

Abstract
Elevated ribosome biogenesis in oncogene‐driven cancers is commonly targeted by DNA‐damaging cytotoxic drugs. Our previous first‐in‐human trial of CX‐5461, a novel, less genotoxic agent that specifically inhibits ribosome biogenesis via suppression of RNA polymerase I (Pol I) transcription, revealed single‐agent efficacy in refractory blood cancers. Despite this clinical response, patients were not cured. In parallel, we demonstrated a marked improvement in the in vivo efficacy of CX‐5461 in combination with PI3K/AKT/mTORC1 pathway inhibitors. Here, we reveal the molecular basis for this improved efficacy observed in vivo, which is associated with specific suppression of translation of mRNAs encoding regulators of cellular metabolism. Importantly, acquired resistance to this cotreatment is driven by translational rewiring that results in dysregulated cellular metabolism and induction of a cAMP‐dependent pathway critical for the survival of blood cancers including lymphoma and acute myeloid leukemia. Our studies thus identify key molecular mechanisms underpinning the response of blood cancers to selective inhibition of ribosome biogenesis and define metabolic vulnerabilities that will facilitate the rational design of more effective regimens for Pol I‐directed therapies.
Keywords: cAMP‐EPAC1/2 pathway, hematological cancers, metformin, ribosome biogenesis and function, RNA Polymerase I inhibitor
Subject Categories: Cancer, Metabolism, Protein Biosynthesis & Quality Control
The elevated metabolic activity and activation of cAMP‐dependent pro‐survival mechanisms that lead to resistance of hematological cancers towards rRNA synthesis inhibition also poses a potentially targetable vulnerability.
Introduction
Many of the commonly used DNA‐damaging cytotoxic drugs including 5‐fluorouracil (5‐FU), etoposide, and oxaliplatin also interfere with ribosome biogenesis and target the “addiction” of transformed cells to elevated rates of ribosome biogenesis, inducing the “impaired ribosome biogenesis checkpoint” (IRBC; Pelletier et al, 2018). Consequently, a number of less genotoxic drugs that selectively target ribosome biogenesis and subsequently induce both the p53‐dependent IRBC and/or p53‐independent nucleolar‐associated activation of the DNA‐damage response have been developed and are showing increasing promise in clinical investigations (Quin et al, 2014; Pelletier et al, 2018). We developed the “first‐in‐class” selective inhibitor of ribosome biogenesis, CX‐5461, which targets RNA polymerase I (Pol I) transcription, suppressing ribosomal RNA (rRNA) synthesis (Bywater et al, 2012). Moreover, we demonstrated its in vivo efficacy in mouse models of both blood and prostate cancer (Bywater et al, 2012; Devlin et al, 2016; Rebello et al, 2016; Hein et al, 2017; Lawrence et al, 2018). Critically, we recently completed a phase I clinical trial of CX‐5461, demonstrating single‐agent efficacy in patients with advanced hematological malignancies (Khot et al, 2019). This dose‐escalation clinical study of 17 patients demonstrated that CX‐5461 induced rapid on‐target inhibition of Pol I transcription, resulting in prolonged partial response in one patient with anaplastic large cell lymphoma and stable disease in five patients with diffuse large B‐cell lymphoma and myeloma (Khot et al, 2019).
In parallel studies, we reasoned that simultaneous targeting of ribosome biogenesis and protein synthesis, the critical processes where oncogenic networks converge to maintain cancer cell growth and survival, would improve the clinical benefit of CX‐5461 and overcome many of the upstream potential drivers of resistance (Chan et al, 2011; Ruggero, 2013; Kang et al, 2016; Saxton & Sabatini, 2017; Janku et al, 2018). Indeed, we demonstrated that combining CX‐5461 with an inhibitor of PI3K/AKT/mTORC1‐dependent mRNA translation, everolimus, markedly improved the pre‐clinical therapeutic efficacy of either drug alone in vivo (Devlin et al, 2016). However, the mechanisms underlying this synergistic effect and the development of ensuing resistance to both CX‐5461 alone or in combination with everolimus are unclear and their definition will be critical in optimizing the clinical efficacy of Pol I‐directed “ribosome‐targeting” therapies.
To interrogate the molecular basis of the response to CX‐5461 and the CX‐5461 plus everolimus combination, we first performed genome‐wide translational profiling to characterize the acute changes in mRNA usage of MYC‐driven B‐cell lymphomas in mice treated with CX‐5461 and the mTORC1 inhibitor, everolimus. CX‐5461 had minimal effect on the lymphoma's translatome after treatment for 2 h; however, the combination therapy specifically inhibited the translation of mRNAs encoding multiple components of the translational apparatus and reduced the association of mRNAs involved in the regulation of energy metabolism with actively translating polysomes. The importance of this selective targeting of translation was emphasized by our finding that acquired resistance to the combination therapy was driven by increased polysomal association of mRNAs encoding key components of the mitochondrial respiration network and the cAMP‐EPAC1/2‐RAP1 survival pathway. We confirmed the functional importance of this reprogramming by demonstrating that both CX‐5461 and combination therapy‐resistant cells are more metabolically active and have elevated levels of cAMP. Importantly, specific inhibition of EPAC1/2 resensitizes resistant cells to the combinatorial ribosome‐targeting therapy. More broadly, EPAC1/2 is also elevated in human acute myeloid leukemia (AML) and inhibition of EPAC1/2 reduced the viability of AML cell lines, demonstrating the critical role of this pro‐survival pathway in lymphoma and AML. These studies reveal a key mechanism by which alterations in the translation of mRNAs encoding metabolic enzymes drive the response of tumor cells to ribosome‐targeting therapies. They identify metabolic vulnerabilities that can be targeted to improve the response of hematological tumors to selective ribosome biogenesis inhibitors, which we predict will also be highly relevant for high proportion of cancers that are characterized by elevated ribosome biogenesis including cancers treated with the numerous standard of care cytotoxic drugs that inhibit this process.
Results
Acute inhibition of ribosome biogenesis and function selectively reduces translation of mRNAs encoding components of the translational apparatus
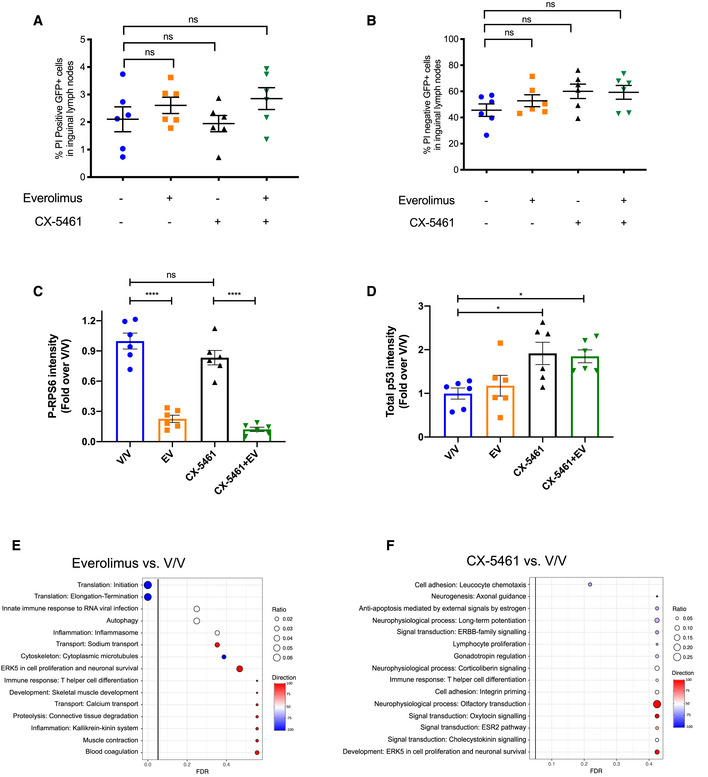
To examine the mechanisms of response to targeting ribosome biogenesis and protein synthesis by CX‐5461 and everolimus, Eμ‐Myc B‐cell lymphoma (MSCV Gfp; clone #4242) cells were transplanted into C57BL/6 mice as we performed previously (Bywater et al, 2012; Devlin et al, 2016). Lymphoma‐bearing mice were treated on day 14 post‐transplant for 2 h with CX‐5461 (35 mg/kg), everolimus (EV; 5 mg/kg), or both (CX‐5461 + EV; 35 mg/kg CX‐5461 and 5 mg/kg EV). We chose this early time point to exclude any confounding effects of drug‐induced cell death on our molecular analyses. Indeed, no change in the percentage of GFP‐positive or propidium iodide‐stained GFP‐positive lymphoma cells was observed in lymph nodes isolated from treated animals (Fig EV1A and B; Appendix Fig S1A), indicating that the downstream analyses were independent of cell death and reduction in tumor burden. The on‐target activity of EV was confirmed by the reduction in RPS6 phosphorylation (P‐RPS6; Ser240/244) in extracts of lymph nodes from individual mice treated with EV or CX‐5461 + EV (Fig 1A, Fig EV1C). In addition, despite the intrinsic variability in the p53 levels in the lymph nodes isolated from different mice, a significant increase of p53 protein was observed in response to CX‐5461 and CX‐5461 + EV treatment as expected (Fig 1A, Fig EV1D).
Figure EV1. In vivo transcriptome‐wide analysis of cellular response to ribosome‐targeting therapy.
-
A, BFlow cytometry analysis of inguinal lymph node cells that are (A) GFP‐positive and propidium iodide‐positive and (B) GFP‐positive and propidium iodide‐negative isolated from C57BL/6 mice with transplanted Eμ‐Myc lymphoma cells treated as indicated for 2 h. 10,000 viable cells of the correct morphology were collected for each sample. Graphs represent mean ± SEM of n = 6.
-
C, DQuantitation of (C) phosphorylated RPS6 levels normalized to total RPS6 and (D) p53 protein levels normalized to actin in Fig 1A. Graphs represent mean ± SEM of n = 6.
-
E, FEnrichment analysis by MetaCore® GeneGO of genes in “translation up” and “translation down” categories identified by anota2seq analysis comparing lymph node cells isolated from mice in (E) everolimus single‐agent treatment group or (F) CX‐5461 single‐agent treatment group with those isolated from mice in vehicle group (n = 6). “Ratio” is the value obtained by dividing the number of genes in an indicated molecular process that is found in our data with the number of genes curated in MetaCore®'s database. “Direction” visualizes the percentage of genes associated with indicated pathways that are up (red) or down (blue).
Figure 1. Acute inhibition of ribosome synthesis and function in vivo reduces translation of components of the translational machinery and decreases the abundance of polysome‐associated, energy metabolism‐related mRNAs.
- Western analysis for on‐target effects for everolimus (EV, P‐RPS6) and CX‐5461 (p53). Each lane represents equal amounts of protein from lymph node tissue isolated from a single mouse that received drug vehicles (everolimus vehicle: 1% methylcellulose; CX‐5461 vehicle: 25 mM NaH2PO4; V/V; mouse #1–6), 5 mg/kg everolimus (EV; mouse #7–12), 35 mg/kg CX‐5461 (mouse #13–18), or both drugs (CX‐5461 + EV; mouse #19–24) for 2 h (n = 6 per treatment group). Actin was used as a loading control.
- A schematic illustration of the polysome profiling analysis workflow: Cytoplasmic lysate was layered on top of a linear 10–40% sucrose gradient, ultracentrifuged (222,228 g, 2¼ h at 4°C using SW41Ti rotor), and fractionated using the Foxy Jr Fraction Collector with constant monitoring of absorbance at 260 (A260) nm by an ISCO UA‐6 Absorbance Detector. Fractions (one fraction per minute: 800 μl per tube) corresponding to polysomal mRNAs that were bound by four or more ribosomes were pooled and analyzed by RNA‐seq followed by data analysis using anota2seq or limma.
- Enrichment analysis by MetaCore® GeneGO of genes in “translation up” (red) and “translation down” (blue) categories identified by anota2seq analysis comparing lymph node cells isolated from mice in CX‐5461 + EV treatment group with the V/V group (n = 6). “Ratio” values were obtained by dividing the number of significant genes in our data assigned to a molecular process by the total number of genes in the process in MetaCore®'s database.
- Activity levels of key biological processes involved in cellular growth, proliferation, and metabolism based on single sample gene set enrichment analysis (ssGSEA) of indicated comparisons. Percentage increase (red) or decrease (blue) in the activity levels of key biological processes involved in cellular growth, proliferation, and metabolism based on ssGSEA of indicated comparisons. Data were obtained from n = 6 mice per treatment group.
Source data are available online for this figure.
