

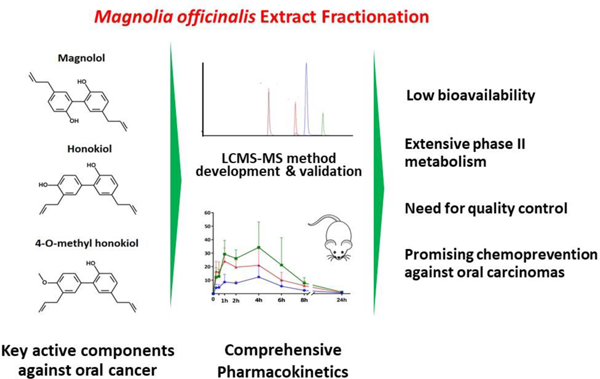
Abstract
Among the three key active components (KACs) of Magnolia officinalis bark extract (ME), 4-O-methylhonokiol (MHNK) and honokiol (HNK) showed higher antiproliferation activities than magnolol (MGN) in the oral squamous cancer cell lines (Cal-27, SCC-9 and SCC-4). Oral bioavailabilities of ME-KACs were poor (<0.2%) in C57BL/6 mice primarily due to their extensive first-pass phase II metabolism and poor solubilities. High plasma concentration of glucuronides upon oral administration and faster rate of glucuronidation by intestinal microsomes indicated intestine as one of the major metabolic organs for ME-KACs. Despite the increase in bioavailabilities of ME-KACs (~8–10-folds) and decrease in AUC0–24 of glucuronides (~10-folds) upon ME solubility enhancement, systemic exposure of ME-KACs failed to improve meaningfully. In conclusion, we propose a quality-controlled and chemically-defined ME mixture, containing an optimized ratio of three KACs, delivered locally in the oral cavity as the most promising strategy for ME use as an oral cancer chemopreventive dietary supplement.
Keywords: First-pass metabolism, 4-O-methylhonokiol, Magnolol, Honokiol, Glucuronides, Magnolia officinalis, Oral squamous cell cancer, Houpo
Graphical Abstract
1-. Introduction
Magnolia Extract (ME) is currently used as an overall wellness nutritional supplement in United States for a variety of maladies including anxiety, depression, diabetes, inflammation, headache, muscle pain, weight loss, asthma, stroke, and bacterial infection etc. 1–8. It is listed in the American Herbal Products Association’s book Herbs of Commerce 9. The extract is prepared from Magnolia officinalis Rehder & E.H.Wilson or M. officinalis var. bark, which is also known as “Houpo” or “Houpu” and has been used in traditional Chinese medicine as an herb for more than 2500 years for the treatment of anxiety, asthma, depression, gastrointestinal disorders and headache10.
In recent preclinical studies, ME and/or its bulk components exhibited inhibition effect against variety of cancers, including breast, ovarian, prostate, colon, stomach, pancreatic, bladder and head & neck cancers both in in vitro and in vivo models 11–19. Oral cancer is a form of head and neck carcinoma that affects tens of thousands of American every year and their incidence has risen notably in recent decades20. Oral cavity carcinomas are accounted for 30% of head and neck cancer - a group of cancer originating from the upper digestive tract - and 90% of oral cancer is squamous cell carcinomas (SCC) 21,22. As the sixth most common cancer in the world, approximately 600,000 new cases are diagnosed every year in the world with the mortality rate of 40–50% 23. However, the 5-year survival rate of these patients has not changed markedly in the past three decades 24. Most oral cancers have premalignant epithelial lesion stages such as leukoplakia and erythroplakia 22 and could be detected early during regular dental exams. As a result, development of effective chemopreventive interventions for oral cavity SCC is promising and urgently needed.
Pure magnolol (MGN) and honokiol (HNK), two of the known key active components of ME, has been shown to exert anti-cancer properties against oral cancer 15,25. For preventing oral cancer, HNK is proposed to work through the mechanisms of apoptotic cell morphologies, cell cycle arrest and autophagy 26 by inhibition in the transcription factor specificity protein (Sp1) 27, JAK/STAT signaling pathway 28 and Wnt/β-catenin signaling pathway 29. MGN was reported to induce Ca2+ influx via protein kinase C-sensitive store-operated Ca2+ entry and cause Ca2+ release from endoplasmic reticulum in a phospholipase C-associated manner in oral cancer 25. Meanwhile, antitumor activity of MHNK in human oral cancer cells was mediated via ROS generation, disruption of mitochondrial potential, cell cycle arrest and modulation of Bcl-2/Bax proteins 30. Despite numerous studies on oral cancer cells and in vivo animal models, no systematic study has been reported so far either identifying the most effective anticancer components of ME mixture or the bioavialabilities of ME components in the target tissue (oral epithelium).
Therefore, in order to advance the understanding of best use of ME dietary supplement for oral cancer chemoprevention, in present study, we aim to 1) identify key active ingredients of magnolia extract (ME-KACs) against oral cancer; and 2) characterize the pharmacokinetics of ME-KACs in C57/BL6 mice using a validated UPLC-MS/MS to simultaneously quantify the ME-KACs and their metabolites in different biological matrices.
2 -. Material and Methods
2.1-. Materials
ME was procured from SK Bioland Ltd (Chungnam, Korea) with 14.3 ± 2.0% MGN, 20.6 ± 0.3% HNK and 16.6 ± 0.9% w/w MHNK. ME was authenticated and technical datasheet supplied by the manufacturer. Additionally, we performed thorough in-house LCMS/MS phytochemical characterization and quantification of the active constituents before cell and animal studies. This source of material has been shown to have pharmacological activity in different disease indications in animals in the published literature 31–33. Baohuoside I was purchased from Chengdu Must Bio-technology Co. Ltd (Chengdu, China). Ammonium acetate, formic acid (LC-MS grade), Uridine-5’-diphosphate-β, D-glucuronic acid ester (UDPGA), 3’-phosphoadenosine-5’-phosphosulfate (PAPS), D-saccharic-1,4-lactone monohydrate, magnesium chloride, magnolol, honokiol were purchased from Sigma (St. Louis, MO). 4-O-methylhonokiol was purchased from LKT Laboratories Inc (Minnesota, USA). Protein assay kit was purchased from BioRad, Hercules, CA. All other materials (typically analytical grade or better) were used as received.
2.2-. Analytical Methods
UPLC-UV-
UPLC-UV was use for characterizing ME and its fractions, samples from solubility study and Caco-2 transport study. The conditions used were: system, Waters Acquity™ with diode array detector (DAD); column, BEH C18 column (100 × 2.1 mm I.D., 1.7 µm, Waters, Milford, MA, USA); mobile phase A (MPA), 0.1% formic acid in water; mobile phase B (MPB), 100% acetonitrile; gradient, 0 – 4.0 min, 5 – 30% MPB, 4.0 – 7.0 min, 30 – 45% MPB, 7.0 – 10.0 min, 45 – 80 % MPB, 10.0 – 12.5 min, 80 – 95% MPB, 12.5 – 13.5 min, 95% MPB, 13.5 – 13.6 min, 95 – 5% MPB, 13.6 – 14.0 min, 5% MPB; flow rate, 0.4 mL/min; column temperature, 45°C; injection volume, 10 µL; analysis wavelength, 254 nm.
UPLC-MS/MS-
UPLC conditions used for method validations and analysis of ME key active components were: system, Waters Acquity™; column, BEH C18 column (50 × 2.1 mm I.D., 1.7 µm, Waters, Milford, MA, USA); mobile phase A (MPA), 2 mM ammonium acetate in water; mobile phase B (MPB), 10% acetonitrile; gradient, 0 – 0.5 min, 5% MPB, 0.5 –3.0 min, 5 – 30% MPB, 3.0 – 8.0 min, 30 – 35% MPB, 8.0 – 14.0 min, 35 – 80% MPB, 14.0 – 16.0 min, 80 – 95% MPB, 16.0 – 17.0 min, 95% MPB, 17.0 – 17.5 min, 95 – 5% MPB, 17.5 – 18.0 min, 5% MPB. MS/MS analysis was performed on an AB Sciex 5500 TripleQ mass spectrometer (Applied Biosystem/ MDS SCIEX, Foster City, CA, USA) equipped with an ESI TurboIonSpray™ source. The instrument dependent parameters for mass spectrum were set as follows: ionspray voltage, −4.5 kV; ion source temperature, 600°C; nebulizer gas (gas 1), nitrogen, 50 psi; turbo gas (gas 2), nitrogen 50 psi; curtain gas, nitrogen, 20 psi. Unit mass resolution was set in both mass-resolving quadruples Q1 and Q3. Compound-dependent parameters were listed in Table 1.
Table 1.
IC50 Values (in µg/mL) of Anti-proliferation Activities of ME, its Fractions and Key Active Components (MGN, HNK, MHNK), Individually and in Different Ratios in Oral Cancer Cell Lines (Cal-27 and SCC-9) Based on the MTT Assay Run in Triplicate.
| Cell lines | Reagent | Treatment period (hr) |
|||
|---|---|---|---|---|---|
| 24 | 48 | 72 | 96 | ||
| Cal-27 | |||||
| MEa | 36.3 ± 2.6 | 25.7 ± 2.3 | 14.2 ± 2.5 | 8.2 ± 1.9 | |
| MGN | 20.7 ± 1.8* | 11.9 ± 0.6* | 5.1 ± 0.6* | 4.0 ± 0.8* | |
| HNK | 20.8 ± 1.3* | 12.2 ± 1.0* | 6.6 ± 0.6* | 4.2 ± 0.4* | |
| MHNK | 14.5 ± 0.9* | 7.7 ± 0.8* | 5.6 ± 1.4* | 3.2 ± 0.3* | |
| Fraction 1 | >100* | ||||
| Fraction 2 | 72.1 ± 2.3* | ||||
| Fraction 3 | 61.8 ± 9.4* | ||||
| Fraction Ab | 22.5 ± 1.4 | ||||
| 1:1:1 (MGN:HNK:MHNK) | 4.8 ± 0.2* | ||||
| 1:2:6 (MGN:HNK:MHNK) | 3.8 ± 0.2* | ||||
| 1:2:3 (MGN:HNK:MHNK) | 3.9 ± 0.3* | ||||
| 5-FU (positive control) | 0.4 ± 0.1* | ||||
| SCC-9 | |||||
| MEa | 14.4 ± 2.9 | ||||
| MGN | 7.9 ± 0.4* | ||||
| HNK | 5.5 ± 0.6* | ||||
| MHNK | 5.2 ± 1.3* | ||||
| Fraction 1 | > 100* | ||||
| Fraction 2 | > 100* | ||||
| Fraction 3 | 99.5 ± 0.6* | ||||
| Fraction Ab | 19.6 ± 1.7 | ||||
| 5-FU (positive control) | 1.9 ± 0.4* | ||||
Ratio of three key active ingredients in ME was 1.0 : 1.4 : 1.2 (MGN : HNK : MHNK)
Ratio of compounds in Fraction A was 1.0 : 0.82 : 0.28 (MGN : HNK : MHNK)
indicates significant different with the respective the respective IC50 value of ME. (p < 0.05). Statistical analysis was performed using one-way ANOVA with Tukey similarity test.
