

Abstract
Elucidating the regulation mechanism of integrin aIIbβ3 is key to understanding platelet biology and thrombotic diseases. Previous in vitro studies have implicated a role of migfilin in the support of platelet aIIbβ3 activation, however, contribution of migfilin to thrombosis and hemostasis in vivo and a detailed mechanism of migfilin in platelets are not known. In this study, through migfilin knock-out (migfilin-/-) mice, we report that migfilin is a pivotal positive regulator of hemostasis and thrombosis. Migfilin-/- mice show a nearly doubled tail-bleeding time and a prolonged occlusion time in FeCl3-induced mesenteric arteriolar thrombosis. Migfilin deficiency impedes platelet thrombi formation on a collagen surface and impairs platelet aggregation and dense-granule secretion. Supported by characteristic functional readings and the phosphorylation status of distinctive signaling molecules in the bidirectional signaling processes of aIIbβ3, the functional defects of migfilin-/- platelets appear to be mechanistically associated with a compromised outside-in signaling, rather than inside-out signaling. A synthesized cell-permeable migfilin peptide harboring filamin A binding sequence rescued the defective function and phosphorylation of signaling molecules of migfilin-/- platelets. Finally, migfilin does not influence the binding of filamin A and β3 subunit of aIIbβ3 in resting platelets, but hampers the re-association of filamin A and β3 during the conduct of outside- in signaling, suggesting that migfilin functions through regulating the interaction dynamics of aIIbβ3 and filamin A in platelets. Our study enhances the current understanding of platelet integrin aIIbβ3-mediated outside-in signaling and proves that migfilin is an important regulator for platelet activation, hemostasis and thrombosis.
Introduction
Platelets are essential for thrombosis and hemostasis. At the sites of vascular injury, platelets instantly activate, adhere to the exposed subendothelial matrix and form a hemostatic or thrombotic plug. Idiosyncratically expressed by platelets, integrin aIIbβ3 is the central adhesion molecule that governs the process of thrombus formation. aIIbβ3 characteristically transmits signals bidirectionally across the plasma membrane, processes termed inside-out and outside-in signaling, respectively. 1 Inside-out signaling conveys the activation information from stimuli and culminates in the high affinity binding of aIIbβ3 to its ligands, which forms bridges to support platelet-vessel wall adhesion and platelet homotypic aggregation. Outside-in signaling, which is initiated by ligand binding and aIIbβ3 clustering, is essential for platelet spreading, clot retraction and thrombus consolidation.2
Platelet aIIbβ3 signaling is regulated by the binding of several molecules to the cytoplasmic domain of integrin β-subunits.3 The cytoplasmic integrin-binding protein talin has been known as the central regulator for aIIbβ3.4 There are also other important integrin-binding proteins such as kindlins and filamin A, which can potentially profoundly influence the function of aIIbβ3.5-7 Thus, kindlin-3 co-operates with talin and positively regulates the activation of integrin aIIbβ3,3 whereas filamin A competitively blocks the talin-integrin interaction5 and negatively regulates aIIbβ3.3,8 Intriguingly, the interactions between integrin β3 and integrin-binding proteins are highly dynamic, even the same pair of interactive partners may serve distinctive roles in different phases of the platelet activation pathway. For example, talin-β3 binding is the essential step for aIIbβ3 activation for inside-out signaling, whilst the subsequent G13-regulated dissociation and re-association between talin and β3 are not only pivotal steps for outside- in signaling but are also the crucial events that differentiate thrombosis from hemostasis.9 This example highlights the importance of clarifying the dynamic interaction between integrin and integrin-binding proteins during aIIbβ3 activation. However, the spatial temporal dynamics for most other integrin-binding proteins remain largely unknown.
Migfilin, also known as FBLIM1/FBLP-1,10 is a putatively expressed protein consisting of an N-terminal filaminbinding domain, a central proline rich domain and three C-terminal LIM domains. Through participation in cytoskeleton reorganization, migfilin is involved in cellular functions such as adhesion, morphological change, and motility.11 Migfilin has also been reported to promote cardiomyocyte differentiation.12 There are multiple binding partners of migfilin, including mitogen inducible gene-2 (MIG-2), VASP, and transcriptional factor CSX/NKX2-5.10 Particularly, migfilin is capable of strong interaction with filamin A, which raises the possibility that migfilin could competitively dissociate filamin A from integrin and thus promote talin-aIIbβ3 interaction.13 Indeed, migfilin peptides are capable of inducing PAC-1 binding in human platelets.14 However, these findings were obtained in in vitro conditions and neither the contribution of migfilin to thrombosis and hemostasis in vivo nor to integrin dynamics during platelet activation are clear.
With migfilin-/-mice, the current study investigated the role of migfilin in platelet activation, hemostasis and thrombosis. The phenotype exhibited by migfilin-/- mice indicated that migfilin is an important positive regulator for hemostasis and thrombosis. Mechanistically, migfilin promotes early outside-in signaling of platelet aIIbβ3, possibly through binding to filamin A and sequestering the aIIbβ3-inhibiting effect of filamin A.
