

Abstract
Drug‐induced liver injury (DILI) sometimes presents with an autoimmune hepatitis‐like phenotype (AI‐DILI), and it is challenging to distinguish it from de novo autoimmune hepatitis (AIH). We conducted a study to identify autoantibodies unique to AI‐DILI by profiling serum autoantibodies. Autoantibodies were quantified using an autoantigen array containing 94 autoantigens from four groups: AI‐DILI (n = 65), DILI controls (n = 67), de novo AIH (n = 17), and healthy controls (HCs; n = 30). In 37 patients with AI‐DILI, samples were also collected 6 months after presentation. AI‐DILI and de novo AIH had similar anti‐neutrophil antibody and anti‐smooth muscle antibody prevalence. Compared to HCs, de novo AIH had an increase in many immunoglobulin G (IgG; 35 [46.1%]) and IgM (51 [70%]) autoantibodies, whereas AI‐DILI had an increase of IgM (40 [54.8%]) but not IgG autoantibodies. DILI controls had a similar IgG and IgM profile compared to HCs. Comparing de novo AIH to AI‐DILI identified 18 (23.7%) elevated IgG but only one (1.4%) IgM autoantibodies, indicating the unique IgG autoantibody profile in de novo AIH. Compared to DILI and HCs, increased IgM autoantibodies in AI‐DILI and de novo AIH were common; however, AI‐DILI induced by different drugs showed different frequencies of IgM autoantibodies, with nitrofurantoin‐related AI‐DILI showing a higher number of increased IgM autoantibodies. AI‐DILI autoantibody levels at diagnosis and at 6 months showed a significant decline in 37 IgM autoantibodies. A model with highly correlated IgG and IgM was fitted into multivariate logistic regression and revealed an area under the curve of 0.87 (95% confidence interval, 0.79‐0.95) to distinguish de novo AIH from AI‐DILI. Conclusion: The unique IgG and IgM autoantibody signature appears to be a promising biomarker for distinguishing AI‐DILI from de novo AIH.
Abbreviations
- AI‐DILI
autoimmune‐like drug‐induced liver injury
- AIH
autoimmune hepatitis
- ALT
alanine aminotransferase
- ANA
anti‐neutrophil antibody
- ASMA
anti‐smooth muscle antibody
- AST
aspartate aminotransferase
- AUC
area under the curve
- BMI
body mass index
- CENP‐B
centromere protein B
- CI
confidence interval
- DILI
drug‐induced liver injury
- DILIN
Drug‐Induced Liver Injury Network
- dsDNA
double‐stranded DNA
- HC
healthy control
- HLA
human leukocyte antigen
- IgG/M
immunoglobulin G/M
- PCA
principal component analysis
- RA
rheumatoid arthritis
- ROC
receiver operator curve
- SCL‐70
topoisomerase 1
- SLE
systemic lupus erythematosus
- SNR
signal‐to‐noise ratio
- ssDNA
single‐stranded DNA
- TB
total bilirubin
- U1‐snRNP
U1 small nuclear ribonucleoprotein
Drug‐induced liver injury (DILI) is a rare complication occurring in individuals receiving prescription medications or herbal and dietary supplements, but it carries significant morbidity and mortality.( 1 , 2 ) The diagnosis is dependent on a careful review of medical history and the temporal relationship with suspected drug exposure along with the thorough exclusion of a competing etiology.
Acute liver injury due to drugs may sometimes present with features similar to that of autoimmune hepatitis (AIH). This autoimmune‐like DILI (AI‐DILI) has been classically observed following use of medications, such as nitrofurantoin, minocycline, methyldopa, and hydralazine.( 3 , 4 , 5 , 6 ) AI‐DILI often has similar histologic findings on liver biopsy( 7 ) and serologic findings( 8 ) as seen in de novo AIH, which can make differentiating these two conditions a challenge. Individuals with AI‐DILI typically have a rapid response to cessation of the causative drug with or without immunosuppression, and when immunosuppressive agents are given, they often can be tapered and stopped relatively quickly. In contrast, idiopathic de novo AIH can be challenging to treat and typically requires long‐term (sometimes life‐long) immunosuppressive therapy. There is currently an unmet need for a reliable biomarker( 9 ) to assist in the clinical differentiation of AI‐DILI and de novo AIH and also to better predict outcomes and monitor for progression/regression of injury.( 10 )
Laboratory tests, such as serum alanine aminotransferase (ALT), alkaline phosphatase, and total bilirubin (TB), are clinical standards for monitoring DILI events. However, ideal biomarkers in DILI would not only identify liver injury but also be specific or selective for an offending drug or drug class.( 9 ) Due to the lack of specific or sensitive biomarkers, scoring systems, like the Roussel Uclaf Causality Assessment Method, continue to be used to assess the likelihood of a DILI event.( 11 ) To date, no clinical biomarker has been shown to successfully differentiate AI‐DILI from de novo idiopathic AIH.
We conducted a proof‐of‐concept study to investigate if there are autoantibodies specific to AI‐DILI by using a multiplex autoantigen array to profile serum autoantibodies across four patient groups: DILI due to medications commonly associated with autoimmune features (AI‐DILI), DILI without AIH features (DILI controls), well‐characterized de novo AIH, and healthy controls (HCs).
Patients and Methods
Subject Recruitment
Patients with AI‐DILI and DILI were recruited from the DILI Network (DILIN) Prospective Study.( 12 ) All cases were reviewed by the DILIN Causality Committee and assigned causality scores, and only cases assessed as definite (>95% likelihood), highly likely (75%‐95%), or probable (51%‐74%) were included in this study. Laboratory and imaging testing to exclude competing etiologies were performed in all cases.
Individuals identified as AI‐DILI had liver injury due to nitrofurantoin, minocycline, methyldopa, or hydralazine; their selected clinical characteristics have been published.( 5 ) DILI controls were patients with acute liver injury due to amoxicillin‐clavulanate, diclofenac, or isoniazid. De novo AIH( 13 ) and HCs were recruited at Indiana University Medical Center. AIH samples were collected at the time of baseline outpatient liver biopsies in the absence of immunosuppression. Patients with AIH were retrospectively identified once diagnosis was complete per standard international AIH classification.( 8 ) HCs were plasma donors with persistently normal liver biochemistries. The DILIN Prospective Study was approved by the institutional review boards at each clinical site, and written consent was obtained from all participants. AIH and HCs were recruited under separate protocols approved by the Indiana University–Purdue University Indianapolis Institutional Review Board.
