |
| DS‐PiB (+) vs DS‐PiB (−) | 0.22 (0.16, 0.29) | 0.14 (0.06, 0.22) | |
| Braak 3 | DS‐PiB (+) vs Controls‐PiB (−) | 0.20 (0.08, 0.32) | 0.15 (0.03, 0.27) |
| DS‐PiB (−) vs Controls‐PiB (−) | −0.006 (−0.12, 0.11) | 0.03 (−0.08, 0.15) | |
| DS‐PiB (+) vs DS‐PiB (−) | 0.21 (0.13, 0.29) | 0.12 (0.02, 0.22) | |
| Braak 4 | DS‐PiB (+) vs Controls‐PiB (−) | 0.17 (0.07,0.27) | 0.12 (0.02, 0.22) |
| DS‐PiB (−) vs Controls‐PiB (−) | −0.002 (−0.10,0.09) | 0.04 (−0.06, 0.13) | |
| DS‐PiB (+) vs DS‐PiB (−) | 0.17 (0.11, 0.24) | 0.08 (0.003, 0.16) | |
| Braak 5 | DS‐PiB (+) vs Controls‐PiB (−) | 0.18 (0.06, 0.30) | 0.14 (0.02,0.27) |
| DS‐PiB (−) vs Controls‐PiB (−) | −0.002 (−0.12,0.12) | 0.03 (−0.09, 0.15) | |
| DS‐PiB (+) vs DS‐PiB (−) | 0.19 (0.12, 0.26) | 0.12 (0.02, 0.22) | |
| Braak 6 | DS‐PiB (+) vs Controls‐PiB (−) | 0.10 (−0.006, 0.20) | 0.08 (−0.02, 0.19) |
| DS‐PiB (−) vs Controls‐PiB (−) | −0.002 (−0.10,0.10) | 0.006 (−0.10, 0.11) | |
| DS‐PiB (+) vs DS‐PiB (−) | 0.10 (0.03, 0.17) | 0.08 (−0.007, 0.17) |
Note : Second column shows the estimated mean differences in AV‐1451 SUVR between groups adjusted for site, and third column shows the differences adjusted for site and age at scan.
TABLE 3.
Estimated coefficients from the regression models
| Region | Intercept and 95% CI | Estimated PiB SUVR coefficient and 95% CI | Estimated PiB2SUVR coefficient and 95% CI |
|---|---|---|---|
| Braak 1 | 0.57(0.43, 0.72) | 0.52(0.44, 0.60) | NA |
| Age adjusted | 0.57(0.41, 0.63) | 0.51(0.40,0.63) | NA |
| Braak 2 | 0.55(0.42, 0.69) | 0.50(0.42, 0.57) | NA |
| Age adjusted | 0.53(0.38, 0.69) | 0.47(0.37, 0.58) | NA |
| Braak 3 | 1.33(0.93, 1.75) | −0.70(−1.22, −0.19) | 0.43(0.27, 0.59) |
| Age adjusted | 1.29(0.86, 1.72) | −0.58(−1.22, 0.06) | 0.40(0.22, 0.58) |
| Braak 4 | 1.11(0.75, 1.47) | −0.33(−0.78, 0.13) | 0.27(0.13, 0.41) |
| Age adjusted | 1.09(0.71, 1.47) | −0.29(−0.85, 0.28) | 0.26(0.10, 0.42) |
| Braak 5 | 1.71(1.32, 2.11) | −1.24(−1.74, −0.73) | 0.59(0.43, 0.74) |
| Age adjusted | 1.62(1.20, 2.04) | −0.98(−1.60, 0.36) | 0.52(0.35, 0.70) |
| Braak 6 | 2.11(1.73, 2.50) | −1.73(−2.22, −1.24) | 0.67(0.52, 0.82) |
| Age adjusted | 1.97(1.57, 2.37) | −1.35(−1.94, −0.76) | 0.58(0.41, 0.75) |
All of the above analyses were repeated for the DS‐PiB(+) only, and the results are presented in supplemental Table 2S and supplemental Figure 1S. None of our analyses were corrected for multiple comparisons.