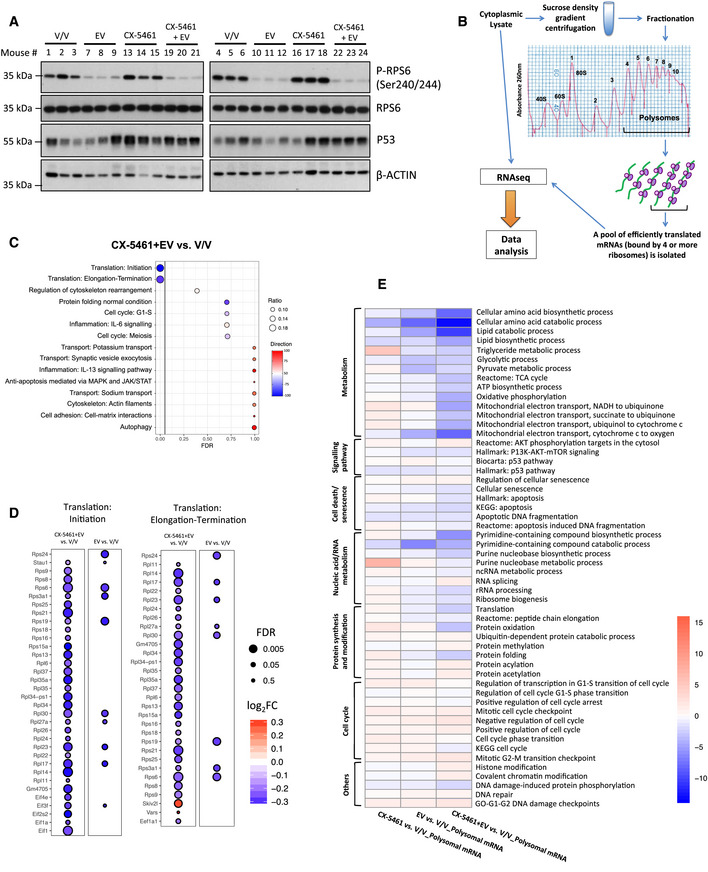
Given the specific alterations in translation patterns observed in response to mTOR inhibition (Hsieh et al, 2012; Morita et al, 2013) and in genetic models of compromised ribosome biogenesis (Khajuria et al, 2018), we performed polyribosome (polysome) profiling to enable whole translatome analysis of mRNAs whose translation efficiency was altered by the acute ribosome‐targeted combination therapy (Fig 1B). This transcriptome‐wide translatomics approach involves the sequencing of polysome‐associated mRNAs separated by sucrose density gradient ultracentrifugation accompanied by cytosolic mRNA sequencing (Fig 1B). To define changes in actively translated mRNAs (indicated by attachment of four or more ribosomes), we compared the translational signatures of lymphoma cells isolated from combination therapy‐treated mice (CX‐5461 + EV) versus vehicle‐treated mice (V/V) using anota2seq analysis (Lorent et al, 2019; Oertlin et al, 2019). After normalization to steady‐state total RNA, the polysomal RNA‐seq data indicated a robust reduction in the translation of transcripts encoding proteins involved in the regulation and activity of mRNA translation (Fig 1C, Table EV1). To further evaluate the significance of this translational rewiring in mediating the synergy between CX‐5461 and EV, we investigated the effects of CX‐5461 and EV treatment as single agents. Not surprisingly, given the role of mTORC1 in translational control (Ma & Blenis, 2009; Hsieh et al, 2012), EV treatment alone affected the translation of mRNAs involved in mRNA translation (Fig EV1E). In contrast, no significant effect on translation was observed upon single‐agent CX‐5461 treatment (Fig EV1F). However, quantitative comparison of the steady‐state and polysome‐associated mRNA counts in the top two enriched processes in Fig 1C and Fig EV1E (“Translation initiation” and “Translation elongation”) revealed that the increased efficacy of combining EV with CX‐5461 compared with EV alone was associated with specific and a more potent suppression of the translation of mRNAs encoding key proteins regulating mRNA translation (Fig 1D).
Acute reduction in translation of the components of the translational apparatus is associated with decreased translation of energy metabolism‐related mRNAs
Critically, despite this potent reduction in the efficiency of translation of components of the translational apparatus, cells treated with CX‐5461 + EV did not indiscriminately reduce the polysome association of all mRNAs globally. To investigate the functional outcome of this specific targeting of the translation apparatus, we performed single sample gene set enrichment analysis (ssGSEA; Barbie et al, 2009; detailed in Materials and Methods) comparing abundance of all mRNAs associated with actively translating polysomes, rather than normalization to steady‐state cytoplasmic mRNA abundance as performed in the anota2seq analysis shown in Fig 1C and D. ssGSEA revealed a selective reduction in the polysome‐association of mRNAs encoding proteins involved in key metabolic processes in response to CX‐5461 + EV treatment as compared to single‐agent treatments. These include proteins involved in energy metabolism, particularly in multiple steps of mitochondrial oxidative phosphorylation. Anota2seq analysis of the expression level of mRNAs with the ontology term “oxidative phosphorylation” identified several key genes in the regulation of oxidative phosphorylation that are significantly translationally downregulated in response to CX‐5461 + EV treatment, including Atp5k (ATP synthase subunit e), mt‐Cytb (component of Cytochrome b‐c1 complex), Cox7a1 (Cytochrome c oxidase subunit 7A1), and mt‐Nd2 (NADH:ubiquinone oxidoreductase core subunit 2; Table EV2). This finding validates our ssGSEA identification of oxidative phosphorylation as being acutely translationally regulated in response to CX‐5461 + EV treatment.
Cotargeting of energy metabolism improved the efficacy of CX‐5461‐mediated inhibition of ribosome biogenesis in vitro
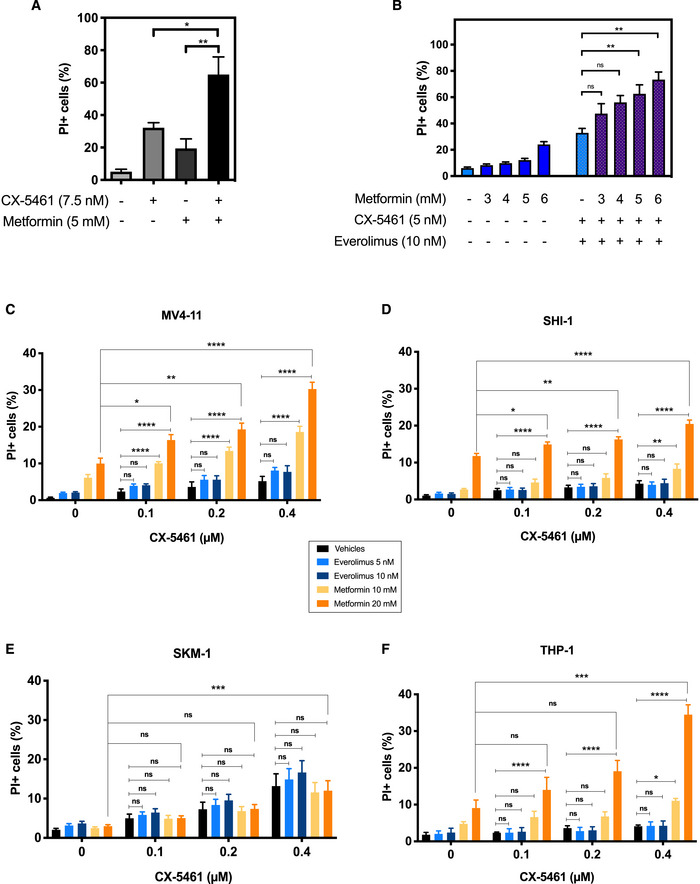
We hypothesized that this CX‐5461 + EV‐induced targeting of metabolism was a key driver of the improved response to the combination. To test this hypothesis, we used metformin, a well‐tolerated antidiabetic drug that lowers cellular energy levels (Foretz et al, 2014), in combination with standard CX‐5461 treatment (Bywater et al, 2012; Devlin et al, 2016) in parental Eμ‐Myc lymphoma cell lines in vitro. Metformin treatment for 48 h robustly increased cell death induced by CX‐5461 (Fig EV2A), consistent with a critical role for inhibition of metabolism in the improved efficacy of CX‐5461 + EV observed in vivo (Fig 1E). Moreover, metformin also markedly improved the therapeutic potency of CX‐5461 + EV (Fig EV2B), emphasizing the importance of the targetable metabolic vulnerability in response to combinatorial Pol I‐directed therapy. Thus, we propose that the reduced translational activity in CX‐5461 + EV combination therapy‐treated cells (Fig 1C), which selectively impaired translation of mRNAs encoding metabolic regulators (Fig 1E, Table EV2), is a key mechanism in the synergistic effect of the two drugs and highlights the intimate coupling of ribosome biogenesis, mRNA translation, and energy metabolism (Morita et al, 2013; Kusnadi et al, 2015; Leibovitch & Topisirovic, 2018).
Figure EV2. Metformin improves the efficacy of CX‐5461 as a single agent or in combination with Everolimus in vitro .
-
APropidium iodide (PI) exclusion analysis of Eμ‐Myc lymphoma cells treated with CX‐5461 in the presence and absence of metformin for 48 h. Graphs represent mean ± SEM of n = 3 experiments, and data were analyzed by one‐way ANOVA.
-
BPI exclusion analysis of Eμ‐Myc lymphoma cells treated with CX‐5461 + EV in the presence and absence of metformin as indicated for 48 h. Graphs represent mean ± SEM of n = 3 experiments, and data were analyzed by Student's t‐test.
-
C–FPI exclusion analysis of (C) MV4‐11, (D) SHI‐1, (E) SKM‐1, and (F) THP‐1 human acute myeloid leukemia cell lines in response to treatments with CX‐5461, everolimus (EV), and metformin as indicated for 72 h. Graphs represent mean ± SEM of n = 4–6, and data were analyzed by two‐way ANOVA.
We have shown that CX‐5461 is efficacious in highly aggressive AML in both p53 WT and p53 null leukemic mice (Hein et al, 2017) that are also highly dependent on oxidative phosphorylation for survival (Chapuis et al, 2019). In order to establish whether energy metabolism is a potential metabolic vulnerability in other hematological cancers that could be exploited to enhance the efficacy of CX‐5461, we evaluated the effects of metformin + CX‐5461 combination in four human AML cell lines, representing a range of common oncogenic drivers of AML: MV4‐11 (MLL‐AF4 gene fusion), SHI‐1 (MLL‐AF6), SKM‐1 (EZH2), and THP‐1 (t(9;11); Hein et al, 2017). As expected, metformin increased the abundance of phosphorylated AMPK and modestly reduced P‐RPS6 levels in these cell lines, and everolimus promoted a robust decrease in the levels of P‐RPS6 but had no effect on AMPK activation (Appendix Fig S1B). In MV4‐11, SHI1 and THP‐1 cells, a significant improvement of efficacy was observed when they were treated with the CX‐5461 + metformin combination in comparison with the single‐agent treatment (Fig EV2C–F). Together, these data demonstrated the therapeutic potential of concurrent inhibition of energy metabolism and ribosome synthesis and function in hematological malignancies.
Elevated energy metabolism is a characteristic of resistance to ribosome‐targeting therapy
Despite the dramatic initial impact of the combination treatment on tumor growth, animals eventually succumb to lymphomagenesis (Bywater et al, 2012; Devlin et al, 2016). Given that translation‐dependent targeting of cellular metabolism is a key mediator of the acute response to combined targeting of ribosome biogenesis and mRNA translation (Fig 1C–E), we hypothesized that metabolic rewiring driven by specific changes in mRNA translation would confer this resistance to therapy. To test this hypothesis, we established early passage Eμ‐Myc lymphoma cell lines from mice that were drug‐naïve (from here onwards referred to as “CTRL” cells), previously treated with everolimus (“EV” cells), CX‐5461 (“CX” cells) or CX‐5461 + EV (“CMB” cells; Fig 2A). These early passage cell lines were derived from lymph node extracts isolated from 12 different mice that were transplanted with Eμ‐Myc lymphoma cells (clone #107) as indicated in Fig 2A and described in the Materials and Methods section.
Figure 2. The early passage Eμ‐Myc lymphoma cells exhibit altered drug response and distinct metabolic profiles.