Autoantibody Profiling Using Autoantigen Microarrays
Ninety‐four autoantigens (Supporting Table S1) were selected based on a literature survey on their reactivates to various autoantibodies in different autoimmune diseases, including systemic lupus erythematosus (SLE), systemic sclerosis, Sjogren's syndrome, idiopathic inflammatory myositis, and rheumatoid arthritis (RA).( 14 , 15 , 16 , 17 ) The antigens were printed in duplicates on nitrocellulose membrane‐coated slides, and the serum samples were measured on this autoantigen microarray at the University of Texas Southwestern Medical Center (https://microarray.swmed.edu/products/category/protein‐array/). Briefly, serum samples were pretreated with deoxyribonuclease‐I and then diluted 1:50 in phosphate‐buffered saline (PBS) with Tween 20 buffer and incubated with the autoantigen arrays. The autoantibodies binding with antigens were detected with cyanine 3 (cy3)‐labeled anti‐human immunoglobulin G (IgG; 1:2,000; Jackson ImmunoResearch Laboratories) and cy5‐labeled anti‐human IgM (1:1,000, Jackson ImmunoResearch Laboratories), using a Genepix 4000B scanner (Molecular Device) with laser wavelengths of 532 and 635 nm. The resulting images were analyzed using Genepix Pro 6.0 software (Molecular Devices). The median of the signal intensity for each spot was calculated and subtracted from local background, and data obtained from duplicate spots were averaged. The background‐subtracted signal intensity of each antigen was normalized to the average intensity of the total human IgG or IgM, which was included on the array as an internal control. Finally, the net fluorescence intensity for each antigen was calculated by subtracting a PBS control that was included for each experiment as a negative control.( 14 , 15 , 16 , 17 ) The signal‐to‐noise ratio (SNR) was used as a quantitative measurement of the true signal above background noise. SNR values equal to or greater than 3 were considered significantly higher than background and therefore true signals.
Statistical Analysis
Database management, validity interrogation, and statistical analysis were performed using the R Project (https://www.R‐project.org). Descriptive statistics of study cohorts were reported as means and SDs or percentages. Comparisons of categorical variables were completed using chi‐square or Fisher’s exact tests, whereas continuous variables were compared using Student t tests and Wilcoxon rank sum tests. Two‐tailed nonparametric tests were used to compare ranks or differences between groups. Cluster maps were generated by Genesis software. R (3.5) was used for principal component analysis (PCA) with the Vegan (2.5) package. Adonis paired tests were performed to test the differences between groups. GraphPad prism (version 8.1) was also used for statistical tests. Leave‐one‐out cross‐validation was performed, which involves using one observation as the validation set and the remaining samples as the training set. This is repeated until all samples are predicted by a stepwise multiple logistic regression model. Prediction performance was evaluated by the area under the curve (AUC) in the receiver operating characteristic (ROC) curve.
Results
A total of 179 patients were included in this study: 65 patients with AI‐DILI, 67 DILI controls, 17 subjects with de novo AIH, and 30 HCs (Table 1). The causality score for AI‐DILI was definite in 13 (20%), highly likely in 43 (66.2%), and probable in 9 (13.8%) patients. The causality scores for DILI without AI features were definite in 39 (58.2%), highly likely in 20 (29.9%), and probable in 8 (11.9%) patients. There was no difference between the distributions of causality scores between the two DILI groups (AI‐DILI mean, 2; SD, 1) versus DILI controls (mean, 2; SD, 0.58).
Table 1.
Selected Characteristics of Four Study Groups
| AI‐DILI (n = 65) | DILI Without AI Features (n = 67) | HCs (n = 30) | De Novo AIH (n = 17) | |
|---|---|---|---|---|
| Age (years), mean (SD) | 48 (21) | 51 (14) | 49 (14) | 50 (14) |
| Female, % | 89 | 54 | 17 | 76 |
| Caucasian, % | 54 | 73 | 93 | 88 |
| BMI (kg/m2), mean (SD) | 28.2 (8.1) | 27.3 (5.7) | 30.1 (6.2) | 29.3 (8.0) |
| ALT (U/L), mean (SD) | 1,154 (845) | 757 (691) | 19 (6) | 262 (254) |
| AST (U/L), mean (SD) | 981 (627) | 592 (700) | 25 (3.7) | 290 (315) |
| ALP (U/L), mean (SD) | 235 (152) | 232 (109) | 61 (14) | 213 (151) |
| Total bilirubin (mg/dL), mean (SD) | 6.7 (6.8) | 6.6 (6.3) | 0.6 (0.2) | 3.2 (6.7) |
| ANA positive, % | 69 | 22 | NA | 73 |
| ASMA positive, % | 60 | 13 | NA | 67 |
Abbreviations: ALP, alkaline phosphatase; NA, not available.
Serum samples from 37 (58%) patients with AI‐DILI were available at the 6‐month follow‐up. Patients with AI‐DILI were on average 48 years old, more often women (89%; P < 0.001), and less‐frequently Caucasian (54%; P < 0.001) compared to all other groups. Liver biochemistries were higher at the time of diagnosis among AI‐DILI (serum ALT, 1,154 U/L; aspartate aminotransferase [AST], 981 U/L) compared to DILI without AI features (ALT, 757 U/L; AST, 592 U/L; P = 0.004 and P = 0.001, respectively) or de novo AIH (ALT, 262 U/L; AST, 290 U/L; P < 0.001 for both). AI‐DILI and DILI controls were no different according to their serum TB levels at presentation (6.7 and 6.6 mg/dL; P = 0.93), but their TB tended to be higher than in de novo AIH (mean TB, 3.2 mg/dL; P = 0.067 and P = 0.07, respectively). There was no significant difference in anti‐neutrophil antibody (ANA) (69% vs. 73%; P = 0.9) or anti‐smooth muscle antibody (ASMA) positivity (60% vs. 67%; P = 0.72) between AI‐DILI and de novo AIH, respectively (Table 1).
Baseline Group Autoantibody Characteristics Compared to HCs
Of the 94 autoantigens tested (Supporting Table S1), 18 IgG autoantibodies and 21 IgM autoantibodies showed a very low signal (SNR, <3) in over 90% of samples among all groups and were excluded from further analysis. Among the remaining autoantibodies, the mean level of each was calculated among individual study groups.
