

Abstract
Three-dimensional tissue culture models are emerging as effective alternatives to animal testing. They are especially beneficial for liver toxicity studies, enabling hepatocytes to display improved levels of liver-specific functions. One common model is hepatocyte spheroids, which are spontaneously formed cell aggregates. Techniques for spheroid formation include the use of ultralow attachment plates and the hanging drop method, both of which are technically challenging and relatively low throughput. Here, we describe a simple-to-use platform that improves spheroid production and is compatible with genotoxicity testing by the comet assay. To achieve this, we created a chip containing a microwell array where dozens of spheroids are contained within a single well of a 96-well plate. The microwells are made from agarose, a nontoxic material suitable for cell growth and spheroid formation. HepG2 cells loaded into customizable microwells formed spheroids through agarose-assisted aggregation within one to two days. In addition, the agarose matrix allows the same platform to be used in DNA damage analysis. Specifically, the comet assay enables quantification of DNA breaks based on the increased migration of damaged DNA through agarose during electrophoresis. Here, we developed a modified comet assay and show that intact HepG2 spheroids cultured in microwells can be electrophoresed to reveal the extent of DNA damage following exposure to inflammatory chemicals (H2O2 and SIN-1). With this SpheroidChip analysis method, we detected a dose-dependent increase in DNA damage and observed rapid repair of H2O2-induced DNA damage. In summary, we utilized an agarose microarray to condense what had required an entire 96-well plate into a single well, enabling analysis techniques that were cumbersome or impossible under conditions of a single spheroid per well of a 96-well plate.
Keywords: Agarose-assisted aggregation, spheroid, hepatocyte, microwell array, comet assay, CometChip
INTRODUCTION
It is known that DNA damage can increase the risk of cancer and disease development. If unrepaired, damage can lead to mutations that drive cancer and disease. Understanding DNA damage and repair kinetics is helpful in predicting the carcinogenicity of chemicals and compounds, which is valuable because it can take years for cancer to arise after exposure to a carcinogen. To study DNA damage and repair, cell culture systems are often used. For many in vitro experiments, cells are removed from their normal environments in tissue and grown as monolayers on treated plastic surfaces prior to chemical exposure. To better mimic a cell’s natural environment for in vitro studies, researchers are now turning to more physiologically relevant 3D cell culture models.1–4 These 3D structures can allow for cell–cell interactions, leading to changes that are important for normal cell behavior. One area where predictive in vitro models are especially useful is for liver toxicity studies, and hepatocyte spheroids are often used for this purpose.5–10
Spheroid culture is one of the simpler methods for 3D culture that can be performed in most laboratories without specialized equipment. Existing platforms for spheroid formation modify the environment in which hepatocytes are cultured to influence cell interactions. For example, commercially available ultralow attachment 96-well plates create a nonadhesive environment that drives hepatocytes to aggregate into a spheroid.11,12 A limitation of this approach is that experiments are low throughput due to the fact that only one spheroid can be cultured in each well of a 96-well plate. Another commonly used approach is the hanging drop method;13,14 however, this method can be technically challenging and labor intensive. More complex techniques have also been developed, such as use of microfabrication techniques to create microcavities in polydimethylsiloxicane or poly(methyl methacrylate) chips to improve spheroid formation throughput,15–26 however these materials still require additional coating or treatment steps to make them suitable surfaces for cell culture purposes. Finally, effective organoid platforms involving coculture of multiple cell types have also been developed, though the complexity of such systems can be a barrier to use.
To improve throughput and to create a simple-to-use device that can readily be integrated with other bioassays, we developed a platform wherein hepatocyte spheroids can be cultured in agarose microwells approximately 100 μm in diameter. For this approach, cells gather in the arrayed agarose microwells at the base of each 96-well and aggregate to form spheroids. Rather than culturing one spheroid per well, as with an ultralow attachment plate, hundreds of spheroids can be cultured per well of a 96-well plate. The efficacy of this approach lies in the fact that agarose is a nontoxic, biocompatible polymer suitable for cell growth, and has been successfully used as a biomaterial in tissue engineering applications.27 Agarose also provides a low attachment environment that drives cell aggregation and is easily shaped into a customizable array of 3D microwell environments for spheroid formation and culture.28–35 As agarose suffices with no need for further surface modification, the approach for agarose-assisted aggregation can be readily adopted.
The comet assay is a commonly used DNA damage assay based on differential electrophoretic mobility for damaged versus undamaged DNA.36–39 As the assay involves electrophoresis through agarose, being able to culture hepatocyte spheroids in agarose microwells facilitates the use of the comet assay. The underlying principle of the comet assay is that intact DNA remains highly supercoiled and thus will not migrate significantly through agarose during electrophoresis, whereas the presence of strand breaks (and other types of DNA damage that can be converted into strand breaks) leads to the release of DNA supercoiling tension and fragmentation, enabling DNA migration in agarose. As a result, when electrophoresed, fragmented DNA is pulled away from the supercoiled nucleus, creating a detectable “comet tail”. The amount of DNA in the tail is proportional to the level of DNA strand breaks, which can be quantified through fluorescence microscopy and subsequent image analysis.40,36–38 One approach for quantifying DNA damage is to use %Tail DNA, which is calculated based on the fluorescence intensity of the comet tail divided by the total comet intensity. Because the end point is the proportion of DNA in the tail, the assay is self-calibrating. This allows for analysis of DNA not only in single cells, as with the traditional comet assay, but also in groups of cells, as has been demonstrated using the CometChip (for which effect analysis has been shown for up to about six cells).41 Here, we describe development and characterization of a spheroid microwell array (SpheroidChip) as an approach for quantifying DNA damage in intact spheroids.
One technical drawback of the traditional comet assay (where cells are suspended in molten agarose) is that it requires single cells. As a result, cultured cells are generally trypsinized prior to being embedded in agarose. However, because DNA repair can be very rapid, significant DNA repair takes place during sample handling. In the case of H2O2-induced oxidative damage, for which the half-life for DNA damage (t1/2) is approximately 30 min,42 repair is mostly complete by the time the samples are ready for comet analysis. Therefore, the ability to perform chemical exposures and the comet assay directly on the agarose chip with intact spheroids has a distinct advantage for studies of DNA damages that are rapidly repaired. This is particularly relevant for DNA damage created by inflammatory chemicals, which has relevance for studies of cancer etiology.
Chronic inflammation is a risk factor for cancer, in part because reactive oxygen species and nitrogen species (RONS) created during inflammation can damage DNA, leading to mutations that promote cancer. Specifically, activated macrophages secrete high levels of nitric oxide (NO•) and superoxide (O2−•). Superoxide can lead to DNA damage, and it can also react with NO• to create peroxynitrite (ONOO−), a highly genotoxic agent.43,44 Most of the RONS-induced DNA lesions are base damages such as 8-oxoguanine, which are rapidly repaired by the base excision repair pathway.45–47 Therefore, when studying RONS exposure, it would be advantageous to quantify DNA damage in intact spheroids because cells could be lysed within seconds after terminating exposure, thus suppressing DNA repair that occurs during sample handling.
Here, we developed the SpheroidChip and investigated whether it could be used to evaluate oxidative damage to DNA with agents that are relevant to inflammation. Using basic tissue culture supplies and a compatible biomaterial, we evaluated whether cells could be cultured and analyzed under more physiologically relevant conditions. We observed that the customizable microwell array improved throughput, as dozens of spheroids could be cultured in a single well of a 96-well plate. In addition, we demonstrated in intact spheroids that all comet assay steps could be condensed within a single platform, allowing for detection of DNA lesions such as those created by inflammatory chemicals, which are rapidly cleared by cells. The ability to measure DNA damage and repair in hepatocyte spheroids in a high-throughput fashion has utility for many applications beyond endogenous chemical exposures, including studies of environmental and pharmaceutical chemicals.
MATERIALS AND METHODS
Cell Culture.
HepG2 cells were cultured in high glucose DMEM (Invitrogen, 11965118) supplemented with 10% FBS (Atlanta Biologicals, S11150), 1% penicillin and streptomycin (Thermo Fisher Scientific, 15140122), and 1% L-glutamine (Thermo Fisher Scientific, A2916801). Cells were detached with 0.25% trypsin-EDTA (Thermo Fisher Scientific, 25200114) and triturated for spheroid seeding when they reached 70–90% confluency.
SU-8 Mold Fabrication.