3. RESULTS
In this cohort, we had 76 male and 80 female participants, with an average age of 39.04 (SD = 9.00). The DS‐PiB (+) participants had an average age of 45.45 (SD = 7.15) years and the DS‐PiB (−) had an average age of 33.61 (SD = 4.80). The average Braak ROIs were higher for DS‐PiB (+) than for the DS‐PiB (−) or Control‐PiB (−). These values were higher for lower Braak ROIs starting at 1.14 (SD = 0.10) for the DS‐PiB (−) in Braak 1 ROI and decreasing to a value of 1.00 (0.06) in Braak 6 ROI. Similarly, the DS‐PiB (+) starts at an average value of 1.38 (SD = 0.27) in Braak 1 ROI SUV (see Table 1). The Control‐PiB(−) mean values are similar to the DS‐PiB (−) mean values (see Table 1). Using the multiple linear regression model, (equation 1), we found differences between DS‐PiB (+) and DS‐PiB (−) as well as between DS‐PiB (+) and Control‐PiB (−), and they are very close in magnitude. These differences are presented in Table 2. It can be observed that the magnitude of the differences between DS‐PiB (+) and DS‐PiB (−) is lower for higher Braak ROIs (ranging from 0.24, 95% CI [0.18, 0.30] in Braak 1 ROI, lower in Braak 4 ROI (0.17, 95% CI [0.11, 0.24]) and much lower in Braak 6 ROI (0.10, 95% [0.03, 0.17]). We found that the differences between DS‐PiB (+) and DS‐PiB (−) in Braak 2 and 3 ROIs were very close to each other. All these differences became lower when the models were adjusted for age at scan (Table 2).
The associations between tau SUVR and PiB SUVR were in the range of moderate effect sizes for most of the coefficients. A linear association was found between tau SUVR in Braak 1 ROI, β = 0.52 (0.44, 0.60) and Braak 2, β = 0.50 (0.42, 0.57), suggesting that for each one unit increase in Global PiB SUVR there is a 0.52 increase in tau SUVR for Braak 1 and a 0.50 increase in tau SUVR for Braak 2. Similar effects were determined when models were additionally adjusted for age. These values were determined from the coefficients of PiB SUVR from model 2 and are presented in Table 3.
Starting with the Braak 3 ROI, we have found an acceleration of tau SUVR deposition with PiB SUVR increase. This acceleration is determined by the coefficient of the quadratic PiB SUVR term. Using model 3, this coefficient gives information with respect to the direction and the steepness of the relationship between these variables. All of the associations that were found were convex (a positive curvature), with lower coefficients for Braak 3 ROI (0.43, 95% CI [0.27, 0.59]) and Braak 4 ROI (0.27, 95% CI [0.13, 0.41]) and higher quadratic coefficients in Braak 5 ROI (0.59, 95% CI [0.43, 0.74]) and Braak 6 ROI (0.67, 95% CI [0.52, 0.82]). Mathematically, this indicates that, as PiB SUVR increases, the tau amyloid changes by (the derivative of the quadratic model 3 with respect to the Global PiB SUVR). For example, for a value of Global‐PIB SUVR equal to 1.5, the estimated TAU SUVR in Braak 3 ROI will be 1.8 (‐0.70+2*0.43*1.5 = 1.8, Table 3). For a value of Global‐PiB SUVR equal to 2.5, the estimated tau SUVR in Braak 6 will be 1.62 (‐1.73+2*0.67*1.25 = 1.62).
The plots showing the linear and the quadratic associations are presented in Figure 1.
FIGURE 1.
Models for AV‐1451 with Global‐PiB SUVR. The plots are showing the linear and quadratic model fit lines for each Braak region (shown on y‐axes) and Global‐PiB SUVR (shown on x‐axes)
4. DISCUSSION
The recent development of tau‐PET ligands has provided the unique opportunity to expand our knowledge of the natural history of AD pathophysiology in DS. These data extend our previous findings, identifying a distinct pattern of predominant striatal Aβ deposition that distinguished the Aβ deposition seen in late‐onset AD and DS 21 into tau pathology in DS. These finding are among the first to report tau pathology using PET in DS and are, to our knowledge, the largest DS cohort using tau PET.