First, we compared AI‐DILI, DILI controls, and de novo AIH with HCs to identify differentially expressed IgG (Supporting Table S2A) and IgM (Supporting Table S2B) autoantibodies. Thirty‐five IgG autoantibodies (46.1%) were observed with more than a 2‐fold increase in the de novo AIH group compared to HCs (P < 0.05) (Fig. 1). However, a similar 2‐fold increase was noted with only a few IgG autoantibodies in the AI‐DILI (four [5.3%]) and DILI control (three [3.9%]) groups (Fig. 1). Among IgM autoantibodies, 53 (69.7%) in de novo AIH and 40 (52.6%) in AI‐DILI were significantly higher compared to HCs (P < 0.05). Among DILI controls, only two (2.6%) IgM autoantibodies had a 2‐fold increase compared to HCs (Fig. 1).
Fig. 1.
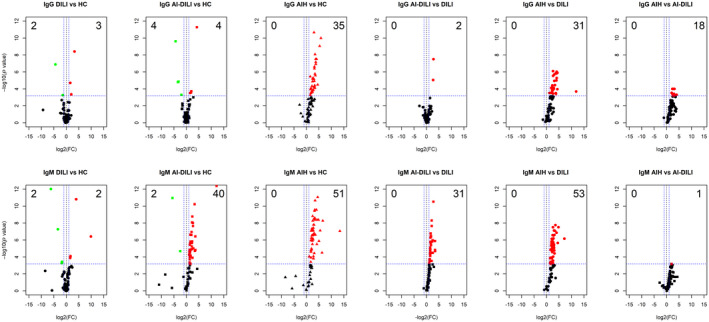
Autoantigen microarray analysis identified differential IgG and IgM autoantibody profiles in four groups of samples (HCs, DILI, AI‐DILI, and de novo AIH). Sera from 30 HCs, 67 DILI controls, 65 AI‐DILI, and 17 de novo AIH were screened for autoantibodies using a high‐throughput 94 autoantigen microarray platform. Volcano plots of the IgG autoantibodies (top row) and IgM autoantibodies (bottom row) displaying each autoantibody as a single point with −log10 (P value) on the y axis versus log2 [FC] on the x axis. Red points indicate a statistically significant increase in mean autoantibody levels for each group comparison; green points indicate a statistically significant decrease for each group comparison. Abbreviation: FC, fold change.
Comparison of Autoantibodies Among De Novo AIH, AI‐DILI, and DILI Controls
Next, we compared IgG and IgM autoantibodies among de novo AIH, AI‐DILI, and DILI control groups to reveal the autoantibody profiles in different disease groups. Again, the de novo AIH group showed the most striking differences in both IgG and IgM, with 31 (40.1%) IgG and 53 (72.6%) IgM autoantibodies significantly increased among de novo AIH compared to DILI controls (Fig. 1; Supporting Table S2A,B). Comparing the AI‐DILI and DILI controls also revealed 33 (45.2%) increased IgM autoantibodies but no increased IgG autoantibodies. Further comparisons between de novo AIH and AI‐DILI groups identified 18 (23.7%) IgG autoantibodies but only one (1.4%) IgM autoantibody that were significantly increased in de novo AIH compared to AI‐DILI (Fig. 1).
When comparing autoantibody profiles of de novo AIH and AI‐DILI (cases) to control groups (DILI and HCs), there was only one (2.4%) IgG autoantibody (single‐stranded DNA [ssDNA]) similarly increased among cases (Fig. 2A) because patients with de novo AIH mostly clustered together with higher levels of IgG against a multitude of autoantibodies (Fig. 1). PCA also showed a statistically significant segregation of individual patients with de novo AIH from AI‐DILI (cases) and control groups (DILI and HCs), but there were no significant differences among AI‐DILI, DILI controls, and HCs (Fig. 2B; Supporting Table S3A). A similar comparison of groups according to IgM autoantibodies revealed 25 (43.9%) autoantibodies increased in cases (Fig. 2C). PCA also showed that de novo AIH and AI‐DILI clustered together (Adonis paired test, P = 0.078) and that they segregated from DILI and HCs (Adonis paired test, P < 0.01) (Fig. 2D; Supporting Table S3B).
Fig. 2.
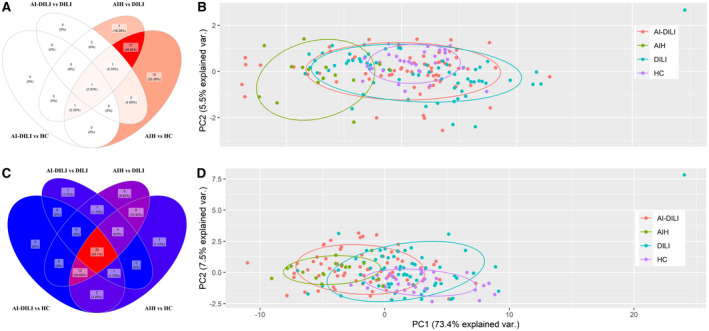
Patients with AI‐DILI showed significant increased IgM autoantibodies but not IgG autoantibodies.(A) Venn diagram showing one elevated IgG autoantigen observed in comparisons of AI‐DILI versus HCs and AI‐DILI versus DILI. Twenty‐four autoantigens were observed between de novo AIH versus HCs and de novo AIH versus DILI controls. (B) PCA of combined IgG autoantibodies showing clustering of de novo AIH compared to the other three groups (pairwise Adonis test with adjusted P values for FDR: de novo AIH versus HCs, P < 0.01; de novo AIH versus DILI, P < 0.01; de novo AIH versus AI‐DILI, P < 0.01; differences between HCs, DILI, and AI‐DILI by autoimmunity status were not statistically significant). (C) Venn diagram showing 25 elevated IgM antigens recognized all by de novo AIH versus HCs, de novo AIH versus DILI, AI‐DILI versus HCs, and AI‐DILI versus DILI. (D) PCA of combined IgM autoantibodies showing clustering of de novo AIH/AI‐DILI difference from HC/DILI (pairwise Adonis test with adjusted P values for FDR: de novo AIH versus HCs, P < 0.01; de novo AIH versus DILI, P < 0.01; AI‐DILI versus HCs, P < 0.01; AI‐DILI versus DILI, P < 0.01; differences between de novo AIH and AI‐DILI (P = 0.078) by autoimmunity status were not statistically significant). Abbreviations: FDR, false discovery rate; var., variance.