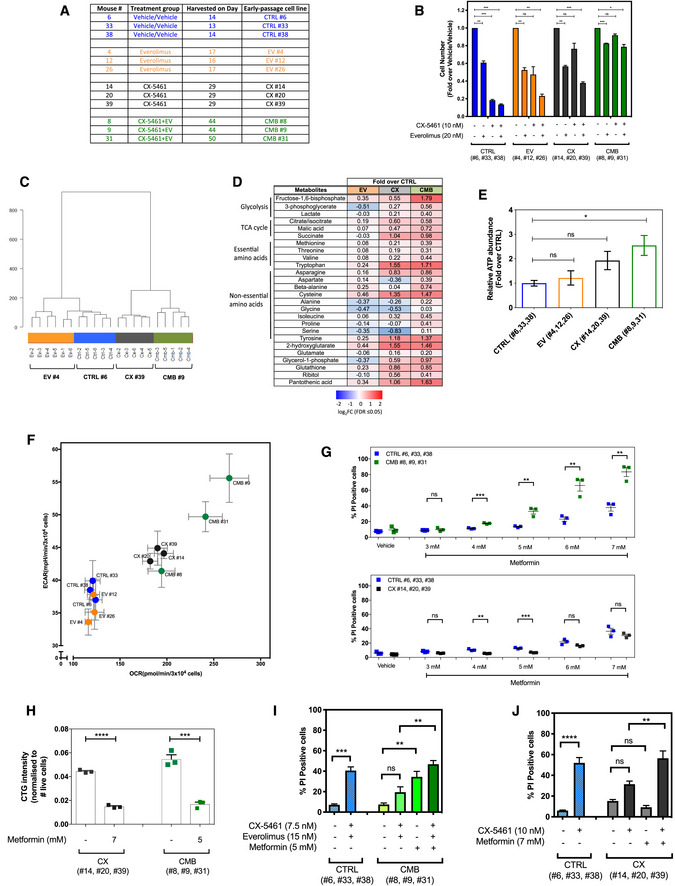
- The early passage Eμ‐Myc lymphoma cells used in this study, which were established from mice that were drug‐naïve (“CTRL” cell lines), previously treated with everolimus (“EV” cell lines) or CX‐5461 (“CX” cell lines) alone, or combination of both (“CMB” cell lines). Harvest day indicates the number of days post‐transplantation of lymphoma cells.
- Cell viability analysis of the indicated early passage Eμ‐Myc lymphoma cells treated with indicated compound(s) for 48 h as determined by Beckman® Coulter counter. Graphs represent mean ± SEM of n = 3. Data were analyzed by two‐way ANOVA.
- Hierarchical clustering analysis of metabolomics data obtained from gas chromatography (GC)–mass spectrometry (MS) analysis of early passage cell lines (n = 5–6).
- Steady‐state abundance of indicated metabolites (fold‐over CTRL cells; FDR ≤ 0.05; n = 5–6).
- Intracellular ATP levels determined using the liquid chromatography–mass spectrometry in CTRL, EV, CX, and CMB early passage cell lines. Graphs represent mean ± SEM of n = 3.
- Basal extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) in the early passage cell lines determined using the Seahorse XF96 Extracellular Flux Analyzer; graphs represent mean ± SEM of 6–8 technical replicates for each biological replicate (n = 3).
- Propidium iodide (PI) exclusion analysis of early passage CX‐5461-everolimus combination therapy‐resistant (CMB), CX‐5461-resistant (CX), and drug‐naïve (CTRL) lymphoma cells treated with indicated concentrations of metformin for 48 h. Graphs represent mean ± SEM of n = 3.
- CellTiterGLO®‐based assay measuring cellular ATP levels of the CX-5461‐resistant (CX) and CX-5461‐everolimus combination therapy‐resistant (CMB) cells treated with metformin as indicated for 48 h. Graphs represent mean ± SEM of n = 3.
- PI analysis of CTRL and CMB cells treated with CX‐5461 and everolimus in the presence and absence of metformin for 48 h as indicated. Graphs represent mean ± SEM of n = 3.
- PI analysis of CTRL and CX cells treated with CX‐5461 in the presence and absence of metformin for 48 h as indicated. Graphs represent mean ± SEM of n = 3.
To examine the drug resistance phenotypes of the early passage cell lines, they were treated with EV, CX‐5461, or CX‐5461 + EV in vitro. The drug‐naive CTRL and EV cell lines retained sensitivity to all treatments. EV cells showed little change in sensitivity to EV treatment, consistent with our previous finding that EV treatment did not provide a significant survival benefit in the Eμ‐Myc B‐cell lymphoma (clone #107) model (Devlin et al, 2016). It is unclear why the EV cells show some resistance to CX‐5461, and we will focus our analysis on CX and CMB cell lines given the efficacy of these treatments. The CX cells were resistant to CX‐5461 and sensitive to CX‐5461 + EV, while the CMB cells’ responses to all the treatments were robustly blunted (Fig 2B). More importantly, the CX and CMB cells maintained drug resistance in vivo when retransplanted into mice and rechallenged with CX‐5461 (Fig EV3A) and CX‐5461 + EV (Fig EV3B), respectively.
Figure EV3. In vitro and in vivo characterization of the early passage cell lines.
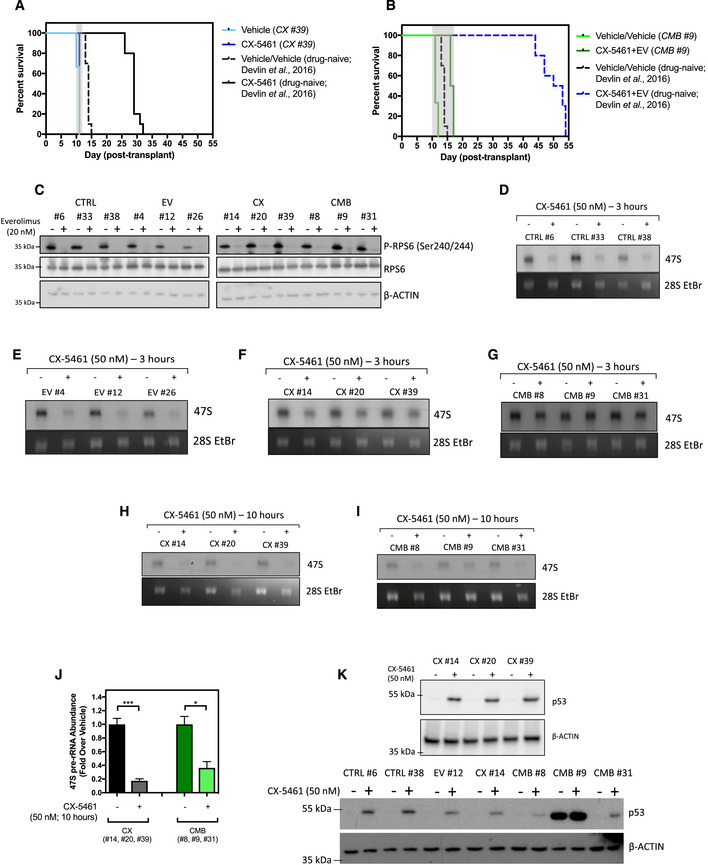
-
AKaplan–Meier curve of C57BL/6 mice transplanted with the CX-5461‐resistant CX #39 early passage Eμ‐Myc lymphoma cells treated with CX‐5461 (35 mg/kg every 3 days; n = 5–6). Survival curves of drug‐naïve cells were actual data reproduced from Devlin et al, 2016.
-
BKaplan–Meier curve of C57BL/6 mice transplanted with CX‐5461 + EV‐resistant CMB #9 early passage cells treated with CX‐5461 (35 mg/kg every 3 days) and everolimus (5 mg/kg daily; n = 5–6). Survival curves of drug‐naïve cells were actual data reproduced from Devlin et al, 2016.
-
CWestern blot analysis of markers for the on‐target effect of 20 nM everolimus treatment for 3 h, specifically phosphorylation of RPS6 at Serine 240/244 residues and total RPS6 (n = 3). Actin was used as a loading control.
-
D–JSynthesis rates of 47S pre‐rRNAs in (D) drug‐naïve (CTRL), (E) everolimus‐resistant (EV), (F) CX-5461‐resistant (CX), and (G) CX‐5461 + EV‐resistant (CMB) cell lines in response to 3‐h 50 nM CX‐5461 treatment were determined by 32P‐orthophosphate “pulse” labeling. Synthesis rate of 47S pre‐rRNAs in (H) CX and (I) CMB cell lines in response to 10‐h 50 nM CX‐5461 was determined by 32P‐orthophosphate “pulse” labeling and (J) its quantitation (n = 3). (D–I) 28S rRNA abundance was used as a loading control. EtBr: ethidium bromide. Data were analyzed by Student's t‐test. Graphs represent mean ± SEM of n = 3. *P ≤ 0.05. ***P ≤ 0.001.
-
KWestern blot analysis for p53 abundance in response to 50 nM CX‐5461 treatment for 3 h. Actin was used as a loading control (n = 2–3).
Source data are available online for this figure.
The in vivo‐derived early passage cell lines maintained the appropriate on‐target responses to these targeted therapies. Everolimus inhibited mTORC1 activity in all cell lines, as reflected by reduced phosphorylation of RPS6 (Ser240/244; Fig EV3C). As expected, standard CX‐5461 treatment routinely used by our group (50 nM for 3 h; Devlin et al, 2016) robustly decreased the rate of 47S pre‐rRNA synthesis in the CTRL and EV cells (Fig EV3D and E). Although the CX and CMB cells at 3 h post‐treatment were less sensitive to rDNA transcription inhibition (Fig EV3F and G) compared to the CTRL and EV cells, longer term CX‐5461 treatment resulted in robust on‐target inhibition of Pol I transcription (Fig EV3H‐J). These findings were also consistent with the ability of CX‐5461 to induce p53 accumulation in the cell lines (Fig EV3K), confirming that loss of the IRBC is not associated with the mechanism of resistance to CX‐5461 and CX‐5461 + EV.
Since acute targeting of cellular metabolism is associated with the cellular response to CX‐5461 + EV treatment (Fig 1E) and targeting energy metabolism improves the efficacy of Pol I‐directed ribosome‐targeted therapy (Fig EV2A–E), we investigated whether metabolic rewiring is associated with acquired resistance, by profiling metabolite levels in CTRL, EV, CX, and CMB in vivo‐derived early passage Eμ‐Myc lymphoma cell lines using gas chromatography–mass spectrometry (GC‐MS; Fig 2C and D) and liquid chromatography–mass spectrometry (LC‐MS; Appendix Fig S2A). Multivariate analysis clearly separated the metabolic phenotype of these cell lines (Fig 2C and Appendix Fig S2A). Strikingly, the CX and CMB cell lines had elevated levels of glycolytic and tricarboxylic acid (TCA) cycle intermediates as well as a number of essential and non‐essential amino acids compared to the EV and CTRL cell lines (Fig 2D). LC‐MS data indicated that the CMB cells had elevated ATP content compared to the drug‐naïve CTRL and EV cells (Fig 2E). Consistent with our metabolomic analysis, Seahorse XF96 analysis highlighted that CX and CMB cells were more metabolically active, as demonstrated by increased glycolysis and oxidative phosphorylation (Fig 2F and Appendix Fig S2B). These results revealed energy, and amino acid metabolism of CMB cells is indeed rewired in response to translation‐dependent inhibition of metabolism by CX‐5461 + EV (Fig 1E). Furthermore, while acute treatment with CX‐5461 did not significantly affect metabolism (Fig 1E), consistent with the protective role of metformin‐sensitive metabolism in limiting response of naïve cells to CX‐5461 (Fig EV2A), acquired resistance to CX‐5461 was also associated with elevated cellular energy metabolism (Fig 2F).
To test whether the rewired metabolism in these CX and CMB cells contributes to drug resistance, we treated the CTRL, CX, and CMB cells with metformin. CMB cells exhibited a greater sensitivity to metformin compared to the CTRL and CX cells (Fig 2G). Strikingly, metformin, at concentrations that resulted in similar levels of cell death and reduction in ATP abundance in both CX and CMB cells (Fig 2H), resensitized CMB cells to CX‐5461 + EV treatment, increasing the percentage of cell death to the levels achieved when CTRL cells were treated with CX‐5461 + EV (Fig 2I). Furthermore, metformin treatment increased CX‐5461‐induced cell death in CX cells (Fig 2J), consistent with the effects of metformin on both ATP levels (Fig 2H) and on mTORC1 signaling to P‐RPS6 following activation of AMPK (Appendix Fig S2C). Thus, reprogramming of metformin‐sensitive metabolism is integral for the acquired resistance to ribosome‐targeting therapy.
Increased polysome association of components of the cAMP pathway is associated with resistance to ribosome‐targeting therapy
To investigate whether there are translational alterations that underpin the energy metabolism‐associated changes observed in cells resistant to chronic ribosome‐targeted therapy, we compared the translatomes of the culture‐adapted drug‐resistant with drug‐naïve cell lines by polysome profiling (as illustrated in Fig 1B). This adaptation to long‐term ribosome‐targeting therapy in vivo is likely to involve alterations in the efficiency of both transcription and translation, and hence to fully explore functional mechanisms of resistance, we applied the standard limma (Ritchie et al, 2015) RNA‐seq analysis pipeline (described in Materials and Methods) to characterize the mRNAs associated with actively translating polysomes. We then performed pathway analysis using MetaCore® GeneGO on the mRNAs enriched on the polysomes of the in vivo‐derived early passage CX and CMB cells to identify potential driver pathways of resistance.
