

Abstract
Mutations in the tumor suppressor gene TP53 are detected in 5–10% of patients with acute myeloid leukemia (AML) and myelodysplastic syndromes. TP53 mutations have been associated with complex karyotypes, therapy-related malignancies, lower response rates to cytotoxic chemotherapy, and an overall adverse prognosis. In this single-center retrospective study, we analyzed the clinicopathologic characteristics and outcomes of 83 patients with TP53-mutated myeloid malignancies treated at Yale Cancer Center between 9/2015 and 5/2019. Complex karyotypes (n=75; 90%) and therapy-related malignancies (n=32; 39%) were common. Median overall survival (OS) was 7.6 months. Intensive chemotherapy did not improve OS compared to lower-intensity treatment for AML patients. Patients who underwent allogeneic hematopoietic stem cell transplant (alloHSCT) had a significantly longer median OS, despite relatively limited follow-up. In conclusion, our data confirm the limited efficacy of intensive chemotherapy approaches for TP53-mutated patients with myeloid neoplasms and suggest that a minority of patients achieve long-term survival with alloHSCT.
Keywords: acute myeloid leukemia, AML, TP53, myelodysplastic syndrome, MDS, stem cell transplant
Introduction:
Acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) are clonal bone marrow disorders characterized by the expansion of immature myeloid precursor cells leading to bone marrow failure. Over the last decade our understanding of the underlying genomic alterations of both AML and MDS has significantly improved and results of genetic testing have been incorporated into the diagnostic workup, treatment selection, and prognostication of patients.(1–4)
The TP53 protein is encoded by the TP53 gene on the short arm of chromosome 17 and mutations in this gene are found in about 50% of human cancers.(5) In its wild-type form TP53 functions as a tumor suppressor protein that triggers cell cycle arrest and apoptosis in the setting of cellular stress such as DNA damage and oncogene activation.(5, 6) TP53 mutations have been associated with adverse outcomes and higher rates of resistance to standard treatments in AML and MDS.(2, 7–10) While these mutations occur in around 10% of patients with de novo AML, they are often associated with complex karyotypes and therapy-related (t)-myeloid neoplasms.(7, 11–17) The precise mechanism for the enrichment of TP53 mutations in patients who received prior cytotoxic therapies and the clonal evolution of TP53-mutated myeloid neoplasms are still incompletely understood. Prior studies have suggested that the inherent chemotherapy resistance of preexisting TP53-mutated clones leads to their expansion in the setting of cytotoxic treatments and contributes to their poor prognosis and treatment response.(12)
Previous studies showed poor outcomes with both intensive chemotherapy and lower-intensity therapies (e.g. hypomethylating agents [HMA]), with increased relapse risk and very low cure rates even after allogeneic hematopoietic stem cell transplant (alloHSCT), leaving the question of optimal treatment unanswered.(10, 18) Given the poor prognosis of this patient population, a better understanding of the clinical and molecular characteristics is needed to derive novel and more effective treatments. In this retrospective cohort study, we describe the clinical, cytogenetic, and molecular characteristics of AML and MDS patients with TP53 mutations and analyze their response to various treatment modalities and overall outcomes.
Methods:
Patient and treatment characteristics:
We conducted a retrospective review of all adult patients with myeloid neoplasms and known pathogenic TP53 mutations detected on next generation sequencing panel testing who were treated at Yale Cancer Center from 9/1/2015 (the date at which we started performing targeted next generation sequencing including the TP53 gene routinely on patients with myeloid malignancies) to 5/31/2019. Last day of follow up was 7/4/2019. We collected data on age, sex, ethnicity, prior malignancies and their treatments, total white blood cell (WBC), hemoglobin, platelet count, disease risk as determined by the treating physician, initial and subsequent lines of therapies, and use of alloHSCT. Responses were recorded as documented by the treating physician in the electronic medical record using modified International Working Group (IWG) criteria 2003 for AML and 2006 for MDS.(19, 20) Lower-intensity treatment (LIT) was defined as azacitidine, decitabine, low-dose cytarabine alone or in combination with other agents. The study protocol was approved by the Institutional Review Board at Yale University.
Molecular analysis:
Next generation sequencing (NGS) of either blood or bone marrow aspirate samples was performed to identify TP53 variants. NGS was performed on the Ion Torrent S5 system (Thermo Fisher Scientific, Waltham, MA, USA) using a custom AmpliSeq 25 gene (9/2015 to 11/2017; the following genes were analyzed: ASXL, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3 [partial sequencing of gene regions with known mutations], HRAS, IDH1 [partial sequencing of gene regions with known mutations], IDH2 [partial sequencing of gene regions with known mutations], JAK2, KIT, KRAS, MLL, MPL, NPM1 [partial sequencing of gene regions with known mutations], NRAS, PHF6, RUNX1, SF3B1, SRSF2, TET2, TP53, WT1) or 49 gene panel (11/2017 to present; included genes: ABL1, ALK, ASXL1, ATRX, BCOR, BCORL1, BRAF [partial sequencing of gene regions with known mutations], BRCC3, CALR [partial sequencing of gene regions with known mutations], CBL, CEBPA, CSF3R, DNMT3A, EED, EP300, ETV6, EZH2, FLT3 [partial sequencing of gene regions with known mutations], GATA1, GATA2, IDH1 [partial sequencing of gene regions with known mutations], IDH2 [partial sequencing of gene regions with known mutations], JAK2, KIT, KRAS, MPL, MYC, NF1, NPM1 [partial sequencing of gene regions with known mutations], NRAS, PDGFRA, PDS5B, PHF6 PRPF8, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG1, STAG2, TET2, TP53, U2AF1, WT1, ZRSR2). Genomic DNA was extracted from the patient samples, quantified, and amplified using multiplex PCR. All coding exons and adjacent splicesites of the TP53 gene were sequenced. Sequence analysis (including alignment, hg19 human reference mapping, and variant calling) was performed using Torrent Suite Software and annotation was performed with Ion Reporter Software (Thermo Fisher Scientific). TP53 variants were only included in the study if they were classified as pathogenic or likely pathogenic using various database sources including Cosmic (https://cancer.sanger.ac.uk/cosmic/), dBSNP (https://www.ncbi.nlm.nih.gov/snp/), Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), and the TP53 database (http://p53.iarc.fr/).