These results demonstrate a significant relationship between in vivo Aβ and tau pathology in DS, similar to those seen in the typical aging population. The differences observed between amyloid‐positive participants with DS and amyloid‐negative participants with DS or amyloid‐negative controls decreased in regions associated with the highest Braak stages. This decreasing difference is likely a result of lower overall tau pathology in Braak stages 5 and 6, given that the majority of the participants in this study were non‐demented and early in the time course of AD pathophysiology. One limitation of the current study is that the majority of the DS participants are classified as cognitively normal and were middle aged (39.04 years); expanding both the age and cognitive function range of this cohort will be essential to understanding the full spectrum of AD.
Although we observed decreasing differences in overall tau pathology in regions associated with higher Braak stages, our quadratic models suggest an acceleration of tau pathology in regions associated with higher Braak stages as amyloid pathology increases. Indeed in Braak ROIs 5 and 6, the quadratic coefficient indicates a steeper increase (0.59 in Braak 5 and 0.67 in Braak 6, Figure 1) associated with accumulating Aβ. Our findings suggest that tau pathology in DS does not differ from that in late‐onset AD, with tau pathology increasing in regions associated with Braak regions in those with significant amyloid pathology. These data track with pathological studies in DS that have not identified clear differences in neurofibrillary tau deposition from late‐onset AD, but these are restricted to patients with late‐stage disease in all groups. 22 , 23 , 24 This model also supports the hypothesis that amyloid pathology is an initiating event leading to the spread of tau from the medial temporal lobes to neocortex, increasing the rate of accumulation as tau levels increase. 25 However, additional longitudinal data from DS participants with AD will be required to determine if our models do in fact represent the true AD pathophysiology in DS and match those identified in pathological studies.
Two technical limitations in the present study are the use of the cerebellum as a reference region for [11C]PiB and the use of the 80‐100 analysis window for [18F]AV‐1451 . We acknowledge the findings reported in the literature of improved sensitivity with PiB using the white matter reference region (Brendel et al., 2015; Chen et al., 2015; Schwarz et al., 2017), particularly related to addressing the shortcomings of technical variability introduced by differences in scanner slice sensitivity and noise. However, we feel strongly that cerebellar gray matter reference region provides a more accurate physiological representation of amyloid and tau burden. Considering that the SUVR metric serves as a proxy for the distribution volume ratio (DVR) or binding potential (BPND), the non‐displaceable distribution volume (V_ND = K1/k2') should be close to equal in these gray matter tissue regions (and thus cancel out in the ratio), and that the overall findings would be expected to be compatible. In addition, it has been suggested that the analyses of [18F]AV‐1451 using the 80‐100 minute window may underestimate the SUVR values, particularly in the high binding range; however, in the same study it was suggested that the 80‐100 minute was best for studying the full range of tau pathology. 26
As we move toward an era of dementia prevention trials in DS, an understanding of the natural history of AD pathophysiology in DS is critical. 27 These data do not yet provide information about the role of tau pathology in the transition to clinical dementia; however, ongoing collection of longitudinal cognitive and imaging data within this cohort will provide data to answer this important question in the further. Furthermore, these data add to our understanding of AD pathology in DS, expanding from the large body of literature related to amyloid PET by exploring tau PET and are an important first step in understanding the natural history of amyloid and tau in the transition to dementia in DS. Furthermore, these biomarker data will provide the necessary framework to identify appropriate participants for clinical trials, track efficacy of interventions, and track dementia progression in this population.
5. CONCLUSIONS AND RELEVANCE
These data demonstrate that similar to late‐onset AD, in vivo Aβ and tau pathology are associated in DS. In addition, these data suggest a higher tau pathology in regions associated with higher Braak stages as amyloid pathology increases. These finding are among the first to report tau pathology using PET in DS and are, to our knowledge, the largest DS cohort using tau PET. The data also contribute to providing the necessary framework to identify appropriate participants for clinical trials, track efficacy of interventions, and track dementia progression in the DS population.