Microfeature molds were created from silicon wafers by lithographically patterning SU-8, an epoxy-based photoresist (MicroChem, SU-8 2100). Briefly, the SU-8 was spun onto the silicon wafers to a desired thickness (100 or 250 μm), followed by soft-baking the photoresist and UV exposure through a photomask. The photomask included dark circular features (microwell pattern) on a transparent background. Areas of photoresist exposed to UV were cross-linked. Following a postexposure bake, the wells (features that were not exposed to UV) were developed in propylene glycol monomethyl ether acetate, revealing an array of patterned microwells with the desired geometries. Polydimethylsiloxane (PDMS) (Dow Corning, SYLGARD 184 Silicone Elastomer Kit) was cast on the SU-8 device to create a reusable stamp with microposts for patterning microwells into agarose (Figure 1C).
Figure 1.

Creation of an agarose microwell array for improved throughput of spheroid formation. (A) Commercial plates for spheroid formation hold one spheroid per macrowell. (B) Proposed SpheroidChip can form dozens of spheroids per macrowell. (C) Molds for creating PDMS stamps are created with photolithography. The PDMS stamps are then used to pattern microwell arrays in agarose.
Microwell Fabrication in Agarose and SpheroidChip Preparation.
The following steps were performed in a cell culture hood to maintain sterility. A 1% normal melting point agarose (Invitrogen, 16500) with 2% of penicillin and streptomycin was prepared in DPBS (Lonza, 17–512F). A PDMS mold was set in the molten agarose over a GelBond film (Lonza, 53761) for 15 min. The PDMS mold was then removed, creating an array of microwells. The agarose chip was clamped between a glass plate and a bottomless 96-well plate (Greiner BioOne, 655000), creating 96 separate environments, or macrowells, for cell loading. HepG2 cell suspensions were loaded by gravity into macrowells for 15–20 min in an incubator (37 °C, 5% CO2). Following loading, the bottomless plate was removed and excess off-grid cells were removed from the agarose chip with aspiration. The bottomless plate was replaced, and 200 μL of warm complete media was added to the macrowells. The chip containing the trapped cells was placed back in the incubator overnight to allow for cell aggregation. Subsequently, the bottomless plate was removed and the spheroids were protected with a layer of 0.5% low melting point agarose (Invitrogen, 16520) and allowed to cool for 3 min at 4 °C. The spheroid chip was then submerged in a dish of media. Media was replaced every 2–3 days until spheroids were needed for experiments.
SpheroidChip Cell Loading Measurements.
The density of the cell suspensions was varied to control the initial number of cells per macrowell. Following cell loading and removal of excess cells via shear force, the cells remaining in the agarose microwells were resuspended in the media. The total number of cells was determined using a Vi-Cell counter (Beckman Coulter). The average number of cells per well was calculated after adjusting for unloaded microwells, determined by manual counting using a bright field microscope.
SpheroidChip Viability Measurements.
HepG2 cell viability and growth in the agarose microwells were monitored in 250 μm diameter wells. Approximately 10,000 cells were loaded into each 96-well macrowell. Following loading, rinsing, and cell trapping using a 0.5% low melting point agarose overlay, the chips were placed in media and cell clusters were allowed to grow on the chip for up to 28 days. Media was replaced every 2–3 days. To determine the average number of viable cells per microwell on a given day, a bottomless 96-well plate was placed over the chip, and Accutase (Innovative Cell Technologies, AT104) was added to each macrowell to disaggregate the spheroids. Cells were recovered by gentle pipetting to break and release cells from the agarose overlay. Once recovered, the spheroids were left suspended in Accutase for an additional 30 min to allow for complete disaggregation. The resulting single cell suspension was counted using a Vi-Cell counter, and percent viability was determined with Trypan blue staining. The total viable cell count was divided by the total number of loaded microwells to estimate the average viable cells per microwell. The diameter of the spheroids was also monitored by taking bright field images of the spheroids, and using ImageJ to measure distance in the images.
Comet Assay (Performed Using the CometChip).
Using an agarose chip with 50 μm diameter microwells, cells were exposed to H2O2 (Sigma-Aldrich, 216763) or SIN-1 (Sigma-Aldrich, M5793). Immediately after treatment, chips were submerged in cell lysis buffer (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris, pH 9.5 with 0.5% Triton X-100) overnight at 4 °C. The following day, the chips were moved to an alkaline buffer (0.3 M sodium hydroxide and 1 mM Na2EDTA) for 40 min at 4 °C to allow for alkaline unwinding of DNA, and then electrophoresed at 4 °C for 30 min at 25 V and 300 mA. The chips were then neutralized in 0.4 M Tris-HCl buffer at room temperature and the DNA was stained with SYBRgold (Invitrogen, S11494). The chips were imaged at 4× using a fluorescent microscope and the %Tail DNA in each comet was determined using a MATLAB in-house image analysis software, described in Wood et al. Supporting Information (available upon request).41 For each experiment, about 100 comets were scored per macrowell, and the median %Tail DNA of triplicate wells were averaged. The steps of the CometChip are summarized in Figure S1.
SpheroidChip Assay.
Using an agarose chip with 100 μm diameter microwells, ∼75000 HepG2 cells were loaded per macrowell. After the excess cells were removed, clusters were allowed to form and grow on the chip for 2 days until they reached a diameter of ∼100 μm. After encapsulation with a 1% low melting point agarose overlay, the spheroids were exposed to H2O2 or SIN-1 directly on- chip (described below). Immediately following chemical exposure, the chips were placed in cell lysis buffer overnight at 4 °C. Following lysis, the agarose chips were attached to an electrophoresis chamber and submerged in alkaline unwinding buffer for 1 h at 4 °C. After alkaline unwinding, the chips were electrophoresed at 4 °C for 1 h at 25 V and 300 mA. The chips were neutralized at room temperature and DNA was stained with SYBRgold for at least an hour. The chips were imaged at 2× using a fluorescent microscope and the %Tail DNA in each spheroid-comet was determined using in-house image analysis software.41 For each experiment, about 40 spheroid-comets were scored per macrowell, and the median %Tail DNA of triplicate wells were averaged. Note, visual inspection was performed in the image analysis software to ensure that spheroid-comets were properly scored (Figure S2).
Chemical Treatments.
For H2O2 experiments, cells were treated with ice cold H2O2 diluted in PBS at 4 °C in the dark for 30 min (SpheroidChip) or 20 min (CometChip). For SIN-1 experiments, cells were treated with SIN-1 diluted in PBS at 37 °C for 30 min (SpheroidChip) or 20 min (CometChip).
H2O2 Repair Experiments.
To measure seven repair time points, an agarose chip made with a GelBond substrate was cut into groups of six or more macrowells per piece. Following H2O2 treatment, a piece of the agarose chip was immediately submerged in lysis buffer at 4 °C (time 0 repair point). The remaining chips were placed in fresh, warm media in the incubator until their repair time point. At the indicated times, the chip was submerged in lysis buffer. Following lysis, CometChip or spheroid-comet analysis was performed as described above.
Spheroid Staining.
Spheroids were cultured in 100 μm diameter microwells (using conditions described above to prepare a SpheroidChip for comet analysis) or 250 μm diameter microwells for 1 or 2 weeks (using conditions described above to prepare a SpheroidChip for viability measurements). Spheroids were fixed by submerging the agarose chip in 10% formalin for 6 h and then transferred to 70% ethanol. The agarose was removed from the GelBond and embedded in paraffin. Tissue sections (4 μm) were prepared and stained with hematoxylin and eosin (H&E) (Thermo Scientific Shandon Varistain 24–4), Ki-67 (BD Pharmigen, 550609), and cleaved caspase-3 (Cell Signaling Technology, 9664).
RESULTS AND DISCUSSION
Fabricating a Micropatterned Agarose Array
To improve the throughput of commercial spheroid formation plates wherein a single spheroid is cultured in each well of a 96-well plate (Figure 1A), we developed a microwell array that allows dozens of spheroids to be cultured in a microarray on the bottom surface of a single well of a 96-well plate (Figure 1B), significantly increasing the number of spheroids that can be cultured in a given surface area. The first step in creating an array of microwells is fabricating molds by lithographically patterning SU-8 photoresist (Figure 1C). PDMS is then poured into the SU-8 mold and cured. Removing the cured PDMS creates a stamp harboring thousands of microposts. This stamp is then pressed in molten agarose. After the molten agarose has cooled, lifting the PDMS stamp reveals microwells that are large enough to hold a single spheroid. Each SU-8 wafer can be used to create multiple PDMS stamps, and each stamp can be reused over dozens of experiments to create many of these agarose chips. Using a PDMS stamp that creates wells 250 μm in diameter spaced 400 μm apart (center to center), approximately 160 microwells can fit into the bottom surface of a single macrowell. The depth of each microwell is approximately 160 μm (thickness was measured using a Dektak surface profiler). The diameter of the microwells can be adjusted to accommodate different spheroid sizes.