Autoantibodies to Discriminate Study Groups
An ROC analysis was conducted to assess the predictive value of individual and combined autoantibodies for separating de novo AIH from AI‐DILI and from distinguishing AI‐DILI from DILI. Among 18 IgG autoantibodies that significantly increased in de novo AIH compared to AI‐DILI, a multiple stepwise logistic model identified the top five IgG autoantibodies for predicting de novo AIH from AI‐DILI (Fig. 3A). The combination of five IgG autoantibodies directed at antigens centromere protein B (CENP‐B), chromatin, mitochondrial antigen, myosin, and nucleosome antigen performed well, with an AUC of 0.88 (95% confidence interval [CI], 0.80‐0.95) for distinguishing de novo AIH from AI‐DILI. Among 31 IgM autoantibodies that significantly increased in AI‐DILI compared to DILI controls, modeling identified four IgM autoantibodies (directed against double‐stranded DNA [dsDNA], topoisomerase 1 [SCL‐70], ssDNA, and U1‐small nuclear ribonucleoprotein [snRNP]‐B/B’) that predicted AI‐DILI from DILI controls (AUC, 0.87; 95% CI, 0.8‐0.93) (Fig. 3B).
Fig. 3.
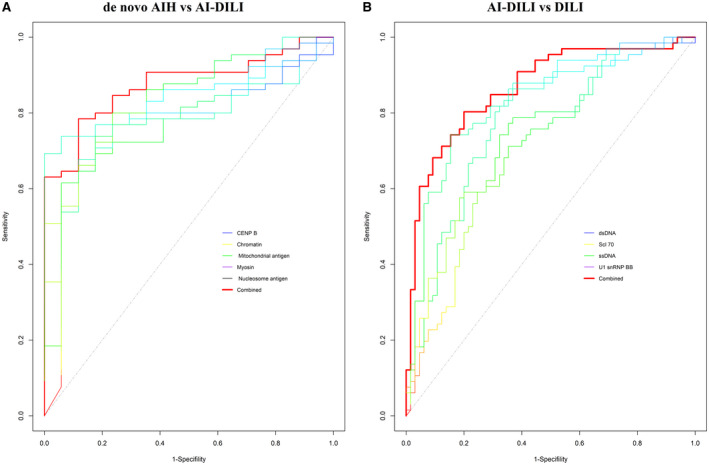
Predictive value of individual and combination of autoantibodies for prediction of AI‐DILI. (A) ROC analysis by multiple stepwise logistic model to distinguish AIH from AI‐DILI. Five IgG “best” autoantibodies were automatically selected in the model from 18 differential AIH versus AI‐DILI. (B) ROC analysis by multiple stepwise logistic model to distinguish AI‐DILI from DILI controls. Four IgM “best” autoantibodies were automatically selected in the model from 31 differential AI‐DILI versus DILI controls.
Association of Autoantibodies in AI‐DILI With Serologic Characteristics
The differential increase in autoantibodies (both IgM and IgG classes) between AI‐DILI and DILI controls was found to be associated with results for several analytes (ALT, AST, ANA, and ASMA) in a multiple regression or multinomial logistic regression model correcting for patient age, sex, and body mass index (BMI) (Fig. 4). Most IgG autoantibodies showed a relatively weaker correlation with the serologic analytes; the exceptions were dsDNA and ssDNA antibodies, which showed strong correlation (P < 0.001) with ANA. However, most of the increased IgM autoantibodies strongly correlated with ANA and ASMA, and a few (U1‐snRNP‐A and U1‐snRNP‐68) also showed a strong correlation (P < 0.001) with AST, which is an indication of liver injury.
Fig. 4.
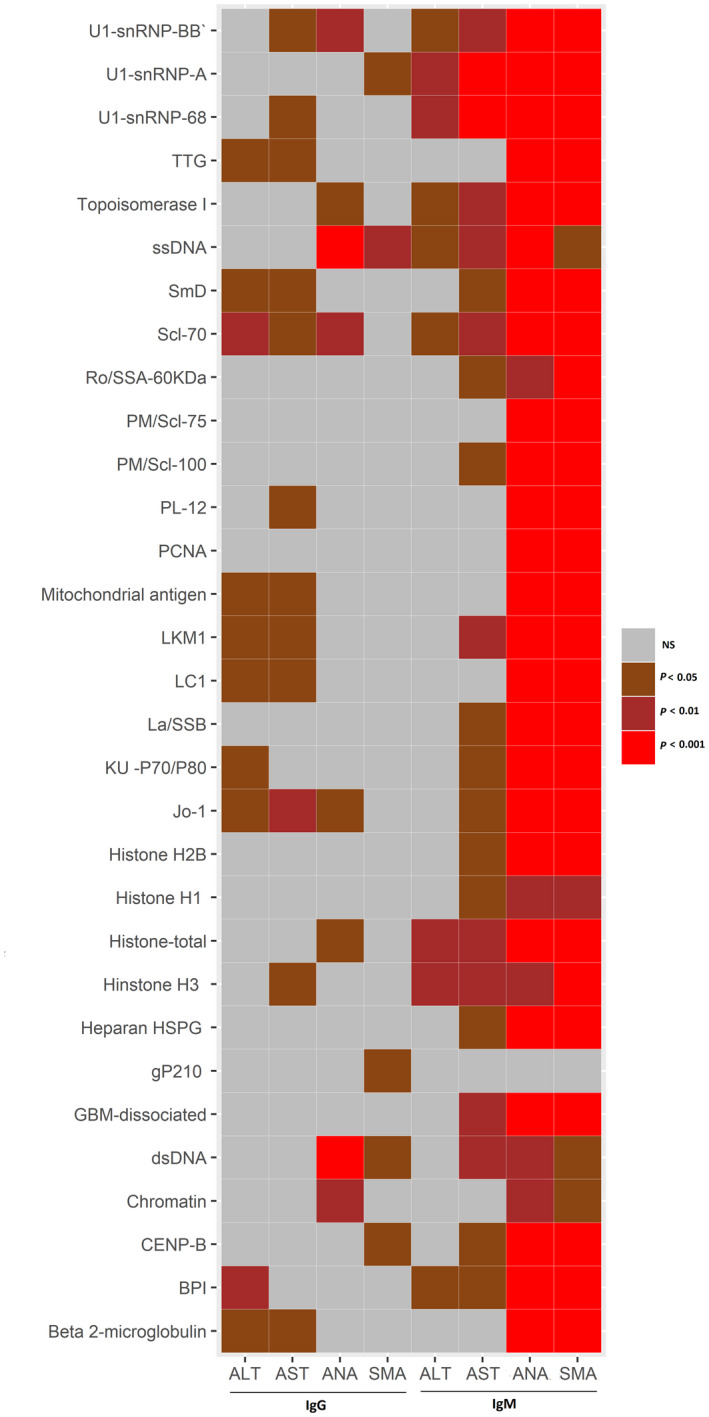
Association between differential autoantibodies and clinical criteria (ALT, AST, ANA, SMA), correcting for age, sex, and BMI. P values were generated by multiple regression model for ALT and AST or multinomial logistics regression model for ANA and SMA, all using age, sex, and BMI as covariates. Autoantibodies: BPI, GBM, gP210, HSPG, Jo‐1, KU, La/SSB, LC1, LKM1, NS, not significant; PCNA, PL‐12, PM, Ro/SSA, SmD, TTG. Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; ANA, antineutrophil antibody; BMI, body mass index; SMA, antismooth muscle antibody.