Methods
Generation of migfilin deficient mice
Migfilin-/- mice were bred as described,15 ablation of migfilin was achieved by the deletion of exon 7. The loss of exon 7 of migfilin in migfilin-/- platelets was confirmed by mRNA expression, using littermate wild-type (WT) mice platelets as a control. All experimental procedures were reviewed and approved by the Animal Care and Use Committee of the Zhejiang University School of Medicine. Total RNA of platelets and heart was extracted using Trizol (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. RNA was reversely transcribed to cDNA using the ReverTraAce qPCR RT kit (Toyobo, Osaka, Japan). PCR was performed using the primers P1: GCTGTTGAGGCCATGAAGAG and P2: TCCTTCCCATGCACTCGATT. Migfilin gene expression was quantified by real-time quantitative PCR based on SYBR Green, using -actin RNA as the loading control. 16 The sequence of the primers used were as follows: migfilin forward primer (mFBLIM1-qRCR-F): TAGCCGTGAGTGAGGAAGTG, reverse primer (mFBLIM1-qRCR-R): CAGAGAGTGAGGCATTGGTCT.
Migfilin peptides
As previously reported,13 WT migfilin (4KPEKRVASSVFITLAP19C) and mutant (MT) peptides (4KPEKRVADSAFITLAP19C) with an additional C-terminal cysteine residue were synthesized by ChinaPeptides (Suzhou, China). WTmigfilin peptide harbors the filamin A binding sequence and the MT-migfilin peptide contains two point mutations (Ser11 was replaced by Asp, and Val13 was replaced by Ala, bold type in the sequence) with substantially reduced ability to bind filamin A (at least 12.5-times weaker than the WT peptide).13 To render them cell permeable, a CR7 transport peptide was conjugated to the migfilin peptides through a disulfide bond.17 The conjugated peptides were purified by high pressure liquid chromatography and conjugation confirmed by electrospray ionization mass spectrometry.
Tail bleeding assay
As previously described,18 tails of anesthetized mice were cut 0.5 cm from the tip and immediately immersed in microtubes filled with 1.7 ml of saline at 37°C. The time for the bleeding to stop (no blood flow for 1 min) and the weight of blood loss were recorded. Tail bleeding assays were stopped at 900 seconds if the bleeding did not stop. The same procedure was used for the evaluation of migfilin peptides in hemostatic function after their intravenous injection.
Platelet preparation
Whole blood was collected from the inferior vena cava into 0.1 vol of ACD buffer (75 mM sodium citrate, 39 mM citric acid, and 135 mM dextrose, pH 6.5), and was diluted 1:2 with modified Tyrode’s buffer (in mM: 20 HEPES, 137 NaCl, 13.8 NaHCO3, 2.5 KCl, 0.36 NaH2PO4, 5.5 glucose, pH 7.4). Diluted whole blood was centrifuged at 180×g for 10 minutes (min) at room temperature, and platelet-rich-plasma was collected into a fresh tube. The platelet-rich-plasma was diluted in ACD buffer, and centrifuged at 700×g for 10 min. Platelet pellet was then re-suspended in modified Tyrode’s buffer.
Detailed information of the reagents, platelet function measurements, Western blotting and statistical analysis is provided in the Online Supplementary Materials and Methods.
Results
Migfilin deficiency impairs hemostasis and thrombosis.
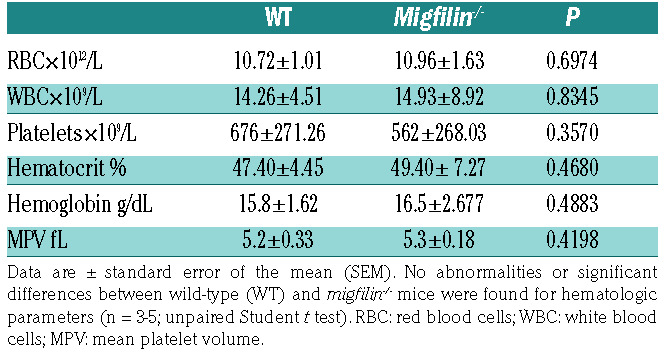
The truncation of exon 7 of migfilin in platelets was validated by PCR measurement of migfilin messenger RNA (Figure 1A). Multiple commercially available antibodies (Santa Cruz Biosciences sc-162823, sc-162822, sc-134724; ABclonal A15850; GeneTex GTX116584) were used to detect migfilin. These antibodies did not clearly reveal the expression of migfilin protein in murine platelets, although they did confirm the presence of migfilin in murine hearts as previously reported (Figure 1B). Realtime quantitative PCR experiments were performed as a surrogate method to quantify migfilin expression in murine platelets. The results not only confirmed the loss of migfilin mRNA in the platelets from KO mice, but also showed that expression of migfilin mRNA is about 1,500-fold lower in platelets than that in heart of WT mice (normalized by actin mRNA) (Figure 1C). Migfilin-/- mice were viable and fertile, and did not exhibit any evident bleeding tendency or thrombotic events over their lifespan. Migfilin-/-mice did not differ significantly from their WT littermates in platelet counts, white cell counts, hematocrits, and hemoglobin concentrations (Table 1). Electron microscopy showed that migfilin-/- platelets had normal discoid morphology (Online Supplementary Figure S1A). No significant differences in the surface expression of the platelet glycoproteins GPVI, CD41 (aIIb subunit) and CD42b (GPIb subunit) were found between WT and migfilin-/- platelets (Online Supplementary Figure S1C).