Culturing HepG2 spheroids in Microwells through Agarose-Assisted Aggregation
To load cells into the microarray on the agarose chip, a bottomless 96-well plate is clamped to the surface of the chip. Suspended cells are loaded into each well of the 96-well plate (e.g., each macrowell) and allowed to settle into the microwells by gravity (Figure 2A). Excess cells are then removed from the chip by aspirating between macrowells (Figure 2B). We have observed that aspirating between macrowells (rather than directly over the macrowell) does not significantly disturb the loaded cells, as most cells remain trapped in the microwells. Using this approach, we have observed that, on average, ∼80% of the microwells contain cells. The trapped cells are then allowed to aggregate overnight (Figure 2C).
Figure 2.
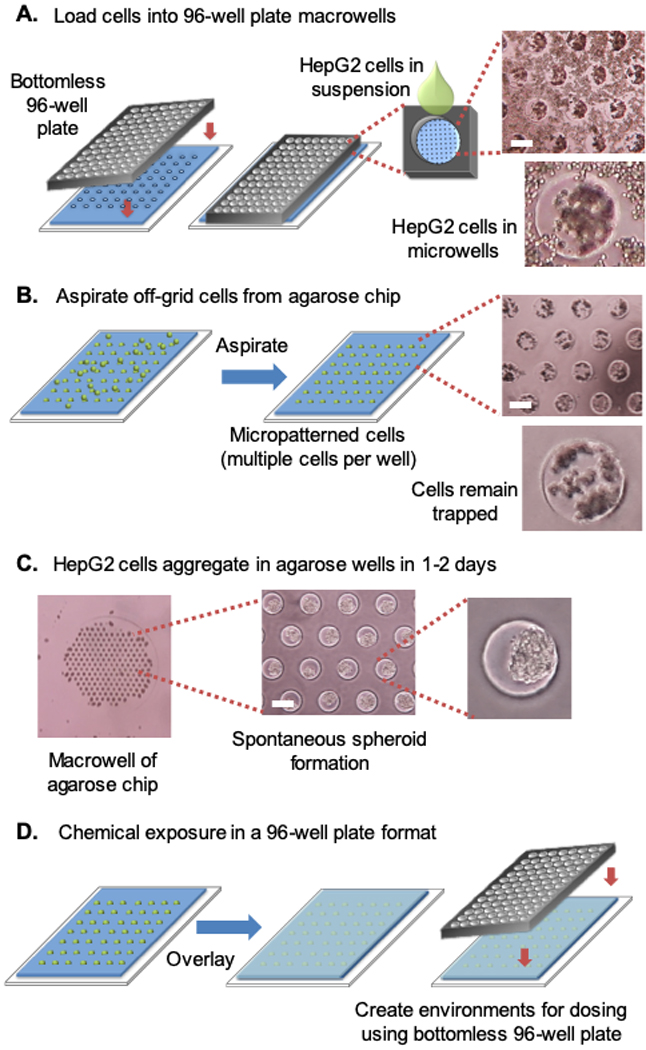
HepG2 cells aggregate once loaded into agarose microwells. The intact spheroids can be handled on the agarose chip in a standard 96-well array. (A) HepG2 cells fall into the agarose microwells by gravity. (B) Excess cells are removed from the agarose chip by aspirating between macrowells. (C) HepG2 spheroids form spontaneously in microwells. (D) Once spheroids have formed, they can be immobilized on the chip with an agarose overlay. A bottomless 96-well plate is then placed over the chip, creating distinct environments for chemical exposure. White scale bar, 250 μm. Note that each microwell is ∼250 μm in diameter.
To keep spheroids within the microwells during media changes, the bottomless plate is removed and a layer of low melting point (LMP) agarose is added, allowing the entire agarose chip to be moved into a fresh plate of media. The bottomless plate can be resecured directly over the encapsulated spheroids, converting the chip into a 96-well plate containing distinct macrowell environments that can then be used to test different conditions (Figure 2D). With multiple spheroids at the base of each macrowell, chemical dosing can quickly be performed in a 96-well format without first pooling single spheroids, as is necessary for spheroids grown in separate wells on commercial 96-well plates.
Calculations and previous data suggest that the agarose overlay does not significantly impede exposures or oxygen diffusion relative to the effect of the media itself. For example, previous studies in our laboratory have shown that large molecules (e.g., proteins) readily permeate the outer agarose layer. Specifically, we have shown that the FPG protein (30 kDa), can access the DNA of encapsulated cells.48 This result is consistent with high porosity of the outer layer of agarose. To further investigate how an agarose overlay might impact oxygen diffusion to the spheroids, we approximated the agarose hydrogel as a thin layer of water and estimated the timescale of oxygen diffusion through a liquid as: . With an LMP overlay thickness of 0.05 – 0.07 cm (estimated according to the volume of agarose overlay required to cover a chip) and a diffusion coefficient of a small molecule in liquid on the order of 10−5 , it would take 1–2 min for oxygen to cross the LMP agarose overlay barrier. This effect on oxygen diffusion is anticipated to be negligible because the typical height of media above HepG2 cells in culture is four- to sixfold thicker than the agarose overlay.
Impact of HepG2 Loading Density on Microwell Trapping
HepG2 cells trapped in the agarose microwells aggregate within approximately 1 day and continue to grow to form regularly shaped spheroids within the microwells (Figure 3A). Day 0 shows approximately 40 HepG2 cells trapped in a 250 μm diameter microwell that aggregate overnight and form spheroids over time. To explore the relationship between the number of cells plated on Day 0 and the number of cells initially trapped into a 250 μm diameter microwell, macrowells were loaded with a known number of cells ranging from 1000 cells to 100,000 cells. After the cells settled into the microwells by gravity, the trapped cells were counted manually under a microscope and/or recovered and counted with a Beckman Coulter Vi-cell. Average microwell cell count was plotted against the macrowell loading density (Figure 3B). We observed that the average number of cells loaded per microwell increased linearly with the macrowell loading density, with an R2 value of 0.98. In the equation below, Mi250 refers to the average number of cells loaded per 250 μm diameter microwell, and Ma refers to the macrowell loading density.
Figure 3.

After loading, HepG2 cells aggregate within microwells, and spheroids continue to grow and remain viable. (A) HepG2 cells in spheroids divide in the agarose microwells after loading. (B) Average cells loaded per 250 μm diameter microwell for various macrowell loading densities. Note, pictured in Day 0 of A is the initial microwell loading when a macrowell density of 10 000 cells per macrowell is used. (C) Average number of viable cells per microwell. The dashed lines correspond to days post loading pictured in A, where blue represents cells at 3 days, and orange represents cells at 7 days. (D) Cell viability measurements obtained with Trypan blue staining. (E) Spheroid diameter over culture time in the agarose chip. (F) H&E staining of spheroids. (G) Ki-67 staining of spheroids. (H) Cleaved caspase-3 staining of spheroids.
| Equation 1 |
Using these best fit parameters, we can calculate the macrowell loading density required to achieve a desired seeding number in 250 μm diameter agarose microwells. For experiments with cells able to undergo cell division, such as HepG2, adjusting microwell loading may be desirable to control spheroid size for experimental use on a later day. Microwell loading density would also be important to consider when applying this platform in nondividing hepatocyte cells, because the initial cell loading per microwell would remain constant for culture duration.
Analysis of Spheroid Growth and Viability
Inflammation of the liver is associated with an increased risk of cancer. During inflammation, high levels of potentially genotoxic RONS are produced. Here, we explored the potential utility of the spheroid platform for studies of inflammatory chemicals. Prior to performing the comet assay, it is first necessary to assess cytotoxicity, because dead cells can harbor broken DNA, which could interfere with studies of exposure-induced DNA damage.49
Our prediction was that increasing spheroid size would negatively impact cell viability in the spheroid interior due to oxygen and nutrient diffusion limitations.50 To learn about the potential cytotoxicity associated with large spheroids, we analyzed cell count, cell viability and spheroid diameter for HepG2 cells over time. We monitored HepG2 growth, viability, and spheroid diameter over the course of 4 weeks in 250 μm diameter microwells to determine an optimal spheroid size for genotoxicity studies. On Day 0, macrowells were loaded with ∼10,000 cells, resulting in ∼40 cells per microwell (calculated using eq 1). After initial HepG2 loading and spheroid formation, cell count increased rapidly in the first week of culture with a doubling time of 1.5 days (Figure 3C; similar to HepG2 grown as a monolayer). Cell viability, as determined by Trypan blue exclusion, also remained high for the first week of culture (Figure 3D; note that the first week of culture is demarked by orange dashed lines), which is consistent with the doubling time of healthy cells. After 1 week in culture, the cell count began to plateau (Figure 3C), corresponding to a notable decline in cell viability (Figure 3D). Other studies with HepG2 have also measured cell proliferation following initial spheroid seeding and a decrease in proliferation with increased culture time.8,15 The decline in cell viability we observed also corresponds to a spheroid diameter approaching and exceeding 200 μm (Figure 3E). Therefore, to ensure that our genotoxicity analysis focuses on conditions reflecting those of healthy cells, we elected to study spheroids at a maximum size of ∼100 μm by limiting the incubation time to 3 days (blue dashed lines), and by using microwells that are 100 μm in diameter.