Autoantibodies in Drug‐Specific AI‐DILI
Few IgG autoantibodies were significantly different between drug‐specific AI‐DILI and HCs or DILI controls (amoxicillin‐clavulanate and diclofenac) (Fig. 5). The most frequently increased IgG autoantibodies were observed in nitrofurantoin‐associated AI‐DILI compared to HCs (5 [6.6%]). On the other hand, mean IgM autoantibodies were significantly higher in drug‐specific AI‐DILI compared to both healthy and DILI controls (Fig. 5). The most frequent IgM autoantibodies (34 [46.6%]) were observed in nitrofurantoin‐associated AI‐DILI compared to HCs, followed by hydralazine (24 [32.9%]) and minocycline (19 [26%]). Differences in IgM autoantibodies between AI‐DILI and DILI controls were minimal except for nitrofurantoin‐associated AI‐DILI (30 [41.1%]).
Fig. 5.
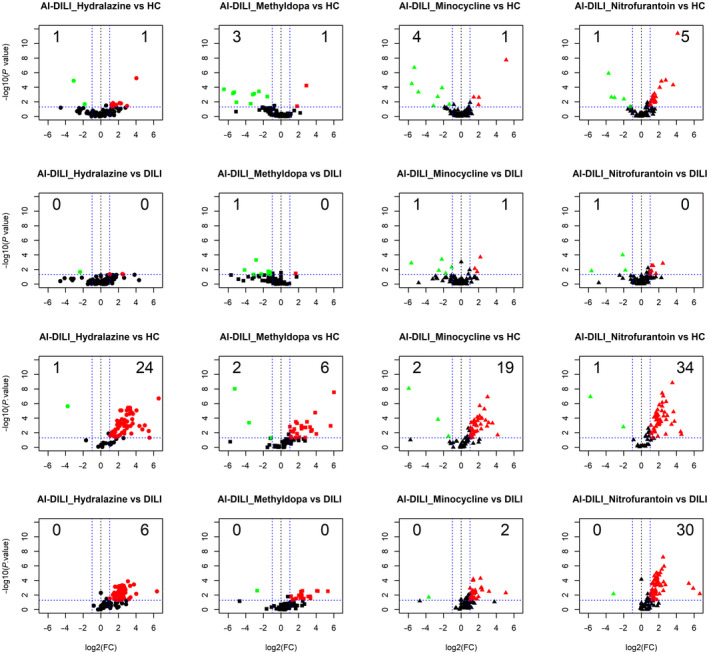
Volcano plots of IgG autoantibodies between medication‐specific AI‐DILI versus HCs (first row), medication‐specific AI‐DILI versus DILI controls (second row), IgM autoantibodies between medication‐specific AI‐DILI versus HCs (third row), IgM autoantibodies medication‐specific AI‐DILI versus DILI controls (fourth row), displaying each autoantibody as a single point with −log10 (P value) on the y axis versus log2 [FC] on the x axis. Red points indicate a statistically significant increase in mean autoantibody levels for each group comparison; green points indicate a statistically significant decrease for each group comparison. Abbreviation: FC, fold change.
AI‐DILI at 6‐Month Follow‐Up
Mean IgG and IgM autoantibody levels were evaluated at the 6‐month follow‐up in 37 patients with AI‐DILI. Among these, all received immunosuppression treatment during the first 6 months following their diagnosis. A majority (97.3%) received corticosteroids as initial treatment, and 32.4% remained on corticosteroids at the 6‐month follow‐up. Eight patients (21.6%) received azathioprine either alone (1/8) or in addition to corticosteroid treatment, and most (7/8) remained on azathioprine at the 6‐month follow‐up. In total, 14 patients (37.8%) were on an immunosuppressant drug at the 6‐month follow‐up. Thirty‐five IgM but no IgG autoantibodies showed significant reductions in mean levels at 6 months compared to baseline values (P < 0.05; fold change, >1.2) (Fig. 6; Supporting Table S5). Among these decreasing IgM autoantibodies, 16 (45.7%) had a trend toward increasing levels in the corresponding IgG class autoantibodies at 6 months (Fig. 6).
Fig. 6.
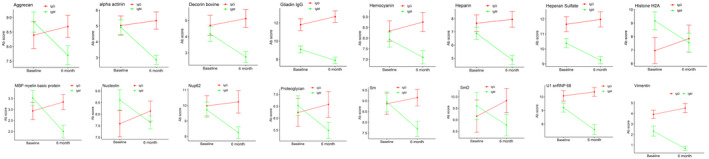
IgM autoantibodies significantly decreased after 6 months of treatment. Thirty‐five mean IgM autoantibody levels were at lower values at 6 months compared with baseline values (P < 0.05; fold change, >1.2) (Supporting Table S5). A majority of IgG autoantibodies showed no significant change between baseline and 6‐month values, but 16 autoantibodies (45.7%) showed a trend toward increase. Data represent mean ± SD. Abbreviation: Ab, antibody; MBP, myelin basic protein; Nup62, nucleoporin 62; Sm, smooth muscle; SmD, small nuclear ribonucleoprotein polypeptide N′.