Figure 1.

Migfilin-/- mice display impaired hemostatic and thrombotic functions. (A) Migfilin mRNA expression in wild-type (WT) platelets and exon 7 (179 bp) deleted migfilin-/- mice platelets. (B) Analysis of migfilin protein expression by Western blotting in heart tissues and platelets from WT and migfilin-/- mice. (C) Quantitative realtime PCR results analysing the expression of migfilin in platelets and heart tissues. Data are represented as a ratio relative to an internal control (β-actin) (mean ± standard error of the mean [SEM], n=3), ***P<0.001, Student t test. (D) Bleeding times for WT (●) and migfilin-/- mice (▲). Means are indicated by horizontal lines. **P<0.01, evaluated with 2-tailed Mann-Whitney U tests. (E) Weight of blood loss from WT (●) and migfilin-/- mice (▲) during bleeding time. Results are expressed as the mean ± SEM (n = 9). *P<0.05, evaluated with 2-tailed Mann-Whitney U tests. (F) Percentages of WT and migfilin-/- mouse bleeding times exceeded 15 minutes (min) (□) or were within 15 min (■). Results were obtained from 21 WT and 22 migfilin-/- mice. (G) Representative images of thrombi in FeCl3-injured mesenteric arterioles at indicated post-injury time points in WT (upper row) and migfilin-/- mice (lower row). a, arteriole; v: venule. Scale bars, 100 mm (left panel). Occlusion times of FeCl3-induced thrombosis in arterioles of WT (n=16) and migfilin-/- mice (n=16). Means are indicated by horizontal lines. **P<0.01, 2-tailed Mann-Whitney test (right panel). (H) Adhesion of platelets from WT and migfilin-/- mice on collagen. Mepacrine-labeled whole blood from WT and migfilin-/- mice was perfused over collagencoated bioflux plates at a shear rate of 40 dynes/cm2 for 5 min. Original magnification 10x. Scale bar, 100 mm (left panel). Area coverage of platelets from WT and migfilin-/- mice (n=10 for both groups) after 5 min perfusion over a collagen surface, **P<0.01, 2-tailed Mann-Whitney test (right panel).
Notably, migfilin-/- mice exhibited tail-bleeding times twice as long as their WT littermates (Figure 1D), consistent with the comparison of the weight of blood loss between migfilin-/- and WT mice (Figure 1E). Moreover, 63.6% of the migfilin-/- mice had bleeding times exceeding 15 min, in comparison, only 14.3% of WT littermates had bleeding times over 15 min (Figure 1F). These data suggest that migfilin positively regulates hemostasis.
In a model of FeCl3-induced mesenteric arteriole thrombosis, the time of forming stable occlusive thrombi in mesenteric arterioles were significantly longer in migfilin-/- mice than in WT mice (35.19±3.656 min vs. 20.63±4.422 min, P<0.01; Figure 1G). To confirm that the observed hemostatic functional defects are due to migfilin deficiency, we synthesized cell-deliverable migfilin peptides as described in the previous study.13 WT-migfilin-CCR7 peptide (range: 0.5-1.5 nM/kg) dose-dependently shortened the prolonged tail bleeding time in migfilin-/- mice (Online Supplementary Figure S2A), whereas MT-migfilin-CCR7 peptide did not show any effect on the bleeding time. WTmigfilin- CCR7 peptide restored the prolonged bleeding time of migfilin-/- mice to a comparable level of WT mice (Online Supplementary Figure S2B-C). WT-migfilin-CCR7, but not MT-migfilin-CCR7, also restored the occlusion time in Fecl3-induced mesenteric arterial injury in migfilin-/- mice (Online Supplementary Figure S2D).
Thrombus formation was also assessed using a microfluidic whole-blood perfusion assay. After a 5-minute perfusion at a shear rate of 1000 s-1, thrombi formed on an immobilized collagen surface by migfilin-/- platelets were significantly smaller (average of a 50% reduction) than those by WT platelets (Figure 1H). In the meanwhile, as shown in the Online Supplementary Video S1, thrombi formed by migfilin-/- platelets displayed a severely compromised stability compared to WT platelets. In order to assess whether the observed phenotype of migfilin-/- platelets is due to a reduced ability of adhesion during the initiation of thrombosis, a recombinant whole-blood system with a reduced concentration of platelets (107/mL) was employed in the perfusion assay. Without the interference from massive platelet aggregation, migfilin-/- and WT platelets had similar coverage area on the collagen surface (Online Supplementary Figure S3). These findings indicate that migfilin promotes thrombosis, possibly through influencing the extension and perpetuation of thrombi.