We estimated the average loading for a 100 μm diameter microwell (Mi100) by scaling eq 1 with the ratio of the area of a 250 μm diameter well to the area of a 100 μm diameter well:
| Equation 2 |
Eq 2 was then used to design loading parameters for comet analysis experiments. For subsequent comet assay studies, based on eq 2, ∼75,000 cells were plated per macrowell to achieve the desired spheroid size (∼100 μm) at the time of comet analysis. The 100 μm microwells ensured that the spheroids remained below the diameter at which diffusion limitations begin impacting cell viability. From our previous analysis of spheroid growth, we expected that it would take approximately 3 days for the 100 μm diameter spheroids to reach the desired size for comet analysis. However, we observed that it only took 2 days for the spheroids to reach this size. It is possible that the smaller diameter microwells placed the loaded cells in closer proximity and promoted agarose-assisted aggregation (not accounted for in our scaled equation).
Finally, given that previous studies have shown that for large spheroids there is often cell death in the centers, we assessed cell viability in our spheroid interiors by creating 4 μm sections and analyzing them using immunohistochemistry for markers of cell division and cell death. Spheroids cultured in the 100 μm diameter microwells (comet analysis conditions, see Figure 3i) were compared with spheroids cultured in 250 μm diameter microwells for 1 (Figure 3ii) or 2 weeks (Figure 3iii). We observed earlier that at 1 week, cell viability still remained high and decreased by week 2 (Figure 3D), and we therefore expected to see signs of cell death in the centers of spheroids that had been cultured for 2 weeks. Representative H&E, Ki-67, and cleaved caspase-3 stained spheroid sections are shown in Figures 3F–H. As predicted, spheroids cultured for 1 week showed organized cell nuclei and cytoplasm (Figure 3Fii), as well as cell proliferation within the core (Figure 3Gii) and minimal staining for apoptosis (Figure 3Hii). In contrast, spheroids cultured for 2 weeks showed extensive signs of necrosis (Figure 3Fiii), diminished cell proliferation (Figure 3Giii), and higher levels of cleaved caspase-3 staining consistent with cell death (Figure 3Hiii). A decline in cell proliferation over time was also observed by Ramaiahgari et al.8 Note that the smaller cell aggregates cultured under comet analysis conditions show healthy cell morphology (Figure 3Fi), continued cell proliferation (Figure 3Gi), and the absence of apoptosis (Figure 3Hi).
Impact of Inflammatory Chemicals on DNA Damage in Spheroids Quantified Using the Comet Assay
To study DNA damage using the previously established CometChip, generally one or a few cells are trapped in patterned 40 or 50 μm diameter wells, encapsulated with agarose, treated with chemicals, and then electrophoresed to reveal the presence of strand breaks (Figure 4A, left). Comets are then scored for %Tail DNA to quantify the amount of DNA damage resulting from chemical treatment. To estimate the amount of DNA damage, the median %Tail DNA is calculated for ∼100 comets. These steps are summarized in Supporting Information Figure S1.
Figure 4.

DNA damage studies on intact spheroids were performed using steps similar to those in the CometChip experiments. (A) Proposed method for SpheroidChip analysis compared to CometChip analysis. (B) Geometry of microwell pattern used for comet analysis. Microwell spacing creates room for comet tails. (C) Comet analysis in CometChip versus SpheroidChip analysis. (D) Examples of H2O2 treated spheroid-comets.
Commercial plates available for spheroid culture (for example, ultralow attachment 96-well plates or hanging drop plates) are not directly compatible with CometChip analysis. Specifically, for the CometChip assay to be performed on spheroids grown in commercial nonadherent plates, spheroids would need to be recovered from the culture plate and disaggregated to form a single cell suspension before the single cells can be encapsulated in agarose in preparation for the gel electrophoresis step that physically separates fragmented DNA. These steps can take over 30 min. Due to rapid repair of H2O2-induced damage (t1/2 is approximately 30 min), sample handling time would preclude the ability to estimate the DNA damage levels present in the spheroids prior to isolation. In contrast, by culturing spheroids directly in agarose arrays with microwells large enough to hold entire cell clusters (Figure 4A, right), a platform can be created where each macrowell contains enough spheroids for analysis of one treatment condition. Because the chip is made of agarose, all steps of the comet assay, including electrophoresis, can be performed on a single device (obviating the need for pooling spheroids or sample disaggregation, and reducing sample handling time).
To learn about the efficacy of the spheroid platform for detecting RONS-induced DNA damage in intact spheroids, we studied H2O2 and SIN-1 as model agents. Specially, we used H2O2 for generating reactive oxygen species and SIN-1 for generating superoxide and nitric oxide, which react with each other to create peroxynitrite, a potent DNA damaging agent.44 One challenge with performing the comet assay on larger cell clusters is incomplete DNA migration away from the head of the comet. Specifically, because large cell clusters have bigger comet “heads”, DNA loops or fragments farthest from the tail will have an added distance to travel before being pulled away from the comet “head” into the comet “tail” where it can be quantified. To reduce the effect of incomplete DNA migration during spheroid-comet analysis, we increased electrophoresis time from 30 min to 1 h (Table 1).
Table 1.
Comparison of Different Comet Analysis Methods
| On-Chip Exposure CometChip | Off-Chip Exposure CometChip | SpheroidChip |
|---|---|---|
| Load single cell suspension directly before chemical exposure | Load single cell suspension after chemical exposure (~40 min lag) | Culture spheroids on chip ~2–3 days in advance, grow to diameter of microwell |
| 20 min chemical treatment | 30 min chemical treatment | |
| Overnight lysis in high salt buffer and detergent | ||
| 40 min alkaline unwinding step at high pH | 1 hour alkaline unwinding at high pH | |
| 30 minute electrophoresis | 1 hour electrophoresis | |
| Image at 4X | Image at 2X | |
Another challenge that the spheroid-comet approach poses is that larger distances are needed between comets so that they do not overlap during analysis. CometChip analysis of HepG2 cells uses 40 μm diameter microwells spaced 250 μm apart. For our initial spheroid platform with 250 μm diameter microwells, the distance between microwells is 150 μm, which is problematic when running the comet assay because comet tails overlap with nearby comet heads. Therefore, we created a chip so that microwells are 600 μm apart (center to center). In addition, the columns are staggered to further suppress comet tail/head overlap with neighboring wells (Figure 4B). It is also important to note that, while spacing the microwells farther apart allows sufficient room for DNA migration during electrophoresis, having large distances between the wells results in cell loss, because most of the cells will not enter microwells and are removed during the aspiration step. If desired, it may nevertheless be possible to recover unloaded cells prior to removing the bottomless 96-well plate and reload these samples into a new set of microwells. We anticipate concentration of cells will remain essentially unchanged because a tiny fraction of the cells in a suspension enter the microwells. With staggered microwells that are 100 μm in diameter and 600 μm apart, there are approximately 60 microwells per macrowell. A typical CometChip comet and a SpheroidChip comet are shown in Figure 4C. While the spheroid-comets are larger than single cell comets, the spheroids are consistent in size and the overall shape and appearance of the comets are similar. Note that when %Tail DNA is calculated, it is based on the proportion of the DNA in the tail, and so each comet is self-calibrated.
To explore the utility of the SpheroidChip for studies of RONS-induced DNA damage, chemical treatment was performed directly on spheroids cultured in the agarose chip, followed by overnight cell lysis, gel electrophoresis, fluorescent imaging, and automated spheroid-comet analysis (Materials and Methods). Examples of spheroid-comets for H2O2-treated spheroids are shown in Figure 4D. Qualitatively, we observed a dose dependent increase in comet tail DNA as the cells were exposed to higher doses of DNA damaging agents, demonstrating that the assay is effective in detecting DNA damage induced by our model inflammatory chemicals.
SpheroidChip Analysis Enables Robust Measurement of DNA Damage
To learn more about the efficacy of the HepG2 SpheroidChip for studies of exposure-induced DNA damage, we compared H2O2-induced damage levels from the SpheroidChip to results from the previously established CometChip. These analysis methods are summarized in Table 1. Our initial analysis was aimed at comparing microwell-to-microwell variation (e.g., comet-to-comet variation). As such, we analyzed the %Tail DNA for 90 comets from each H2O2 dose within a single experiment (see Figure 5A, where each dot represents a single comet). For both CometChip and SpheroidChip analysis, there is a clear dose- dependent increase in the median %Tail DNA. This plateaus at higher concentrations of H2O2 for the CometChip, which is consistent with comet assay saturation (typically occurring once %Tail DNA values of 60–80% are reached). We see a similar trend for SpheroidChip (Figure 5B), although higher levels of H2O2 are required to reach equivalent levels of DNA damage. The requirement for increased levels of H2O2 to achieve similar %Tail DNA values is consistent with a reduction in assay sensitivity, presumably due to DNA that does not emerge from the well due to the large comet head.