Supporting information
Supporting Information
Tudorascu DL, Laymon CM, Zammit M, et al. Relationship of amyloid beta and neurofibrillary tau deposition in Neurodegeneration in Aging Down Syndrome (NiAD) study at baseline. Alzheimer's Dement. 2020;6:e12096 10.1002/trc2.12096
REFERENCES
- 1. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer‐type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology. 1992;42(9):1681‐1688. [DOI] [PubMed] [Google Scholar]
- 3. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound‐B. Ann Neurol. 2004;55(3):306‐319. [DOI] [PubMed] [Google Scholar]
- 4. Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF‐tau radioligand [F‐18]‐T807. J Alzheimers Dis. 2013;34(2):457‐468. [DOI] [PubMed] [Google Scholar]
- 5. Ossenkoppele R, Schonhaut DR, Scholl M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016;139(Pt 5):1551‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mishra S, Gordon BA, Su Y, et al. AV‐1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure. Neuroimage. 2017;161:171‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maass A, Lockhart SN, Harrison TM, et al. Entorhinal tau pathology, episodic memory decline, and neurodegeneration in aging. J Neurosci. 2018;38(3):530‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Head E, Lott IT, Wilcock DM, Lemere CA. Aging in Down syndrome and the development of Alzheimer's disease neuropathology. Curr Alzheimer Res. 2016;13(1):18‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci. 1998;21:479‐505. [DOI] [PubMed] [Google Scholar]
- 10. Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin‐1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27(23):6174‐6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Handen BL, Cohen AD, Channamalappa U, et al. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012;8(6):496‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Braak H, Braak E. Alzheimer's disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol. 1990;49(3):215‐224. [PubMed] [Google Scholar]
- 13. Mann DM, Iwatsubo T. Diffuse plaques in the cerebellum and corpus striatum in Down's syndrome contain amyloid beta protein (A beta) only in the form of A beta 42(43). Neurodegeneration. 1996;5(2):115‐120. [DOI] [PubMed] [Google Scholar]
- 14. Rafii MS, Lukic AS, Andrews RD, et al. PET imaging of tau pathology and relationship to amyloid, longitudinal MRI, and cognitive change in Down syndrome: results from the Down Syndrome Biomarker Initiative (DSBI). J Alzheimers Dis. 2017;60(2):439‐450. [DOI] [PubMed] [Google Scholar]
- 15. Silverman W, Schupf N, Zigman W, et al. Dementia in adults with mental retardation: assessment at a single point in time. Am J Ment Retard. 2004;109(2):111‐125. [DOI] [PubMed] [Google Scholar]
- 16. Cohen AD, Mowrey W, Weissfeld LA, et al. Classification of amyloid‐positivity in controls: comparison of visual read and quantitative approaches. Neuroimage. 2013;71:207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tziortzi AC, Searle GE, Tzimopoulou S, et al. Imaging dopamine receptors in humans with [11C]‐(+)‐PHNO: dissection of D3 signal and anatomy. Neuroimage. 2011;54(1):264‐277. [DOI] [PubMed] [Google Scholar]
- 18. Svarer C, Madsen K, Hasselbalch SG, et al. MR‐based automatic delineation of volumes of interest in human brain PET images using probability maps. Neuroimage. 2005;24(4):969‐979. [DOI] [PubMed] [Google Scholar]
- 19. Braak H, Braak E. Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol Aging. 1995;16(3):271‐278. [DOI] [PubMed] [Google Scholar]
- 20. Scholl M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89(5):971‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cohen AD, McDade E, Christian B, et al. Early striatal amyloid deposition distinguishes Down syndrome and autosomal dominant Alzheimer's disease from late‐onset amyloid deposition. Alzheimers Dement. 2018;14(6):743‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bateman R, Aisen P, De Strooper B, et al. Autosomal‐dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res Ther. 2010;3(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid β‐Peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis. 1996;3(1):16‐32. [DOI] [PubMed] [Google Scholar]
- 24. Mann DMA, Jones D, Prinja D, Purkiss MS. The prevalence of amyloid (A4) protein deposits within the cerebral and cerebellar cortex in Down's syndrome and Alzheimer's disease. Acta Neuropathologica. 1990;80(3):318‐327. [DOI] [PubMed] [Google Scholar]
- 25. Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45(3):358‐368. [DOI] [PubMed] [Google Scholar]
- 26. Baker SL, Lockhart SN, Price JC, et al. Reference tissue‐based kinetic evaluation of 18F‐AV‐1451 for tau imaging. J Nucl Med. 2017;58(2):332‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ness S, Rafii M, Aisen P, Krams M, Silverman W, Manji H. Down's syndrome and Alzheimer's disease: towards secondary prevention. Nature reviews. 2012;11(9):655‐656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information