Statistical analysis:
Student’s t-test and Fisher’s exact tests were performed to compare baseline characteristics between AML and MDS patients for continuous and categorical variables, respectively. The same tests were employed to compare the baseline characteristics of AML patients treated with intensive chemotherapy or low-intensity treatment as well as between alloHSCT recipients and non-alloHSCT patients. Fisher’s exact test was used to compare the rates of complex karyotype in the TP53-mutated and TP53-wild type patient population. Overall survival (OS) was defined from the time of diagnosis. Patients were censored at the time of last follow up or the end of the study period (7/4/2019) whichever was earlier. OS was measured using Kaplan Meier methods. OS among AML patients treated with induction chemotherapy vs low-intensity treatment and for patients who underwent alloHSCT vs those who did not were compared using Cox proportional hazard models, which were stratified for variables with statistically significant differences between the groups to be compared. For all analyses, p-values <0.05 were considered to be statistically significant. All analyses were conducted using Stata 14 (Stata Inc., College Station, TX, USA).
Results:
Patient characteristics
We identified 83 patients with TP53-mutated myeloid neoplasms (45 AML, 31 MDS, four chronic myelomonocytic leukemia, and three JAK2 V617F mutation-positive myeloproliferative neoplasms [JAK2-positive MPN]). Median age at diagnosis was 69 years (range [R], 27–88 years), 52% were female, and 88% were Caucasian. Prior malignancy was noted in 35 patients (42%) and 32 (18 AML, 14 MDS) patients were classified as therapy-related myeloid neoplasms. Median WBC on presentation was 3 × 109 /L (R, 0.2 – 74×109/L) and 38 patients (45.8%) had circulating blasts in the peripheral blood with a median of 0% peripheral blasts (R, 0–43%). At diagnosis, complex (n=75; 90%) and monosomal karyotypes (n=54; 65%) were highly prevalent. Among MDS patients, high and very high disease risk by IPSS-R (low risk: 3 patients [9.7%], intermediate risk: 2 [6.5%], high risk: 5 [16.1%], very high risk: 21 [67.7%]) and intermediate-2 and high-risk by IPSS were common (low risk: 1 [3.2%], intermediate-1 risk: 7 [22.6%], intermediate-2 risk: 16 [51.6%], high risk: 7 [22.6%]). Further patient characteristics are shown in Table 1.
Table 1:
Patient characteristics, treatments and outcome
| Characteristics | All patients (Median or N; [range or %]) | AML (median or N; [range or %]) | MDS (median or N; [range or %]) | P-value |
|---|---|---|---|---|
| Median age (years) | 69 [27–88] | 68 [27–88] | 71 [46–85] | 0.48 |
| Sex | ||||
| Female | 43 [51.8%] | 20 [44.4%] | 17 [54.8%] | |
| Male | 40 [48.2%] | 25 [55.6%] | 14 [45.2%] | 0.37 |
| Race | ||||
| White/Caucasian | 73 [88.0%] | 42 [93.3%] | 25 [80.7%] | |
| Asian | 1 [1.2%] | 1 [2.2%] | 0 [0%] | |
| African American | 3 [3.6%] | 0 [0%] | 3 [9.7%] | |
| Hispanic | 6 [7.2%] | 2 [4.4%] | 3 [9.7%] | 0.07 |
| Prior malignancy | 35 [42.2%] | 19 [42.2%] | 15 [48.4%] | 0.60 |
| Disease classification | ||||
| AML | 45 [54.2%] | |||
| De novo | 8 [9.6%] | |||
| Therapy-related | 18 [20.5%] | |||
| AML-MRC | 17 [21.7%] | |||
| Prior MPN | 3 [3.6%] | |||
| MDS | 31 [37.3%] | |||
| MDS-EB1 | 5 [6.0%] | |||
| MDS-EB2 | 6 [7.2%] | |||
| t-MDS | 14 [16.9%] | |||
| other MDS subtypes | 7 [8.4%] | |||
| CMML | 4 [4.8%] | |||
| CMML-1 | 2 [2.4%] | |||
| CMML-2 | 2 [2.4%] | |||
| JAK2-positive MPN | 3 [3.6%] | |||
| Disease characteristics at presentation | ||||
| WBC (x109/L) | 3 [0.2–73.9] | 2.5 [0.2–73.9] | 3.2 [1.6–26.3] | 0.61 |
| peripheral blast % | 0 [0–43] | 4 [0–43] | 0 [0–18] | 0.0009 |
| Platelet | 43.5 [1–642] | 33 [4–317] | 56 [1–368] | 0.40 |
| Hgb (g/dL) | 8.4 [6.2–13.4] | 8.0 [6.2–11.5] | 8.6 [6.4–12.6] | 0.006 |
| bone marrow cellularity (%) | 70 [10–95] | 70 [20–95] | 70 [10–90] | 0.16 |
| bone marrow blasts (%) | 16 [1–90] | 30 [2–90] | 7 [1–17] | <0.0001 |
| Initial treatment | ||||
| Induction chemotherapy | 20 [22.3%] | 20 [45.0%] | 0 [0%] | |
| Low-intensity (HMA-based regimens, LDAC) | 35 [42.2%] | 14 [31.1%] | 19 [61.3%] | |
| hydroxyurea | 6 [7.2%] | 4 [8.9%] | 0 [0%] | |
| targeted therapy | 3 [3.6%] | 1 [2.2%] | 2 [6.5%] | |
| BSC | 13 [15.7%] | 5 [11.1%] | 6 [19.4%] | |
| Others (ruxolitinib, lenalidomide) | 7 [8.4%] | 1 [2.2%] | 4 [12.9%] | <0.0001 |
| Rate of CR/CRi with initial treatment | ||||
| CR with IC | 9 [45.0%] | 9 [45.0%] | N/A | |
| CR/CRi with LIT | 9 [28.1%] | 2 [14.3%] | 6 [31.6%] | |
| Allogeneic hematopoietic stem cell transplant (alloHSCT) | ||||
| Received alloHSCT | 11 [13.3%] | 7 [15.6%] | 4 [12.9%] | |
| Relapse after alloHSCT | 3 [27.7%] | 2 [28.6%] | 1 [25.0%] | |
| Median OS among | Not reached after median follow up of 12.9 months | |||
| alloHSCT recipients | ||||
| Outcome | ||||
| deceased | 59 [71.1%] | 34 [75.6%] | 21 [67.7%] | |
| alive at last follow up | 21 [25.3%] | 10 [22.2%] | 8 [25.8%] | |
| lost to follow up | 3 [3.6%] | 1 [2.2%] | 2 [6.5%] | |
| 1-year OS rate | 30.5% [95% CI: 20.0–41.6%] | |||
| Median OS (months) | 7.6 months [95% CI: 5.7–10.0 months] | 6.7 months [95% CI: 1.9–9.4 months] | 10.0 months [95% CI: 5.7 −12.9 months] |
Molecular analysis:
We identified 101 unique, pathogenic or likely pathogenic mutations in the TP53 gene with 17 patients harboring more than one pathogenic variant (16 patients with two variants, one patient with four variants). Missense variants were most commonly encountered (n=82 variants, 81%) followed by splicesite (n=9, 9%), frameshift (n=6, 6%), and nonsense variants (n=4, 4%). Median variant allele frequency (VAF) at initial genetic testing was 32.4% (R: 3.6–94.6). We detected a wide variety of variants with TP53 c.844C>T being the most frequently detected genetic abnormality (five patients, 7%). Ninety-four percent of the pathogenic variants detected affected the DNA binding site. Twenty-four patients (28.9%) had deletions of chromosome 17 detected on karyotype analysis.