Discussion
The identification of reliable biomarkers for DILI diagnosis and prognosis has been an active area of research, yet there are no currently available tests. In an earlier study, DILIN investigators observed that patients with hepatocellular‐type DILI had different levels of serum fructose‐bisphosphonate aldolase B, which correlated with increasing serum ALT and AST and returned to normal at follow‐up.( 18 ) Other DILIN investigations have also shown promise for microRNA and serum cytokine profiling in characterizing patients with DILI.( 19 , 20 ) A more recent international collaborative effort showed that glutamate dehydrogenase offered potential for identifying DILI whereas cytokeratin18, osteopontin, and macrophage colony‐stimulating factor receptor were associated with acute DILI outcomes.( 21 ) However, to our knowledge, no biomarker studies to date have attempted to distinguish AI‐DILI from de novo AIH.
In this proof‐of‐concept study, we provide evidence that autoantibody profiling using autoantigen arrays is a potential approach for developing diagnostic testing that could distinguish DILI with autoimmune features (AI‐DILI) from de novo idiopathic AIH. Similar high‐throughput autoantibody profiling has been used in autoimmune diseases, such as SLE, to dissect heterogeneous clinical manifestations.( 22 ) Our data revealed that de novo AIH is characterized by a group of both IgG and IgM autoantibodies while AI‐DILI is only by IgM, which could be used as a feature to distinguish AI‐DILI from de novo AIH. Using modeling, a panel of five IgG autoantibodies directed at CENP‐B, chromatin, antimitochondrial antigen, myosin, and nucleosome antigen was able to distinguish de novo AIH from AI‐DILI with high accuracy (AUC, 0.88) (Fig. 3). Our data also revealed that four IgM autoantibodies directed at dsDNA, SCL‐70, ssDNA, U1‐snRNP‐BB were able to predict AI‐DILI from DILI (AUC, 0.87). Limited association between cases and IgG autoantibody class may be linked to eventual immunosuppressive therapy found in both de novo AIH and AI‐DILI. Other approaches to differentiate patients with AI‐DILI versus de novo AIH have been pursued but have not included a systematic approach to discern differences in autoantibodies. In previous studies, histologic findings, such as advanced stages of fibrosis, minimal portal neutrophils, and subpopulations of leukocytes in the portal infiltrates, have been reported to help in some cases( 7 , 23 , 24 ) but are yet to replicate broadly among large populations.
Collective presence of IgG antibodies specific to CENP‐B, chromatin, mitochondrial antigen, myosin, and nucleosome antigen performed well to differentiate de novo AIH from AIH‐DILI (Fig. 3A). Antibodies to CENP‐B, chromatin, mitochondrial antigen, and nucleosome antigen have been observed with variable prevalence in prior AIH studies, sometimes corresponding with clinical characteristics.( 25 , 26 , 27 , 28 ) No studies have associated the formation of anti‐myosin with AIH, but it has been observed in patients with hepatitis C and alcohol‐related liver disease.( 29 , 30 ) Formation of autoantibodies may be dependent on the human leukocyte antigen (HLA; also major histocompatibility complex) locus and associated risk alleles, as observed in patients within a Spanish DILI registry.( 31 ) HLA genotype has been shown to influence progression of autoimmune disease among those with the presence of specific autoantibodies, possibly by an antigen‐specific T‐cell response (such as in RA and anti‐citrullinated protein antibodies( 32 )). However, top HLA risk alleles (HLA‐class II, DR beta 1 [DRB1]*03:01 and DRB1*04:01) in de novo AIH have not been shown to be enriched in drug‐induced AIH( 5 ) and may represent differences in autoantibody formation between the two clinical entities.
Other autoantibody patterns emerged when comparing profiles within all study groups to HCs and each other (Fig. 1). Compared to HCs, de novo AIH had the highest frequency of both IgM and IgG autoantibody differences. Interestingly, differences between AI‐DILI and HCs were largely associated with IgM autoantibodies, whereas DILI cases without AI features had few differences. Similar findings were observed when comparing de novo AIH and AI‐DILI to DILI controls. However, most striking was the mean increase of 18 IgG autoantibodies and only one IgM autoantibody between de novo AIH and AI‐DILI. These autoantibody class observations may provide insight into underlying mechanisms of disease pathogenesis and aspects of disease chronicity in AIH. B cells can play an active role in AIH development through antigen presentation and modulation of T lymphocytes( 33 , 34 ) and are likely important in AIH severity and immunoglobulin (IgG and IgM) concentrations.( 35 ) The increased frequency of IgG autoantibody formation in de novo AIH relative to AI‐DILI may be in part due to chronic hepatic inflammation often present long before AIH diagnosis and may represent autoantibody class switching.( 36 , 37 ) This illuminates a possible key difference between de novo AIH and AI‐DILI because cessation of the inciting drug and/or treatment with immunosuppression attenuates the elevated IgM (innate immune system) autoantibodies in AI‐DILI (Fig. 6) and prevents robust formation of IgG autoantibodies. We did observe an increasing but nonsignificant trend in 16 IgG autoantibodies in a subset of AI‐DILI cases 6 months after diagnosis. Unfortunately, we were unable to compare this with autoantibodies in AIH after treatment.
Few IgG and many IgM autoantibodies were different among drug‐specific AI‐DILI and control groups. The increase in abundance of autoantibodies in AI‐DILI relative to DILI further establishes the connection of AI‐DILI as an immune‐mediated reaction as opposed to DILI. The panel of four autoantibodies with good prediction of AI‐DILI from DILI (Fig. 3B) includes autoantibodies that are often present in other connective tissue diseases, such as SLE, SS, and mixed connective tissue disease.( 22 , 38 , 39 ) We also observed IgM autoantibodies (not the IgG form) directed at antigens differing between AI‐DILI and DILI often correlated well with other autoantibodies (ANA, ASMA) but not liver test abnormalities (Fig. 4). Using a multiclass confusion matrix of 16 autoantibodies (both IgG and IgM form), we were also able to discern the three groups (AI‐DILI, de novo AIH, and DILI) but with a wider range of accuracy (de novo AIH, 76.5%; AI‐DILI, 100%; DILI, 93.9%). Among specific compounds implicated in AI‐DILI, the IgG autoantibody signature of nitrofurantoin was clearly different from hydralazine, methyldopa, and minocycline, yet most drugs had increased IgM autoantibodies compared to controls (Fig. 5). While the mechanism behind this differential pattern within AI‐DILI is not readily apparent, we can hypothesize that differences among the AI‐DILI drugs could be specific to agent, biochemical expression of injury, or a specific immunogenetic reaction. Immunoglobulin class switching in nitrofurantoin‐induced DILI may also occur because the latency of nitrofurantoin‐related AI‐DILI may often exceed 6 months.( 5 ) Changes in mean IgG and IgM autoantibody levels were evaluated in a subset of AI‐DILI (37 patients). Thirty‐five IgM autoantibodies were significantly lower among these patients at 6 months, and no statistically significant changes in mean IgG autoantibody levels were found (Fig. 6). The reduction in IgM seems most consistent with a signal of (autoimmune‐mediated) inflammation reduction in the setting of drug cessation and/or immunosuppressive therapy.