2.3-. Identification of ME Key Active Components
Activity-Guided Fractionation-
Column chromatography was used to separate ME into Fractions 1, 2, 3 that contained none or trace amounts of the three key major components or KACs (HNK, MGN, MHKN), and Fraction A contains all of three major components. The contents of ME components in fraction A were as follows MGN: 36.9 ± 12.8% (w/w), HNK: 30.4 ± 10.6% (w/w), MHNK 10.5 ± 0.9% (w/w). Elution solvents were 30%, 50%, 80%, 100% MeOH for Fraction 1, 2, A, 3, respectively. The anti-proliferation activities of each fractions were determined using MTT assay in oral cancer cell lines Cal-27, SCC-9 and SCC-4 to identify the key active components of ME.
Anti-proliferation Assay-
The anti-cancer activities of ME, individual KACs and ME fractions were determined in Cal-27, SCC-9 and SCC-4 (oral squamous cell carcinoma cell lines) using MTT assay and reported as IC50 values. The initial concentrations of cell studies for MTT assay was selected based on likely in vivo exposure levels upon dosing, and gradually increase until an IC50 value can be determined. The cells were cultured in DMEM medium (Hyclone, USA), supplemented with 10% of fetal bovine serum in 5% CO2 at 37°C. The assays were performed followed MTT method in 96-well plate. Briefly, 6000 cells were seeded into each well and allowed to adhere for 24 hours before treatment. The growth medium in each well was replaced by the medium containing test compound(s)/fractions at suitable concentration. The cell viability was measured at the desired end point using colorimetry and cell growth curve was plotted.
2.4-. Method Development and Validation
Standard Samples-
Calibration standard samples were prepared in 80% MeOH by diluting MGN, HNK, and MHKN stock solution to final concentration of 10000.0, 5000.0, 2500.0, 1250.0, 625.0, 312.5, 156.3, 78.1, 39.1, 19.5, 9.8 and 4.9 nM, respectively. The calibration standard samples were prepared by spiking 10 µL of blank mouse blood with samples in 80% MeOH (10 µL) and 200 µL of internal standard (IS - Baohuoside I) in ethyl acetate.
Quality Control Samples-
The quality control (QC) samples for each compound were prepared at three different concentrations (high, medium and low) in the same way as the blood samples for calibration were prepared. All the analyzed and QC samples were prepared in the same day with UPLC-MS/MS analysis.
Method Validation-
Details of method validation procedure is given in the method section of Supplementary Material. Briefly, calibration curves were prepared and linearity, lower limit of detection (LLOD), lower limit of quantification (LLOQ), and other validation parameters were established as published before 34. Due to unavailability of analytical standards for metabolites, standard curves of the respective parent compounds were used for metabolites quantification.
2.5-. Solubility Determination
For each medium, weigh approximately 100 mg ME into glass tubes. Add 0.5 mL of each medium into its respective tube. The tubes were then shaken at 100 rpm overnight in the shaker. Samples were then collected and analyzed by UPLC-UV.
2.6-. Caco-2 Monolayer Transport Study
Cell culture:
Caco-2 TC7 cells were originally a kind gift of Prof. Monique Rousset of INSERMU178 (Villejuif, France). The Caco-2 cells have been grown and maintained in our laboratory for about three decades until now 35. A cell monolayer was prepared by seeding 400,000 cells per insert (Nunc, surface area = 4.2 cm2, 3 µm pore size). Cells were maintained at 37 oC under 90% humidity and 5% CO2. Monolayers were used between 19 and 22 days after seeding. The integrity of each monolayer was checked by measuring the transepithelial electrical resistance (Millicell ERS) before the experiment. The normal TEER values obtained were between 500 and 750 Ω·cm2. Cell monolayers with TEER values of < 400 Ω·cm2 were not used. HBSS (9.8 g/mL) supplemented with NaHCO3 (0.37 g/L), HEPES (5.96 g/L), and glucose (3.5 g/L) was used for all experiments after the pH had been adjusted to a desired value (pH = 7.4).
Apical to Basolateral Transport: -
The experiment protocol was followed as described previously 36. Briefly, ME solution in HBSS buffer was loaded onto the apical (donor) side and blank HBSS buffer was loaded on the basolateral (receiver) side. Five donor samples (400 µL) and five receiver samples (400 µL) were taken, followed by the addition of 400 µL of fresh ME solution to the donor side or 400 µL of fresh blank buffer to the receiver side. Samples were collected at 0, 1, 2, 3, 4 hours to obtain 3–4 data points within the linear range of apical to basolateral epithelial transport in order to calculate the transport values. Both ME solutions and blank buffer are maintained at 37ºC throughout the study. 200 µL of IS in MeOH was added to each sample. Sample was stored in −80ºC till analyzed using UPLC-UV. The apparent permeability coefficient (P) was determined by the equation
where, dQ/dt is the drug permeation rate (in mMol/s), A is the surface area of the epithelium (in cm2), and Co is the initial concentration in the donor compartment at time 0 (in μMol).
2.7-. In vitro Metabolism Studies
Preparation of Mouse Liver and Intestinal Microsomes and S9 -
Pooled male C57BL/6 (from 10 mice, 25–30 grams, 12 week old) intestinal S9 fraction, liver microsomes and liver S9 fractions were prepared as described previously 37. The resulting microsome & S9 fraction were suspended in 250 mM sucrose solution, separated into microcentrifuge tubes, and stored at −80°C until use. Protein concentrations of microsomes were determined according to the manufacturer’s instructions, using bovine serum albumin as the standard.
Measurement of UGT Activities-
The glucuronidation procedures were used as those published previously from our group 38. Genistein was used as a positive control. The procedure for glucuronidation were as follows: (1) Liver S9 fraction/liver microsome/intestine S9 fraction (final concentration = 0.0053 mg protein/mL), magnesium chloride (0.88 mM), saccharolactone (4.4 mM), alamethicin (0.022 mg/mL), 5.0/1.0/0.5 µM HNK/MGN/MHNK, 50 mM KPI buffer (pH 7.4), and UDPGA (3.5 mM, add last); (2) incubate the mixture (final volume = 200 µL) at 37 oC for 60 min; (3) end the reaction with addition of 200 µL methanol. The samples were extracted and analyzed using UPLC-MS/MS.
Measurement of CYP Activities-
The incubation procedures for CYP reaction using liver microsomes were the same as those published previous by our laboratory 38. Briefly, the steps were: (1) mix microsomes (final concentration ≈ 0.02 mg protein/mL), NADP (2.61 mM), 6-DP (6.6 mM), MgCl2 (6.6 mM), 5.0/1.0/0.5 µM MHNK in a 50 mM potassium phosphate buffer (pH 7.4), and 6-DPD in 5 mM sodium citrate solution (40 U/mL, add last); (2) incubate the mixture (final volume = 200 μl) at 37°C for 1 h; (3) end the reaction with the addition of 200 µL MeOH. The samples were extracted and subjected to UPLC-MS/MS system for analysis. Formation rates of 6β-hydroxytestosterone from 1 μM testosterone was used as positive control.
2.8-. Pharmacokinetics Study
Animals -
C57BL/6 mice (20–25 g, 8–10 weeks old) were purchased from Taconic Biosciences, Inc. (New York, USA) and acclimatized in an environmentally controlled room (temperature: 25 ± 2°C, humidity: 50 ± 5%, 12 hours dark-light cycle) for at least one week before the experiment.
ME Nanosuspension -
ME formulation for I.V. tail vein injection and oral administration was prepared using Solvent anti-solvent precipitation technique 39 by dissolving ME in 2% DMSO and 2% EtOH, followed by addition of 10% PEG 400 and 10% Tween 80 in sequence. Drug solution was vortexed carefully and diluted with saline to the desired volume. The clear drug solution was aseptically filtrated through 0.2 µM membrane filter. The particle size was determined by diluting ME formulation 500 times in distilled water using Litesizer™ 500 Particle analyzer, Anton Paar, Virginia, USA. The ME formulation particle size was 104.76 ± 27.24 nm.
Animal Dosing and Sample Collection-
Pharmacokinetic studies were performed as per the animal protocol approved by the University of Houston’s Institutional Animal Care and Uses Committee (IACUC). A dose of 60 mg/kg was used because it is equivalent to 276 mg for a 60 kg adult 40 which is closer to the recommended 200 – 400 mg daily dosages of many ME supplements available in the market. A dose of 5mg/kg was used for tail vein injection because ME is poorly soluble and cannot be administered at 60 mg/kg dose safely to the animals. For oral pharmacokinetic studies, ME unformulated (in corn oil) or formulated as nanosuspension was administered by oral gavage at a dose of 60 mg/kg. Blood samples (about 20 µL) were collected in heparinized tubes at 5, 15, 30, 60, 120, 240, 360, 480, and 1440 min, respectively using mouse tail sectioning method. The tubes were heparinized (treated with % of heparin solution and air dried) before sample collection. 20µl of blood sample was collected at each time point using established lab procedure. The blood samples were stored at −80°C until analysis. The samples were thawed and vortexed carefully before the blood was taken for processing and analysis. There was no or very little blood coagulation found upon storage in −80oC.
Samples Analysis-
The whole blood sample (10 µL) was spiked with 10 µL of 80% MeOH and 100 µL of 100 nM internal standard (IS - Baohuoside I) in ethyl acetate. The mixture was vortexed for 1 min. After centrifugation at 15,000 rpm for 15 min, the supernatant was transferred to a new tube and evaporated to dryness under a stream of air flow. The residue was reconstituted in 100 µL of 80% methanol and centrifuged at 15,000 rpm for 15 min. 10 µL of supernatant was injected into the UPLC-MS/MS system for quantitative analysis.
Pharmacokinetic Modeling-
MGN, HNK and MHNK pharmacokinetic parameters were calculated by the non-compartmental method, using WinNonlin 5.2 (Pharsight Corporation, Mountain View, California).
2.9-. Statistical Analysis
All the data in this paper were presented as means ± RSD, if not specified otherwise. Significance differences were assessed by using unpaired Student’s t-test and One-way ANOVA with Tukey similarity test. P value of <0.05 was considered as statistically significant.
3-. Results
3.1-. Identification of Key Active Components of ME-
We applied the approach of inactive species elimination by separating out phytochemical fractions that did not contain any of the key active components (KACs) of ME: magnolol, honokiol and 4-O-methylhonokiol. ME was separated into two Fractions 1 and 2 that did not contain any of the three KACs, Fraction 3 with trace amounts of major compounds and Fraction A contained all three components (36.9 ± 12.8% MGN, 30.4 ± 10.6% HNK, 10.5 ± 0.9% MHNK w/w) (Figure 1).