Fifty-two patients had isolated somatic TP53 variants (63%). Among the other 31 patients DNMT3A (n=8 patients), JAK2 (n=6), and TET2 (n=5) variants were the most common somatic mutations. Of note, targetable driver mutations such as FLT3 (n=0), IDH1 (n=3), and IDH2 (n=1) were very rare. Notably, patients with complex karyotype were more likely to have isolated TP53 variants compared to non-complex karyotype patients (complex karyotype + isolated TP53 variants: 51 patients [68.0%] vs non-complex karyotype + isolated TP53 variants: 2 patients [25.0%]; Fisher’s exact test: p=0.024) [Table 2].
Table 2:
Cytogenetic characteristics at time of diagnosis
| Characteristics | Median or N | Range or % |
|---|---|---|
| Karyotype | ||
| complex | 75 | 90.4% |
| monosomal | 54 | 65.1% |
| 17p deletion | 24 | 28.9% |
| Characteristics of TP53 variants | ||
| patients with multiple TP53 abnormalities | 17 | 20.5% |
| 4 TP53 variants | 1 | 1.2% |
| 2 TP53 variants | 16 | 19.3% |
| missense variant | 82 patients (56 unique variants) | |
| splicesite variant | 9 patients (7 unique variants) | |
| frameshift variant | 6 patients (6 unique variants) | |
| nonsense variant | 4 patients (3 unique variants) | |
| Concurrent variants | ||
| none | 53 | 62.7% |
| TP53 + 1 additional variant | 18 | 21.7% |
| TP53 + 2 variants | 9 | 10.8% |
| TP53 + ≥3 variants | 3 | 3.6% |
| Specific concurrent variants | ||
| DNMT3A | 8 | 9.6% |
| JAK2 | 6 | 7.2% |
| TET2 | 5 | 6.0% |
| U2AF1 | 4 | 4.8% |
| NRAS | 4 | 4.8% |
| EZH2 | 3 | 3.6% |
| IDH1 | 3 | 3.6% |
| PTPN11 | 2 | 2.4% |
| KIT | 2 | 2.4% |
| One patient each with mutations in IDH2, NPM1, ZRSR2, CBL, SF3B1, PHF6, EP300, NF1, ETV6, SRSF2, CALR | 1 | 1.2% |
| Association of complex karyotype and presence or absence of concurrent variants | ||
| complex karyotype without other variants | 51 | 68.0% |
| complex karyotype with other variants | 24 | 32.0% |
| non-complex karyotype without other variants | 2 | 25.0% |
| non-complex karyotype with other variants | 6 | 75.0% |
Repeated genetic testing was available in 29 out of 83 patients (34.9%). As indications and timing for repeated assessment varied from patient to patient, we were unable to assess systematically whether changes in TP53 VAF or the emergence of new concurrent mutations was associated with disease relapse or progression. Ten patients cleared the initially detected TP53 variants (seven AML, two MDS, one JAK2-positive MPN patient) during follow up testing with induction chemotherapy, decitabine, alloHSCT, or ruxolitinib being the most recent form of treatment in two, three, four, and one case, respectively. However, even in these ten patients with a molecular response, four patients relapsed, and three patients died. At a median duration of follow-up of 16.4 months median relapse-free and overall survival for patients who had cleared the initial TP53 variant had not been reached. In all three patients with repeated genetic testing available at the time of relapse, either re-emergence of the initial TP53 variant that had become undetectable with treatment or an increase in VAF were noted. Interestingly, two patients had a new FLT3 and KRAS mutation at the time of relapse, respectively, while no new concurrent mutations were detected in the other patient. In one AML patient, donor-lymphocyte infusion was able to induce a second cytogenetic complete remission (CR).
Treatment pattern and survival analyses
Median follow up of the entire cohort was 6.4 months (R: 0.2–55.3 months). Median OS in the combined study population was 7.6 months (95% CI: 5.7–10.0 months) with a 1-year and 2-year OS rate of 22.6% (95% CI: 14.2–32.2%) and 7.5% (95% CI: 3.1%−14.6%), respectively. In the total patient population, 20 patients (24.1%) were treated with intensive chemotherapy, 35 patients (42.2%) received low-intensity treatment, while 19 patients (22.9%) received best supportive care or hydroxyurea only as their initial treatment. Of note, CPX-351 and venetoclax in combination with AZA (six patients) or low-dose cytarabine (one patient) were used in nine (10.8%) and seven (8.4%) patients, respectively. Fourteen patients (16.9%) were treated on clinical trials in the frontline setting.
In univariate analyses among AML patients, patients with de novo AML were more likely to be treated with induction chemotherapy, while patients with prior malignancy and secondary AML were more likely to receive low-intensity treatment. In Cox proportional hazard models stratifying for those covariates, induction chemotherapy did not improve OS compared to patients who received low-intensity treatment (hazard ratio: 0.63 [95% CI: 0.2–2.1]; median OS 8.8 months [95% CI: 1.9–15.5] vs 9.4 months [95% CI: 1.8–13.6] and 1-year OS rate 25.0% [95% CI: 9.1%−44.9%] vs 14.3% [95% CI: 2.3%−36.6%]; p=0.46). Rates of CR were 45.0% (nine out of 20 patients) and 14.3% (two out of 14 patients) for patients treated with induction chemotherapy and low-intensity therapy, respectively.