Figure 5.

DNA damage analysis in spheroids was compared to the previously developed CometChip. (A, B) Microwell to microwell variation for CometChip or SpheroidChip analysis. The median %Tail DNA is indicated for each H2O2 dose. (C, D) Macrowell to macrowell variation in a single CometChip or single SpheroidChip experiment. Each bar represents a unique macrowell. (E, F) Experiment to experiment variation for CometChip or SpheroidChip experiments. Each bar represents a unique experiment. For CometChip, representative data from four experiments is shown. For SpheroidChip, representative data from three experiments are shown.
In terms of comet-to-comet variation, there is variability for both the CometChip and the SpheroidChip for the untreated conditions. The %Tail DNA values begin to plateau once DNA damage passes 60% (indicated by the dotted lines on Figure 5), with the assay reaching full saturation at around 80%. The distribution of damage levels among comets remains broad under conditions of linear increase (before levels of 60%Tail DNA), but as H2O2-induced DNA damage reaches saturation, the variation declines. Therefore, to compare variation in comets analyzed using CometChip or SpheroidChip methods, we compared results at doses before the assays approached saturation. For CometChip, the median approaches a level of 60%Tail DNA at 30 μM H2O2 (Figure 5A, dotted line), while the median for SpheroidChip approaches 60%Tail DNA at 150 μM H2O2 (Figure 5B, dotted line). At and before these doses, comet-to-comet variability is similar between CometChip and SpheroidChip.
We also analyzed macrowell-to-macrowell variation (Figure 5C, D) and experiment-to-experiment variation (Figure 5E, F). Before the assay detection limit is reached (e.g., the assay reaches saturation above ∼60%Tail DNA), similar variation is observed using both analysis methods. Specifically, the coefficient of variation (CV) in CometChip macrowells ranges from 8.7 to 15.5% at levels below saturation (Table 2). The CV in SpheroidChip macrowells ranges from 3 to 15.1% at levels below saturation (Table 2). With regard to experiment- to-experiment variation, between experiments, the CV for the SpheroidChip tends to be larger than the CometChip (note that the CV also decreases as the saturation limit is approached). In addition, although the average %Tail DNA for the CometChip is higher than that for spheroid-comets analyzed at the same dose, there is nevertheless a clear dose response. Taken together, the SpheroidChip can be used to discern a dose response, making it a particularly useful platform where the goal is to compare variables that impact DNA damage.
Table 2.
Macrowell and Experiment Variation (Coefficient of Variation, in %)
| H2O2 dose (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 10 | 30 | 50 | 75 | 100 | 150 | 200 | |
| CometChip macrowell CV | 13.6% | 15.5% | 8.7% | 0.2% | 3.8% | 0.6% | 0.7% | 0.8% |
| SpheroidChip macrowell CV | 10.0% | 13.0% | 3.0% | 8.0% | 15.1% | 12.1% | 2.5% | 2.6% |
| CometChip experiment CV | 13.2% | 17.2% | 10.9% | 8.4% | 5.7% | 8.7% | 5.1% | 4.1% |
| SpheroidChip experiment CV | 34.1% | 31.9% | 16.7% | 16.2% | 22.9% | 22.2% | 16.4% | 11.1% |
For the traditional comet assay, there are reports that the sensitivity of the assay increases with increasing numbers of comets, as is also observed with the CometChip.51 To estimate how many spheroid-comets are needed for robust analysis of DNA damage, we scored pools of 10, 30, 40, 50, or 100 comets selected at random from our combined data for each H2O2 dose. This random sampling was repeated three times for each pool size, and the results were averaged and compared to randomly selected comets scored during CometChip analysis. As expected based on previous studies,51 varying the number of spheroid-comets analyzed did not significantly change the final calculated %Tail DNA values among the different analysis pools (Figure 6A, B). To discern whether assay sensitivity changed with additional spheroid-comets scored, we performed a paired, two-tailed Student’s t-test to compare each H2O2 dose to the corresponding nontreated negative control (Figure 7A–J). When 10 comets are scored from the CometChip (Figure 7A), we could detect a significant level of DNA damage in cells treated with H2O2 doses as low as 30 μM. Increasing analysis pools to 30, 40, 50, or 100 comets allowed for differences to be detected in the lowest H2O2 dose tested (10 μM). When 10 spheroid-comets are scored (Figure 7F), we detected a significant level of damage in cells treated with H2O2 doses as low as 50 μM. By increasing the analysis pool to 40 spheroid-comets, (the average number of spheroid- comets captured per macrowell on the SpheroidChip), we could detect a significant level of DNA damage in cells treated with H2O2 doses as low as 30 μM (Figure 7H). To detect DNA damage induced by smaller doses of H2O2, 50 comets or more may be needed (Figure 7I, J). Overall, compared to CometChip, SpheroidChip analysis required a greater number of comets to be scored to achieve statistically significant differences, however assay sensitivity increased with additional spheroid-comets scored. For studies where the objective is to detect lower levels of damage, two to three macrowells can be easily pooled for analysis, enabling analysis of 50–100 comets.
Figure 6.

Comparison of average median comet %Tail DNA when various comet pool sizes were analyzed for CometChip (A) or SpheroidChip (B). For each pool size, mean and standard deviation are shown of three randomly selected groups from the combined comet data.
Figure 7.

Analysis of the impact of comet pool size. H2O2 treated cells and nontreated controls from Figure 6 were compared with a paired, two-tailed, Student’s t-test, *P < 0.05. For each pool size, means and standard deviation of three randomly selected groups are shown for CometChip (A–E) or SpheroidChip (F–J).
Comparison of “On-chip” and “Off-chip” Exposures of Cells in the Comet Assay
One challenge for comet assays is that DNA repair can happen very quickly. H2O2- induced damage is repaired rapidly in HepG2 cells (Figure 8A), such that repair is nearly complete within the first hour. (Note that all repair experiments are performed on cells already loaded onto the agarose chip; see Materials and Methods for more details.) For cells that are cultured and chemically treated on plastic tissue culture coated dishes prior to CometChip loading for DNA analysis (referred to here as “off-chip” exposure), DNA repair can happen between the time that trypsinization has begun and the time when cells are lysed. For HepG2 cells, it takes ∼40 min to trypsinize and load the CometChip prior to lysis. During this lag time, DNA repair could potentially go to completion before cells are lysed. A second option is to treat cells after they have been transferred to agarose (referred to as “on-chip” exposure). On-chip experiments allow cells to be lysed immediately after chemical exposure via rapid submersion in lysis buffer, increasing the possibility that DNA damage can be quantified before it is repaired.
Figure 8.

Performing chemical exposure directly on cells encapsulated in agarose chips allows for analysis of rapidly repaired DNA damage. H2O2 or SIN-1 treated cells and nontreated controls were compared. (A) CometChip repair kinetics of HepG2 cells treated with 30 μM of H2O2. Treatment and repair is performed on-chip. (B) Comparison of on-chip versus off-chip CometChip dose response in H2O2 treated cells. For on- chip experiments, statistically significant increases in DNA damage were observed at all doses. The average of the experiments shown in Figure 5E is plotted. For off-chip experiments, there is a ∼40 min lag time prior to comet analysis and statistical significance was observed at a dose of 200 μM. (C) Comparison of on-chip versus off-chip CometChip dose response in SIN-1 treated cells. For on-chip experiments, statistically significant increases in DNA damage were observed at 4 mM and above. For off-chip experiments, there is a ∼40 min lag time prior to comet analysis and statistical significance was observed at 30 mM and above. (D) Dose response for H2O2 treated spheroids, consisting of the average of the experiments shown in Figure 5F. A statistically significant increase in DNA damage was observed at 75 μM and above. On-chip CometChip results from B are replotted on same graph for comparison. (E) Dose response for SIN-1 treated spheroids. A statistically significant increase in DNA damage was observed at 2 mM and above. On-chip CometChip results from C are replotted on same graph for comparison. (F) SpheroidChip repair kinetics of HepG2 spheroids treated with 100 μM of H2O2. Treatment and repair is performed on-chip. Statistical analysis was performed with a one-way ANOVA with post hoc analysis using Dunnett’s multiple comparison test, *P < 0.05.