Figure 1.
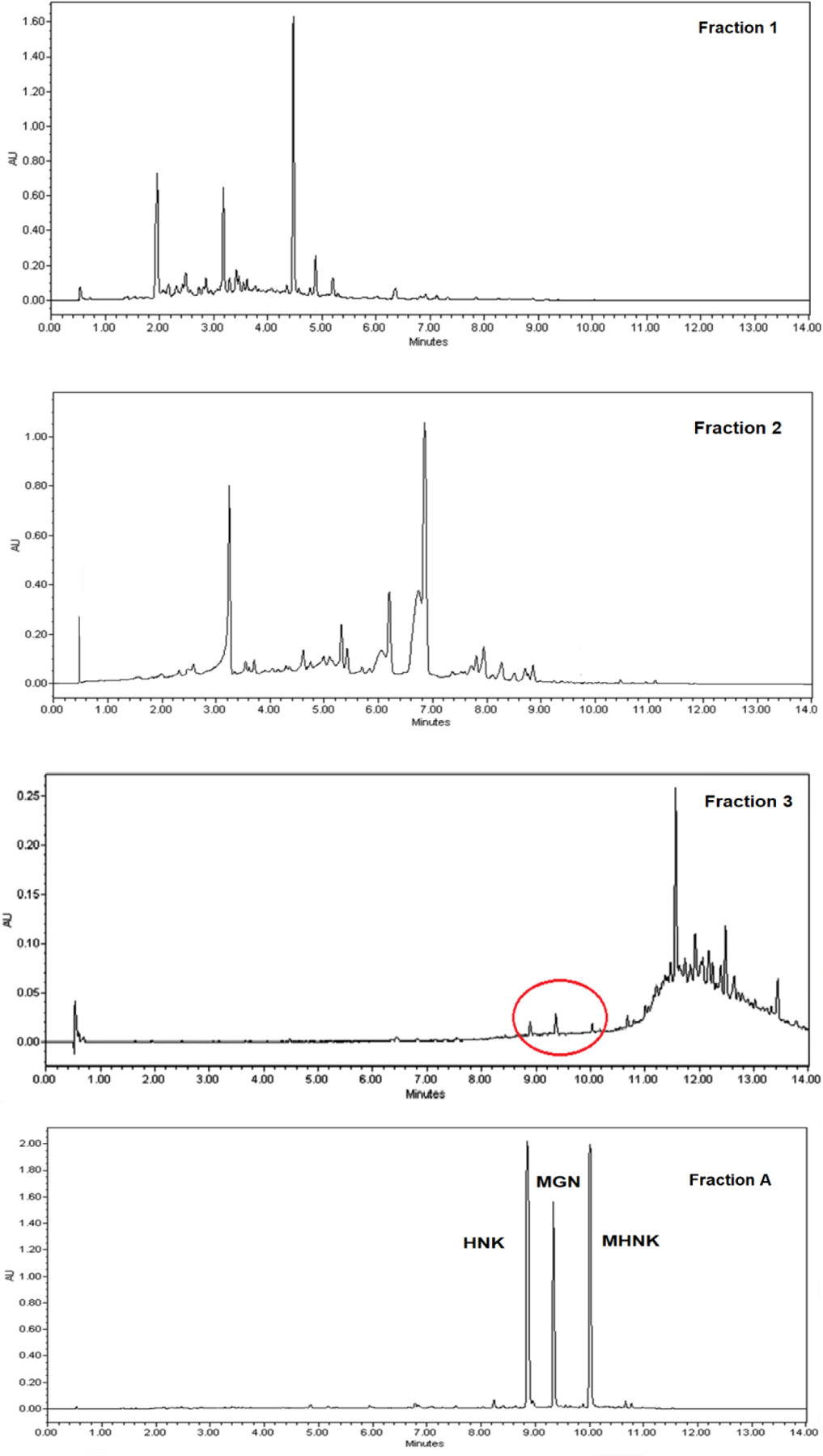
UPLC-UV chromatograms of ME fraction 1, 2, 3, A
The anti-proliferation activities were determined as IC50 values from the MTT assay using oral cancer cell lines SCC-9, Cal-27 (Table 1) and SCC-4 (Supplementary Material, Table S6). Treatment for 72 and 96 hours with ME demonstrated the significant better inhibition effect on the cell lines (p<0.05). The ME and its compounds showed significantly higher antiproliferation activities when the cell lines were treated for 72 and 96 hours than 48 and 24 hours with the mixture/single compounds (p<0.05). Eventually, 72-hour treatment was selected because it provided enough resolution for the purpose of screening different ratios of KACs in various cell lines. The results showed that fraction A was the most active fraction in comparison to other fractions and the whole extract (p < 0.05). In Cal-27 cell line, fraction A was about three times more active than fraction 1, 2 and 3 as based on their IC50 values (22.5 µg/mL compared to 100 µg/mL, 72.1 µg/mL and 61.8 µg/mL, respectively). We also determined the inhibition effect of each compound (MGN, HNK and MHNK) identified in fraction A against oral cancer cell lines. Each KAC showed good anticancer activity but with different inhibition potential (based on IC50 values).
Among ME components, HNK and MHNK were significantly better in inhibition effect against SCC-9 cells than MGN (p < 0.05). However, all three compounds did not show different antiproliferation against Cal-27 cell line. MHNK was surprisingly one of the most active compounds of ME against oral cancer cells.
As expected, when three compounds were mixed in different ratios, different levels of inhibition activities against oral cancer cell line were observed. Among the three different ratios of KACs used in the cell studies, the ratio 1:1:1 was close to the ratio of MGN, HNK and MHNK in commercial ME (1:1.4:1.2). The other two ratios 1:2:3 and 1:2:6 were used to test if the effect of increase in MHKN content in ME on its cancer chemopreventive properties, since MHNK was found to be one of the most active components in the mixture. Interestingly, we found that the 1:1:1 mixture of the three active compounds was three folds more active than ME. Additionally, best activity (lowest IC50) was observed, when content of MHNK was increased in the mixture. The IC50 value of 1:2:6 (MGN: HNK: MHNK) mixture (3.8 µg/mL) and the 1:2:3 mixture (3.9 µg/mL) were significantly better than the 1:1:1 ratio (4.8 µg/mL) (p < 0.05) (Table 1). CompuSyn software was used to test the synergy and no synergistic effect was observed among ME compounds against oral cancer (data not shown).
3.2-. LC-MS/MS Analytical Method Development and Validation-
Method development and validation for the simultaneous quantification of HNK, MGN and MHNK in biological matrix, though extremely challenging, was successfully achieved. It was reported recently that HNK could not be detected with the mass spec detector 41, however we were able to obtain good peak shapes, short analysis time and high sensitivity for all compounds including HNK (Figure 2). Good peak separation was observed for all compounds and their phase II metabolites.
Figure 2.
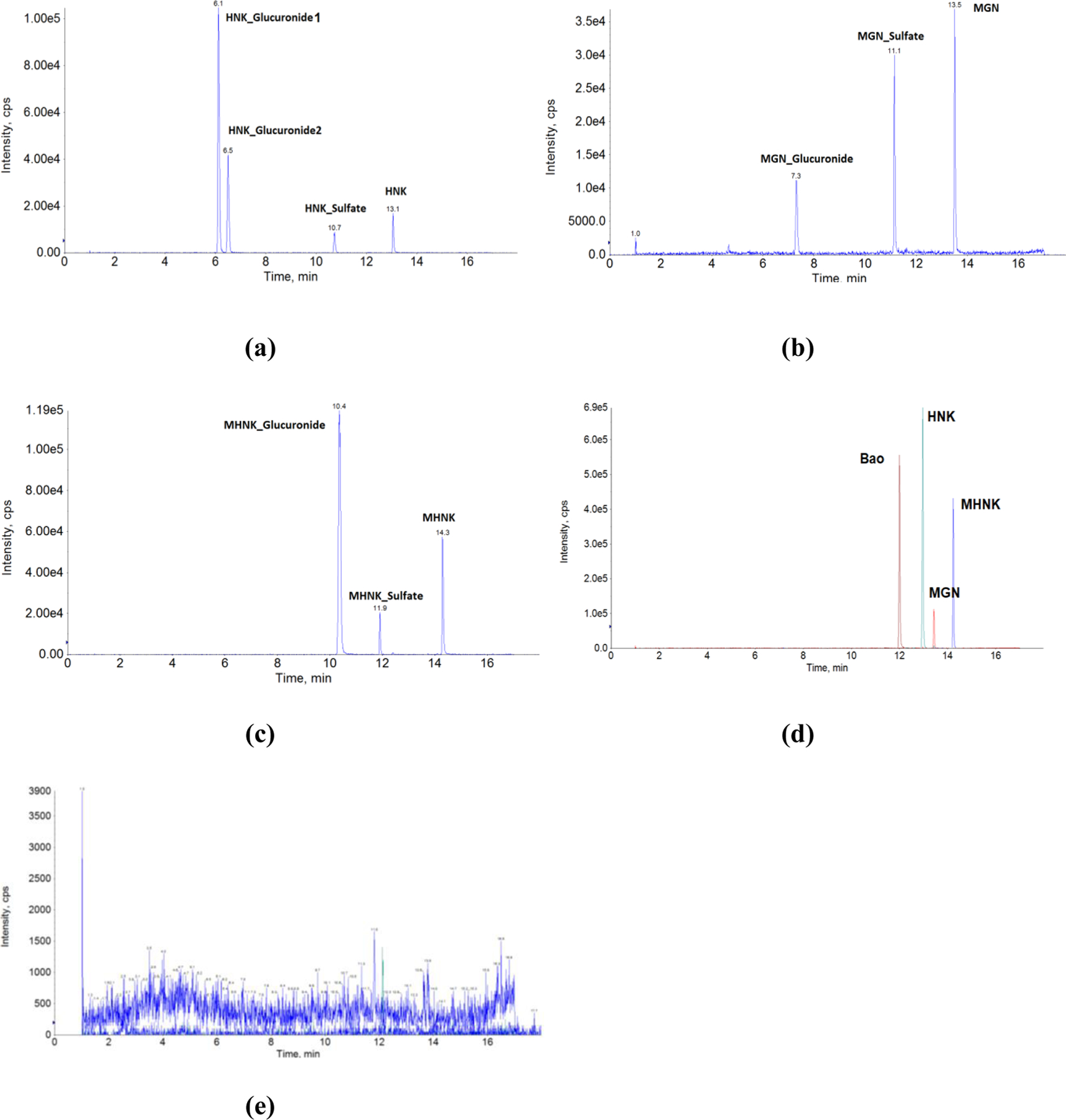
UPLC-MS/MS chromatograms in matrix (mouse blood) of (a) HNK and it phase II metabolites, (b) MGN and its phase II metabolites, (c) MHNK and its phase II metabolites, (d) ME KACs and the internal standard (Baohuoside I), (e) blank matrix.