None of the MDS patients received induction chemotherapy as frontline treatment. Among the 19 MDS patients who received HMA-based therapies, the median OS was 12.1 months (95% CI: 5.8 months – not reached) with six patients achieving CR and one patient achieving CR with incomplete cell count recovery. Median OS for all 26 HR-MDS patients (defined as IPSS score ≥1.5 points or IPSS-R >4.5 points) was 6 months (95% CI: 4.8–12.1 months) and 9.6 months (95% CI: 4.8 months – not reached) among HR-MDS patients treated with HMA. Response rates and outcome for AML and MDS patients by treatment regimen are shown in Table 3.
Table 3:
Treatment regimens and outcomes
| Disease | ORR (%) | CR/CRi for AML (%) CR/mCR for MDS (%) |
Median OS (mos, 95% CI) |
|---|---|---|---|
| AML | |||
| Induction chemotherapy (n=20 pts) | 9 (45.0%) | 9 (45.0%) | 8.8 (1.9–15.5) |
| LIT (HMA-based regimens, LDAC) [n=14 pts] | 2 (14.3%) | 2 (14.3%) | 9.4 (1.8–13.6) |
| MDS | |||
| HMA-based regimens [n=19 pts] | 7 (36.8%) | 6 (31.6%) | 12.1 (5.8-not reached) |
Median and 1-year OS among AML patients were 6.7 months (95% CI: 1.9–9.4) and 16% (95% CI: 7.0%−28.2%), respectively. Among MDS patients median OS and 1-year OS were 10 months (95% CI 5.7–12.9) and 31.1% (95% CI 15.6–48.0%), respectively. Figure 1 shows the survival curves for the overall population of AML and MDS patients as well as for AML patients stratified by initial treatment. Patients who were not actively treated (hydroxyurea or best supportive care only) had a dismal prognosis with a median OS of 0.8 months (95% CI: 0.3–2.2 months).
Figure 1: Kaplan-Meier survival estimates by diagnosis and treatment.
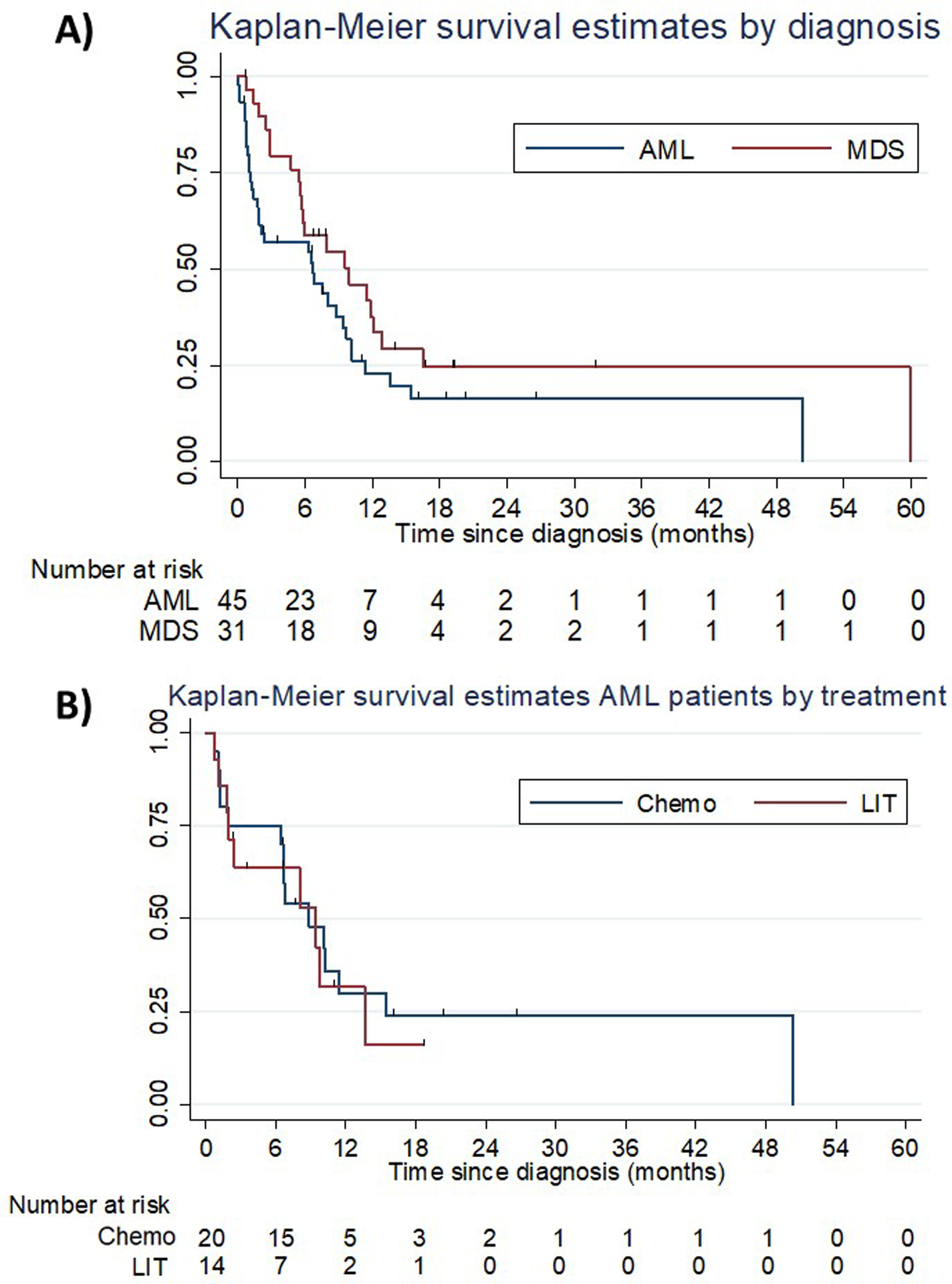
(A) illustrates Kaplan-Meier survival estimates for AML and MDS patients. Median and 1-year OS among AML patients were 6.7 months (95% CI: 1.9–9.4) and 16% (95% CI: 7.0%−28.2%), respectively. Among MDS patients median OS and 1-year OS were 10 months (95% CI 5.7–12.9) and 31.1% (95% CI 15.6–48.0%), respectively. (B) shows Kaplan-Meier survival curves for AML patients who received intensive chemotherapy and low-intensity therapy as frontline treatment. There was no statistically significant difference between the two treatment modalities (hazard ratio: 0.63 [95% CI: 0.2–2.1]; p=0.46; induction chemotherapy: median OS 8.8 months [95% CI: 1.9–15.5] and 1-year OS rate 25.0% [95% CI: 9.1%−44.9%]; low-intensity therapy: median OS: 9.4 months [95% CI: 1.8–13.6]; 1-year OS: 14.3% [95% CI: 2.3%−36.6%]).