Migfilin-/-platelets have a decreased capability of aggregation due to a hampered dense granule secretion
To further evaluate the function of migfilin in platelets, aggregation and dense granule secretion in response to common platelet stimuli were measured. Compared to WT platelets, migfilin-/- platelets displayed a prominently decreased aggregation rates in response to low dose collagen (0.4 mg/mL), and a mildly decreased aggregation rates in response to thrombin (0.018 U/mL) (Figure 2A). Platelet aggregation induced by ADP (10 mM; 20 mM), U46619 (0.3 M; 0.6 M) or higher concentrations of thrombin (0.025 U/mL) and collagen (0.8 mg/mL) was not affected by migfilin deficiency. Notably, migfilin-/- platelets exhibited a markedly decreased ATP secretion in response to all stimuli including U46619, thrombin and collagen, even when the aggregation difference was no longer present (Figure 2A). Apyrase (0.25 U/mL) hydrolyzed secreted ADP and eliminated the aggregation difference between WT and migfilin-/- platelets induced by low concentrations of thrombin or collagen (Figure 2B). Supplementation with a low concentration of ADP (1 mM), insufficient to induce aggregation on its own, reversed the inhibitory effect of migfilin deficiency on collagen- and thrombin-stimulated platelet aggregation (Figure 2B). In addition, thrombin- and collagen-induced secretion of serotonin from migfilin-/- platelets was largely reduced compared to that of WT platelets, even though similar levels of serotonin were harbored inside resting migfilin-/- and WT platelets (Online Supplementary Figure S4). These data suggest that an impaired dense granule secretion is the central functional defect exhibited by migfilin-/- platelets.
Table 1.
Hematologic analysis.
WT-migfilin-CCR7 (5 mM), but not MT-migfilin-CCR7 (5 mM), rescued the impaired aggregation and ATP release of washed migfilin-/- platelets in response to thrombin or collagen, and eliminated the difference between WT and migfilin-/- platelets (Figure 3A-B). This result supports that the observed platelet phenotypes in the current study are bona fide migfilin effects.
Interestingly, an integrin aIIbβ3 inhibitor tirofiban (4 g/mL) eliminated the differences of aggregation and ATP secretion between migfilin-/- and WT platelets in response to thrombin and collagen (Online Supplementary Figure S6A-B), suggesting that migfilin-regulated platelet function is aIIbβ3-dependent. However, neither the secretion of -granules nor the conformational change of aIIbβ3 on a single platelet, indicated by P-selectin expression and JON/A binding, respectively, was affected by migfilin deficiency or exogenously applying of migfilin peptide (Online Supplementary Figure S7). These findings therefore suggest that migfilin may not be involved in inside-out signaling of aIIbβ3, but rather participate in the process post ligand-aIIbβ3 engagement.
Figure 2.

Migfilin-/- platelets display impaired aggregation responses due to defective dense granule secretion. (A) Platelets were stimulated with collagen, thrombin, U46619, and ADP (in the presence of fibrinogen). Aggregation and ATP release was assessed with a Chrono-log lumiaggregometer under stirring at 1,200 rpm. Traces are representative of at least three independent experiments. Results are expressed as mean ± standard error of the mean (SEM) from at least four independent experiments. Statistical significance was evaluated with paired Student t test. (*P<0.05, **P<0.01). (B) Aggregation of washed WT or migfilin-/- platelets stimulated with collagen (0.4 mg/mL) or thrombin (0.018 U/mL) in the presence of vehicle (left panel), or apyrase (0.25 U/mL) (middle panel), or a low concentration of ADP (1 μM) (right panel). Percentage of platelet aggregation from at least four independent experiments is depicted on the right, and the results are shown as mean ± SEM (*P<0.05, **P<0.01, ***P<0.001, ns: no significant difference, paired Student t test).
Migfilin positively regulates early aIIbβ3 outside-in signaling in platelets
Since the effects of migfilin in platelets are aIIbβ3- dependent, outside-in signaling mediated by aIIbβ3 was evaluated. First, early outside-in signaling was assessed by two functional assays, i.e., spreading of platelets on immobilized fibrinogen and Mn2+ (0.5mM)-caused platelet aggregation. Both spread areas on fibrinogen and the aggregation rates by manganese were largely reduced in migfilin-/- platelets (Figure 4A-B). WT-migfilin-CCR7, but not MT-migfilin-CCR7 peptide, fully rescued platelet spreading on fibrinogen and Mn2+ induced platelet aggregation (Figure 4A-B). The late outside-in signaling was assessed by a clot retraction experiment.21-23 Contrary to the spreading results, neither the rate of clot retraction nor the final volumes of clots differed between WT and migfilin-/- platelets (Figure 4C), indicating an intact late outside- in signaling upon migfilin deficiency.
Figure 3.