This analysis (Table EV3) identified the cyclic‐adenosine 3′,5′‐monophosphate (cAMP) signaling pathway as the top hit when comparing CMB versus CTRL (Fig 3A), with increased abundance of polysome‐associated mRNAs encoding essential components of this pathway, including adenylate cyclase and cAMP‐guanine nucleotide exchange factors (cAMP‐GEFs) 1 and 2, which is also known as exchange protein directly activated by cAMP (EPAC) 1 and 2. Adenylate cyclase converts ATP into cAMP, which then activates protein kinase A (PKA) or EPAC1/2, the GEFs for activation of Ras‐related protein 1 (RAP1), the two best characterized direct downstream targets of cAMP (Bos, 2006; Gloerich & Bos, 2010). To confirm functional elevation of cAMP signaling, we applied liquid chromatography–MS (LC‐MS), Western blot, and pull‐down analysis to demonstrate elevated cAMP abundance (Fig 3B), increased EPAC2 and GTP‐bound RAP1 levels in the CMB cell lines compared to CTRL (Fig 3C, Appendix Fig S3A–C). Consistent with this finding, our polysome profiling dataset (Table EV3) indicated that CMB cells exhibit an increase of polysome association of Rap1 mRNA (Appendix Fig S3D, adjusted P value = 0.0081). In CX cells, cAMP signaling pathway was identified to be upregulated (Fig EV4A), with increased GTP‐bound RAP1 (Fig 3C) and polysome association of Rap1 mRNA (Appendix Fig S3D, adjusted P value = 0.0385).
Figure 3. cAMP‐dependent pathway mediates resistance to CX‐5461‐everolimus cotreatment.
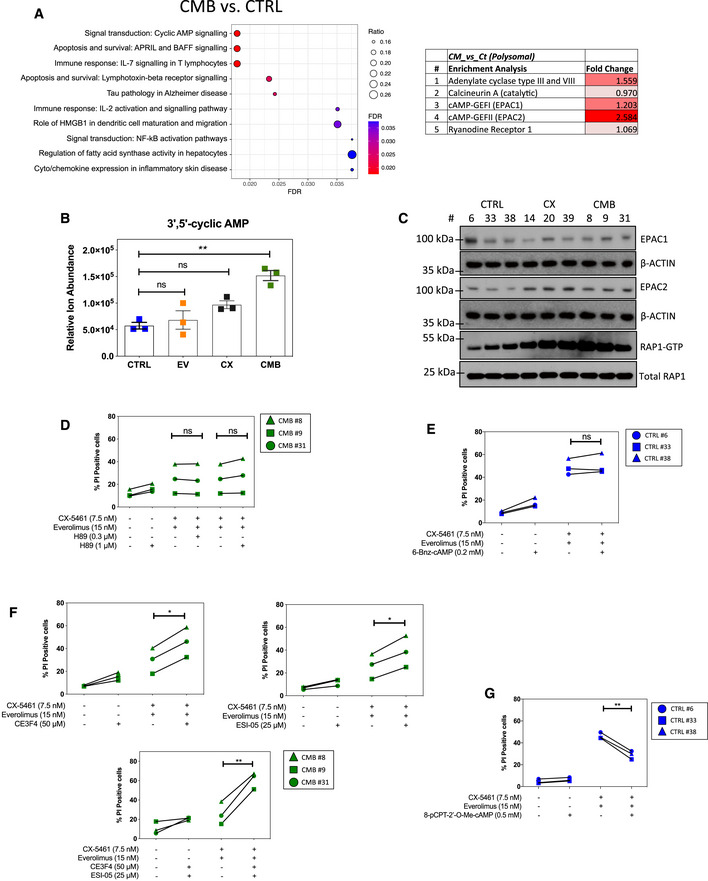
- Enrichment analysis by GeneGO MetaCore® of the polysomal RNA‐seq data comparing CX‐5461 + EV‐resistant (CMB) and drug‐naïve cells (CTRL; n = 3; false discovery rate (FDR) ≤ 0.05; fold change (FC) ≥ 1.5 or ≤ ‐1.5).
- Abundance of intracellular 3'5’‐cyclic AMP as measured by a liquid chromatography (LC)–mass spectrometry (MS) analysis. Graphs represent mean ± SEM of n = 3 (5–6 technical replicates each). Data were analyzed by one‐way ANOVA. CTRL vs. EV, P = 0.8951; CTRL vs. CX, P = 0.0871; CTRL vs. CMB, P = 0.0012.
- Western analysis of EPAC1 and EPAC2 abundance, as well as active GTP‐bound RAP1 levels in the indicated early passage cells during their log‐phase growth period (n = 3). Actin and total RAP1 were used as loading controls. Quantitations of the blots are shown in Appendix Fig S3A–C.
- Propidium iodide (PI) exclusion assays of CMB cells treated with CX‐5461 and everolimus as indicated in the presence or absence of a selective PKA inhibitor H89 for 48 h.
- PI exclusion analysis of early passage drug‐naïve (CTRL) lymphoma cells treated with CX‐5461 and everolimus in the presence or absence of selective PKA activator 6‐Bnz-cAMP for 48 h.
- PI exclusion analysis of the CMB cells treated with CX‐5461 and everolimus as indicated in the presence or absence of EPAC1 inhibitor CE3F4 or EPAC2 inhibitor ESI‐05 for 48 h.
- PI exclusion analysis of early passage drug‐naïve (CTRL) cells treated with CX‐5461 and everolimus in the presence or absence of the selective EPAC activator 8‐pCPT-2‐O-Me-cAMP for 48 h.
Figure EV4. Polysome profiling analysis of the early passage Eμ‐Myc lymphoma “CX” cell lines and investigating potential roles of RAPGEF3 (EPAC1) and RAPGEF4 (EPAC2) in human blood cancers.
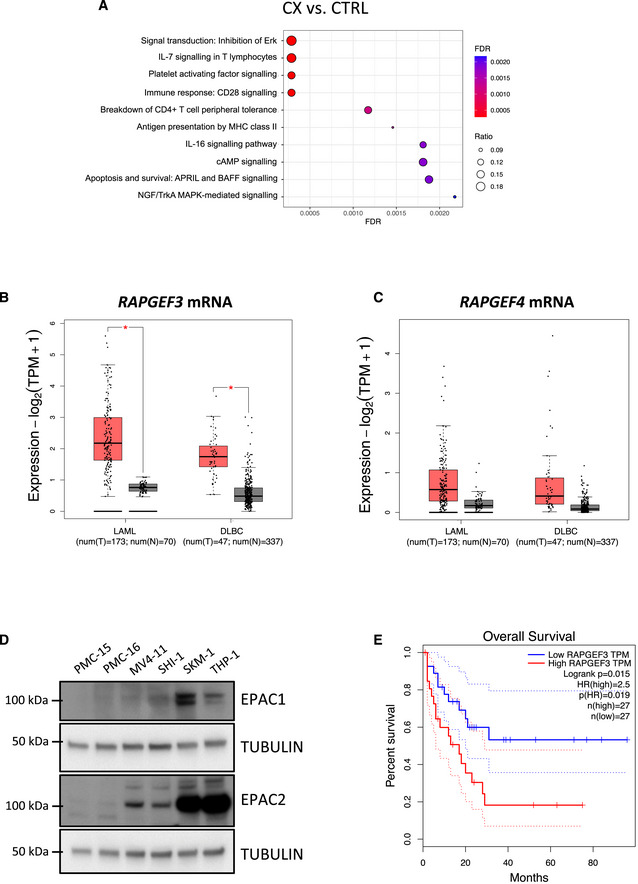
-
AEnrichment analysis by GeneGO MetaCore® of the polysomal RNA‐seq data comparing CX‐5461‐resistant (CX) and drug‐naïve cells (CTRL; n = 3; false discovery rate (FDR) ≤ 0.05; fold change (FC) ≥ 1.5 or ≤ ‐1.5).
-
B, CMedian mRNA expression levels of (B) RAPGEF3 (mRNA encoding EPAC1 protein) and (C) RAPGEF4 (mRNA encoding EPAC2 protein) generated by gene expression profiling interactive analysis (GEPIA) bioinformatics web tool based on The Cancer Genome Atlas (TCGA) database. LAML: acute myeloid leukemia (tumor, n = 173; normal, n = 70). DLBC: diffuse large B‐cell lymphoma (tumor, n = 47; normal, n = 337). Red: tumor, black: normal. The central band corresponds to the median. The lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles). The upper and lower whiskers extend from the hinges to the largest and smallest values no further than 1.5 * interquartile range from the hinges, respectively. Data points are plotted individually. Data were analyzed by one‐way ANOVA; *P < 0.05. tpm, transcript count per million.
-
DWestern analysis of EPAC1 and EPAC2 abundance in white blood cell samples (isolated from healthy donors #PMC-15 and #PMC-16) versus MV4‐11, SHI‐1, SKM1, and THP1 human AML cell lines (representative of n = 2).
-
ESurvival curves of AML patients with low and high EPAC1 (RAPGEF3; n = 27 each; cut‐off: 50%) generated by GEPIA based on TCGA database.
Source data are available online for this figure.
Importantly, the expression levels of mRNAs from the genes encoding EPAC1 and 2 (RAPGEF3 and RAPGEF4) are elevated in human hematological cancers including AML and diffuse large B‐cell lymphoma (DLBCL; Fig EV4B and C). Indeed, EPAC1 and EPAC2 protein expression was elevated in the human AML cell lines MV4‐11, SKM‐1, SHI‐1, and THP‐1 compared to white blood cells from healthy patients (Fig EV4D), and high expression of EPAC1 was associated with poorer survival (Fig EV4E). Furthermore, using the EPAC1‐specific inhibitor CE3F4 (Courilleau et al, 2012) and the EPAC2‐specific inhibitor ESI‐05 (Tsalkova et al, 2012; Rehmann, 2015), the viability of all four AML cell lines tested was reduced in response to EPAC1/2 inhibition (Fig EV5A and B). Together, these data demonstrate the potential for a pro‐survival role of the cAMP‐EPAC1/2 pathway (Grandoch et al, 2009) in the etiology of hematological malignancies with deregulated MYC activity such as AML and B‐cell lymphoma and in mediating cell survival in response of targeting the cell's translational machinery.
Figure EV5. Inhibiting EPAC1/2 in human acute myeloid leukemia (AML) cell lines and in vivo‐derived Eμ‐Myc lymphoma cell lines.
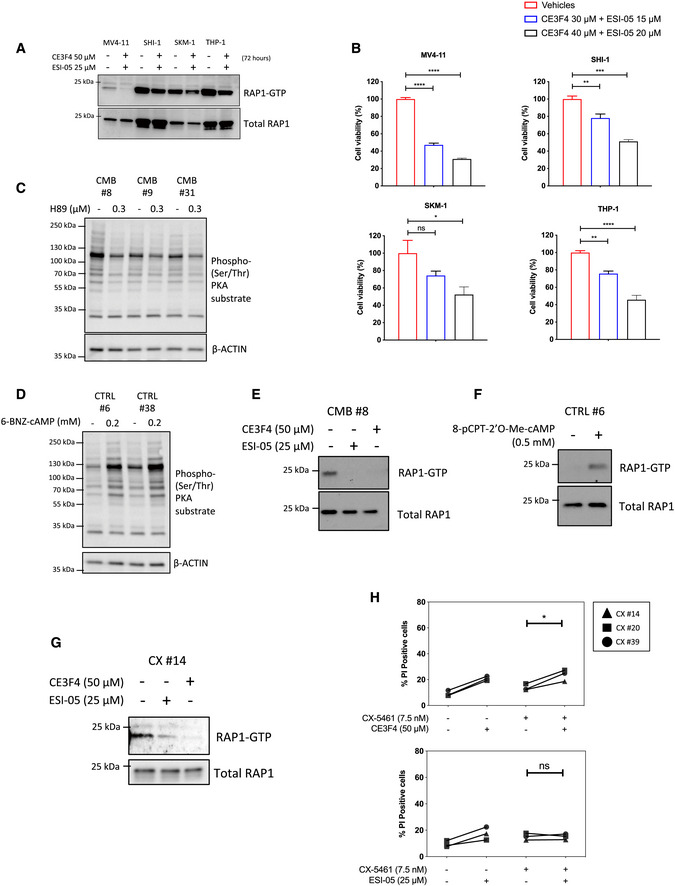
-
AWestern analysis of the activated, GTP‐bound RAP1 of the indicated human AML cell lines following treatments with the EPAC1 inhibitor CE3F4 and EPAC2 inhibitor ESI‐05 for 72 h (representative of n = 2).
-
BCellTiterGLO®‐based viability assay of MV4‐11, SHI‐1, SKM‐1, and THP‐1 human AML cells treated as indicated for 72 h. Graphs represent mean ± SEM of n = 3. Data were analyzed by one‐way ANOVA.