Subgroup analysis of patients proceeding to alloHSCT
Notably, among the 11 patients who proceeded to alloHSCT, only three patients relapsed after a median of 8.4 months following alloHSCT. All patients were in CR at the time of transplant with five patients receiving myeloablative and six patients receiving a reduced-intensity conditioning (RIC) regimen, respectively. All patients except for two patients got transplanted after achieving CR with initial treatment with either induction chemotherapy (five patients) or HMA (four patients). The other two patients had residual disease after induction chemotherapy but achieved CR with subsequent HMA treatment. All except for one patient were transplanted about six months after starting initial treatment. Three out of six patients treated with RIC and zero out of five with myeloablative conditioning regimens relapsed. HSCT recipients were more likely to achieve CR with initial treatment (CR/CRi vs non-CR/CRi; p<0.001), to have received intensive chemotherapy (intensive chemotherapy vs other; p=0.003), and to be younger (<65 years vs ≥65 years of age; p=0.04) compared to patients who did not proceed to HSCT. In a Cox proportional hazard model stratified by age (<65 years vs ≥65 years of age), initial treatment (intensive chemotherapy vs other) and response category (CR/CRi vs other), there was a statistically significant difference in the median OS for patients who underwent alloHSCT compared to those who did not (hazard ratio: 0.08; median OS: not reached [95% CI: 6.6 months – not reached] vs 6 months [95% CI: 2.9 – 8.8 months]; p = 0.002). One patient with FLT3-mutated AML received midostaurin maintenance therapy following alloHSCT. No other patients received any maintenance treatment post-alloHSCT. Kaplan-Meier survival curves are shown in Figure 2. The median duration of follow up among HSCT patients was 12.9 months. Of note, two HSCT patients were alive and relapse-free at 50.3 months and 31.9 months after diagnosis, respectively.
Figure 2: Kaplan-Meier survival estimates by transplant recipient status.
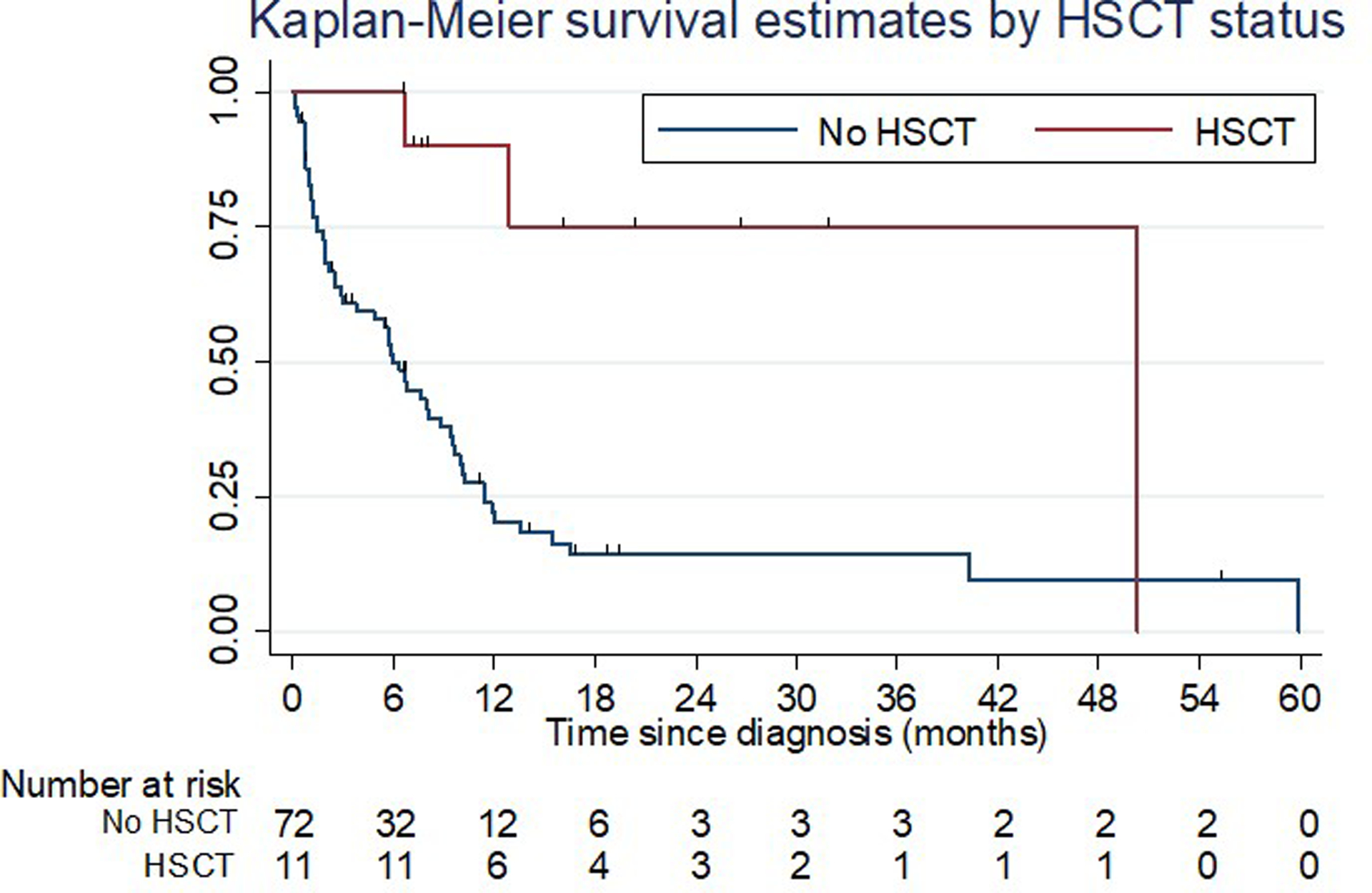
Figure 2 shows Kaplan-Meier survival curves for patients who proceeded to alloHSCT (n=11 patients) and those who did not (n=72 patients). In a stratified Cox proportional hazards model, patients who received alloHSCT had a significant survival benefit compared to non-transplant patients (median OS: not reached [95% CI: 6.6 months – not reached] vs 6 months [95% CI: 2.9 −8.8 months]; hazard ratio: 0.08; p = 0.002)
Discussion:
In this single-center, retrospective study we confirmed the dismal prognosis of patients with TP53-mutated myeloid neoplasms and its association with both complex karyotype and therapy-related disease. While this has been shown previously,(11, 14) little is known about concurrent mutations at both diagnosis and along the disease course. In our study we showed that the majority of patients had isolated TP53 mutations and that other classic AML driver mutations such as FLT3 or NPM1 were rare at the time of diagnosis. The lower frequency of NPM1 and FLT3 mutations in therapy-related MDS and AML and higher rates of mutations in RAS-BRAF signaling pathways have been noted previously and suggest an alternative pathway of leukemogenesis.(12, 21–23) Interestingly, in two out of three patients with genetic testing performed at the time of disease relapse, new FLT3 and KRAS mutations were detected. We can only speculate if these new mutations may have driven disease relapse independently of the TP53 mutations, as both patients also had re-emergence of the initially present TP53 clone that had disappeared with treatment. Further studies on the interaction of various mutations along the disease course are needed.