Method Optimization-
The method was set up by optimizing UPLC and MS/MS condition to obtain the best sensitivity. Methanol, acetonitrile, 2 mM ammonium acetate (pH = 7.6), 0.1% formic acid (pH = 2.5), and 100% water were tested as potential mobile phases. The ionization of MGN, HNK and MHNK in the instrument was the best with acetonitrile and 2 mM ammonium acetate as the mobile phases. A gradient elution was used to avoid cross peaks. In order to obtain a sharp and symmetrical peak, the column temperature was set at 45 oC and the flow rate was 0.45 mL/min. Under this optimized elution scheme, a specific MRM scan was used to improve the analysis specificity. The compounds and instrument dependent parameters were optimized by tuning these three analytes in negative scan mode and the results were shown in Supplementary Material, Table S1.
The problem we found during method development was the low solubilities of those compounds (especially MHNK) in aqueous solutions. In order to solve this problem, samples were extracted with ethyl acetate and reconstituted with 80% MeOH before analyzed by UPLC-MS/MS. We also tried MeOH and ACN as the extracted solvents and it showed comparable results (data were not included). Ethyl acetate was chosen because of cleaner extracted supernatant and very fast evaporation under air flow.
Method Validation-
A highly specific, sensitive, reproducible, and robust LC-MS/MS method was developed for MGN, HNK and MHNK in blank mouse blood. The method validation data were reported in the Supplementary Material of this paper.
Specificity, linearity and sensitivity-
The standard curves were linear in the concentration range of 0.96 – 500 nM for MGN, HNK and 2.4 – 1000 nM for MHNK. The lower limit of quantification (LLOQ) were 7.8 nM for MGN, HNK and MHNK (Supplementary Material, Table S2).
Accuracy and precision -
Accuracy, intra-day and inter-day precision was determined by measuring six replicates of QC samples at low, medium and high concentration levels: 7.8 nM (LLOQ), 125 nM (MQC) and 500 nM (HQC), respectively in mouse blood. The precision and accuracy were shown in the Supplementary Material, Table S2. The intra-day accuracy of MGN, HNK and MHNK were 94.1 – 99.4, 98.4 – 110.2, 93.0 – 116.7% while the precision range were 5.0 – 7.6, 4.3 – 9.6, 8.4 – 10.6%, respectively. Those of the inter-day accuracy were 94.5 – 105.4, 93.2 – 107.0, 90.6 – 11.3% and the precision were 7.1 – 10.1, 3.7 – 12.3, 1.1 – 12.9%, respectively. These results demonstrated that the precision and accuracy values were in the acceptance range (±15% for MQC, HQC and ±20% for LLOQ) 42.
Recovery -
The mean extraction recoveries determined using three replicates of QC samples at three concentration levels (the same concentrations as QC sample) in mouse blood were shown in the Supplementary Material, Table S2. Ethyl acetate was used to extract HNK, MGN, and MHNK in the biological samples. The recovery of HNK were 110.7 ± 11.0%, 95.0 ± 7.5%, 90.2 ± 12.8% for low, medium and high concentration, respectively. Those of MGN were 120.7 ± 4.2%, 117.8 ± 23.0% and 111.9 ± 7.4%. MHNK recovery range were 84.9 ± 1.5%, 79.6 ± 3.3%, 76.0 ± 7.4%. The result showed the recoveries were not less than 70% for these three analytes at low, medium, and high concentrations.
Matrix effect-
The relative peak areas of these three analytes after spiking evaporated blood samples at three concentration levels were comparable to similarly prepared aqueous standard solutions (ranged from 85 to 115%). The results suggested that there was no measurable matrix effect that interfered with ME key active components’ determination in mouse blood.
Stability-
The stabilities of ME KACs in blood samples were evaluated by analyzing three replicates of quality control samples at three different concentrations after short-term (25 oC, 4 h), post-processing (20 oC, 8 h), long-term cold storage (−80 oC, 6 months), and after going through three freeze-thaw cycles (−80 to 25 oC). All the samples displayed 85–115% recoveries after various stability tests (Supplementary Material, Table S3).
3.3-. Absorption, Metabolism and Pharmacokinetic Studies
ME-KACs exhibited good transport across Caco-2 cells monolayer.
The permeabilities of ME KACs were investigated using Caco-2 cell monolayer transport model. The vectorial transport result showed that the apical to basolateral permeability (Pab) of MGN, HNK and MHNK were 9.0 ± 0.9 × 10−6, 13.4 ± 0.7 × 10−6 and 7.7 ± 0.4 × 10−6 cm/sec, respectively. The permeability of HNK was significantly better than MGN and MHNK (p<0.05) (Figure 3).
Figure 3.
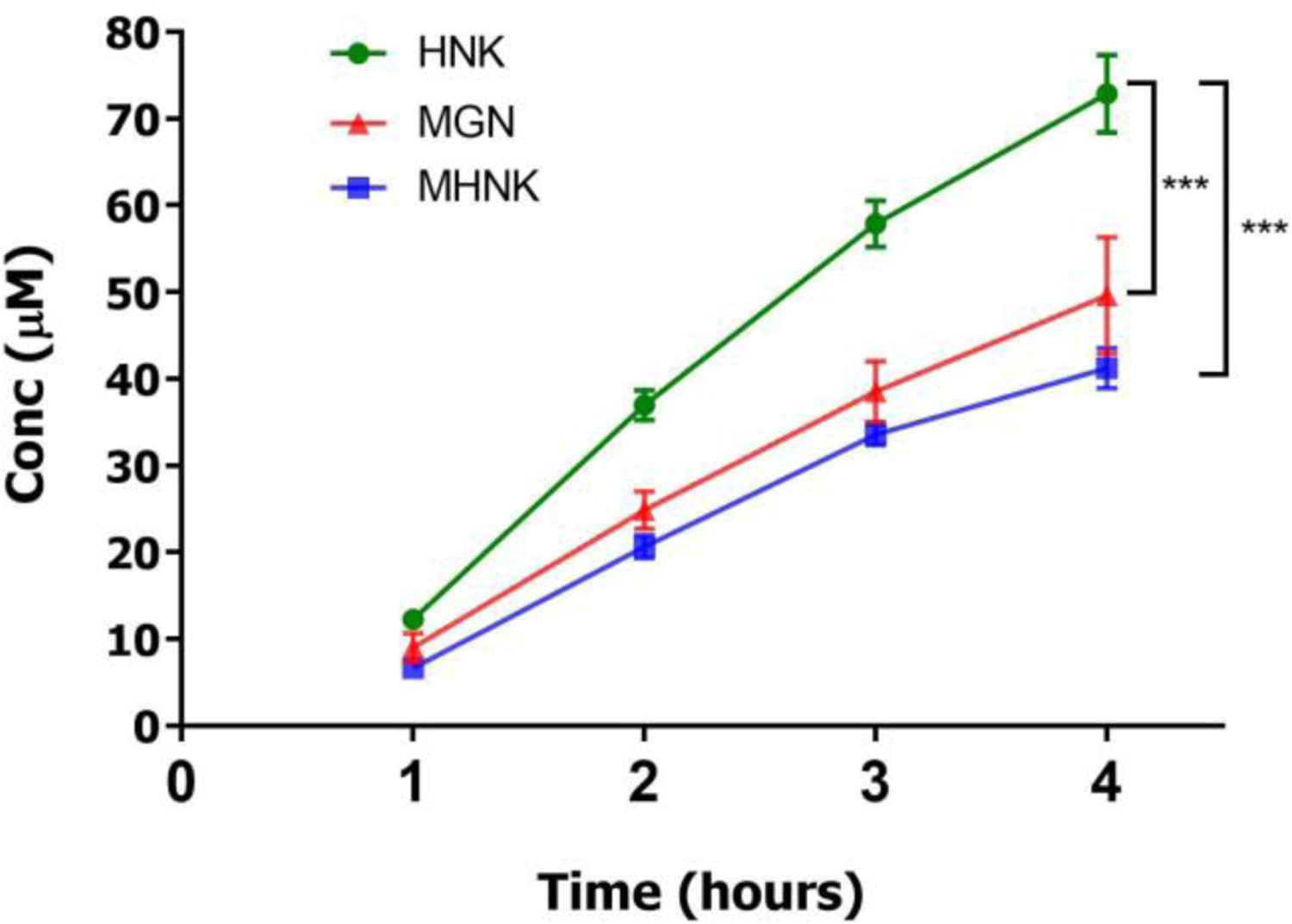
Transport of ME at 0.05 mg/mL concentration in HBSS buffer (pH 7.4) from apical to basolateral side of Caco-2 cell monolayer seeded on transwell membrane at 37 oC for 4 hours. ME solution was added on the apical side and blank buffer was added on the basolateral side. Samples were collected every 60 mins and equal volume of ME solution and blank buffer were added on apical and basolateral side respectively. Experiment was run in n=3 wells and each data point was presented as mean ± SD. Rates of apical to basolateral transport of magnolol, MGN; honokiol, HNK; and 4-O-methylhonokiol, MHNK was calculated using linear regression of first three data points (linear range). The asterisk (*) indicates indicate significant difference (p < 0.05, unpaired Student T-test).
MHNK got metabolized to HNK by CYP450.
MHNK has been reported to transform into HNK by rat CYP450 43. We confirmed this metabolic pathway in mice using different concentrations of MHNK (5, 1 and 0.5 μM) in mouse liver microsomes, liver S9 fraction and intestine S9 fraction. Concentrations of MHNK decreased and those of HNK increased in a time-dependent and concentration-dependent manner in the reaction mixtures (Figure 4) (p < 0.05). The results indicated that MHNK were metabolized by Cytochrome P450 and HNK was one of its metabolites. After 60 min, 51 ± 23.9% of the MHNK was metabolized to HNK in the liver microsomes reaction mixture at the substrate concentration of 0.5 µM (Figure 4c). There was no HNK detected in the intestinal S9 fraction reaction (Figure 4a). The sum of MHNK and HNK did not equal to 100% because both MHNK and HNK are metabolized to other phase I metabolites, which has been shown previously 43. These results confirm the conversion to HNK by CYP enzyme as one of the metabolic elimination pathways of MHNK, which was also supported by the observed in vivo PK profiles of HNK and MHNK.
Figure 4.