WT-migfilin-CCR7 (5 mM) rescued the impaired aggregation and ATP release of washed migfilin-/- platelet in response to thrombin or collagen. (A) Platelets were stimulated with collagen (0.4 mg/mL, 0.8 mg/mL), thrombin (0.018 U/mL, 0.025 U/mL) in the presence of WT-migfilin-CCR7 (5 mM) peptide or MT-migfilin-CCR7 (5 mM) peptide. Platelet aggregation and ATP release were assessed with a Chrono-log lumi-aggregometer under stirring at 1,200 rpm. Traces were representative of at least three independent experiments. (B) WT (□) and migfilin-/- (■) platelets treated with WT-migfilin-CCR7 and MT-migfilin-CCR7 respectively were stimulated by collagen and thrombin. Results are expressed as mean ± standard error of the mean (SEM) from at least three independent experiments. Statistical significance was evaluated with paired Student t test (*P<0.05, **P<0.01, ns: no significant difference).
Figure 4.

Migfilin deficiency reduces outside-in signaling in platelets. (A) Spreading of WT and migfilin-/- platelets on immobilized fibrinogen in the presence or absence of migfilin peptides (5 mM). Images are representative of three independent experiments with similar results. Original magnification 100x. Scale bars, 10 mm (left panel). Quantification of the areas of spread (mm2) of WT and migfilin-/- platelets (mean ± standard error of the mean [SEM], ***P<0.001, ns: no significant difference, Student t test) (right panel). (B) Representative curves and quantification of Mn2+ (0.5 mM)-induced aggregation of washed WT and migfilin-/- platelets in the presence or absence of migfilin peptide. Platelets were stimulated in the cuvettes of a Chrono-log lumiaggregometer in the presence of fibrinogen (25 mg/mL) and under stirring at 1,200 rpm. Experiments were repeated at least four times and the results are shown as mean ± SEM (*P<0.05, ns: no significant difference, paired Student t test). (C) Platelets from WT or migfilin-/- mice were re-suspended with human platelet-poor plasma at a concentration of 4x108/mL, and recombined plasma was stimulated to coagulate with thrombin (0.4 U/mL), then photographed at different time points. Experiments were repeated at least three times. (D) Measurement of early aIIbβ3 outside-in signaling in WT and migfilin-/- platelets spread on fibrinogen or stimulated with Mn2+ (0.5 mM) in the presence of fibrinogen (25 mg/mL) in suspension. At indicated time points, platelets were lysed and analyzed by Western blotting with antibodies recognizing phosphorylated β3 Tyr747, phosphorylated β3 Tyr759, phosphorylated SFK Tyr416, phosphorylated Syk Tyr525/526, Beta3, Src, and Syk. Experiments were repeated at least three times. (E) WT and migfilin-/- platelets were spread on fibrinogen for 60 minutes (min) or stimulated with Mn2+ (0.5 mM) in the presence of fibrinogen (25 mg/mL) for 10 min, in the presence or absence of migfilin peptides (5 mM). Platelets were lysed and analyzed by Western blotting with antibodies recognizing phosphorylated phosphorylated β3 Tyr747, phosphorylated β3 Tyr759, phosphorylated SFK Tyr416, phosphorylated Syk Tyr525/526, Beta3, Src and Syk. Experiments were repeated at least three times.
Upon spread on immobilized fibrinogen or provoked by Mn2+ stimulation, the typical early outside-in signal molecules, including β3 cytoplasmic sites (Tyr747, Tyr759), Srcfamliy kinase c-Src (Tyr416) and Syk kinase (Tyr525/526), exhibited a significantly reduced phosphorylation in migfilin-/- platelets, compared to WT platelets (Figure 4D and Online Supplemntary Figure S8A). The phosphorylation defects of these signal molecules in migfilin-/- platelets were restored by exogenous WT-migfilin-CCR7 peptide (5 mM) (Figure 4E and Online Supplemntary Figure S8B). In contrast, late outside-in signaling events, such as phosphorylation of ERK and p38,24 were not drastically changed (Online Supplemntary Figure S9). Together, these data suggest that migfilin contributes to the early but not late outside-in signaling of platelet aIIbβ3.