We compared the efficiency of on-chip and off-chip exposures to inflammatory chemicals (H2O2 and SIN-1) with the CometChip (summarized in Table 1 and Figure 8B, C). Treated cells and nontreated controls were compared with a one-way ANOVA and posthoc analysis using Dunnett’s multiple comparisons test. HepG2 cells treated on-chip with either H2O2 (Figure 8B) or SIN-1 (Figure 8C) displayed a clear dose response, wherein H2O2 damage plateaued at dose of around 100 μM, while SIN-1 damage plateaued at a dose of around 20 mM. For on-chip H2O2 CometChip experiments (Figure 8B, blue line), a statistically significant increase in DNA damage was observed at all doses. For on-chip SIN-1 CometChip experiments (Figure 8C, blue line), a statistically significant increase in DNA damage was observed at doses of 4 mM and above. However, when cells were treated off-chip with the same doses, there was a negligible increase in measurable DNA damage induced until a dose of 200 μM for H2O2 (Figure 8B, green line) and 30 mM for SIN-1 (Figure 8C, green line), which is consistent with repair having mostly gone to completion prior to analysis. In a recent comet assay study,52 it was shown that DNA damage was readily detected when disaggregated cells were encapsulated in agarose prior to treatment, but that much lower levels of damage were observed when spheroids were treated prior to disaggregation and encapsulation, which agree with the results shown here.
Spheroid Platform Enables Dose-Response and Repair Studies for Inflammatory Chemicals
Analysis of the dose response for H2O2 or SIN-1 was also performed using intact spheroid-comet analysis. Spheroids are created by culturing cells in agarose microwells, which are subsequently exposed directly on-chip to the chemicals. Treated cells and nontreated controls were compared with a one-way ANOVA and posthoc analysis using Dunnett’s multiple comparisons test. Background levels of damage in untreated control spheroids was similar to that of cells cultured on the CometChip. As shown qualitatively in Figure 4D, a dose response was observed when intact spheroids were treated with H2O2. Here, higher doses of H2O2 also resulted in higher levels of DNA damage (Figure 8D, lower purple line). Note that the on-chip CometChip results have been replotted in Figure 8D (blue line) for comparison, and the magnitude of damage is higher in CometChip-analyzed cells across all doses. For H2O2- treated spheroids, a statistically significant increase in DNA damage was observed at doses of 75 μM and above (Figure 8D). This difference in assay sensitivity compared to CometChip was consistent with the known comet assay challenges of tail migration during electrophoresis when analyzing larger cell clusters.41 For SIN-1, a statistically significant increase in DNA damage was observed at doses of 2 mM and above (Figure 8E). Thus, there are both advantages and disadvantages to SpheroidChip analysis. Intact spheroid analysis is less sensitive compared to CometChip, but allows for experiments to be performed under more physiologically relevant conditions. In addition, spheroid experiments can be performed on-chip, capturing DNA damage that off-chip experiments do not. Similar to what has been observed using CometChip, these on-chip conditions enable studies of DNA repair in the HepG2 spheroids as well (Figure 8F). Taken together, the advantage of the agarose SpheroidChip platform is that on-chip experiments can be easily performed because cells are already cultured under the same conditions as those needed for measuring DNA damage, providing a way to measure rapidly repaired damage.
Future Directions: Measuring DNA Damage Induced by Metabolically Activated Chemicals
The work described here focuses on the detection of DNA breaks that are directly induced by inflammatory chemicals. In addition to direct-acting DNA damaging agents, there are also chemicals that require metabolic activation (e.g., benzo[a]pyrene or aflatoxin) to form reactive intermediates that can then damage DNA. Our current SpheroidChip model uses HepG2, a human liver hepatocellular carcinoma cell line. Our rational for using HepG2 cells was that they are well characterized for their utility in studying indirect-acting DNA damaging agents, such as benzo[a]pyrene, and are widely accessible to researchers.53–57 Interestingly, prolonged culture of HepG2 spheroids on nonadherent surfaces has been shown to lead to enhanced metabolic capacity.8,9,58–60 Although beyond the scope of this particular study, it will be interesting to learn the extent to which SpheroidChip is beneficial for enhancing metabolism for studies of indirect-acting DNA damaging agents. As such, the current study forms the basis of future studies of agents that require metabolic activation. Along these lines, ongoing studies are focused on evaluating not only the metabolic capacity of HepG2 cells in the spheroids, but also the metabolic capacity of HepaRG cells, which are known for their increased metabolic capacity.61–63 HepaRG spheroids have also been shown to maintain high levels of metabolic activity, making them a promising candidate for future SpheroidChip DNA damage studies.11,9,14
CONCLUSION
As a step toward more physiologically relevant assays, we have created the SpheroidChip, which can be used to create hepatocyte spheroids and to study DNA damage and repair. The platform is low cost, does not require specialized equipment for spheroid formation, and can be assembled using basic tissue culture supplies and a reusable PDMS stamp. The geometry of the microwells can also be modified to drive self-assembly of spheroids with different diameters. Culturing cells in the customizable patterned array of agarose microwells allows spheroid formation in a high-throughput manner with effective use of a given surface area, and creates distinct environments for spontaneous cell aggregation. By condensing what had originally required an entire 96-well plate into a single well, it is now possible to use comet analysis techniques that previously were difficult to perform on spheroids due to culture throughput and methods. In addition, the ability to culture hundreds of spheroids in each well of a 96-well plate may be useful for other end points that require large cell sample volumes, such as mass spectrometry, flow cytometry, Western blots, or ‘omics studies.
Following high-throughput spheroid culture on our platform, the spheroid-comets analyzed on the chip displayed trends of DNA damage and repair similar to those in the standard CometChip assay. The agarose chip described here is thus both amenable for spheroid culture and for analysis of DNA damage. This dual-purpose platform simplifies sample handling and shortens assay time, making it possible to study DNA damage that is rapidly repaired. In addition, because samples do not need to be disaggregated prior to studies of DNA damage and repair, cells can be studied under more physiological conditions (e.g., where cell–cell contact is retained). Taken together, these studies demonstrate the utility of this novel platform for culturing hepatocytes through agarose-assisted aggregation and for analyzing DNA damage and points to the potential for similar approaches to be utilized for other organoid systems.
Supplementary Material
ACKNOWLEDGMENT
We thank Kurt Broderick and the MIT Microsystems Technology Laboratory for guidance in microfabrication techniques. We thank Joanna Joy and Caroline Atkinson at the Department of Comparative Medicine for their assistance with spheroid sectioning and staining. C.C. acknowledges funding by the Training Grant in Environmental Toxicology (T32-ES007020), the MIT Nitric Oxide Program Project Grant (P01-CA026731), the National Institute of Environ- mental Health Sciences Superfund Basic Research Program, National Institute of Health (P42 ES027707), the National Institute of Environmental Health Sciences Core Center Grant, and the National Institute of Health (P30-ES002109). L.P.N. acknowledges funding by Integrated Laboratory Systems (R44-ES024698).
Funding Sources
1. Training Grant in Environmental Toxicology (T32-ES007020)
2. MIT Nitric Oxide Program Project Grant (P01-CA026731)
3. National Institute of Environmental Health Sciences Superfund Basic Research Program, National Institute of Health (P42 ES027707)
4. National Institute of Environmental Health Sciences Core Center Grant, National Institutes of Health (P30-ES002109)
5. SBIR CometChip: development of a high throughput DNA damage assay in hepatocytes, Integrated Laboratory Systems (R44-ES024698)
ABBREVIATIONS
- (PDMS)
Polydimethylsiloxane
- (LMP)
Low melting point
- (ROS)
Reactive oxygen species
- (RNS)
Reactive nitrogen species
- (RONS)
Reactive oxygen and nitrogen species
Footnotes
The authors declare the following competing financial interest(s): Bevin P. Engelward is a co-inventor for the CometChip, which has been patented and licensed to Trevigen.