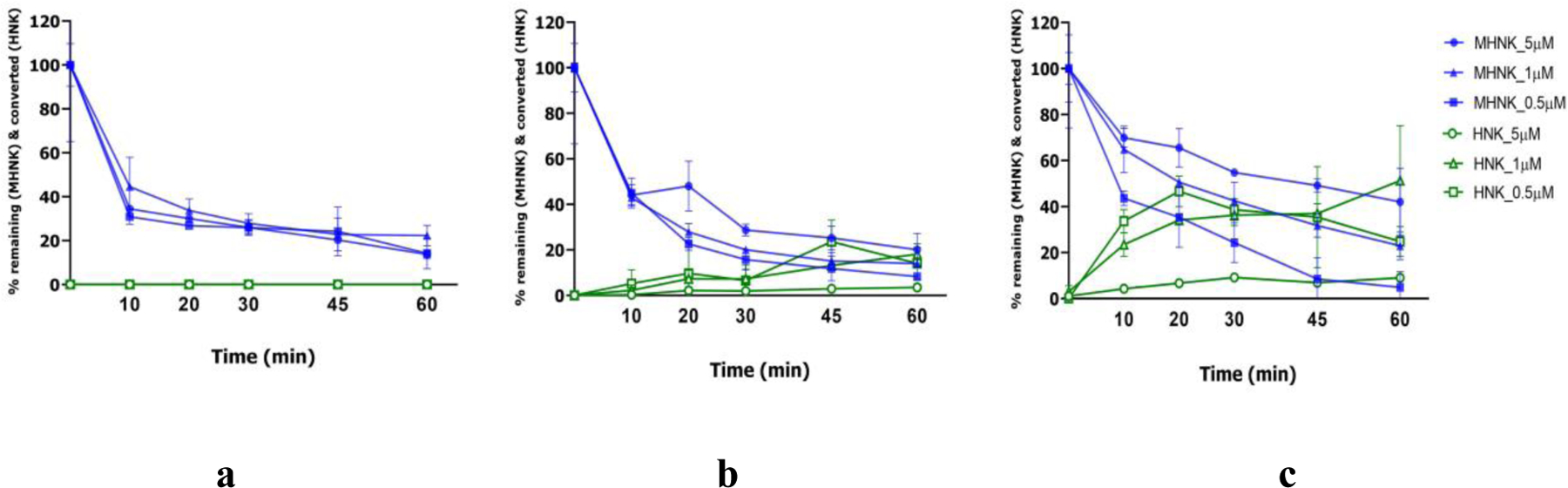
Percentage fraction of 4-O-methylhonokiol, MHNK remaining (blue) and converted to honokiol, HNK (green) in C57BL/6 mouse intestinal S9 fraction (a), liver S9 fraction (b) and liver microsomes (c) at 0.02 mg/mL final protein concentration and three MHNK concentrations (5, 1 and 0.5 μM) in the phase I CYP metabolism reaction mixture (200 µL) incubated at 37oC for 60 mins. Each experiment was run in triplicates and results were presented as mean ± SD.
Pharmacokinetics of ME KACs in mice showed low bioavailability.
We studied I.V. and oral pharmacokinetic behaviors of ME KACs in C57/BL6 mice. Mean blood concentration-time curves of MGN, HNK and MHNK and their phase-II metabolites after I.V. administration of 5 mg ME/kg (equal to 0.72 mg/kg MGN, 1.03 mg/kg HNK and 0.83 mg/kg MHNK) were illustrated in Figures 6a and 6b. The AUC0−∞ values of MGN, HNK and MHNK were 1232.2 ± 649.6, 1150.6 ± 719.3 and 623.2 ± 358.8 hr*µg/L, respectively (Table 2). The three KACs of ME were rapidly eliminated from systemic circulation. Half-lives of MGN, HNK and MHNK were 1.0 ± 0.3, 0.7 ± 0.04 and 2.9 ± 2.1 hours, respectively (Table 2).
Figure 6.
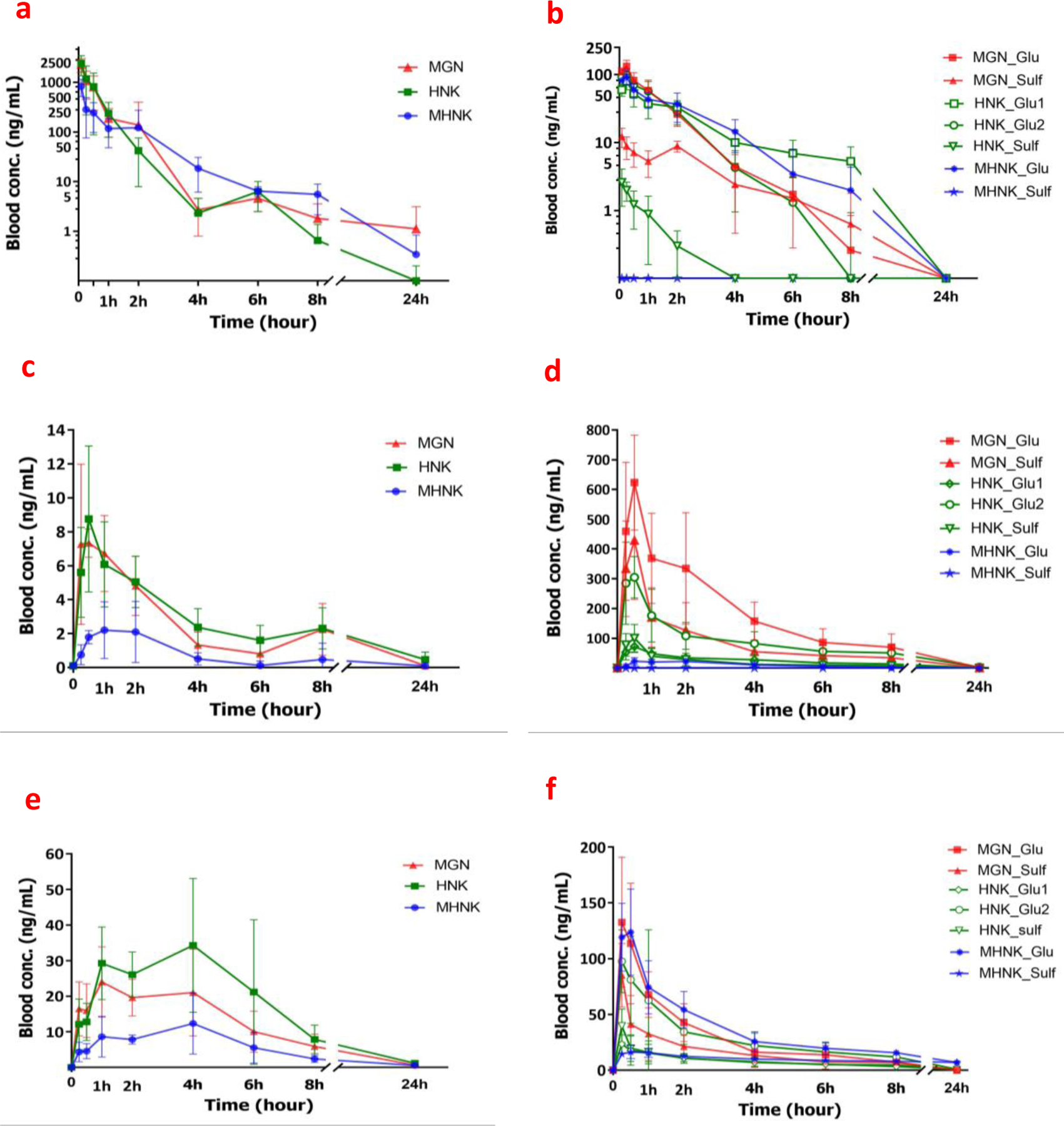
Concentrations of ME key active components (magnolol, MGN; honokiol, HNK; and 4-O-methylhonokiol, MHNK) and their phase-II metabolites (glucuronide, Glu; and sulfate, Sulf) in whole blood of C57BL/6 mice (n = 5) after I.V. administration of 5 mg/kg ME in nanosuspension (a, b), oral administration of 60 mg/kg ME dissolved in corn oil (c, d), and oral administration of 60 mg/kg ME nanosuspension (e, f). Each data point was represented as mean ± SD of n=5 mice.
Table 2.
Pharmacokinetic Parameters of MGN, HNK, MHNK and Their Respective Metabolites (Glucuronides and Sulfates) After I.V. Administration of 5 mg ME/kg (n = 5)
| Parameters | Tmax (hr) | Cmax (ng/mL) | AUC0–24 (hr*ng/mL) | AUC0−∞ (hr*ng/mL) | Half-life (hr) | V (L/kg) | CL (L/hr/kg) | MRT0–24 (hr) |
|---|---|---|---|---|---|---|---|---|
| MGN | - | 2284.4 ± 700.5 | 1232.2 ± 649.6 | 1234.1 ± 651.2 | 1.0 ± 0.3 | 1.0 ± 0.4 | 0.8 ± 0.6 | 0.8 ± 0.5 |
| MGN_Glucuronide | 0.2 ± 0.1 | 137.0 ± 21.6 | 171.7 ± 39.4 | 173.3 ± 39.6 | 0.9 ± 0.2 | - | - | 1.1 ± 0.2 |
| MGN_Sulfate | 0.1 ± 0.1 | 17.4 ± 3.5 | 41.7 ± 12.1 | 49.0 ± 14.6 | 2.2 ± 0.7 | - | - | 2.1 ± 0.5 |
| HNK | - | 2400.5 ± 1041.4 | 1150.6 ± 719.3 | 1151.3 ± 719.1 | 0.7 ± 0.04 | 1.3 ± 1.0 | 1.2 ± 0.9 | 0.6 ± 0.1 |
| HNK_Glucuronide1 | 0.2 ± 0.1 | 102.7 ± 17.5 | 156.9 ± 36.6 | 158.3 ± 37.1 | 0.8 ± 0.3 | - | - | 1.2 ± 0.3 |
| HNK_Glucuronide2 | 0.3 ± 0.1 | 83.0 ± 15.7 | 164.2 ± 39.1 | 186.4 ± 57.3 | 2.7 ± 1.1 | - | - | 2.1 ± 0.3 |
| HNK_Sulfate | 0.1 ± 0.1 | 2.8 ± 1.2 | 2.3 ± 1.1 | 4.0 ± 2.3 | 2.0 ± 1.0 | - | - | 1.0 ± 0.4 |
| MHNK | - | 824.3 ± 309.5 | 623.2 ± 358.8 | 631.1 ± 360.0 | 2.9 ± 2.1 | 5.5 ± 1.7 | 2.0 ± 1.6 | 1.5 ± 0.6 |
| MHNK_Glucuronide | 0.2 ± 0.1 | 95.6 ± 17.4 | 180.5 ± 62.2 | 191.3 ± 63.7 | 1.4 ± 0.6 | - | - | 1.7 ± 0.6 |
| MHNK_Sulfate | - | - | - | - | - | - | - | - |
In the oral PK studies of with 60 mg/kg ME in corn oil (equivalent to 8.58 mg/kg MGN, 12.36 mg/kg HNK and 9.95 mg/kg MHNK), bioavailability of ME active components were very low (< 0.2%) (Figure 6c and Table 3). The low solubility (Supplementary Material, Table S4) and/or extensive metabolism of these compounds (Figure 4 and Figure 5) were found to be the major contributing factors towards their poor bioavailabilities.
Table 3.