-
C, DWestern analysis of PKA activity using the phosphorylated PKA substrate antibody on whole cell extracts isolated from the indicated early passage cell lines treated for 24 h with (C) H89 (n = 3) or (D) 6‐BNZ-cAMP (representatives of n = 3). Actin was used as a loading control.
-
EWestern analysis of the activated, GTP‐bound RAP1 of the early passage CX‐5461 + EV‐resistant (CMB) cell extracts isolated following treatments with indicated EPAC inhibitors for 24 h (representative of n = 2).
-
FWestern analysis of activated, GTP‐bound RAP1 using early passage drug‐naive (CTRL) Eμ‐Myc lymphoma cell extracts isolated following treatments with the selective EPAC activator 8‐pCPT-2‐O-Me-cAMP for 24 h (n = 1).
-
GWestern analysis of the activated, GTP‐bound RAP1 of the early passage CX‐5461-resistant (CX) cell extracts isolated following treatments with indicated EPAC inhibitors for 24 h (n = 1).
-
HPI exclusion analysis of the CX cells treated with CX‐5461 as indicated in the presence or absence of EPAC1 inhibitor CE3F4 or EPAC2 inhibitor ESI‐05 for 48 h. Data were analyzed by paired one‐way ANOVA. Graphs represent mean ± SEM of n = 3. Black triangle: CX-5461‐resistant (CX) cells clone #14, black square: CX cells clone #20, black circle: CX cells clone #39.
To investigate whether altered cAMP signaling is fundamental for resistance to single‐agent and/or combinatorial ribosome‐targeted therapy, we utilized specific inhibitors/activators of PKA and EPAC (Poppe et al, 2008; Brown et al, 2014). Inhibition of PKA by H89 (Poppe et al, 2008; Fig EV5C) did not alter CMB cells’ response to CX‐5461 + EV treatment (Fig 3D). Similarly, 6‐Bnz‐cAMP, which robustly activates PKA signaling, as detected by a phospho‐PKA substrate antibody (Fig EV5D), was unable to protect CTRL cells from CX‐5461 + EV‐induced cell death (Fig 3E; Poppe et al, 2008; Lo et al, 2012). Importantly, inhibition of EPAC1 and EPAC2 function, as measured by the reduced abundance of GTP‐bound RAP1 by CE3F4 (Courilleau et al, 2012) and ESI‐05 (Tsalkova et al, 2012; Rehmann, 2015), respectively, resensitized CMB cells to CX‐5461 + EV treatment (Fig 3F and Fig EV5E). Moreover, dual inhibition of EPAC1 and EPAC2 by combining CE3F4 and ESI‐05 resulted in a more significant resensitization to CX‐5461 + EV treatment (Fig 3F), consistent with the proposed functional redundancy of the two isoforms (Robichaux & Cheng, 2018; Berkey et al, 2020). These data indicate that EPAC1/2 activity, but not PKA, is required for the development of resistance to CX‐5461 + EV. Consistent with these findings, activation of EPAC1/2 in the CTRL cells using 8‐pCPT‐2′‐O‐Me‐cAMP (Enserink et al, 2002; Rehmann et al, 2003), which increases RAP1‐GTP abundance (Fig EV5F), was able to reduce CX‐5461 + EV‐mediated cell death (Fig 3G). This confirms that elevated EPAC1/2 activity contributes to protection from CX‐5461 + EV‐mediated cell death and therefore the development of an energy‐ (Fig 2I) and cAMP‐dependent resistant mechanism in the CX‐5461 + EV‐resistant CMB cells. EPAC1/2 inhibitors did not resensitize CX cells to CX‐5461 treatment (Fig EV5G and H), indicating that while the resistant mechanisms induced following single‐agent and combinatorial ribosome‐directed treatments entail rewired metabolism, there are subtle differences in the resulting requirements for cAMP signaling. This may be reflected in the lack of increased cAMP observed in CX in comparison with CTRL cells (Fig 3B).
Reprogramming of mRNA translation underpins the metabolic rewiring required for resistance to ribosome‐targeting therapy
To determine whether the energy‐ and cAMP‐dependent changes observed in drug‐resistant cells are driven by specific reprogramming of mRNA translation, we evaluated the expression patterns of mRNAs encoding proteins that are known to be involved in the regulation of mRNA translation (genes in the “Translation” category in the Gene Ontology Resource databases; Fig 4A) in CTRL, CX, and CMB cells using the same polysome profiling dataset as Fig 3A. Importantly, CMB cells displayed distinct patterns of actively translating polysome‐associated mRNAs encoding the majority of ribosomal proteins, translation initiation (such as eIF4G1 and eIF2A), and elongation factors (such as eEF1A1 and eEF2) as compared to the CTRL and CX cells (Fig 4A).
Figure 4. Polysome profiling analysis identifies a link between translational reprogramming and resistance to CX‐5461 and CX‐5461 + everolimus.
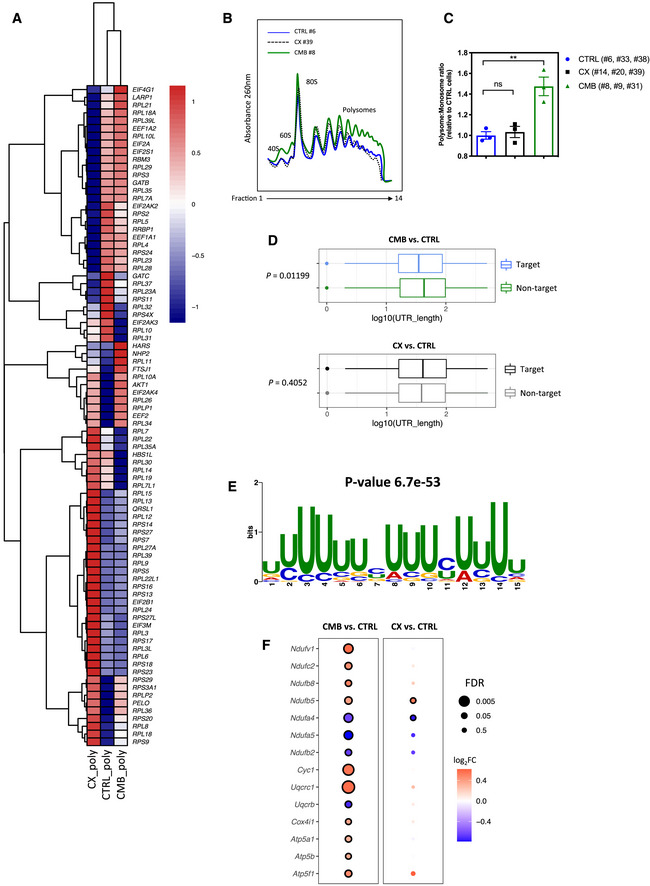
- A heatmap illustrating the abundances of polysome‐associated mRNAs that are associated with the mRNA translation/protein synthesis based on the Gene Ontology Resource Database in drug‐naïve (CTRL) or CX-5461‐everolimus combination therapy‐resistant (CMB) cell lines (n = 3). The colors illustrate the expression values of indicated mRNAs that were normalized using voom (red: high expression; blue: low expression). Values represent the median gene expression levels across replicate samples.
- Polysome profiles demonstrating increased ribosome abundance in CMB cells as compared to CTRL and CX-5461‐resistant (CX) cells (representatives of n = 3).
- Quantitation of polysome:monosome ratio in the indicated early passage Eμ‐Myc lymphoma cells as determined by measuring the area under the curve using ImageJ software. Graph represents mean ± SEM of n = 3. Data were analyzed by one‐way ANOVA. ns, not significant; **P ≤ 0.01.
- Length of the 5'UTR of mRNAs that are more actively translated by the CMB cells (as compared to CTRL cells) vs. non‐target mRNAs (n = 3). The central band corresponds to the median. The lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles). The upper and lower whiskers extend from the hinges to the largest and smallest values no further than 1.5 * interquartile range from the hinges, respectively. Data beyond the end of the whiskers are plotted individually. One‐sided Wilcoxon tests were used to determine significance.
- The most significantly enriched motif in the 5'UTR of mRNAs that are more actively translated in the CMB cells compared to CTRL cells based on Multiple Em for Motif Elicitation (MEME) analysis (n = 3).
- Polysomal abundance of mRNAs (selected based on a nominal P value < 0.01 cut‐off in either the CMB vs. CTRL or CX vs. CTRL as analyzed by limma) encoding components of mitochondrial oxidative phosphorylation based on polysome profiling data. Red denotes upregulation, and blue denotes downregulation (n = 3; genes with an adjusted P value (false discovery rate; FDR) ≤ 0.05 were considered to be significant and are denoted with a black border).
Source data are available online for this figure.
CMB cells were also characterized by significantly elevated levels of actively translating polysomes as shown in the increased polysome:monosome ratio as compared to CTRL and CX (Fig 4B and C). Further characterization of the 5′UTR sequences of mRNAs that are enriched in the polysomes of CMB cells revealed that they are more likely to have shorter UTRs (P = 0.01199), whereas in CX cells no significant differences were observed (P = 0.4052; Fig 4D). Moreover, motif analysis of the 5′UTR region of mRNAs that are actively translated in the CMB cells identified a uridine‐rich motif similar to the pyrimidine‐rich translational element (PRTE; Fig 4E). Short, PRTE‐containing mRNAs are a characteristic of mTOR targeted mRNAs (Hsieh et al, 2012; Gandin et al, 2016), and our findings are consistent with upregulation of translation of mRNAs normally targeted by mTOR inhibition as a source of resistance to ribosome‐directed therapies.
Since we observed that downregulation of oxidative phosphorylation is associated with the acute treatment in vivo (Fig 1E, Table EV2), we compared the polysome association of the oxidative phosphorylation‐associated mRNAs in the CTRL, CX, and CMB cell lines based on the same polysome profiling dataset used to generate Fig 3A (Table EV3). Indeed, CMB cells strikingly upregulated the abundance of polysome‐associated mRNAs encoding multiple components of the mitochondrial electron transport chain, such as NDUFV1 and NDUFC2 (Complex I), CYC1 and UQCRC1 (Complex III), and multiple subunits of the ATP synthase (Complex V) as compared to CTRL cells (Fig 4F). These differences may explain CMB cells’ higher metformin sensitivity compared to both CX and CTRL cells (Fig 2G). Collectively, these results indicate that such intrinsic differences in translational reprogramming events occurred in CMB cells as compared to CX cells gave rise to a distinct drug resistance program, which may, at least in part, explain the differences in metformin sensitivity (Fig 2G) and dependence on energy‐dependent cAMP levels and EPAC1/2 pathway activity (Fig 3F and Fig EV5H) in these in vivo‐derived early passage cell lines.
CX‐5461 + EV‐resistant cells are sensitized to metformin‐mediated targeting of the cAMP‐EPAC1/2‐RAP1 pathway in vivo
The data presented above demonstrated that targeting oxidative phosphorylation or the cAMP‐EPAC1/2‐RAP1 signaling pathway resensitized the CMB cells to CX‐5461 + EV treatment. While there is currently no inhibitor of the cAMP‐EPAC pathway in the clinic or in clinical trials, metformin is already being used in the clinic and can indeed reduce the intracellular abundance of cAMP (Miller et al, 2013). We hypothesized that metformin's ability to sensitize CMB cells to CX‐5461 + EV treatment is associated with inhibition of cAMP‐EPAC1/2‐RAP1 signaling. The CMB and CX cells were treated with metformin as for Fig 2G and H. This treatment robustly lowered RAP1‐GTP levels, but only in the CMB cells (Fig 5A), supporting a model (Synopsis Fig) where resistance to CX‐5461 + EV is, at least in part, mediated through increased energy metabolism and cAMP‐EPAC1/2‐RAP1 pathway that can be suppressed using metformin (Figs 2I and 5A).
Figure 5. Metformin inhibits cAMP‐EPAC1/2 signaling and resensitizes combination therapy‐resistant early passage Eμ‐Myc lymphoma cells to CX‐5461‐everolimus cotreatment in vivo .