Previous studies have suggested that both the size of the clonal population and the specific type of TP53 mutations affect outcomes and treatment responses.(24) Recent data from MDS patients have also shown that the prognostic significance of TP53 mutations is different depending on whether it is found in the setting of a complex vs a non-complex karyotype and whether it is present as a monoallelic compared to a biallelic abnormality.(25) In our study the majority of TP53 mutations were missense variants affecting the DNA-binding domain. Preclinical studies have shown that these missense mutations affecting the DNA binding site exert a dominant-negative effect that confers a selective advantage for hematopoietic cells under conditions of cellular stress.(26, 27) Given the small sample size, we were unable to assess whether particular TP53 variants had an impact on treatment response and outcome in our cohort. Larger studies are needed to identify subgroups based on the location and biological effect of a particular mutation to allow for targeted therapies of these separate functional variants.(5, 18, 24) Recently, novel agents that target mutant TP53 have been successfully tested in hematologic and solid malignancies.(28) APR-246 is a small molecule that induces apoptosis in TP53-mutated cancer cells as a single agent as well as in combination with azacitidine.(28, 29) In a cohort of 12 patients with refractory myeloid malignancies (3 AML-MRC and 9 MDS patients), 11 out of 11 evaluable patients treated with APR-246 and azacitidine achieved a response with nine CRs and a median OS and progression-free survival that have not been reached at seven months of follow up.(29) While highly promising, these results need to be confirmed in larger trials which are ongoing (NCT03072043, NCT03588078).
There is controversial data on the impact of the size of the clonal population harboring TP53 mutations on OS. It is unclear whether the mere presence of a TP53 mutant clone is sufficient to confer an adverse prognosis, or if a certain VAF is required.(30, 31) In MDS patients, TP53 mutations have also been found to be an adverse prognostic marker independent of IPSS and IPSS-R.(32, 33) Our study was limited by its small sample size and heterogeneity precluding assessment of a correlation between VAF and OS.
With a median OS of only 7.6 months for the entire study population, our study is well in line with prior reports.(2, 8, 9, 11) While TP53 mutations have been linked to higher rates of resistance to cytotoxic chemotherapy (10) and only about 50% of patients achieved CR with induction chemotherapy in our study, patients who were not actively treated fared significantly worse. A prior study of decitabine extended to the 10-day schedule reported impressive response rates in AML patients with TP53 mutations.(34) However, none of the responding patients cleared all leukemia-specific mutations, which led to eventual disease relapse likely due to the expansion of a decitabine-resistant subclone.(34) Furthermore, these results have yet to be replicated by other studies.(34–36) While analysis is limited by the fact that only eight patients (4 AML and 4 MDS) received decitabine in our study, one AML patient and three MDS patients achieved a CR. Furthermore, we could show that intensive chemotherapy did not lead to a survival benefit in AML patients compared to lower intensity treatment.
The role for alloHSCT in TP53-mutated myeloid neoplasms is controversial given the high rate of disease relapse and the significant procedure-related morbidity and mortality.(37–39) However, we did observe a significant survival benefit for patients proceeding to alloHSCT with two patients being alive and relapse-free at 50.3 months and 31.9 months after diagnosis, respectively. While the duration of follow up for most of the alloHSCT patients was short and extended follow up is necessary, our findings support the consideration of alloHSCT (ideally in first CR) for eligible patients given that disease relapse is common and alloHSCT is the only potentially curative therapeutic option.(18, 34, 37) All of our patients were transplanted in CR and the majority of those patients achieved CR with the initial treatment. Assessment of TP53 mutational status may also have a role in the selection of conditioning regimens and additional studies to identify patient subsets who are most likely to benefit from alloHSCT are needed given the high rate of relapse and non-relapsed mortality.(37, 39–43) Prior studies have suggested that the presence of a complex karyotype in MDS and secondary AML patients with TP53 mutations is associated with higher rates of relapse and poor survival (median OS 4.8 months; 2-year mortality >80%), while patients without complex karyotype had a better prognosis (73% 5-year OS [95% CI: 51–100%]).(41, 44) The impact of the TP53 VAF at time of transplant, karyotype abnormalities (e.g. del(5q)) and the presence of co-mutations (e.g. JAK2 or RAS pathway) could also be used as a prognostic marker although data are controversial.(39, 44, 45) While validation in larger datasets is necessary, our data suggest that alloHSCT can be a viable option in selected patients. Identifying biomarkers predicting outcomes after alloHSCT remains a very important research question.
Emerging data from the RELAZA-2 trial suggested a survival benefit for minimal residual disease (MRD)-guided preemptive treatment with azacitidine for up to 24 cycles after intensive chemotherapy or allo-HSCT.(46) Repeated genetic testing after alloHSCT can be a prognostic factor and failure to clear mutations prior to transplant has been demonstrated to negatively impact survival in MDS patients.(39, 43) While mutational analysis by next-generation sequencing is limited by its lower sensitivity compared to PCR- or flow cytometry-based techniques, it can potentially be used to assess MRD in the post-transplant setting.(47, 48) Despite the results of RELAZA-2, whether MRD-positivity should lead to pre-emptive treatment with HMA (+/− venetoclax) or immune checkpoint inhibitors remains controversial and is actively studied in multiple trials.
Furthermore, data from the QUAZAR-AML-001 trial of maintenance therapy with CC-486, an oral formulation of azacitidine, showed a survival benefit of 9.9 months survival benefit compared to placebo (CC-486: 24.7 months [95% CI, 18.7–30.5] vs 14.8 months for placebo [95% CI, 11.7–17.6]; p=0.0009) in AML patients ≥55 years in CR following IC and not deemed to be transplant candidates.(49) Additional trials with longer follow up are needed to further assess the role of HMA maintenance therapy in both non-transplant and alloHSCT patients.