Pharmacokinetic Parameters of MGN, HNK, MHNK and Their Respective Metabolites (Glucuronides and Sulfates) After Oral Administration With 60 mg ME/kg dose in Corn Oil (n = 5)
| Parameters | Tmax (hr) | Cmax (ng/mL) | AUC0–24 (hr*ng/mL) | AUC0−∞ (hr*ng/mL) | Half-life (hr) | MRT0–24 (hr) |
|---|---|---|---|---|---|---|
| MGN | 0.9 ± 0.7 | 9.4 ± 3.3 | 23.1 ± 5.3 | 37.8 ± 16.5 | 3.8 ± 2.3 | 2.3 ± 0.6 |
| MGN_Glucuronide | 0.9 ± 0.7 | 697.5 ± 79.0 | 1830.5 ± 352.4 | 2320.1 ± 959.5 | 4.6 ± 3.0 | 3.0 ± 0.6 |
| MGN_Sulfate | 0.6 ± 0.3 | 477.7 ± 169.5 | 789.3 ± 195.2 | 1039.3 ± 223.3 | 4.5 ± 2.2 | 2.3 ± 0.4 |
| HNK | 1.0 ± 0.7 | 9.3 ± 3.8 | 26.4 ± 6.7 | 41.5 ± 17.8 | 4.2 ± 2.1 | 2.7 ± 0.5 |
| HNK_Glucuronide1 | 0.6 ± 0.3 | 80.4 ± 12.6 | 302.4 ± 90.8 | 338.6 ± 78.5 | 3.6 ± 1.4 | 4.0 ± 1.0 |
| HNK_Glucuronide2 | 0.5 ± 0.3 | 368.9 ± 69.4 | 1242.8 ± 359.5 | 1268.3 ± 347.4 | 4.5 ± 2.2 | 4.8 ± 0.5 |
| HNK_Sulfate | 0.6 ± 0.3 | 113.5 ± 36.2 | 192.8 ± 64.8 | 260.8 ± 60.7 | 4.6 ± 1.3 | 2.6 ± 0.6 |
| MHNK | 1.3 ± 0.6 | 3.7 ± 0.9 | 9.6 ± 4.0 | 10.8 ± 3.0 | 1.4 ± 0.6 | 1.8 ± 0.3 |
| MHNK_Glucuronide | 1.0 ± 0.6 | 30.9 ± 8.1 | 96.4 ± 30.8 | 107.4 ± 44.9 | 2.3 ± 1.9 | 2.7 ± 0.4 |
| MHNK_Sulfate | - | - | - | - | - | - |
Figure 5.
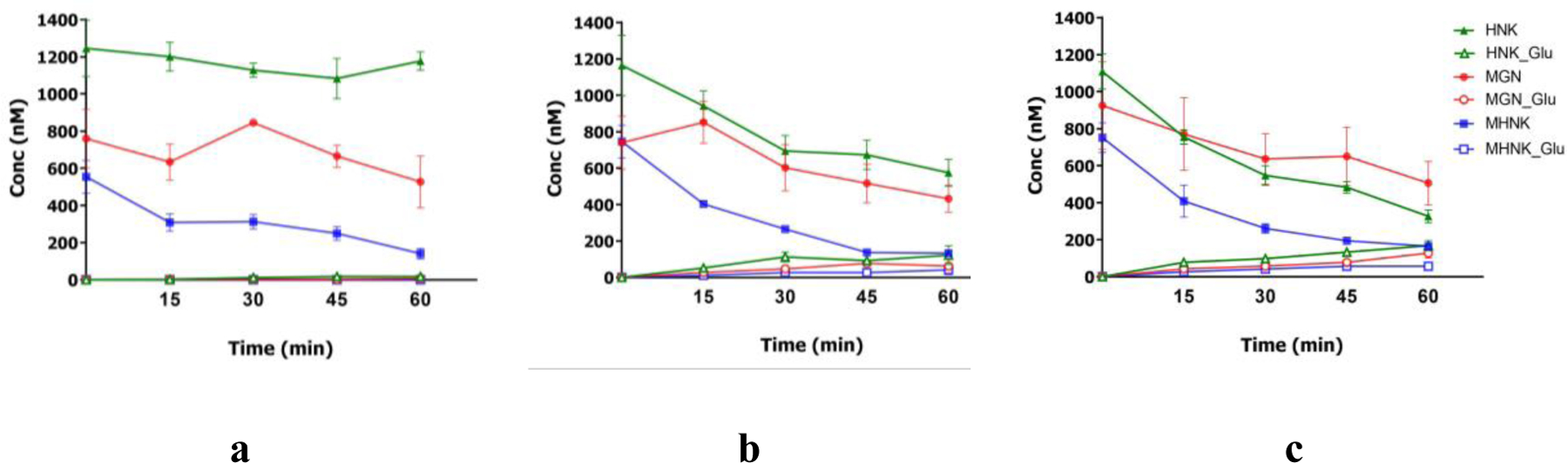
Concentrations of magnolol, MGN; honokiol, HNK; and 4-O-methylhonokiol, MHNK and their respective glucuronides in mouse intestine S9 fraction (a), liver S9 fraction (b) and liver microsome (c) at 0.0053 mg/mL final protein concentration in phase II UGT metabolism reaction mixture (200 µL) incubated at 37oC in 60 mins. Each experiment was run in triplicates and results were presented as mean ± SD.
ME-KACs were extensively metabolized by UGTs.
Since, glucuronides were found to be the major metabolites of KACs in in vivo pharmacokinetic studies of ME (Figure 6), their metabolism by UDP-glucuronosyltransferase (UGT) was investigated. Results from in vitro glucuronidation reaction showed that ME-KACs were extensively and rapidly metabolized by mouse liver and intestinal microsomes/S9 fractions. Concentration of MHNK, HNK and MGN declined rapidly in a concentration-dependent and time-dependent manner in the reaction mixtures (p < 0.05). No significant change was observed in the concentration of HNK, MGN or MHNK in the absence of the cofactor (data not shown). The rates of glucuronidation of ME KACs were highest in liver microsomes, followed by liver S9 fraction and intestine S9 fraction (p < 0.05; one-way ANOVA with Tukey similarity test) (Figure 5).
ME nanosuspension significantly increased bioavailability of ME components.
The oral pharmacokinetic of ME (at same dose of 60 mg/kg) in solubility enhancing formulation (using PEG 400 and Tween 80) was performed. The blood concentration-time profiles of ME-KACs and their phase-II metabolites were illustrated in Figure 6e and 6f. The pharmacokinetics profiles of MGN, HNK and MHNK in formulated ME were significantly different (higher Cmax, Tmax and longer half-life) (p < 0.05) from the ones in unformulated ME (Figure 6c and 6e).
The bioavailability of ME-KACs in formulation was significantly increased as compared to ME-KACs in corn oil, whereas the metabolism decreased. AUCs of all three KACs increased about 8–10-folds (Tables 3 and 4), whereas of their glucuronides decreased by approximately 10-folds. However, even with the significant solubility increase after the application of formulation and change in parent and glucuronide systemic exposure, the absolute bioavailability of MGN, HNK and MHNK were still low (< 2%). The estimated bioavailability values were 1.2%, 1.8% and 1.1% for MGN, HNK and MHNK, respectively (Table 4).
Table 4.
Pharmacokinetic Parameters of MGN, HNK, MHNK and Their Respective Metabolites (Glucuronides and Sulfates) After Oral Administration With 60 mg ME/kg Dose in Nanosuspension (n = 5).
| Parameters | Tmax (hr) | Cmax (ng/mL) | AUC0–24 (hr*ng/mL) | AUC0−∞ (hr*ng/mL) | Half-life (hr) | MRT0–24 (hr) |
|---|---|---|---|---|---|---|
| MGN | 2.2 ± 1.5 | 29.1±10.4* | 176.4 ± 27.3* | 181.9±28.8* | 5.0 ± 1.5 | 5.1 ± 0.6* |
| MGN_Glucuronide | 0.4 ± 0.1 | 157.4±54.4* | 260.4 ± 69.4* | 283.4±74.6* | 2.2 ± 0.2 | 2.1 ± 0.3* |
| MGN_Sulfate | 0.3 ± 0.0 | 429.3 ± 138.0 | 803.8 ± 151.7 | 1041.3 ± 233.6 | 3.1 ± 0.8 | 2.8 ± 0.6 |
| HNK | 3.5 ± 1.8* | 42.5 ± 14.8* | 253.5 ± 45.4* | 263.4±46.9* | 5.3 ± 1.5 | 5.5 ± 0.6* |
| HNK_Glucuronide1 | 0.3 ± 0.1 | 15.0 ± 6.7* | 34.5 ± 10.1* | 37.8 ± 11.2* | 2.1 ± 0.6 | 2.5 ± 0.4* |
| HNK_Glucuronide2 | 0.3 ± 0.1 | 64.0 ± 19.7* | 130.5 ± 39.1* | 164.2±47.9* | 3.7 ± 1.4 | 2.6 ± 0.3* |
| HNK_Sulfate | 0.3 ± 0.0 | 39.8 ± 15.1* | 118.2 ± 25.3* | 121.4 ± 24.9* | 5.1 ± 0.7 | 5.2 ± 0.4* |
| MHNK | 3.0 ± 2.0 | 14.3 ± 6.6* | 79.9 ± 15.8* | 86.3 ± 15.7* | 7.1±2.5* | 5.9 ± 0.7* |
| MHNK_Glucuronide | 0.5 ± 0.3 | 57.1 ± 8.6* | 102.5 ± 31.5 | 106.2 ± 33.4 | 1.6 ± 0.4 | 1.8 ± 0.4* |
| MHNK_Sulfate | 0.5 ± 0.3 | 2.0 ± 1.6* | 1.1 ± 0.6 | 1.7 ± 0.9* | - | 0.7 ± 0.4* |
indicates significant differences with the respective parameter obtained from oral PK study of ME in Corn oil (in Table 3) (p < 0.05). Statistical analysis was performed using Student t-test.
ME components underwent extensive phase II metabolism and enteric and enterohepatic recycling
After I.V. injection of ME as well as oral administration, ME phase II metabolites (glucuronide and sulfate conjugates) showed up very fast (5 min in I.V and 15 min in oral gavage) (Figure 6). There were higher amounts of ME metabolites measured in the oral gavage of ME in corn oil as compared to I.V. administration (Figures 6b and 6d). Cmax for glucuronides were achieved quickly, in about 10 min after I.V. administration, 40 min after oral administration in corn oil, and 20 min after oral administration as nanosuspension. Tmax for glucuronides were found to be comparable to the parent compounds (Table 2, 3 and 4).
The Cmax of KAC-glucuronides after oral administration was much higher than the parents suggesting that ME-KACs are extensively metabolized in intestinal cells. The AUC0–24 for sulfate did not change but significantly reduced (~ 10-folds) for glucuronides when ME was orally administered as nanosuspension as compared to in corn oil, however it did not significantly increase the absolute bioavailabilities. PK profiles of ME KACs showed the recycling peak for MGN, HNK and MHNK at about 6–8 hours (Figures 6a and 6c) after I.V. or oral (in corn oil) administration and at about 4 hours (Figure 6e) after oral administration as nanosuspension.