Migfilin promotes outside-in signaling through inhibiting filamin A-β3 interaction
Previous studies have established that migfilin promotes the activation of aIIbβ3 via binding to filamin and dissociate the latter from integrin β3 cytoplasmic tail. Limited by the expression level of migfilin in platelets, filamin A-β3 binding was used as a surrogate marker for a more detailed molecular mechanism of migfilin in the context of aIIbβ3 outside-in signaling. Shown in the Figure 5A, when platelets spread on immobilized fibrinogen were examined by confocal microscopy, both WT and migfilin-/- platelets exhibit a strong colocalization signal of β3 and filamin A at resting states. Upon the commencing of spreading, the co-localization signal of β3 and filamin A diminishes in WT platelets (nadir appears at 60 min) and gradually recovers. In contrast, the temporal changes of the colocalization signal is much less obvious in migfilin-/- platelets (Figure 5A-B). In addition, binding of filamin A to integrin β3 tails is clearly seen in resting WT platelets, Mn2+ stimulation induces a rapid and complete dissociation of filamin A and β3, followed by a later re-association of filamin A and β3. In resting migfilin-/- platelets, the initial association between filamin A and 3 is unchanged, however, upon Mn2+ stimulation, an obviously deterred dissociation of filamin A and β3 occurs (Figure 5C). Because filamin A also binds GPIba,25 platelet responses to von Willebrand factor (VWF) in the presence of ristocetin was measured. Neither spreading on immobilized VWF nor binding between filamin A and GPIba upon ristocetin stimulation revealed difference between WT and migfilin-/- platelets (Online Supplemntary Figure S10),26 thus excluding the possibility that migfilin works through filamin AGPIbα binding. Therefore, migfilin appears to modulate the interaction between filamin A and β3 and thus promote outside-in signaling of aIIbβ3 (Figure 6).
Discussion
The results presented here demonstrate that migfilin is an important regulator for the in vivo system of hemostasis and thrombosis. In vitro, migfilin deficiency impedes thrombus formation on collagen surface and impairs various platelet functions, including aggregation, dense-granule secretion, and spreading on immobilized fibrinogen. These defective functions of migfilin-/- platelets appear to be the results of a compromised outside-in signaling, rather than inside-out signaling. Furthermore, migfilin promotes the dissociation of filamin A from β3 subunit of aIIbβ3 in the milieu of early outside-in signaling.
The results presented here demonstrate that migfilin is an important regulator for the in vivo system of hemostasis and thrombosis. In vitro, migfilin deficiency impedes thrombus formation on collagen surface and impairs various platelet functions, including aggregation, dense-granule secretion, and spreading on immobilized fibrinogen. These defective functions of migfilin-/- platelets appear to be the results of a compromised outside-in signaling, rather than inside-out signaling. Furthermore, migfilin promotes the dissociation of filamin A from β3 subunit of aIIbβ3 in the milieu of early outside-in signaling.
Previous studies found that a relatively high concentration of cell permeable migfilin peptides (50 mM) promotes PAC-1 binding and induces significant aggregation (10 mM peptide) in washed human platelets,13,14 thus suggesting that migfilin actively participates in the inside-out signaling associated with aIIbβ3. However, supported by the functional measurements and phosphorylation events representative of inside-out and outside-in signaling, our current study demonstrates that the involvement of migfilin is restricted to outside-in signaling of aIIbβ3. In particular, using migfilin peptide synthesized in the same way as described in the previous study,13 the current study demonstrate that platelet aggregation is only induced by the peptide at a concentration above 10 mM. At a lower concentration (5 mM), the migfilin peptide does not induce platelet aggregation (Online Supplementary Figure S5), yet this concentration of migfilin peptide fully rescues the impaired function and aIIbβ3 outside-in signaling in migfilin-/- platelets. These results not only support that the observed platelet phenotypes in the current study are bona fide migfilin-dependent, but also reveal that platelets are sensitive to the amount of intracellular migfilin. Hence, combining the observations obtained in migfilin-/- platelets and those using migfilin peptides, it is possible to envisage that the natural amount of migfilin participates in outsidein signaling, while excessive migfilin might influence inside-out signaling. This may also justify the extremely low expression of migfilin in platelets. Intriguingly, the use of migfilin peptide could be further translated into a rescued hemostatic and thrombotic function in vivo in migfilin-/- mice.
Figure 5.

Migfilin promotes outside-in signaling through regulating filamin A-β3 interaction. (A) Representative confocal microscopic images of colocalization of Beta 3 (β3) and filamin A in WT and migfilin-/- platelets spread on fibrinogen at the indicated time points. Experiments were repeated independently at least three times with similar results. Scale bars, 10 mm. (B) Colocalization of filamin A and β3 was quantified by Pearson Correlation Coefficient calculation using NIH Image J software. Results are expressed as mean ± standard error of the mean (SEM) from at least three independent experiments. Statistical significance was evaluated with the Student t test (*P<0.05, **P<0.01). (C) Washed WT platelets were stimulated with Mn2+ (0.5 mM) in the presence of fibrinogen (25 mg/mL) in suspension and lysed at indicated time points. Platelet lysates were immunoprecipitated with antibody against β3 and then immunoblotted with antibodies against filamin A and β3, respectively. Representative result from at least three independent experiments.
Figure 6.

A model for the role of the migfilin in regulating filamin A-β3 interaction during platelet activation. In resting platelets, filamin A binding to the β3 tail prevents interactions among talin, kindlin, and integrin. When a platelet is activated, inside-out signals, through displacement of filamin A and enabling subsequent talin and kindlin binding to β3, culminate in the high affinity conformational change of aIIbβ3. Upon binding of activated aIIbβ3 to fibrinogen, migfilin (or WT-migfilin-CCR7 peptide) prevents the reassociation of filamin A to β3 and supports a sustained outside-in signaling. Without migfilin, filamin A binds back to β3 prematurely and inhibits further transmission of integrin outside-in signals.