Our study has several limitations. The small sample size in this single center retrospective cohort study precluded assessment of the outcomes and treatment effects of specific TP53 variants. Several analyses such as the correlation between VAF and outcomes and the IPSS-R risk group and OS should be re-evaluated in larger studies. Second, repeated molecular testing during the disease course and especially at the time of relapse or progression was available only in a small subset of patients. We were therefore unable to assess whether changes in the VAF or the appearance of new concurrent mutations drive disease relapse and progression. Third, factors such as patient comorbidities and preferences that may have influenced treatment decision-making were not available for analysis. Fourth, patients were included base on TP53 mutations detected on our NGS gene panel and patients who only had TP53 deletions (e.g. del17p) without TP53 mutations would have been missed. In prior studies, isolated TP53 deletions have been found in 1% of AML patients and associated with an especially poor prognosis.(9, 50) Finally, in the absence of paired samples (i.e. non-involved specimen [e.g. skin biopsy] and leukemic cells) we were unable to assess whether any of the detected TP53 mutations were germline in nature or if all patients had acquired somatic mutations. Nevertheless, our study adds to the growing body of evidence that patients with TP53-mutated myeloid neoplasms are a heterogenous population with an overall poor prognosis but the potential to benefit substantially from alloHSCT.
Conclusions:
In this retrospective case series, we confirm the poor prognosis for patients with TP53-mutated myeloid neoplasms. Median OS was only 7.6 months, and intensive chemotherapy did not appear to improve OS compared to LIT for AML patients. Although limited by the small sample size and the relatively short duration of follow up, patients who underwent alloHSCT had significantly longer median OS and alloHSCT should be considered for eligible patients. Novel therapies are urgently needed to improve outcomes of this patient population.
Acknowledgements:
A.M.Z. is a Leukemia and Lymphoma Society Scholar in Clinical Research and is also supported by a NCI’s Cancer Clinical Investigator Team Leadership Award (CCITLA). This research was in part supported by the National Cancer Institute of the National Institutes of Health under Award Number P30 CA016359. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Declaration of conflicts of interest:
S.D.G. has consulted for and receives research funding from Celgene; personal fees from Abbvie, Jazz Pharmaceuticals, Kyowa Hakko Kirin, Tolero Pharmaceuticals, and Daiichi Sankyo outside of submitted work. N.A.P. consulted for and received honoraria from Alexion, Pfizer, Agios Pharmaceuticals, Blueprint Medicines, Incyte, Novartis, Celgene, Bristol-Myers Squib and CTI biopharma. N.A.P. received research funding (all to the institution) from Boehringer Ingelheim, Astellas Pharma, Daiichi Sankyo, Sunesis Pharmaceuticals, Jazz Pharmaceuticals, Pfizer, Astex Pharmaceuticals, CTI biopharma, Celgene, Genentech, AI Therapeutics, Samus Therapeutics, Arog Pharmaceuticals and Kartos Therapeutics. A.M.Z. received research funding (institutional) from Celgene, Abbvie, Astex, Pfizer, Medimmune/AstraZeneca, Boehringer-Ingelheim, Trovagene, Incyte, Takeda, Novartis, Aprea, and ADC Therapeutics. A.M.Z had a consultancy with and received honoraria from AbbVie, Otsuka, Pfizer, Celgene, Jazz, Incyte, Agios, Boehringer-Ingelheim, Novartis, Acceleron, Astellas, Daiichi Sankyo, Cardinal Health, Taiho, Seattle Genetics, BeyondSpring, Trovagene, Takeda, Ionis, and Epizyme. A.M.Z received travel support for meetings from Pfizer, Novartis, and Trovagene. None of these relationships were related to the development of this manuscript. The other authors have no relevant conflicts of interest to declare.
References
- 1.Bullinger L, Döhner K, Döhner H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J Clin Oncol. 2017;35(9):934–46. [DOI] [PubMed] [Google Scholar]
- 2.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–27; quiz 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89–102. [DOI] [PubMed] [Google Scholar]
- 6.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137(3):413–31. [DOI] [PubMed] [Google Scholar]
- 7.Prokocimer M, Molchadsky A, Rotter V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood. 2017;130(6):699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Görlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686–98. [DOI] [PubMed] [Google Scholar]
- 9.Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31(3):705–11. [DOI] [PubMed] [Google Scholar]
- 10.Kadia TM, Jain P, Ravandi F, Garcia-Manero G, Andreef M, Takahashi K, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer. 2016;122(22):3484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–21. [DOI] [PubMed] [Google Scholar]
- 12.Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haase D, Stevenson KE, Neuberg D, Maciejewski JP, Nazha A, Sekeres MA, et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia. 2019;33(7):1747–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mrózek K, Eisfeld A-K, Kohlschmidt J, Carroll AJ, Walker CJ, Nicolet D, et al. Complex karyotype in de novo acute myeloid leukemia: typical and atypical subtypes differ molecularly and clinically. Leukemia. 2019;33(7):1620–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bewersdorf JP, Ardasheva A, Podoltsev NA, Singh A, Biancon G, Halene S, et al. From clonal hematopoiesis to myeloid leukemia and what happens in between: Will improved understanding lead to new therapeutic and preventive opportunities? Blood Rev. 2019:100587. [DOI] [PubMed] [Google Scholar]
- 16.Zeidan AM, Al Ali N, Barnard J, Padron E, Lancet JE, Sekeres MA, et al. Comparison of clinical outcomes and prognostic utility of risk stratification tools in patients with therapy-related vs de novo myelodysplastic syndromes: a report on behalf of the MDS Clinical Research Consortium. Leukemia. 2017;31(6):1391–7. [DOI] [PubMed] [Google Scholar]
- 17.Abou Zahr A, Kavi AM, Mukherjee S, Zeidan AM. Therapy-related myelodysplastic syndromes, or are they? Blood Rev. 2017;31(3):119–28. [DOI] [PubMed] [Google Scholar]
- 18.Leung GMK, Zhang C, Ng NKL, Yang N, Lam SSY, Au CH, et al. Distinct mutation spectrum, clinical outcome and therapeutic responses of typical complex/monosomy karyotype acute myeloid leukemia carrying TP53 mutations. Am J Hematol. 2019;94(6):650–7. [DOI] [PubMed] [Google Scholar]
- 19.Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108(2):419–25. [DOI] [PubMed] [Google Scholar]
- 20.Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21(24):4642–9. [DOI] [PubMed] [Google Scholar]