For honokiol, two monoglucuronide and one monosulfate, whereas for magnolol, one monoglucuronide and one monosulfate were the major metabolites measured in mouse blood (Figures 2b and 2c). On the other hand, only one monoglucuronide was found in measurable concentration for 4-O-methylhonokiol (Figure 2d). There was no diglucuronide of disulfate conjugate detected in mouse blood. The position of glucuronidation for two HNK glucuronides can only be confirmed using NMR.
There could be two possible reasons for the detection of only one sulfate of HNK. First, most UGT isoforms have a large substrate binding pocket capable of forming multiple glucuronides from the same substrate as published earlier 44–46. On the other hand, SULT enzyme has much smaller binding pocket and are more prone to the effects of steric hindrance 47. Second, the other HNK sulfate though formed was very small in quantity and may be below the detection limit of LCMS method used.
4-. Discussion
This is the first study with detailed pharmacokinetic profiling and identification of key active components (KACs) of Magnolia officinalis bark extract (ME) responsible for oral health in humans. Magnolol (MGN), honokiol (HNK) and 4-methylhonokiol (MHNK) were found to be the three major KACs of ME that showed good antiproliferative properties against oral cancer and therefore could be responsible for promoting and maintaining oral health in general. However, the low oral bioavailability of ME due to poor solubility and extensive first pass metabolism of KACs severely limits ME potential to be used as dietary supplement for oral cancer chemoprevention. Since, all KACs showed good membrane permeabilities, ME is a good candidate for development of local delivery in the oral cavity to maximize its oral health benefits. Moreover, MHNK, one of the most active KACs of ME against oral cancer, should be included for the quality control of ME supplement products.
Non-standardized ratio of the KACs in the various marketed ME formulation can lead to variable benefits among population. MGN, HNK and MHNK were found to be the three KACs of ME with significant activities against oral cancer cell lines (Table 1) as evident by the 3–5 folds higher (p < 0.01) anti-proliferation activity (IC50 values) of fraction A (containing most of MGN, HNK and MHNK) as compared to the other fractions (with insignificant amounts of KACs) (Figure 1). While MGN and HNK are usually reported as the leading compounds in the published literature, MHNK surprisingly demonstrated similar efficacy to HNK and increasing its content in ME significantly improved the activity. The mixture with highest amount of MHNK (1: 2: 3 :: MGN: HNK: MHNK) was about 5 times more active (p < 0.01) than the commercial ME (1: 1.4: 1.2 :: MGN: HNK: MHNK) (Table 2). On the product label of most marketed ME supplements, the content of only MGN, HNK or both can be usually found without any mention of MHNK. Our study has demonstrated that due to the differences in oral cancer chemopreventive activities of the KACs, ME mixture should be standardized for the ratio of all 3 KACs and MHNK content should be an added quality control parameter along with MGN and HNK contents on the product label.
We found that the biggest challenge in the development of Magnolia Extract (ME) as a chemopreventive agent against oral carcinoma is its likely poor target tissue (oral) concentration. The latter is caused by extensive first-pass metabolism and poor solubility leading to low systemic exposure upon oral administration. The moderate effective permeability values of KACs in the Caco-2 cells (Figure 3) were comparable with the reported ones in rats 43 and suggested that intestinal permeation was not the contributing factor towards the poor bioavailability of ME. On the other hand, extensive hepatic and intestinal glucuronidation (Figure 4–5) is the primary elimination pathway for ME-KACs in mice, as evident by the short half-lives of MGN (1 hr), HNK (0.7 hr) and MHNK (3 hrs) after I.V. dosing of ME (Figure 6a, Table 2).
The in vitro transport and metabolism studies and in vivo pharmacokinetic studies showed that MGN and HNK exhibited similar characteristics and profiles. These results were similar to the results cited in a review paper by Maruyama et al 48. The T1/2, Cl, Vd values of MGN, HNK and MHNK from our I.V. and oral PK studies were comparable to the published studies of PK of magnolol and honokiol together and 4-O-methylhonookiol alone 43,49. Larger volume of distribution of MHNK in both I.V. and oral pharmacokinetic studies as compare to MGN and HNK suggested that MHNK may have broader tissue distribution (Tables 2 and 3).
Based on our detailed and large body of work on the disposition of phenolic compounds 50–53 we can confidently expect ME-KACs, which has phenolic groups present in their chemical structures, to undergo enteric and enterohepatic recycling. The biliary and luminal excretion of the glucuronides and sulfates of ME-KAC, followed by subsequent hydrolysis and reabsorption of ME-KACs in colon can explain the observed plasma double peak phenomena and longer half-lives of KACs and their glucuronides after oral administration of ME; as well as account for the lower blood glucuronide levels upon oral than I.V. administration.
At this stage, potentials for misuse of ME-KACs as a sedative/sleeping aid due to their GABA-ergic/cannabimimetic activities 54 is small due to the strong evidence of limited systemic exposure of KACs when ME is used as oral dietary supplement for weight control or other indications. However, the pharmacokinetics and brain exposure potential will need re-evaluation if more potent derivates of KACs are synthesized or ME products with higher amounts of KAC is marketed. This again supports our proposal to develop well-standardized and chemically defined ME as dietary supplement for various indications, instead of using currently marketed products with unknown ratios of active constituents that can result in variable pharmacological activities in different target organs.
Further investigation showed that though improving the solubilities of KACs by solubility-enhancing formulation of ME could significantly increase the AUC0–24 of ME-KACs (about 8–10 folds), however the overall bioavailabilities still remained very low (< 2%) (Figure 6e, Table 4). This was probably because even after enhanced solubilities in nanosuspension, the overall metabolism of KACs remained significantly high. Therefore, we concluded that enhancing solubility alone might not be enough to overcome ME poor bioavailability challenge and different local delivery approaches of ME in the oral cavity, such as mucoadhesive buccal patch 55,56 or chewing gums, need to be explored to deliver pharmacologically relevant concentrations of ME-KACs at the site of action. There have been successful attempts to include ME in the chewing gum for the treatment of oral malodor bacteria, caries and gingivitis 57–59.
Based on the above observations, we believe that a novel approach of chemically-defining ME such that the in vivo concentration ratio of ME-KACs can mimic their most effective in vitro ratio, is the most effective way to achieve desired health outcomes. Moreover, targeted drug delivery at the site of action is a desired strategy for making the drug locally bioavailable at the site of action, thereby by preventing the first-pass metabolism and unwanted systemic exposure and off-organ on-target side effects and increasing drug exposure in target organ at a reduced input dose. Combining these two approaches can further lead to the development of a more efficacious dietary supplement for oral health.
This paper contributes towards further understanding of the pharmacological effects of Magnolia officinalis extract (ME) and propose a novel approach to optimize ME composition to derive an optimized product to be used for oral cancer chemoprevention. The comprehensive pharmacokinetic profiling unveiled that the metabolic instability of ME-KACs upon per-oral administration is substantial barrier to overcome for reaching pharmacologically relevant tissue concentrations in oral cavity, even with solubility-enhancing formulation approaches. However, the chemopreventive potential of ME against oral cancer should be enhanced by a local delivery approach targeting the oral cavity itself. Therefore, we propose to investigate in the future the potential of using a locally-delivered chemically-defined standardized ME mixture for oral health promotion and oral cancer chemoprevention.
Supplementary Material
Methods
S1. Method Validation
S1.1. Calibration curve and LLOD
S1.2. Precision and accuracy
S1.3. Extraction recovery and matrix effect
S1.4. Stability
Results
Figure S1. The MS/MS chromatograms of magnolol (a), honokiol (b) and 4-O-methylhonokiol (c). The results showed the molecular ion and its MS2 fragment at m/Z 265 → 247 for MGN, 265 → 224 for HNK and 279 → 249 for MHKN, respectively.
Table S1. Compound-dependent parameters in UPLC-MS Analysis
Table S2. Intra-day, inter-day accuracy and precision in the analysis of magnolol, honokiol and 4-O-methylhonokiol
Table S3. Recovery and matrix effect of MGN, HNK and MHNK at low, medium and high concentrations in mouse blood
Table S4. Stability results of ME components in different conditions
Table S5. Solubility (µg/mL) of ME key active components in different buffer
Table S6. Anti-proliferation (MTT) assay results of ME and its key active components against SCC4 cell line (IC50 value in µg/mL)
Acknowledgement
We would like to acknowledge SK Bioland Ltd. Company (Chungnam, Korea) for providing Magnolia extract.
Funding
This project is supported by NIH Grant CA205633 and GM070737.