REFERENCES
- (1).Edmondson R; Broglie JJ; Adcock AF; Yang L. Three- Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. Assay Drug Dev. Technol 2014, 12 (4), 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Huh D; Hamilton GA; Ingber DE From 3D Cell Culture to Organs-on-Chips. Trends Cell Biol. 2011, 21 (12), 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).LeCluyse EL; Witek RP; Andersen ME; Powers MJ Organotypic Liver Culture Models: Meeting Current Challenges in Toxicity Testing. Crit. Rev. Toxicol 2012, 42 (6), 501–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Laschke MW; Menger MD Life Is 3D: Boosting Spheroid Function for Tissue Engineering. Trends Biotechnol. 2017, 35 (2), 133–144. [DOI] [PubMed] [Google Scholar]
- (5).Koide N; Sakaguchi K; Koide Y; Asano K; Kawaguchi M; Matsushima H; Takenami T; Shinji T; Mori M; Tsuji T. Formation of Multicellular Spheroids Composed of Adult Rat Hepatocytes in Dishes with Positively Charged Surfaces and Under Other Nonadherent Environments. Exp. Cell Res 1990, 186 (2), 227–235. [DOI] [PubMed] [Google Scholar]
- (6).Landry J; Bernier D; Ouellet C; Goyette R; Marceau N. Spheroidal Aggregate Culture of Rat Liver Cells: Histotypic Reorganization, Biomatrix Deposition, and Maintenance of Functional Activities. J. Cell Biol 1985, 101 (3), 914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fennema E; Rivron N; Rouwkema J; van Blitterswijk C; De Boer J. Spheroid Culture as a Tool for Creating 3D Complex Tissues. Trends Biotechnol. 2013, 31 (2), 108–115. [DOI] [PubMed] [Google Scholar]
- (8).Ramaiahgari SC; Den Braver MW; Herpers B; Terpstra V; Commandeur JNM; Van De Water B; Price LSA 3D in Vitro Model of Differentiated HepG2 Cell Spheroids with Improved Liver- like Properties for Repeated Dose High-Throughput Toxicity Studies. Arch. Toxicol 2014, 88 (5), 1083–1095. [DOI] [PubMed] [Google Scholar]
- (9).Takahashi Y; Hori Y; Yamamoto T; Urashima T; Ohara Y; Tanaka H. 3D Spheroid Cultures Improve the Metabolic Gene Expression Profiles of HepaRG Cells. Biosci. Rep 2015, 35, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bell CC; Hendriks DFG; Moro SML; Ellis E; Walsh J; Renblom A; Puigvert LF; Dankers ACA; Jacobs F; Snoeys J; et al. Characterization of Primary Human Hepatocyte Spheroids as a Model System for Drug-Induced Liver Injury, Liver Function and Disease. Sci. Rep 2016, 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ramaiahgari SC; Waidyanatha S; Dixon D; DeVito MJ; Paules RS; Ferguson SS Three-Dimensional (3D) HepaRG Spheroid Model With Physiologically Relevant Xenobiotic Metabolism Competence and Hepatocyte Functionality for Liver Toxicity Screening. Toxicol. Sci 2017, 159 (1), 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vinci M; Gowan S; Boxall F; Patterson L; Zimmermann M; Court W; Lomas C; Mendiola M; Hardisson D; Eccles SA Advances in Establishment and Analysis of Three- Dimensional Tumor Spheroid-Based Functional Assays for Target Validation and Drug Evaluation. BMC Biol. 2012, 10 (1), 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tung YC; Hsiao AY; Allen SG; Torisawa YS; Ho M; Takayama S. High-Throughput 3D Spheroid Culture and Drug Testing Using a 384 Hanging Drop Array. Analyst 2011, 136 (3), 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gunness P; Mueller D; Shevchenko V; Heinzle E; Ingelman-Sundberg M; Noor F. 3D Organotypic Cultures of Human HepaRG Cells: A Tool for in Vitro Toxicity Studies. Toxicol. Sci 2013, 133 (1), 67–78. [DOI] [PubMed] [Google Scholar]
- (15).Sakai Y; Nakazawa K. Technique for the Control of Spheroid Diameter Using Microfabricated Chips. Acta Biomater. 2007, 3, 1033–1040. [DOI] [PubMed] [Google Scholar]
- (16).Nakazawa K; Izumi Y; Fukuda J; Yasuda T. Hepatocyte Spheroid Culture on a Polydimethylsiloxane Chip Having Microcavities. J. Biomater. Sci., Polym. Ed 2006, 17 (8), 859–873. [DOI] [PubMed] [Google Scholar]
- (17).Lee DH; Park JY; Lee EJ; Choi YY; Kwon GH; Kim BM; Lee SH Fabrication of Three-Dimensional Microarray Structures by Controlling the Thickness and Elasticity of Poly (Dimethylsiloxane) Membrane. Biomed. Microdevices 2010, 12 (1), 49–54. [DOI] [PubMed] [Google Scholar]
- (18).Wong SF; No DY; Choi YY; Kim DS; Chung BG; Lee SH Concave Microwell Based Size-Controllable Hepatosphere as a Three-Dimensional Liver Tissue Model. Biomaterials 2011, 32 (32), 8087–8096. [DOI] [PubMed] [Google Scholar]
- (19).Fukuda J; Nakazawa K. Orderly Arrangement of Hepatocyte Spheroids on a Microfabricated Chip. Tissue Eng. 2005, 11 (7–8), 1254–1262. [DOI] [PubMed] [Google Scholar]
- (20).Fukuda J; Sakai Y; Nakazawa K. Novel Hepatocyte Culture System Developed Using Microfabrication and Collagen/Polyethylene Glycol Microcontact Printing. Biomaterials 2006, 27 (7), 1061–1070. [DOI] [PubMed] [Google Scholar]
- (21).Shimizu K; Kusamori K; Nishikawa M; Mizuno N; Nishikawa T; Masuzawa A; Katano S; Takahashi Y; Takakura Y; Konishi S. Poly (N-Isopropylacrylamide)-Coated Microwell Arrays for Construction and Recovery of Multicellular Spheroids. J. Biosci. Bioeng 2013, 115 (6), 695–699. [DOI] [PubMed] [Google Scholar]
- (22).Lee GH; Lee JS; Oh HJ; Lee SH Reproducible Construction of Surface Tension-Mediated Honeycomb Concave Microwell Arrays for Engineering of 3D Microtissues with Minimal Cell Loss. PLoS One 2016, 11 (8), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Markovitz-Bishitz Y; Tauber Y; Afrimzon E; Zurgil N; Sobolev M; Shafran Y; Deutsch A; Howitz S; Deutsch M. A Polymer Microstructure Array for the Formation, Culturing, and High Throughput Drug Screening of Breast Cancer Spheroids. Biomaterials 2010, 31 (32), 8436–8444. [DOI] [PubMed] [Google Scholar]
- (24).No DY; Lee S; Choi YY; Park D; Jang JY; Kim D; Lee H. Functional 3D Human Primary Hepatocyte Spheroids Made by Co-Culturing Hepatocytes from Partial Hepatectomy Specimens and Human Adipose-Derived Stem Cells. PLoS One 2012, 7 (12), e50723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Goral VN; Au SH; Faris RA; Yuen PK Methods for Advanced Cell Culture in Microwells Utilizing Air Bubbles. Lab Chip 2015, 15, 1032–1037. [DOI] [PubMed] [Google Scholar]
- (26).Yamazaki H; Gotou S; Ito K; Kohashi S; Goto Y; Yoshiura Y; Sakai Y; Yabu H; Shimomura M; Nakazawa K. Micropatterned Culture of HepG2 Spheroids Using Microwell Chip with Honeycomb-Patterned Polymer Film. J. Biosci. Bioeng 2014, 118 (4), 455–460. [DOI] [PubMed] [Google Scholar]
- (27).Zarrintaj P; Manouchehri S; Ahmadi Z; Saeb MR; Urbanska AM; Kaplan DL; Mozafari M. Agarose-Based Biomaterials for Tissue Engineering. Carbohydr. Polym 2018, 187, 66–84. [DOI] [PubMed] [Google Scholar]
- (28).Napolitano AP; Dean DM; Man AJ; Youssef J; Ho DN; Rago AP; Lech MP; Morgan JR Scaffold-Free Three- Dimensional Cell Culture Utilizing Micromolded Nonadhesive Hydrogels. BioTechniques 2007, 43 (4), 494–500. [DOI] [PubMed] [Google Scholar]
- (29).Napolitano AP; Chai P; Dean DM; Morgan JR Dynamics of the Self-Assembly of Complex Cellular Aggregates on Micromolded Nonadhesive Hydrogels. Tissue Eng. 2007, 13 (8), 2087–2094. [DOI] [PubMed] [Google Scholar]
- (30).Gevaert E; Dolle L; Billiet T; Dubruel P; Grunsven L; Van Apeldoorn A; Cornelissen R. High Throughput Micro-Well Generation of Hepatocyte Micro-Aggregates for Tissue Engineering. PLoS One 2014, 9 (8), e105171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mirab F; Kang YJ; Majd S. Preparation and Characterization of Size-Controlled Glioma Spheroids Using Agarose Hydrogel Microwells. PLoS One 2019, 14 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liu T; Chien CC; Parkinson L; Thierry B. Advanced Micromachining of Concave Microwells for Long Term On-Chip Culture of Multicellular Tumor Spheroids. ACS Appl. Mater. Interfaces 2014, 6 (11), 8090–8097. [DOI] [PubMed] [Google Scholar]