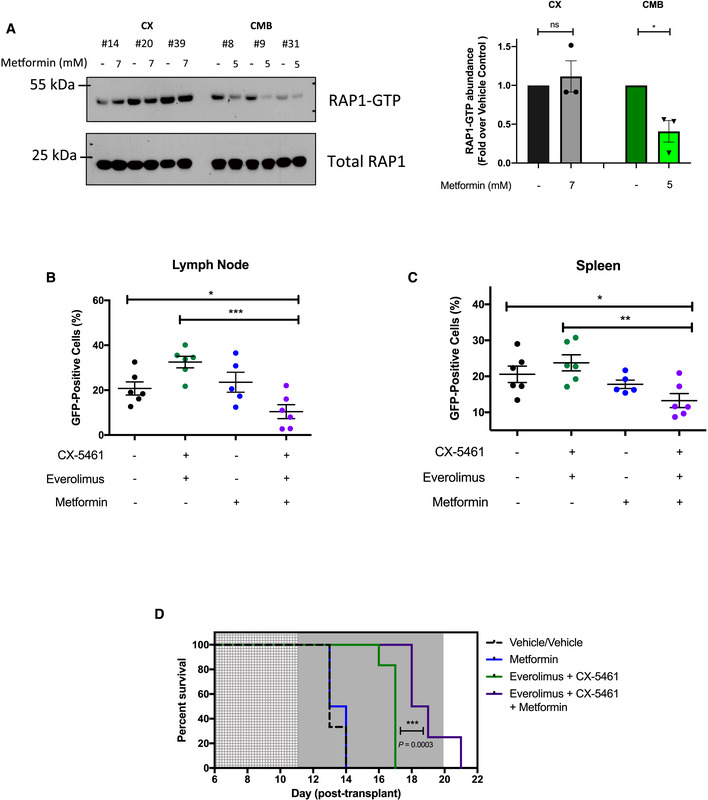
-
AWestern analysis demonstrating the effects of metformin treatment for 48 h on the levels of active GTP‐bound RAP1 in CX and CMB cells (n = 3) and its quantitation.
-
B, CProportion of green fluorescent protein (GFP)‐positive CMB (clone #8) cells in (B) lymph node and (C) spleen of transplanted C57BL/6 mice treated as indicated for 6 h on day 12 post‐transplant. Graphs represent mean ± SEM of six mice per group.
-
DKaplan–Meier curve of C57BL/6 mice transplanted with CX‐5461‐everolimus‐resistant (CMB #8) early passage Eμ‐Myc lymphoma cells treated with vehicles (everolimus vehicle: 1% methylcellulose; CX‐5461/metformin vehicle: 25 mM NaH2PO4; n = 6); CX‐5461 (35 mg/kg every twice weekly) and everolimus (5 mg/kg daily; n = 8), metformin (400 mg/kg twice daily; n = 6), or CX‐5461, everolimus and metformin (35 mg/kg twice weekly, 5 mg/kg daily and 400 mg/kg twice daily, respectively; n = 8). Light gray: 5‐day metformin pre‐treatment period, dark gray: treatment period. Data were analyzed by a log‐rank (Mantel–Cox) test. Vehicle vs. CX-5461‐everolimus: P = 0.0006, Vehicle vs. CX-5461‐everolimus‐metformin: P = 0.0001. CX-5461‐everolimus vs. CX-5461‐everolimus‐metformin: P = 0.0003.
To investigate whether metformin could also resensitize the CMB cells to CX‐5461 + EV treatment in vivo, CMB lymphoma‐bearing C57BL/6 mice were pre‐treated with metformin (Veiga et al, 2018) or vehicle for 3 days (600 mg/kg twice daily on the first and second day; 500 mg/kg twice daily on the third day) and then treated for 6 h with CX‐5461 and EV as single agents or in combination. As expected, CX‐5461 + EV did not alter the abundance of GFP‐positive drug‐resistant CMB cells in the lymph node (Fig 5B) or spleen (Fig 5C) compared to vehicle pre‐treated mice. Importantly, the single‐agent metformin had no effect on the number of GFP‐positive cells, whereas they were significantly reduced in the lymph nodes (Fig 5B) and spleens (Fig 5C) of mice receiving the CX‐5461 + EV plus metformin treatment. Thus, metformin was able to resensitize the CMB cells to CX‐5461 + EV treatment in vivo.
To determine whether the triple combination therapy could enhance survival of mice bearing this highly aggressive disease, C57BL/6 mice transplanted with the CMB cells were pre‐treated for 5 days with metformin (500 mg/kg twice daily) then at day 11 post‐transplant, treated with either: (i) vehicle; (ii) CX‐5461 + EV (35 mg/kg every Monday–Wednesday–Friday and 5 mg/kg daily, respectively); iii) metformin (400 mg/kg twice daily); or (iv) CX‐5461 + EV+metformin triple combination therapy. The triple combination significantly increased the survival window compared to all the groups including the CX‐5461 + EV treatment (Fig 5D). Together, these data show that the resensitization of the highly aggressive, drug‐resistant CMB cells to CX‐5461 + EV treatment in the presence of metformin in vitro (Fig 2I) was reproducible in vivo (Fig 5D), thus providing proof‐of‐principle that combined inhibition of translational machinery and energy metabolism for the treatment of highly aggressive MYC‐driven lymphoma can provide a therapeutic window.
Discussion
Oncogene‐driven cancers are characterized by elevated ribosome biogenesis that can be targeted with specific inhibitors of ribosomal RNA synthesis. We reported that CX‐5461 potently enhances the efficacy of PI3K/AKT/mTORC1 inhibitors in treating MYC‐driven lymphoma (Devlin et al, 2016). Here, we use a combination of polysome profiling and metabolomics analysis of tumors from acutely treated mice and early passage cells from acquired resistant tumors to interrogate the mechanisms underpinning this ribosome‐directed therapeutic approach. We demonstrate that the synergistic efficacy of combining CX‐5461 with the mTORC1 inhibitor everolimus is associated with significant suppression of the translation of mRNAs encoding key proteins controlling mRNA translation itself (Fig 1C), leading to selective inhibition of the translation of mRNAs encoding proteins critical for the regulation of energy metabolism, with the subsequent increased lymphoma cell death. It will be of interest in future studies to interrogate the mechanisms of this acute, specific targeting of the translational apparatus further including analysis of the role of the eIF2α‐dependent integrated stress response (Roux & Topisirovic, 2018).
Since p53‐null Eμ‐Myc B‐cell lymphomas are resistant to CX‐5461 + EV treatment (Devlin et al, 2016), we initially postulated that mutation or loss of p53 might underlie the resistance mechanism. Indeed, loss of TP53 is observed in ~40% of human Burkitt lymphoma cases (Schmitz et al, 2012, 2014). However, we observed that the p53 responses were maintained in acquired resistant cells (Fig EV3K). Instead, our studies define a new model (Synopsis Fig) of molecular response of oncogene‐driven cancer to ribosome‐targeted therapy.
Given the long‐term treatment of lymphoma‐bearing mice required to induce the transition of lymphomas to therapy resistance, it is likely that altered transcription may contribute to the functional changes in mRNA translation. We therefore focused on analyzing the levels of mRNAs associated with actively translating polysomes. The in vivo‐derived, drug‐resistant early passage tumor cells are characterized by translational reprogramming as shown by distinct patterns polysome association levels of mRNAs encoding proteins involved in protein synthesis (Fig 4A) compared to the drug‐naïve cells. This reprogramming of translation resulted in increased polysome abundance in CMB cells (Fig 4B). Moreover, there was increased polysome association of mRNAs containing short, pyrimidine‐rich 5′UTRs (Fig 4D and E) typical of mTOR‐sensitive mRNAs encoding components of the translational apparatus (Hsieh et al, 2012; Gandin et al, 2016). We propose that this is due to the robust upregulation of eIF4G1 in our CMB models (Fig 4A). eIF4G1 was previously shown to promote eIF4E‐dependent translation initiation of mRNAs containing short 5′UTRs termed as translation initiator of short 5′UTR (TISU) element (Gandin et al, 2016).
Importantly, gene enrichment analysis revealed that the polysome association of mRNAs encoding components of the mitochondrial respiratory electron transport chain (Fig 4F) and the cAMP‐EPAC1/2‐RAP1 pathway (Fig 3A) was significantly elevated. The resulting increase in ATP and cAMP production (Figs 2E and 3B) further activates EPAC signaling to RAP1, an energy‐dependent pro‐survival pathway that provides protection from drug‐induced cell death (Grandoch et al, 2009). Thus, the transition to resistance to combination therapy with CX‐5461 and everolimus is associated with reactivation of the translation of mRNAs encoding proteins that are involved in the processes of mRNA translation and cellular metabolism that are suppressed by acute treatment of lymphoma‐bearing mice. Specifically, anota2seq analysis identified mRNA translation to be significantly downregulated in CX‐5461 + EV‐treated mice (Fig 1C), but the CX‐5461 + EV‐resistant CMB cells exhibit higher polysomal abundance (Fig 4B and C) and altered expression profile of mRNAs encoding proteins implicated in protein synthesis (Fig 4A). Furthermore, we have demonstrated that acute targeting of translation results in compromised metabolism (Fig 1E, Table EV2). The drug‐resistant cells then re‐establish elevated metabolism (Fig 2) as shown by Seahorse and metabolomics data (Figs 2C‐F) and oxidative phosphorylation (Fig 4F).
Functionally, specific inhibition of cAMP‐EPAC1/2‐RAP1 signaling resensitizes resistant cells to combination therapy demonstrating that this pro‐survival pathway is a critical mediator of resistance (Figs 3D–G). Importantly, targeting elevated energy metabolism with the antidiabetic drug metformin sensitized both CX cells to CX‐5461 and CMB cells to CX‐5461 + EV. In CX cells, drug resensitization by metformin was likely to be associated with AMPK‐mediated inhibition of mTORC1 (Fig 2J and Appendix Fig S2C). In CMB cells, metformin was able to inhibit EPAC1/2‐RAP1 signaling and markedly improved the efficacy of combination therapy in resistant disease in vitro and in vivo (Fig 5A,C–E). Moreover, we highlighted the association of elevated EPAC1/2‐RAP1 signaling with poor prognosis in AML and DLBCL (Figs EV4B, EV4C and EV4E), as well as the potential of improving CX‐5461 treatment in AML (Figs EV2C–F).
Our findings demonstrate that elevated metabolic activity and energy production are not merely a hallmark of cancer associated with uncontrolled growth, but can be harnessed to activate the pro‐survival signaling through cAMP‐EPAC1/2‐RAP1, a new metabolic vulnerability that can be exploited to further improve the efficacy of ribosome‐targeting therapy. This makes a convincing case for the importance of developing inhibitors to EPAC function with improved pharmacological properties as an additional tool for targeting oncogene‐driven cancer. Thus, we believe this approach of cotargeting metabolism and Pol I‐directed ribosome‐targeting therapy provides a new paradigm for improving the efficacy of metabolic cancer therapies, as well as both traditional chemotherapeutics that target ribosome biogenesis and the newly developed low genotoxic approaches including CX‐5461 and BMH‐21 (Pelletier et al, 2018).
Finally, this study reinforces the recent finding that genetically compromised ribosome biogenesis results in specific rewiring of translation that underlies impaired erythroid differentiation (Khajuria et al, 2018). More broadly, diseases of genetically compromised ribosome biogenesis, such as Diamond Blackfan anemia, are similarly characterized by specific alterations in mRNA translation (Khajuria et al, 2018) and by significantly increased incidence of cancer, a paradox termed “Dameshek's riddle” (De Keersmaecker et al, 2015). This riddle is reinforced by observations of somatically acquired mutations and deletions in ribosomal proteins in T‐cell acute lymphoblastic leukemia as well as solid tumors, such as gastric and ovarian cancer (De Keersmaecker et al, 2013, 2015; Kandoth et al, 2013; Novetsky et al, 2013). Despite intense investigation, the mechanisms by which genetically compromised ribosome biogenesis leads to increased cancer susceptibility in patients with ribosomopathies remain a mystery. Our findings raise the possibility that at least part of the answer to Dameshek's riddle lies in specific rewiring of translation in response to chronically compromised ribosome biogenesis, whereby the subsequent translationally driven elevated metabolism and pro‐survival mechanisms promote malignant transformation later in life.
Materials and Methods
Cell culture and reagents
Eμ‐Myc B‐cell lymphoma cells (MSCV Gfp) were cultured in Anne Kelso DMEM, supplemented with 10% heat‐inactivated (h.i.) fetal bovine serum (FBS), 100 μM L‐asparagine (Merck), penicillin/streptomycin/glutamine (Thermo Fisher Scientific), and 0.5% beta‐mercaptoethanol.
Early passage Eμ‐Myc B‐cell lymphoma cell lines (listed in Fig 2A) were isolated from the mice transplanted with Eμ‐Myc lymphoma (MSCV Gfp; clone #107) cells who had succumbed to disease relapse following treatment with vehicles, everolimus, CX‐5461, or CX‐5461 + EV at the end of a survival experiment reported previously (fig 2B in Devlin et al, 2016). Specifically, lymph nodes isolated at endpoint of the in vivo experiment were manually disrupted by pressing with the plunger of a 3‐ml syringe and the cell suspension was passed through 40‐μm cell filters to obtain single‐cell suspensions. These cells were then cultured in Anne Kelso DMEM supplemented with 20% h.i. FBS (Gibco), 100 μM L‐asparagine (Merck), penicillin/streptomycin/glutamine (Thermo Fisher Scientific), and 0.5% beta‐mercaptoethanol until exponential growing phase was obtained (2–3 passages). The study of resistance mechanisms (Figs 2, 3, 4) is all performed in these early passage cell lines until Fig 5 where hypotheses generated are tested in vivo.