References
- 1.Wang L et al. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): a review. Biochem. Pharmacol 92, 73–89 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu Q et al. Antidepressant-like effects of the mixture of honokiol and magnolol from the barks of Magnolia officinalis in stressed rodents. Prog. Neuro-Psychopharmacology Biol. Psychiatry (2008). doi: 10.1016/j.pnpbp.2007.11.020 [DOI] [PubMed] [Google Scholar]
- 3.Weeks BS Formulations of dietary supplements and herbal extracts for relaxation and anxiolytic action: Relarian™. Medical Science Monitor (2009). [PubMed] [Google Scholar]
- 4.Sohn EJ et al. Effects of magnolol (5,5′-diallyl-2,2′-dihydroxybiphenyl) on diabetic nephropathy in type 2 diabetic Goto-Kakizaki rats. Life Sci (2007). doi: 10.1016/j.lfs.2006.09.037 [DOI] [PubMed] [Google Scholar]
- 5.Kim H Neuroprotective herbs for stroke therapy in traditional eastern medicine. Neurological Research (2005). doi: 10.1179/016164105X25234 [DOI] [PubMed] [Google Scholar]
- 6.Lu YC, Chen HH, Ko CH, Lin YR & Chan MH The mechanism of honokiol-induced and magnolol-induced inhibition on muscle contraction and Ca2+ mobilization in rat uterus. Naunyn. Schmiedebergs. Arch. Pharmacol (2003). doi: 10.1007/s00210-003-0802-8 [DOI] [PubMed] [Google Scholar]
- 7.Yimam M et al. UP601, a standardized botanical composition composed of Morus alba, Yerba mate and Magnolia officinalis for weight loss. BMC Complement. Altern. Med (2017). doi: 10.1186/s12906-017-1627-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Syu WJ, Shen CC, Lu JJ, Lee GH & Sun CM Antimicrobial and cytotoxic activities of neolignans from Magnolia officinalis. Chem. Biodivers (2004). doi: 10.1002/cbdv.200490046 [DOI] [PubMed] [Google Scholar]
- 9.McGuffin MME Herbs of Commerce. (American Herbal Products Association, 2000). [Google Scholar]
- 10.Poivre M & Duez P Biological activity and toxicity of the Chinese herb Magnolia officinalis Rehder & E. Wilson (Houpo) and its constituents. J. Zhejiang Univ. B 18, 194–214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho JH et al. Multifunctional effects of honokiol as an anti-inflammatory and anti-cancer drug in human oral squamous cancer cells and xenograft. Biomaterials 53, 274–284 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Prasad R & Katiyar SK Honokiol, an Active Compound of Magnolia Plant, Inhibits Growth, and Progression of Cancers of Different Organs. Adv Exp Med Biol 928, 245–265 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Alonso-Castro AJ et al. Magnolia dealbata seeds extract exert cytotoxic and chemopreventive effects on MDA-MB231 breast cancer cells. Pharm Biol 52, 621–627 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Lee SJ et al. Inhibitory effects of the aqueous extract of Magnolia officinalis on the responses of human urinary bladder cancer 5637 cells in vitro and mouse urinary bladder tumors induced by N-butyl-N-(4-hydroxybutyl) nitrosamine in vivo. Phyther. Res 23, 20–27 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Singh T et al. Honokiol inhibits the growth of head and neck squamous cell carcinoma by targeting epidermal growth factor receptor. Oncotarget 6, 21268–21282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee YJ et al. Therapeutic applications of compounds in the Magnolia family. Pharmacol Ther 130, 157–176 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Biersack B Current state of phenolic and terpenoidal dietary factors and natural products as non-coding RNA/microRNA modulators for improved cancer therapy and prevention. Non-coding RNA Res 1, 12–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X et al. Honokiol: A promising small molecular weight natural agent for the growth inhibition of oral squamous cell carcinoma cells. Int. J. Oral Sci 3, 34–42 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Q et al. Magnolia extract is effective for the chemoprevention of oral cancer through its ability to inhibit mitochondrial respiration at complex I. 2, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.American Cancer Society. Cancer facts & figures 2019. Atlanta Am. Cancer Soc 20–21 (2019). [Google Scholar]
- 21.Masthan KM, Babu NA, Sankari SL & Priyadharsini C Leukoplakia: A short review on malignant potential. J Pharm Bioallied Sci 7, S165–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dionne KR, Warnakulasuriya S, Zain RB & Cheong SC Potentially malignant disorders of the oral cavity: current practice and future directions in the clinic and laboratory. Int J Cancer 136, 503–515 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Ferlay J et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136, E359–86 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Gillison ML et al. Randomized Phase III Trial of Concurrent Accelerated Radiation Plus Cisplatin With or Without Cetuximab for Stage III to IV Head and Neck Carcinoma: RTOG 0522. J. Clin. Oncol 32, 2940–2950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsieh SF et al. The effect of magnolol on Ca2+ homeostasis and its related physiology in human oral cancer cells. Arch. Oral Biol 89, 49–54 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Huang KJ, Kuo CH, Chen SH, Lin CY & Lee YR Honokiol inhibits in vitro and in vivo growth of oral squamous cell carcinoma through induction of apoptosis, cell cycle arrest and autophagy. J. Cell. Mol. Med 22, 1894–1908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DW et al. Anti-proliferative effect of honokiol in oral squamous cancer through the regulation of specificity protein 1. Int. J. Oncol 43, 1103–1110 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Huang JS et al. Honokiol inhibits sphere formation and xenograft growth of oral cancer side population cells accompanied with JAK/STAT signaling pathway suppression and apoptosis induction. BMC Cancer 16, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao CJ et al. Honokiol eliminates human oral cancer stem-like cells accompanied with suppression of Wnt/ β -Catenin signaling and apoptosis induction. Evidence-based Complement. Altern. Med 2013, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao S, Chen F & Gao C Antitumor activity of 4-O-Methylhonokiol in human oral cancer cells is mediated via ROS generation, disruption of mitochondrial potential, cell cycle arrest and modulation of Bcl-2/Bax proteins. J. B.U.ON 22, 1577–1581 (2017). [PubMed] [Google Scholar]
- 31.Lee YK et al. Protective effect of the ethanol extract of Magnolia officinalis and 4-O-methylhonokiol on scopolamine-induced memory impairment and the inhibition of acetylcholinesterase activity. J. Nat. Med 63, 274–282 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee YJ et al. A comparison between extract products of Magnolia officinalis On memory impairment and amyloidogenesis in a transgenic mouse model of Alzheimer’s disease. Biomol. Ther 20, 332–339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JW et al. Inhibitory effect of ethanol extract of Magnolia officinalis and 4-O-methylhonokiol on memory impairment and neuronal toxicity induced by beta-amyloid. Pharmacol. Biochem. Behav 95, 31–40 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Gao S, Yang Z, Yin T, You M & Hu M Validated LC-MS/MS method for the determination of maackiain and its sulfate and glucuronide in blood: Application to pharmacokinetic and disposition studies. J. Pharm. Biomed. Anal 55, 288–293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Zhu Y & Hu M Mechanisms and Kinetics of Uptake and Efflux of L-Methionine in an Intestinal Epithelial Model (Caco-2). J. Nutr (1994). doi: 10.1093/jn/124.10.1907 [DOI] [PubMed] [Google Scholar]
- 36.Hu M, Chen J & Lin H Metabolism of flavonoids via enteric recycling: mechanistic studies of disposition of apigenin in the Caco-2 cell culture model. J Pharmacol Exp Ther 307, 314–321 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Doerge DR, Chang HC, Churchwell MI & Holder CL Analysis of soy isoflavone conjugation in vitro and in human blood using liquid chromatography-mass spectrometry. Drug Metab Dispos 28, 298–307 (2000). [PubMed] [Google Scholar]
- 38.Zhu W, Xu H, Wang SWJ & Hu M Breast Cancer Resistance Protein (BCRP) and Sulfotransferases Contribute Significantly to the Disposition of Genistein in Mouse Intestine. AAPS J 12, 525–536 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wais U, Jackson AW, He T & Zhang H Nanoformulation and encapsulation approaches for poorly water-soluble drug nanoparticles. Nanoscale 8, 1746–1769 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Nair A & Jacob S A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm (2016). doi: 10.4103/0976-0105.177703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Wang D, Yang G, Shi Q & Feng F Comparative pharmacokinetics and brain distribution of magnolol and honokiol after oral administration of Magnolia officinalis cortex extract and its compatibility with other herbal medicines in Zhi-Zi-Hou-Po Decoction to rats. Biomed Chromatogr 30, 369–375 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Cder, F. D. A. US Food and Drug Administration. Guidance for industry: bioanalytical method validation guidance for industry bioanalytical method validation. 1–22 (2018).
- 43.Yu HE et al. Pharmacokinetics and metabolism of 4-O-methylhonokiol in rats. Phytother Res 28, 568–578 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Singh R, Wu B, Tang L & Hu M Uridine diphosphate glucuronosyltransferase isoform-dependent regiospecificity of glucuronidation of flavonoids. J. Agric. Food Chem 59, 7452–7464 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang L, Singh R, Liu Z & Hu M Structure and concentration changes affect characterization of UGT isoform-specific metabolism of isoflavones. in Molecular Pharmaceutics 6, 1466–1482 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang L et al. Use of glucuronidation fingerprinting to describe and predict mono- and dihydroxyflavone metabolism by recombinant UGT isoforms and human intestinal and liver microsomes. Mol. Pharm 7, 664–679 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu B, Basu S, Meng S, Wang X & Hu M Regioselective Sulfation and Glucuronidation of Phenolics: Insights into the Structural Basis. Curr. Drug Metab 12, 900–916 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maruyama Y & Kuribara H Overview of the Pharmacological Features of Honokiol. CNS Drug Rev 6, 35–44 (2010). [Google Scholar]
- 49.Sheng YL et al. UPLC-MS/MS-ESI assay for simultaneous determination of magnolol and honokiol in rat plasma: application to pharmacokinetic study after administration emulsion of the isomer. J Ethnopharmacol 155, 1568–1574 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Yang Z, Kulkarni K, Zhu W & Hu M Bioavailability and Pharmacokinetics of Genistein: Mechanistic Studies on its ADME. Anticancer. Agents Med. Chem (2012). doi: 10.2174/187152012803833107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang G et al. Glucuronidation: driving factors and their impact on glucuronide disposition. Drug Metabolism Reviews 49, 105–138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu W, Kulkarni K & Hu M Gender-dependent differences in uridine 5’-diphospho- glucuronosyltransferase have implications in metabolism and clearance of xenobiotics. Expert Opinion on Drug Metabolism and Toxicology 9, 1555–1569 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Wu B, Kulkarni K, Basu S, Zhang S & Hu M First-pass metabolism via UDP-glucuronosyltransferase: A barrier to oral bioavailability of phenolics. Journal of Pharmaceutical Sciences 100, 3655–3681 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schifano F et al. Is there a potential of misuse for Magnolia officinalis compounds/metabolites? Hum. Psychopharmacol 32, 1–7 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Holpuch AS et al. Evaluation of a mucoadhesive fenretinide patch for local intraoral delivery: A strategy to reintroduce fenretinide for oral cancer chemoprevention. Carcinogenesis 33, 1098–1105 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Desai KGH, Mallery SR, Holpuch AS & Schwendeman SP Development and in vitro-in vivo evaluation of fenretinide-loaded oral mucoadhesive patches for site-specific chemoprevention of oral cancer. Pharm. Res (2011). doi: 10.1007/s11095-011-0489-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tian M & Greenberg M Confectionery containing magnolia bark extract effective against bacteria responsible for oral malodor. in Recent Highlights in Flavor Chemistry & Biology (Proceedings of the 8th Wartburg Symposium) 383–388 (2008).
- 58.Greenberg M, Urnezis P & Tian M Compressed mints and chewing gum containing magnolia bark extract are effective against bacteria responsible for oral malodor. J. Agric. Food Chem 55, 9465–9469 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Campus G et al. Effect of a sugar-free chewing gum containing magnolia bark extract on different variables related to caries and gingivitis: A randomized controlled intervention trial. Caries Res 45, 393–399 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods
S1. Method Validation
S1.1. Calibration curve and LLOD
S1.2. Precision and accuracy
S1.3. Extraction recovery and matrix effect
S1.4. Stability
Results
Figure S1. The MS/MS chromatograms of magnolol (a), honokiol (b) and 4-O-methylhonokiol (c). The results showed the molecular ion and its MS2 fragment at m/Z 265 → 247 for MGN, 265 → 224 for HNK and 279 → 249 for MHKN, respectively.
Table S1. Compound-dependent parameters in UPLC-MS Analysis
Table S2. Intra-day, inter-day accuracy and precision in the analysis of magnolol, honokiol and 4-O-methylhonokiol
Table S3. Recovery and matrix effect of MGN, HNK and MHNK at low, medium and high concentrations in mouse blood
Table S4. Stability results of ME components in different conditions
Table S5. Solubility (µg/mL) of ME key active components in different buffer
Table S6. Anti-proliferation (MTT) assay results of ME and its key active components against SCC4 cell line (IC50 value in µg/mL)