Migfilin-/- platelets exhibit a reduced phosphorylation level of the molecules corresponding to early outside-in signaling such as β3, Src, and Syk, as well as a hampered ability to spread on immobilized fibrinogen. On the contrary, the late outside-in signaling events, such as phosphorylation of ERK, and p38,24 were not drastically changed, nor did migfilin-deficiency critically influence clot retraction. Corroborating these findings, migfilin peptides harboring the filamin A binding sequence rescue the phosphorylation events and sequential functions associated with early but not late outside-in signaling. Our results may have suggested that filamin A constitute the major functional partner of migfilin in platelets. Yet the inability to visualize migfilin protein in platelets has presented itself as a large hurdle to further dissection of the mechanism of migfilin. Particularly, one question persists: how can migfilin have such an important role when the protein is present in such a low amount? Indeed, based on the published literature,13 stoichiometry of migfilin binding to filamin A (IgFLNa21 domain) is either 1:1 or 1:2. However, since migfilin-filamin A interaction is much stronger than filamin A-integrin interaction,29,14 the amount of migfilin required to trigger integrin activation is conceivably much lower than the proposed stoichiometry of migfilin-filamin A binding. Alternatively, migfilin may be enriched spatially to the integrin adhesion sites during activation,30,31 which eliminates the needs of high expression in the cytoplasm. Moreover, migfilin interacts with multiple important proteins such as kindlin, Src and VASP,30-33 the confluence of synergistic effects executed by these important platelet regulators may result in an amplified platelet activation.7,34,35 We acknowledge that the mechanism of migfilin in platelet is only partially unveiled by the current study. A further elaboration of the migfilin interaction network in platelets is warranted.
A previous study has elegantly unveiled the important role of filamin A-aIIbβ3 interaction in pro-platelet formation and explained the pathogenesis of macrothrombocytopenia in filaminopathies (conditions connected to heterozygous filamin A mutations).36 Intriguingly, although the current study has shown that migfilin regulates filamin A-aIIbβ3 interaction in platelets, deletion of migfilin seems to neither influence the platelet count nor the platelet morphology. It may be explained by the fact that loss of migfilin essentially enhances the interaction between filamin A and aIIbβ3, which contrasts the phenotype caused by filamin A mutations, whose ability to interact with aIIbβ3 is reduced. The heterogenous phenotypes of platelets and megakaryocytes associated with filamin A mutations indicate that compared to migfilin, changes of filamin A has far more extensive impact on megakaryocytes and platelets.6,37
In conclusion, migfilin is an important positive regulator for hemostasis and thrombosis. Through regulating the binding dynamics of filamin A and aIIbβ3 (Figure 6), migfilin promotes early outside-in signaling of aIIbβ3 and supports platelet dense granule secretion. Further studies of migfilin will possibly lead to the discovery of new regulators important for platelet function and effective management of thrombosis.
Supplementary Material
Funding Statement
Funding This study was supported by grants from National Natural Science Foundation of China (81670131 and 81870106), Zhejiang Provincial Natural Science Foundation (LZ18H080001).
References
- 1.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104(6):1606-1615. [DOI] [PubMed] [Google Scholar]
- 2.Li Z, Delaney MK, O'Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30(12):2341-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metcalf DG, Moore DT, Wu Y, et al. NMR analysis of the alphaIIb beta3 cytoplasmic interaction suggests a mechanism for integrin regulation. Proc Natl Acad Sci U S A. 2010;107(52):22481-22486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tadokoro S, Shattil SJ, Eto K, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302(5642):103-106. [DOI] [PubMed] [Google Scholar]
- 5.Kiema T, Lad Y, Jiang P, et al. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell. 2006;21(3): 337-347. [DOI] [PubMed] [Google Scholar]
- 6.Berrou E, Adam F, Lebret M, et al. Gain-offunction mutation in filamin A potentiates platelet integrin alphaIIbbeta3 activation. Arterioscler Thromb Vasc Biol. 2017;37(6): 1087-1097. [DOI] [PubMed] [Google Scholar]
- 7.Moser M, Nieswandt B, Ussar S, et al. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008; 14(3):325-330. [DOI] [PubMed] [Google Scholar]
- 8.Bouvard D, Pouwels J, De Franceschi N, et al. Integrin inactivators: balancing cellular functions in vitro and in vivo. Nat Rev Mol Cell Biol. 2013;14(7):430-442. [DOI] [PubMed] [Google Scholar]
- 9.Shen B, Zhao X, O'Brien KA, et al. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature. 2013;503(7474):131-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu C. Migfilin and its binding partners: from cell biology to human diseases. J Cell Sci. 2005;118(Pt 4):659-664. [DOI] [PubMed] [Google Scholar]
- 11.Tu Y, Wu S, Shi X, et al. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113(1):37-47. [DOI] [PubMed] [Google Scholar]