Human acute myeloid lymphoma cell lines (MV4‐11 (MLL‐AF4; p53 wild‐type (WT), SKM‐1 (EZH2; p53‐mutant (R248Q)), SHI‐1 (MLL‐AF6; p53WT), THP‐1 (t(9;11); p53‐null R174 fs)) were obtained from German Collection of Microorganisms and Cell Cultures and cultured in RPMI 1640 medium plus HEPES supplemented with 20% h.i. FBS, 4 mM Glutamax (Thermo Fisher Scientific), and 1% v/v antibiotics/antimycotics (Thermo Fisher Scientific). Everolimus (S1120) was purchased from Selleckchem. CX‐5461 was purchased from SYNkinase (SYN‐3031). Metformin was purchased from Merck (D150959). CEF34 was purchased from Cayman Chemicals (17767). ESI‐05 and 8‐pCPT‐2‐O‐Me‐cAMP were purchased from Biolog (catalogue number M092 and C041, respectively). 6‐Bnz‐cAMP was purchased from Tocris Bioscience (5255). H89 was purchased from Merck (B1427)
Animal experiments
All animal experiments were performed with approval from the Animal Experimentation Ethics Committee at the Peter MacCallum Cancer Centre (Ethics number E462 and E557). For in vivo drug studies, 2 × 105 green fluorescent protein (GFP)‐expressing Eμ‐Myc lymphoma cells were injected into the tail vein of 6‐ to 8‐week‐old male C57BL/6 mice (Walter and Eliza Hall Institute, Australia). Disease onset/progression was monitored by analysis of GFP+ cells in peripheral blood (tail bleed) on day 9 post‐injection. Mice were treated with pharmacologic inhibitors from 10 days post‐transplant (survival studies). In acute studies, the details on duration and number of days post‐transplant, as well as metformin pre‐treatment (if applicable) are included in the text or figure legends. Everolimus was administered daily at 5 mg/kg via oral gavage in 5% dimethyl sulfoxide (DMSO) in 1% methylcellulose. CX‐5461 was administered twice weekly (unless otherwise indicated in figure legends) at 35 mg/kg via oral gavage in 25 mM NaH2PO4 (pH 4.5). Metformin was administrated twice daily (2 × 500 mg/kg unless otherwise indicated in figure legends) via oral gavage in 25 mM NaH2PO4 (pH 4.5). For survival studies, mice were euthanized at the ethical endpoint upon disease relapse as indicated by signs of distress including enlarged lymph nodes, ruffled fur, inactivity, hunched posture, labored breathing, and equal to or greater than 20% weight loss. For acute studies, mice were euthanized at the pre‐determined time points. Cardiac blood, lymph nodes (inguinal, axillary, brachial), and spleen were collected. One inguinal lymph node was crushed in PBS with 2% h.i. FBS to generate a single‐cell suspension for flow cytometry analysis. White blood cells and lymph node cells were stained for B220 (CD45R; Thermo Fisher Scientific 17‐0452‐82) and PI before flow cytometry analysis using BD CantoII or BD Fortessa. Flow cytometry data were analyzed with Flowlogic software (Inivai Technologies). The remaining lymph nodes were snap‐frozen. For Western analysis in Fig 1A, the lymph nodes were homogenized using a Precellys 24/Cryolys cryomill (Bertin Technologies; 6,800 rpm; 2 × 30‐s pulse, 45‐s interval between pulses; 0°C) before protein extraction.
Protein analysis and Western blotting
Protein was extracted with SDS‐lysis buffer (0.5 mM EDTA, 20 mM HEPES, 2% (w/v) SDS pH 7.9), and protein concentrations were determined with the Bio‐Rad DC protein assay. Proteins were resolved by SDS–PAGE, transferred to PVDF membranes, and immunoblotted with primary and horseradish peroxidase‐conjugated secondary antibodies (Appendix Table S1). Active RAP1 pull‐down experiments were performed using the RAP1 Activation Assay kit (Abcam ab212011) according to manufacturer's instructions. The positive controls were included as shown in Source Data. Protein was visualized by Amersham enhanced chemiluminescence (ECL) Western Blotting Detection Reagent (GE Healthcare Life Sciences), and X‐ray film (Fujifilm SuperRX) or ChemiDoc™ Touch Imaging System (Bio‐Rad).
Cytoplasmic lysates for polysome profiling
To obtain cytoplasmic lysate from suspension cells, 30 × 106 cells per sample were harvested by removing the culture media following centrifugation (400 g, 4 min, 4 °C) and washed with ice‐cold hypotonic buffer (5 mM Tris pH 7.5; 1.5 mM KCl; 2.5 mM MgCl2; and 100 μg/ml cycloheximide (Sigma)). The supernatant was removed by centrifugation (400 g, 4 min, 4°C). Hypotonic lysis buffer (5 mM Tris pH 7.5; 1.5 mM KCl; 2.5 mM MgCl2; 0.5% Triton X; 0.5% sodium deoxycholate; 1× EDTA‐free protease inhibitor; 2 mM DTT; 10 μl RNAsin (Promega); 100 μg/ml cycloheximide) was added to the cell pellet. The samples were mixed by pipetting and centrifuged (16,000 g, 7 min, 4°C). The supernatant (cytoplasmic lysate) was transferred to a fresh microfuge tube. Ten percent of the cytoplasmic lysate was transferred into a microfuge tube containing 500 μl TRIzol® reagent to be used for normalization of the polysome‐associated mRNA sequencing data.
For in vivo translational profiling experiments, cytoplasmic lysate was obtained by grinding snap‐frozen tissue samples (40–50 mg per sample) in mortar and pestle submerged in dry ice‐100% ethanol slurry to a fine powder. The sample was transferred to an ice‐cold dounce homogenizer and a modified lysis buffer (containing 10‐fold higher concentration of cycloheximide (i.e., 1,000 μg/ml) compared to that used for cultured cells; RNAsin was replaced with 10 mM ribonucleoside vanadyl complex (New England Biolabs S1402S) was added to the ground tissue. The sample was then homogenized in the dounce homogenizer (30–60 strokes). The homogenate was transferred to a chilled microfuge tube and centrifuged (16,000 g, 7 min, 4°C). The supernatant (cytoplasmic lysate) was transferred to a fresh microfuge tube. 10% of the cytoplasmic lysate was set aside into a microfuge containing 500 μl TRIzol® reagent to be used for cytoplasmic mRNA isolation and analysis and the subsequent normalization of polysome‐associated mRNA sequencing data, while the remaining lysate was layered on top of a sucrose gradient.
Polysome profiling and RNA isolation
The cytoplasmic lysate was layered on top of a linear 10–40% sucrose gradient, ultracentrifuged (SW41 rotor, 222,228 g, 2¼ h at 4°C using SW41Ti rotor), and fractionated using the Foxy Jr Fraction Collector with constant monitoring of absorbance at 260 nm by an ISCO UA‐6 Absorbance Detector (Teledyne). Cytoplasmic (input) RNAs and polysomal RNAs from pooled fractions corresponding to four or more ribosomes were isolated using a TRIzol®‐based method and purified using the Qiagen RNeasy® Minikit according to manufacturer's instructions and analyzed by RNA‐seq. RNA concentration was determined using a NanoDrop Spectrophotometer (Thermo Scientific), and integrity was evaluated using the RNA Nano Kit and 2100 bioanalyzer (Agilent Technologies). cDNA libraries were generated using a ribodepletion method (TruSeq Ribo Profile Mammalian Kit, Illumina) from 1 μg of RNA sample. Fragment sizes were evaluated using the DNA 1000 kit and 2100 bioanalyzer (Agilent Technologies). Libraries were subjected to single‐end sequencing (SE50 bp; Hiseq2500, Illumina) to generate ~30 million reads per sample using standard protocols.
RNA‐seq analysis
Sequencing reads were aligned with Tophat2, and reads counts were obtained with htseq (Anders et al, 2015) with the mouse genome (Ensembl release 78) as a reference, using an in‐house bioinformatics pipeline, seqliner (http://bioinformatics.petermac.org/seqliner/). Differential translation was determined using anota2seq analysis as described in (Lorent et al, 2019; Oertlin et al, 2019). Only coding genes with a count of at least one in any of the samples were considered. Samples were normalized using the “voom” option in anota2seq. An omnibus analysis was first performed in anota2seq to enrich for genes affected by any of the treatments, using a groupRvmPAdj cut‐off of 0.15. Differential translation analysis was subsequently performed using anota2seq on this reduced set. Only genes with an apvRvm adjusted P value < 0.05 and an apvEff (fold change) greater than 1.25 were considered to be significant. Enrichment analysis of the significant genes was performed with MetaCore® GeneGO (Clarivate Analytics) using the default parameters.
Comparison of pathway activity after acute in vivo treatment by single sample gene set enrichment analysis (ssGSEA)
Pathway annotations
Pathway information was obtained from two sources, the Molecular Signatures database (MSigDB) version 5.2 and the Gene Ontology (GO). From MSigDB the H (Hallmark), C2 (Curated) and C5 (Gene Ontology) gene sets for mouse orthologs were downloaded from http://bioinf.wehi.edu.au/software/MSigDB/. GO annotations were downloaded from http://www.geneontology.org/ on the June 28, 2017. Only non‐electronic (non‐IEA) annotations were included in the analysis. The hierarchical tree structure of GO terms was used to correct for incompleteness and inconsistencies in GO annotations, by using the annotations of genes in lower‐order offspring terms to complete the gene list of higher‐order ancestor GO terms, creating a set of comprehensive GO gene sets.
Calculating activity levels
We selected pathways representative of key cellular biological processes involved in growth, proliferation, and metabolism. We measured the level of activity of pathways at the level of both transcription (cytosolic mRNA) and translation (mRNA bound to polysomes) in individual samples using single sample gene set enrichment analysis (ssGSEA; Barbie et al, 2009) from the GSVA (Hanzelmann et al, 2013) package (version 1.20.0). Here, genes in each sample were ranked according to their expression levels, and a score for each pathway was generated based on the empirical cumulative distribution function, reflecting how highly or lowly genes of a pathway are ranked. Since ssGSEA relies on the ranking of genes to determine the score pathways, all genes, regardless of their levels of expression, were considered in the analysis and no pre‐filtering was performed. Samples were normalized using voom normalization, and normalization of ssGSEA was performed with the standard method implemented in GSVA.
Comparison of the activity of pathways between treatments and sample types
For each condition, we first averaged the ssGSEA scores of each pathway across polysomal mRNA samples and scaled to a range of 1–10 by linear transformation to avoid negative scores. We calculated the difference in ssGSEA means in the relevant contrasts for each sample type separately as a percentage change in expression of that pathway relative to condition 1.
| (1) |
where cond1 (condition 1) represents the reference control condition, and cond2 (condition 2) represents the case condition (CMB, EV or CX, depending on the comparison).
To perform differential analysis of polysomal data in resistant studies (i.e., to compare mRNAs that were bound to polysomes of drug‐resistant vs. drug‐naïve control cells; Figs 3 and 4), we used limma version 3.28.21 (Ritchie et al, 2015) from Bioconductor. The data were normalized using trimmed mean of M values (TMM; Robinson & Oshlack, 2010) followed by voom (Law et al, 2018). Only coding genes with more than 25 reads were considered. Genes were considered to be significant if their P values were less than 0.01. Enrichment analysis of genes was performed with MetaCore® GeneGO (Clarivate Analytics) using the default parameters, with additional adjusted P values (after Benjamini and Hochberg correction; FDR) cut‐off of less than 0.05, and fold change cut‐off of greater than 1.5 or less than −1.5. Details of the 5′UTR analysis are provided in Appendix Supplementary Methods.
Cell viability assay
Cells were seeded in 96‐well plates. Twenty‐four hours later, cells were treated with pharmacological compounds as indicated in the figure legends or text. Cells were stained with 1 μg/ml PI (Merck P4170) and analyzed using the FACSVerse (BD Biosciences). Flow cytometry data were analyzed with Flowlogic software (Inivai Technologies). Cell number and percent live cells were determined using the Z2 Coulter Counter (Beckman Coulter 383550) or CellTiterGLO®‐based assay followed by luminescence reading by Cytation 3 Cell Imaging Multimode Reader (BioTek).
32P‐orthophosphate labeling
Eμ‐Myc lymphoma cells (3 × 106) were cultured in 3 ml media in the presence of 0.5 mCi of 32P‐orthophosphate for 15 min. Cells were harvested on ice and RNA extracted using the QIAGEN RNeasy Minikit according to the manufacturer's instructions. RNA (5 μg) was run overnight on a 1.2% MOPS/formaldehyde agarose gel. The gel was dried using a Model 583 Gel Drier (Bio‐Rad), exposed overnight to a phospho‐imager screen (Molecular Dynamics), and scanned using a Typhoon Trio Variable Mode Imager (GE Healthcare). Band intensities were quantitated using Image Quant TL software (GE Healthcare).