- 21.Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39(3):348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2008;22(2):240–8. [DOI] [PubMed] [Google Scholar]
- 23.Welch JS. Patterns of mutations in TP53 mutated AML. Best Pract Res Clin Haematol. 2018;31(4):379–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nature Reviews Clinical Oncology. 2017;15:13. [DOI] [PubMed] [Google Scholar]
- 25.Bernard E, Nannya Y, Yoshizato T, Hasserjian RP, Saiki R, Shiozawa Y, et al. TP53 State Dictates Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Blood. 2019;134(Supplement_1):675-. [Google Scholar]
- 26.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50(10):1381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boettcher S, Miller PG, Sharma R, McConkey M, Leventhal M, Krivtsov AV, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science. 2019;365(6453):599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lehmann S, Bykov VJ, Ali D, Andren O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. 2012;30(29):3633–9. [DOI] [PubMed] [Google Scholar]
- 29.Sallman DA, DeZern AE, Steensma DP, Sweet KL, Cluzeau T, Sekeres MA, et al. Phase 1b/2 Combination Study of APR-246 and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Acute Myeloid Leukemia (AML). Blood. 2018;132(Suppl 1):3091-. [Google Scholar]
- 30.Prochazka KT, Pregartner G, Rücker FG, Heitzer E, Pabst G, Wölfler A, et al. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Haematologica. 2019;104(3):516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sallman DA, Komrokji R, Vaupel C, Cluzeau T, Geyer SM, McGraw KL, et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia. 2016;30(3):666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belickova M, Vesela J, Jonasova A, Pejsova B, Votavova H, Merkerova MD, et al. TP53 mutation variant allele frequency is a potential predictor for clinical outcome of patients with lower-risk myelodysplastic syndromes. Oncotarget. 2016;7(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bejar R, Papaemmanuil E, Haferlach T, Garcia-Manero G, Maciejewski JP, Sekeres MA, et al. Somatic Mutations in MDS Patients Are Associated with Clinical Features and Predict Prognosis Independent of the IPSS-R: Analysis of Combined Datasets from the International Working Group for Prognosis in MDS-Molecular Committee. Blood. 2015;126(23):907-. [Google Scholar]
- 34.Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med. 2016;375(21):2023–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roboz GJ, Mandrekar SJ, Desai P, Laumann K, Walker AR, Wang ES, et al. Randomized trial of 10 days of decitabine ± bortezomib in untreated older patients with AML: CALGB 11002 (Alliance). Blood advances. 2018;2(24):3608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Short NJ, Kantarjian HM, Loghavi S, Huang X, Qiao W, Borthakur G, et al. Treatment with a 5-day versus a 10-day schedule of decitabine in older patients with newly diagnosed acute myeloid leukaemia: a randomised phase 2 trial. Lancet Haematol. 2019;6(1):e29–e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aldoss I, Pham A, Li SM, Gendzekhadze K, Afkhami M, Telatar M, et al. Favorable impact of allogeneic stem cell transplantation in patients with therapy-related myelodysplasia regardless of TP53 mutational status. Haematologica. 2017;102(12):2030–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bejar R, Stevenson KE, Caughey B, Lindsley RC, Mar BG, Stojanov P, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol. 2014;32(25):2691–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N Engl J Med. 2017;376(6):536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciurea SO, Chilkulwar A, Saliba RM, Chen J, Rondon G, Patel KP, et al. Prognostic factors influencing survival after allogeneic transplantation for AML/MDS patients with TP53 mutations. Blood. 2018;131(26):2989–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Middeke JM, Fang M, Cornelissen JJ, Mohr B, Appelbaum FR, Stadler M, et al. Outcome of patients with abnl(17p) acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. Blood. 2014;123(19):2960–7. [DOI] [PubMed] [Google Scholar]
- 42.Kharfan-Dabaja MA, Komrokji RS, Zhang Q, Kumar A, Tsalatsanis A, Perkins J, et al. TP53 and IDH2 Somatic Mutations Are Associated With Inferior Overall Survival After Allogeneic Hematopoietic Cell Transplantation for Myelodysplastic Syndrome. Clin Lymphoma Myeloma Leuk. 2017;17(11):753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duncavage EJ, Jacoby MA, Chang GS, Miller CA, Edwin N, Shao J, et al. Mutation Clearance after Transplantation for Myelodysplastic Syndrome. N Engl J Med. 2018;379(11):1028–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshizato T, Nannya Y, Atsuta Y, Shiozawa Y, Iijima-Yamashita Y, Yoshida K, et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood. 2017;129(17):2347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poiré X, Labopin M, Maertens J, Yakoub-Agha I, Blaise D, Ifrah N, et al. Allogeneic stem cell transplantation in adult patients with acute myeloid leukaemia and 17p abnormalities in first complete remission: a study from the Acute Leukemia Working Party (ALWP) of the European Society for Blood and Marrow Transplantation (EBMT). J Hematol Oncol. 2017;10(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Platzbecker U, Middeke JM, Sockel K, Herbst R, Wolf D, Baldus CD, et al. Measurable residual disease-guided treatment with azacitidine to prevent haematological relapse in patients with myelodysplastic syndrome and acute myeloid leukaemia (RELAZA2): an open-label, multicentre, phase 2 trial. Lancet Oncol. 2018;19(12):1668–79. [DOI] [PubMed] [Google Scholar]
- 47.Thol F, Gabdoulline R, Liebich A, Klement P, Schiller J, Kandziora C, et al. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood. 2018;132(16):1703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bewersdorf JP, Shallis RM, Boddu PC, Wood B, Radich J, Halene S, et al. The minimal that kills: Why defining and targeting measurable residual disease is the “Sine Qua Non” for further progress in management of acute myeloid leukemia. Blood Rev. 2019:100650. [DOI] [PubMed] [Google Scholar]
- 49.Wei AH, Döhner H, Pocock C, Montesinos P, Afanasyev B, Dombret H, et al. The QUAZAR AML-001 Maintenance Trial: Results of a Phase III International, Randomized, Double-Blind, Placebo-Controlled Study of CC-486 (Oral Formulation of Azacitidine) in Patients with Acute Myeloid Leukemia (AML) in First Remission. Blood. 2019;134(Supplement_2):LBA-3-LBA-. [Google Scholar]
- 50.Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016;531(7595):471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]