- (33).Gong X; Lin C; Cheng J; Su J; Zhao H; Liu T; Wen X; Zhao P. Generation of Multicellular Tumor Spheroids with Microwell-Based Agarose Scaffolds for Drug Testing. PLoS One 2015, 10 (6), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Thomsen AR; Aldrian C; Bronsert P; Thomann Y; Nanko N; Melin N; Rücker G; Follo M; Grosu AL; Niedermann G; et al. A Deep Conical Agarose Microwell Array for Adhesion Independent Three-Dimensional Cell Culture and Dynamic Volume Measurement. Lab Chip 2018, 18 (1), 179–189. [DOI] [PubMed] [Google Scholar]
- (35).Tang Y; Liu J; Chen Y. Agarose Multi-Wells for Tumour Spheroid Formation and Anti-Cancer Drug Test. Microelectron. Eng 2016, 158, 41–45. [Google Scholar]
- (36).Ostling O; Johanson KJ Microelectrophoretic Study of Radiation-Induced DNA Damages in Individual Mammalian Cells. Biochem. Biophys. Res. Commun 1984, 123 (1), 291–298. [DOI] [PubMed] [Google Scholar]
- (37).Singh NP; McCoy MT; Tice RR; Schneider EL A Simple Technique for Quantitation of Low Levels of DNA Damage in Individual Cells. Exp. Cell Res 1988, 175 (1), 184–191. [DOI] [PubMed] [Google Scholar]
- (38).Olive PL; Banat́h JP The Comet Assay: A Method to Measure DNA Damage in Individual Cells. Nat. Protoc 2006, 1 (1), 23–29. [DOI] [PubMed] [Google Scholar]
- (39).Collins AR The Comet Assay for DNA Damage and Repair: Principles, Applications, and Limitations. Mol. Biotechnol 2004, 26 (3), 249–261. [DOI] [PubMed] [Google Scholar]
- (40).Olive PL; Vikse CM; Banat́h JP Use of the Comet Assay to Identify Cells Sensitive to Tirapazamine in Multicell Spheroids and Tumors in Mice. Cancer Res. 1996, 56 (19), 4460–4463. [PubMed] [Google Scholar]
- (41).Wood DK; Weingeist DM; Bhatia SN; Engelward BP Single Cell Trapping and DNA Damage Analysis Using Microwell Arrays. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (22), 10008–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ge J; Prasongtanakij S; Wood DK; Weingeist DM; Fessler J; Navasummrit P; Ruchirawat M; Engelward BP Cometchip: A High-Throughput 96-Well Platform for Measuring DNA Damage in Microarrayed Human Cells. J. Visualized Exp 2014, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Dedon PC; Tannenbaum SR Reactive Nitrogen Species in the Chemical Biology of Inflammation. Arch. Biochem. Biophys 2004, 423 (1), 12–22. [DOI] [PubMed] [Google Scholar]
- (44).Kiziltepe T; Yan A; Dong M; Jonnalagadda VS; Dedon PC; Engelward BP Delineation of the Chemical Pathways Underlying Nitric Oxide-Induced Homologous Recombination in Mammalian Cells. Chem. Biol 2005, 12 (3), 357–369. [DOI] [PubMed] [Google Scholar]
- (45).Wallace SS Base Excision Repair: A Critical Player in Many Games. DNA Repair 2014, 19, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Krokan HE; Bjørås M. Base Excision Repair. Cold Spring Harbor Perspect. Biol 2013, 5 (4), a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wilson DMI; Bohr VA The Mechanics of Base Excision Repair, and Its Relationship to Aging and Disease. DNA Repair 2007, 6 (4), 544–559. [DOI] [PubMed] [Google Scholar]
- (48).Ge J; Wood DK; Weingeist DM; Prasongtanakij S; Navasumrit P; Ruchirawat M; Engelward BP Standard Fluorescent Imaging of Live Cells Is Highly Genotoxic. Cytometry, Part A 2013, 83 (6), 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Olive PL; Frazer G; Banath JP Radiation- Induced Apoptosis Measured in TK6 Human B Lymphoblast Cells Using the Comet Assay. Radiat. Res 1993, 136 (1), 130–136. [PubMed] [Google Scholar]
- (50).Mehta G; Hsiao YA; Ingram M; Luker GD; Takayama S. Opportunities and Challenges for Use of Tumor Spheroids as Models to Test Drug Delivery and Efficacy. J. Controlled Release 2012, 164 (2), 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ge J; Chow DN; Fessler JL; Weingeist DM; Wood DK; Engelward BP Micropatterned Comet Assay Enables High Throughput and Sensitive DNA Damage Quantification. Mutagenesis 2015, 30 (1), 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Elje E; Hesler M; Rundeń-Pran E; Mann P; Mariussen E; Wagner S; Dusinska M; Kohl Y. The Comet Assay Applied to HepG2 Liver Spheroids. Mutat. Res., Genet. Toxicol. Environ. Mutagen 2019, 845, 403033. [DOI] [PubMed] [Google Scholar]
- (53).Seo JE; Tryndyak V; Wu Q; Dreval K; Pogribny I; Bryant M; Zhou T; Robison TW; Mei N; Guo X. Quantitative Comparison of in Vitro Genotoxicity Between Metabolically Competent HepaRG Cells and HepG2 Cells Using the High-Throughput High-Content CometChip Assay. Arch. Toxicol 2019, 93, 1433–1448. [DOI] [PubMed] [Google Scholar]
- (54).Park SY; Lee SM; Ye SK; Yoon SH; Chung MH; Choi J. Benzo[a]Pyrene-Induced DNA Damage and P53 Modulation in Human Hepatoma HepG2 Cells for the Identification of Potential Biomarkers for PAH Monitoring and Risk Assessment. Toxicol. Lett 2006, 167 (1), 27–33. [DOI] [PubMed] [Google Scholar]
- (55).Knasmüller S; Mersch-Sundermann V; Kevekordes S; Darroudi F; Huber WW; Hoelzl C; Bichler J; Majer BJ Use of Human-Derived Liver Cell Lines for the Detection of Environmental and Dietary Genotoxicants; Current State of Knowledge. Toxicology 2004, 198 (1–3), 315–328. [DOI] [PubMed] [Google Scholar]
- (56).Valentin-Severin I; Le Hegarat L; Lhuguenot JC; Le Bon AM; Chagnon MC Use of HepG2 Cell Line for Direct or Indirect Mutagens Screening: Comparative Investigation between Comet and Micronucleus Assays. Mutat. Res., Genet. Toxicol. Environ. Mutagen 2003, 536 (1–2), 79–90. [DOI] [PubMed] [Google Scholar]
- (57).Yusuf AT; Vian L; Sabatier R; Cano JP In Vitro Detection of Indirect-Acting Genotoxins in the Comet Assay Using Hep G2 Cells. Mutat. Res., Genet. Toxicol. Environ. Mutagen 2000, 468 (2), 227–234. [DOI] [PubMed] [Google Scholar]
- (58).Mueller D; Koetemann A; Noor F. Organotypic Cultures of HepG2 Cells for In Vitro Toxicity Studies. J. Bioeng. Biomed. Sci 2011. S2. [Google Scholar]
- (59).Terashima J; Goto S; Hattori H; Hoshi S; Ushirokawa M; Kudo K; Habano W; Ozawa S. CYP1A1 and CYP1A2 Expression Levels Are Differentially Regulated in Three-Dimensional Spheroids of Liver Cancer Cells Compared to Two-Dimensional Monolayer Cultures. Drug Metab. Pharmacokinet 2015, 30 (6), 434–440. [DOI] [PubMed] [Google Scholar]
- (60).Shah UK; Mallia J. de O.; Singh N; Chapman KE; Doak SH; Jenkins GJS A Three-Dimensional in Vitro HepG2 Cells Liver Spheroid Model for Genotoxicity Studies. Mutat. Res., Genet. Toxicol. Environ. Mutagen 2018, 834, 35–41. [DOI] [PubMed] [Google Scholar]
- (61).Gripon P; Rumin S; Urban S; Le Seyec J; Glaise D; Cannie I; Guyomard C; Lucas J; Trepo C; Guguen-Guillouzo C. Infection of a Human Hepatoma Cell Line by Hepatitis B Virus. Proc. Natl. Acad. Sci. U. S. A 2002, 99 (24), 15655–15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Parent R; Marion MJ; Furio L; Treṕo C; Petit MA Origin and Characterization of a Human Bipotent Liver Progenitor Cell Line. Gastroenterology 2004, 126 (4), 1147–1156. [DOI] [PubMed] [Google Scholar]
- (63).Aninat C; Piton A; Glaise D; LeCharpentier T; Langoueẗ S; Morel F; Guguen-Guillouzo C; Guillouzo A. Expression of Cytochromes P450, Conjugating Enzymes and Nuclear Receptors in Human Hepatoma HepaRG Cells. Drug Metab. Dispos 2006, 34 (1), 75–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.