Limitations of this study include its modest sample sizes, lack of external validation, and our application of an existing microarray rather than customizing an autoantigen array. Autoantibody formation also has not closely associated with specific cellular reactions, including type of injury or inflammation (necrosis or apoptosis), and can be present even before clinically relevant disease. Further, some autoantibodies may play an active role in mediating injury in an inflammation‐dependent and independent manner, as observed in RA.( 40 ) Future investigation into the effector function of these autoantibodies may provide a deeper understanding of the pathophysiologic processes involved in AIH and AI‐DILI.
Another challenge in our study included the missing follow‐up clinical and serologic data among all study groups as this issue limits our ability to assess autoantibody changes in response to treatment or time. Well‐characterized AI‐DILI is a rare event, and thus collecting biosamples from a larger cohort is challenging. Collaboration among international consortia, including the recently established Prospective European DILIN, is essential for cross‐validating observations made from pilot studies such as ours. Instead of candidate autoantigen arrays, the application of highly sensitive platforms for a global survey of autoantibodies might be a more attractive approach to predict clinical disease with possibly a more sensitive/specific panel of autoantibodies.
In summary, our proof‐of‐concept study suggests that autoantibody profiling is a promising approach for distinguishing DILI with autoimmune features from de novo AIH. More studies are needed to further validate and extend our observations.
Supporting information
Supplementary Material
Acknowledgment
This paper represents an ancillary study from the DILIN and authors the DILIN for providing biosamples and associated clinical data. Data analyses were done by investigators from the University of Texas Southwestern and from Indiana University. We thank Dr. Herbert Bonkovsky from Wake Forest University for his critical review of the manuscript and Ms. Julianne Nanzer for her editorial assistance.
Potential conflict of interest: Dr. Chalasani consults for NuSirt, AbbVie, Allergan, Madrigal, Genentech, Foresite, Galectin, and Zydus; he has received grant support from Intercept and Exact Sciences. The other authors have nothing to report.
Contributor Information
Quan‐Zhen Li, Email: Quan.li@utsouthwestern.edu.
Naga Chalasani, Email: nchalasa@iu.edu.
References
- 1. Ostapowicz G, Lee WM. Acute hepatic failure: a Western perspective. J Gastroenterol Hepatol 2000;15:480‐488. [DOI] [PubMed] [Google Scholar]
- 2. Chalasani NP, Hayashi PH, Bonkovsky HL, Navarro VJ, Lee WM, Fontana RJ; Practice Parameters Committee of the American College of Gastroenterology . ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug‐induced liver injury. Am J Gastroenterol 2014;109:950‐966. [DOI] [PubMed] [Google Scholar]
- 3. Ghabril M, Bonkovsky HL, Kum C, Davern T, Hayashi PH, Kleiner DE, et al.; US Drug‐Induced Liver Injury Network . Liver injury from tumor necrosis factor‐alpha antagonists: analysis of thirty‐four cases. Clin Gastroenterol Hepatol 2013;11:558‐564.e553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Russo MW, Hoofnagle JH, Gu J, Fontana RJ, Barnhart H, Kleiner DE, et al. Spectrum of statin hepatotoxicity: experience of the drug‐induced liver injury network. Hepatology 2014;60:679‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Boer YS, Kosinski AS, Urban TJ, Zhao Z, Long N, Chalasani N, et al.; Drug‐Induced Liver Injury Network . Features of autoimmune hepatitis in patients with drug‐induced liver injury. Clin Gastroenterol Hepatol 2017;15:103‐112.e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andrade RJ, Lucena MI, Fernandez MC, Pelaez G, Pachkoria K, Garcia‐Ruiz E, et al.; Spanish Group for the Study of Drug‐Induced Liver Disease . Drug‐induced liver injury: an analysis of 461 incidences submitted to the Spanish registry over a 10‐year period. Gastroenterology 2005;129:512‐521. [DOI] [PubMed] [Google Scholar]
- 7. Suzuki A, Brunt EM, Kleiner DE, Miquel R, Smyrk TC, Andrade RJ, et al. The use of liver biopsy evaluation in discrimination of idiopathic autoimmune hepatitis versus drug‐induced liver injury. Hepatology 2011;54:931‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hennes EM, Zeniya M, Czaja AJ, Pares A, Dalekos GN, Krawitt EL, et al.; International Autoimmune Hepatitis Group . Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008;48:169‐176. [DOI] [PubMed] [Google Scholar]
- 9. Teschke R, Schulze J, Eickhoff A, Danan G. Drug induced liver injury: can biomarkers assist RUCAM in causality assessment? Int J Mol Sci 2017;18:803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan L, Kaplowitz N. Mechanisms of drug‐induced liver injury. Clin Liver Dis 2013;17:507‐518, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Danan G, Benichou C. Causality assessment of adverse reactions to drugs–I. A novel method based on the conclusions of international consensus meetings: application to drug‐induced liver injuries. J Clin Epidemiol 1993;46:1323‐1330. [DOI] [PubMed] [Google Scholar]
- 12. Chalasani N, Bonkovsky HL, Fontana R, Lee W, Stolz A, Talwalkar J, et al.; United States Drug Induced Liver Injury Network . Features and outcomes of 899 patients with drug‐induced liver injury: the DILIN prospective study. Gastroenterology 2015;148:1340‐1352.e1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lammert C, McKinnon EJ, Chalasani N, Phillips EJ. Novel HLA class I alleles outside the extended DR3 haplotype are protective against autoimmune hepatitis. Clin Transl Gastroenterol 2019;10:e00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, et al. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest 2005;115:3428‐3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li QZ, Zhou J, Wandstrat AE, Carr‐Johnson F, Branch V, Karp DR, et al. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin Exp Immunol 2007;147:60‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li QZ, Zhou J, Lian Y, Zhang B, Branch VK, Carr‐Johnson F, et al. Interferon signature gene expression is correlated with autoantibody profiles in patients with incomplete lupus syndromes. Clin Exp Immunol 2010;159:281‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li QZ, Karp DR, Quan J, Branch VK, Zhou J, Lian Y, et al. Risk factors for ANA positivity in healthy persons. Arthritis Res Ther 2011;13:R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bell LN, Vuppalanchi R, Watkins PB, Bonkovsky HL, Serrano J, Fontana RJ, et al.; US Drug‐Induced Liver Injury Network (DILIN) Research Group . Serum proteomic profiling in patients with drug‐induced liver injury. Aliment Pharmacol Ther 2012;35:600‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Russo MW, Steuerwald N, Norton HJ, Anderson WE, Foureau D, Chalasani N, et al. Profiles of miRNAs in serum in severe acute drug induced liver injury and their prognostic significance. Liver Int 2017;37:757‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bonkovsky HL, Barnhart HX, Foureau DM, Steuerwald N, Lee WM, Gu J, et al. US Drug‐Induced Liver Injury Network and the Acute Liver Failure Study Group . Correction: Cytokine profiles in acute liver injury‐results from the US Drug‐Induced Liver Injury Network (DILIN) and the Acute Liver Failure Study Group. PLoS One 2019;14:e0212394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Church RJ, Kullak‐Ublick GA, Aubrecht J, Bonkovsky HL, Chalasani N, Fontana RJ, et al. Candidate biomarkers for the diagnosis and prognosis of drug‐induced liver injury: an international collaborative effort. Hepatology 2019;69:760‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu H, Luo H, Yan M, Zuo X, Li QZ. Autoantigen microarray for high‐throughput autoantibody profiling in systemic lupus erythematosus. Genomics Proteomics Bioinformatics 2015;13:210‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bjornsson E, Talwalkar J, Treeprasertsuk S, Kamath PS, Takahashi N, Sanderson S, et al. Drug‐induced autoimmune hepatitis: clinical characteristics and prognosis. Hepatology 2010;51:2040‐2048. [DOI] [PubMed] [Google Scholar]
- 24. Foureau DM, Walling TL, Maddukuri V, Anderson W, Culbreath K, Kleiner DE, et al. Comparative analysis of portal hepatic infiltrating leucocytes in acute drug‐induced liver injury, idiopathic autoimmune and viral hepatitis. Clin Exp Immunol 2015;180:40‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Himoto T, Murota M, Yoneyama H, Deguchi A, Kurokochi K, Senda S, et al. Clinical characteristics of patients with autoimmune hepatitis seropositive for anticentromere antibody. Hepatol Res 2010;40:786‐792. [DOI] [PubMed] [Google Scholar]
- 26. Muratori P, Efe C, Muratori L, Ozaslan E, Schiano T, Yoshida EM, et al. Clinical implications of antimitochondrial antibody seropositivity in autoimmune hepatitis: a multicentre study. Eur J Gastroenterol Hepatol 2017;29:777‐780. [DOI] [PubMed] [Google Scholar]
- 27. Li L, Chen M, Huang DY, Nishioka M. Frequency and significance of antibodies to chromatin in autoimmune hepatitis type I. J Gastroenterol Hepatol 2000;15:1176‐1182. [DOI] [PubMed] [Google Scholar]
- 28. Yokokawa J, Kanno Y, Abe K, Saito H, Monoe K, Katsushima F, et al. Anti‐nucleosome autoantibodies as markers for autoimmune hepatitis and their correlation with disease activity. Hepatol Res 2014;44:420‐428. [DOI] [PubMed] [Google Scholar]
- 29. Parfieniuk‐Kowerda A, Swiderska M, Szulzyk T, Jaroszewicz J, Lapinski TW, Flisiak R. Serum concentrations of Th17‐associated interleukins and autoimmune phenomena are associated with the degree of liver damage in alcoholic liver disease. J Gastrointestin Liver Dis 2017;26:269‐274. [DOI] [PubMed] [Google Scholar]
- 30. Łapiński TW, Rogalska‐Płońska M, Parfieniuk‐Kowerda A, Świderska M, Flisiak R. The occurrence of autoantibodies in patients with chronic HCV infection, including patients dialyzed and after kidney transplantation. Clin Exp Hepatol 2016;2:161‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stephens C, Castiella A, Gomez‐Moreno EM, Otazua P, López‐Nevot M, Zapata E, et al. Autoantibody presentation in drug‐induced liver injury and idiopathic autoimmune hepatitis: the influence of human leucocyte antigen alleles. Pharmacogenet Genomics 2016;26:414‐422. [DOI] [PubMed] [Google Scholar]
- 32. van Heemst J, van der Woude D, Huizinga TW, Toes RE. HLA and rheumatoid arthritis: how do they connect? Ann Med 2014;46:304‐310. [DOI] [PubMed] [Google Scholar]
- 33. Liu X, Jiang X, Liu R, Wang L, Qian T, Zheng Y, et al. B cells expressing CD11b effectively inhibit CD4+ T‐cell responses and ameliorate experimental autoimmune hepatitis in mice. Hepatology 2015;62:1563‐1575. [DOI] [PubMed] [Google Scholar]
- 34. Beland K, Marceau G, Labardy A, Bourbonnais S, Alvarez F. Depletion of B cells induces remission of autoimmune hepatitis in mice through reduced antigen presentation and help to T cells. Hepatology 2015;62:1511‐1523. [DOI] [PubMed] [Google Scholar]
- 35. Zgair AK. Involvement of (IgG and IgM)‐secreting B lymphocytes in severity of autoimmune hepatitis type 1. Med Microbiol Immunol 2013;202:229‐237. [DOI] [PubMed] [Google Scholar]
- 36. Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol 2007;94:157‐214. [DOI] [PubMed] [Google Scholar]
- 37. Diamant E, Melamed D. Class switch recombination in B lymphopoiesis: a potential pathway for B cell autoimmunity. Autoimmun Rev 2004;3:464‐469. [DOI] [PubMed] [Google Scholar]
- 38. Kattah NH, Kattah MG, Utz PJ. The U1‐snRNP complex: structural properties relating to autoimmune pathogenesis in rheumatic diseases. Immunol Rev 2010;233:126‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gourh P, Safran SA, Alexander T, Boyden SE, Morgan ND, Shah AA, et al. HLA and autoantibodies define scleroderma subtypes and risk in African and European Americans and suggest a role for molecular mimicry. Proc Natl Acad Sci U S A 2020;117:552‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fang Q, Ou J, Nandakumar KS. Autoantibodies as diagnostic markers and mediator of joint inflammation in arthritis. Mediators Inflamm 2019;2019:6363086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material