- 12.Akazawa H, Kudoh S, Mochizuki N, et al. A novel LIM protein Cal promotes cardiac differentiation by association with CSX/NKX2-5. J Cell Biol. 2004;164(3):395-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ithychanda SS, Das M, Ma YQ, et al. Migfilin, a molecular switch in regulation of integrin activation. J Biol Chem. 2009;284(7):4713-4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das M, Ithychanda SS, Qin J, et al. Migfilin and filamin as regulators of integrin activation in endothelial cells and neutrophils. PLoS One. 2011;6(10):e26355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao G, Cheng H, Cao H, et al. Critical role of filamin-binding LIM protein 1 (FBLP- 1)/migfilin in regulation of bone remodeling. J Biol Chem. 2012;287(25):21450-21460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong H, Ni J, Xu Z, et al. Shp2 in myocytes is essential for cardiovascular and neointima development. J Mol Cell Cardiol. 2019;137 (71-81). [DOI] [PubMed] [Google Scholar]
- 17.Chen L, Wright LR, Chen CH, et al. Molecular transporters for peptides: delivery of a cardioprotective epsilonPKC agonist peptide into cells and intact ischemic heart using a transport system, R(7). Chem Biol. 2001;8(12):1123-1129. [DOI] [PubMed] [Google Scholar]
- 18.Mehta AY, Mohammed BM, Martin EJ, et al. Allosterism-based simultaneous, dual anticoagulant and antiplatelet action: allosteric inhibitor targeting the glycoprotein Ibalpha-binding and heparin-binding site of thrombin. J Thromb Haemost. 2016;14(4):828-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moik DV, Janbandhu VC, Fassler R. Loss of migfilin expression has no overt consequences on murine development and homeostasis. J Cell Sci. 2011;124(Pt 3):414-421. [DOI] [PubMed] [Google Scholar]
- 20.Haubner BJ, Moik D, Schuetz T, et al. In vivo cardiac role of migfilin during experimental pressure overload. Cardiovasc Res. 2015;106(3):398-407. [DOI] [PubMed] [Google Scholar]
- 21.Chari-Turaga R, Naik UP. Integrin alphaIIbbeta3: a novel effector of Galpha13. Cell Adh Migr. 2011;5(1):4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong H, Shen B, Flevaris P, et al. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin "outside- in" signaling. Science. 2010;327(5963): 340-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flevaris P, Stojanovic A, Gong H, et al. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007;179(3):553-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flevaris P, Li Z, Zhang G, et al. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood. 2009;113(4):893-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Falet H. New insights into the versatile roles of platelet FlnA. Platelets. 2013; 24(1):1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin H, Stojanovic A, Hay N, et al. The role of Akt in the signaling pathway of the glycoprotein Ib-IX induced platelet activation. Blood. 2008;111(2):658-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ren Q, Ye S, Whiteheart SW. The platelet release reaction: just when you thought platelet secretion was simple. Curr Opin Hematol. 2008;15(5):537-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reed GL, Fitzgerald ML, Polgar J. Molecular mechanisms of platelet exocytosis: insights into the "secrete" life of thrombocytes. Blood. 2000;96(10):3334-3342. [PubMed] [Google Scholar]
- 29.Lad Y, Jiang P, Ruskamo S, et al. Structural basis of the migfilin-filamin interaction and competition with integrin beta tails. J Biol Chem. 2008;283(50):35154-35163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z, Lu D, Wang X, et al. Kindlin-2 phosphorylation by Src at Y193 enhances Src activity and is involved in Migfilin recruitment to the focal adhesions. FEBS Lett. 2015;589(15):2001-2010. [DOI] [PubMed] [Google Scholar]
- 31.Brahme NN, Harburger DS, Kemp-O'Brien K, et al. Kindlin binds migfilin tandem LIM domains and regulates migfilin focal adhesion localization and recruitment dynamics. J Biol Chem. 2013;288(49):35604-35616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao J, Zhang Y, Ithychanda SS, et al. Migfilin interacts with Src and contributes to cell-matrix adhesion-mediated survival signaling. J Biol Chem. 2009;284(49):34308-34320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Tu Y, Gkretsi V, et al. Migfilin interacts with vasodilator-stimulated phosphoprotein (VASP) and regulates VASP localization to cell-matrix adhesions and migration. J Biol Chem. 2006;281(18):12397-12407. [DOI] [PubMed] [Google Scholar]
- 34.Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124(13):2013-2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benz PM, Laban H, Zink J, et al. Vasodilator- Stimulated Phosphoprotein (VASP)-dependent and -independent pathways regulate thrombin-induced activation of Rap1b in platelets. Cell Commun Signal. 2016;14(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donada A, Balayn N, Sliwa D, et al. Disrupted filamin A/alphaIIbbeta3 interaction induces macrothrombocytopenia by increasing RhoA activity. Blood. 2019;133(16):1778-1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berrou E, Adam F, Lebret M, et al. Heterogeneity of platelet functional alterations in patients with filamin A mutations. Arterioscler Thromb Vasc Biol. 2013; 33(1):e11-18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.