Bioenergetics analysis using the Seahorse XF96 Extracellular Flux Analyzer
All bioenergetics analyses were performed using the Seahorse Bioscience XF96 extracellular flux analyzer (Seahorse Bioscience, Billerica, USA). Details of the analysis are provided in Appendix Supplementary Methods.
Metabolomics analysis
Details of the gas chromatography–mass spectrometry (GC‐MS) and liquid chromatography–mass spectrometry (LC‐MS) are provided in Appendix Supplementary Methods.
Statistical analysis
Statistical tests were performed as described in Figure legends with GraphPad Prism Software (version 8) or as outlined in relevant polysome profiling or metabolomics methods sections detailing their respective bioinformatics analysis. In AML studies, visualization of expression analysis was generated using the gene expression profiling interactive analysis (GEPIA) bioinformatics tool (Tang et al, 2017).
Author contributions
E.P.K, R.D.H., K.M.H., J.K., and R.B.P conceptualized. A.S.T. involved in data curation. E.P.K, A.S.T., C.C., D.L.G., O.L., J.R.D., D.P.D., M.J.M, K.M.H., J.K., and R.B.P. formally analyzed the manuscript. G.A.M., R.D.H., and R.B.P. acquired funding. E.P.K., C.C., J.R.D., D.P.D, and J.K involved in investigation. O.L., D.P.D., and M.J.M involved in methodology. C.C., D.L.G., J.R.D, K.T.C, D.P.D, M.J.M., G.A.M., G.T., E.S., G.P., and R.D.H provided resources. O.L provided software. K.M.H., J.K., and R.B.P supervised the study. E.P.K. wrote original draft. E.P.K., A.S.T., G.T., K.M.H., J.K., and R.B.P involved in writing, review, and editing.
Conflict of interest
G.A. McArthur reports receiving commercial research grants from Celgene, Novartis, and Ventana and is a consultant/advisory board member for Provectus. R.D. Hannan is a consultant/advisory board member for Pimera, Inc. No potential conflicts of interest were disclosed by the other authors.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
The authors thank Kerry Ardley, Susan Jackson, and Rachael Walker for technical assistance with animal experiments. They also thank the Peter MacCallum Cancer Centre Animal Facility, Molecular Genomics Core, FACS Facility, Laboratory Services, and Media Kitchen. The authors would like to acknowledge Dr Maurits Evers (ANU) for his input into the visualization of the next‐generation sequencing data. This work was supported by project grants and fellowships (M.J.M. Principal Research Fellowship #1154540, R.D.H. Project Grant #1158732; Principal Research Fellowship #1116999, G.A.M Principal Research Fellowship #1106576, and R.B.P. Project Grants #1053792 and #1102609; Senior Research Fellowship #1058586) and a program grant (R.D.H., G.A.M, and R.B.P. NHMRC Program Grant #1053792) from the National Health and Medical Research Council (NHMRC) of Australia. E.K. was supported by a University of Melbourne International Fee Remission Research Scholarship (MIFRS), a Melbourne International Research Scholarship (MIRS), and a Cancer Therapeutics (CTx) PhD Top Up Scholarship. A.S.T was supported by a University of Melbourne MIFRS and a Melbourne International Engagement Award (MIEA). E.S. is supported by a Victorian Cancer Agency Mid‐Career Research Fellowship (MCRF19007).
The EMBO Journal (2020) 39: e105111
Data availability
The datasets and computer code produced in this study are available in the following databases:
RNA‐Seq data: Gene Expression Omnibus GSE154614 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154614)
Modeling computer scripts: https://github.com/cancer-evolution/CX5461_translation_reprogramming
References
- Anders S, Pyl PT, Huber W (2015) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C (2009) Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature 462: 108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkey SC, Herrera JJ, Odem MA, Rahman S, Cheruvu SS, Cheng X, Walters ET, Dessauer CW, Bavencoffe AG (2020) EPAC1 and EPAC2 promote nociceptor hyperactivity associated with chronic pain after spinal cord injury. Neurobiol Pain 7: 100040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL (2006) Epac proteins: multi‐purpose cAMP targets. Trends Biochem Sci 31: 680–686 [DOI] [PubMed] [Google Scholar]
- Brown LM, Rogers KE, Aroonsakool N, McCammon JA, Insel PA (2014) Allosteric inhibition of Epac: computational modeling and experimental validation to identify allosteric sites and inhibitors. J Biol Chem M114: 569319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K et al (2012) Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer‐specific activation of p53. Cancer Cell 22: 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JC, Hannan KM, Riddell K, Ng PY, Peck A, Lee RS, Hung S, Astle MV, Bywater M, Wall M (2011) AKT promotes rRNA synthesis and cooperates with c‐MYC to stimulate ribosome biogenesis in cancer. Sci Signal 4: ra56 [DOI] [PubMed] [Google Scholar]
- Chapuis N, Poulain L, Birsen R, Tamburini J, Bouscary D (2019) Rationale for targeting deregulated metabolic pathways as a therapeutic strategy in acute myeloid leukemia. Front Oncol 9: 405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courilleau D, Bisserier M, Jullian J‐C, Lucas A, Bouyssou P, Fischmeister R, Blondeau J‐P, Lezoualćh F (2012) Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP‐binding protein Epac. J Biol Chem M112: 422956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker K, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, Gianfelici V, Geerdens E, Clappier E, Porcu M (2013) Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T‐cell acute lymphoblastic leukemia. Nat Genet 45: 186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker K, Sulima SO, Dinman JD (2015) Ribosomopathies and the paradox of cellular hypo‐to hyperproliferation. Blood 125: 1377–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin JR, Hannan KM, Hein N, Cullinane C, Kusnadi E, Ng PY, George AJ, Shortt J, Bywater MJ, Poortinga G et al (2016) Combination therapy targeting ribosome biogenesis and mRNA translation synergistically extends survival in MYC‐driven lymphoma. Cancer Discov 6: 59–70 [DOI] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL (2002) A novel Epac‐specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol 4: 901 [DOI] [PubMed] [Google Scholar]
- Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B (2014) Metformin: from mechanisms of action to therapies. Cell Metab 20: 953–966 [DOI] [PubMed] [Google Scholar]
- Gandin V, Masvidal L, Hulea L, Gravel S‐P, Cargnello M, McLaughlan S, Cai Y, Balanathan P, Morita M, Rajakumar A (2016) nanoCAGE reveals 5'UTR features that define specific modes of translation of functionally related MTOR‐sensitive mRNAs. Genome Res 26: 636–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich M, Bos JL (2010) Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol 50: 355–375 [DOI] [PubMed] [Google Scholar]
- Grandoch M, Bujok V, Fleckenstein D, Schmidt M, Fischer J, Weber AA (2009) Epac inhibits apoptosis of human leukocytes. J Leukoc Biol 86: 847–849 [DOI] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics 14: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein N, Cameron DP, Hannan KM, Nguyen NN, Fong CY, Sornkom J, Wall M, Pavy M, Cullinane C, Diesch J et al (2017) Inhibition of Pol I transcription treats murine and human AML by targeting the leukemia‐initiating cell population. Blood 129: 2882–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485: 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F, Yap TA, Meric‐Bernstam F (2018) Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol 15: 273 [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA (2013) Mutational landscape and significance across 12 major cancer types. Nature 502: 333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Kusnadi EP, Ogden AJ, Hicks RJ, Bammert L, Kutay U, Hung S, Sanij E, Hannan RD, Hannan KM (2016) Amino acid‐dependent signaling via S6K1 and MYC is essential for regulation of rDNA transcription. Oncotarget 7: 48887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajuria RK, Munschauer M, Ulirsch JC, Fiorini C, Ludwig LS, McFarland SK, Abdulhay NJ, Specht H, Keshishian H, Mani DR et al (2018) Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell 173: 90–103.e119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khot A, Brajanovski N, Cameron DP, Hein N, Maclachlan KH, Sanij E, Lim J, Soong J, Link E, Blombery P (2019) First‐in‐human RNA polymerase I transcription inhibitor CX‐5461 in patients with advanced hematological cancers: results of a phase I dose escalation study. Cancer Discov 9: 1036–1049 [DOI] [PubMed] [Google Scholar]
- Kusnadi EP, Hannan KM, Hicks RJ, Hannan RD, Pearson RB, Kang J (2015) Regulation of rDNA transcription in response to growth factors, nutrients and energy. Gene 556: 27–34 [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK (2014) voom: precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biol 15: R29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MG, Obinata D, Sandhu S, Selth LA, Wong SQ, Porter LH, Lister N, Pook D, Pezaro CJ, Goode DL (2018) Patient‐derived models of abiraterone‐and enzalutamide‐resistant prostate cancer reveal sensitivity to ribosome‐directed therapy. Eur Urol 74: 562–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibovitch M, Topisirovic I (2018) Dysregulation of mRNA translation and energy metabolism in cancer. Adv Biol Regul 67: 30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo KWH, Kan HM, Ashe KM, Laurencin CT (2012) The small molecule PKA‐specific cyclic AMP analogue as an inducer of osteoblast‐like cells differentiation and mineralization. J Tissue Eng Regen Med 6: 40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorent J, Kusnadi EP, van Hoef V, Rebello RJ, Leibovitch M, Ristau J, Chen S, Lawrence MG, Szkop KJ, Samreen B (2019) Translational offsetting as a mode of estrogen receptor α‐dependent regulation of gene expression. EMBO J 38: e101323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J (2009) Molecular mechanisms of mTOR‐mediated translational control. Nat Rev Mol Cell Biol 10: 307 [DOI] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ (2013) Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494: 256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Gravel S‐P, Chénard V, Sikström K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C (2013) mTORC1 controls mitochondrial activity and biogenesis through 4E‐BP‐dependent translational regulation. Cell Metab 18: 698–711 [DOI] [PubMed] [Google Scholar]
- Novetsky AP, Zighelboim I, Thompson DM Jr, Powell MA, Mutch DG, Goodfellow PJ (2013) Frequent mutations in the RPL22 gene and its clinical and functional implications. Gynecol Oncol 128: 470–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertlin C, Lorent J, Murie C, Furic L, Topisirovic I, Larsson O (2019) Generally applicable transcriptome‐wide analysis of translation using anota2seq. Nucleic Acids Res 47: e70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Thomas G, Volarevic S (2018) Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer 18: 51–63 [DOI] [PubMed] [Google Scholar]
- Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang X‐B, Christensen AE, Schwede F, Genieser H‐G, Bos JL, Doskeland SO et al (2008) Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods 5: 277 [DOI] [PubMed] [Google Scholar]
- Quin JE, Devlin JR, Cameron D, Hannan KM, Pearson RB, Hannan RD (2014) Targeting the nucleolus for cancer intervention. Biochim Biophys Acta 1842: 802–816. [DOI] [PubMed] [Google Scholar]
- Rebello RJ, Kusnadi E, Cameron DP, Pearson HB, Lesmana A, Devlin JR, Drygin D, Clark AK, Porter L, Pedersen J (2016) The dual inhibition of RNA Pol I transcription and PIM kinase as a new therapeutic approach to treat advanced prostate cancer. Clin Cancer Res 22: 5539–5552 [DOI] [PubMed] [Google Scholar]
- Rehmann H, Schwede F, Døskeland SO, Wittinghofer A, Bos JL (2003) Ligand‐mediated activation of the cAMP‐responsive guanine nucleotide exchange factor Epac. J Biol Chem 278: 38548–38556 [DOI] [PubMed] [Google Scholar]
- Rehmann H (2013) Epac‐inhibitors: facts and artefacts. Sci Rep 3: 3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux WG III, Cheng X (2018) Intracellular cAMP sensor EPAC: physiology, pathophysiology, and therapeutics development. Physiol Rev 98: 919–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol 11: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Topisirovic I (2018) Signaling pathways involved in the regulation of mRNA translation. Mol Cell Biol 38: e00070‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D (2013) Translational control in cancer etiology. Cold Spring Harb Perspect Biol 5: a012336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168: 960–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E (2012) Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490: 116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM (2014) Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb Perspect Med 4: a014282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z (2017) GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45: W98–W102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL, Cheng X (2012) Isoform‐specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci USA 109: 18613–18618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga SR, Ge X, Mercer CA, Hernández‐Alvarez MI, Thomas HE, Hernandez‐Losa J, Cajal SRY, Zorzano A, Thomas G, Kozma SC (2018) Phenformin‐induced mitochondrial dysfunction sensitizes hepatocellular carcinoma for dual inhibition of mTOR. Clin Cancer Res 24: 3767–3780 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
The datasets and computer code produced in this study are available in the following databases:
RNA‐Seq data: Gene Expression Omnibus GSE154614 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154614)
Modeling computer scripts: https://github.com/cancer-evolution/CX5461_translation_reprogramming