

Abstract
Jawed vertebrate adaptive immunity relies on the RAG1/RAG2 (RAG) recombinase, a domesticated transposase, for assembly of antigen receptor genes. Using an integration‐activated form of RAG1 with methionine at residue 848 and cryo‐electron microscopy, we determined structures that capture RAG engaged with transposon ends and U‐shaped target DNA prior to integration (the target capture complex) and two forms of the RAG strand transfer complex that differ based on whether target site DNA is annealed or dynamic. Target site DNA base unstacking, flipping, and melting by RAG1 methionine 848 explain how this residue activates transposition, how RAG can stabilize sharp bends in target DNA, and why replacement of residue 848 by arginine during RAG domestication led to suppression of transposition activity. RAG2 extends a jawed vertebrate‐specific loop to interact with target site DNA, and functional assays demonstrate that this loop represents another evolutionary adaptation acquired during RAG domestication to inhibit transposition. Our findings identify mechanistic principles of the final step in cut‐and‐paste transposition and the molecular and structural logic underlying the transformation of RAG from transposase to recombinase.
Keywords: DNA bending, evolution, recombination‐activating gene, transposition, V(D)J recombination
Subject Categories: DNA Replication, Repair & Recombination; Immunology; Structural Biology
Cryo‐EM structures reveal mechanisms of the final step in cut‐and‐paste transposition and the molecular logic underlying RAG domestication from transposase to adaptive immune system recombinase.

Introduction
Transposons are genetic elements that can move from one genomic location to another through the action of transposon‐encoded transposases (Craig, 2015). Transposon‐derived sequences constitute a large fraction of many genomes and have numerous biological functions including regulators of gene expression and chromatin structure and mediators of genetic instability and disease (Feschotte & Pritham, 2007; Payer & Burns, 2019). While most transposase genes have been inactivated during evolution, some have undergone “molecular domestication” and become adapted to perform a new function for the host (Sinzelle et al, 2009; Jangam et al, 2017; Payer & Burns, 2019; Koonin et al, 2020). A paradigmatic example of transposon molecular domestication is the RAG1/RAG2 (RAG) recombinase, which is essential for jawed vertebrate adaptive immunity by virtue of its function as the endonuclease that initiates V(D)J recombination. This reaction assembles antibody and T‐cell receptor genes in developing lymphocytes and in many species is responsible for generating the vast pre‐immune repertoire of antigen receptors expressed by B and T cells (Lewis, 1994).
DNA cleavage by RAG requires an asymmetric “12/23” pair of recombination signal sequences (RSSs) composed of heptamer and nonamer elements separated by a spacer of 12 or 23 bp (Gellert, 2002; Swanson, 2004; Fig 1A). DNA cleavage proceeds through a series of structurally well‐characterized steps that begin with DNA binding and nicking immediately adjacent to the heptamer to generate the hairpin‐forming complex (HFC) (Kim et al, 2015, 2018; Ru et al, 2015, 2018; Chen et al, 2020a; Fig 1B). In the HFC, the 3′‐OH liberated by nicking attacks the opposite strand to complete double‐strand breakage, resulting in blunt RSS signal ends and hairpin‐sealed coding ends. The ends are then processed and joined by DNA repair enzymes to complete the recombination reaction. The RAG catalytic domain is encoded within RAG1 and adopts an RNaseH fold similar to that of DDE family transposases and retroviral integrases (Kim et al, 2015). The parallels between RAG and DDE transposases/integrases extend to the chemistry of DNA cleavage and to the ability of RAG to perform transposition in vitro, resulting in the staggered insertion of a pair of signal ends into target DNA (Fugmann et al, 2000; Gellert, 2002). Notably, RAG‐mediated transposition is powerfully suppressed in vivo (i.e., in living cells) (Chatterji et al, 2006; Reddy et al, 2006; Curry et al, 2007; Little et al, 2015), presumably to prevent insertional mutagenesis and other types of genomic instability proposed to be associated with RAG‐mediated transposition (Gellert, 2002).
Figure 1. RAG‐mediated transposition and model for the evolution of the RAG recombinase.

- Diagram of the 12RSS and 23RSS, which differ based on the length of a poorly conserved spacer between relatively well‐conserved heptamer and nonamer elements (consensus sequences shown in dark letters).
- V(D)J recombination and transposition involve steps of substrate DNA recognition, nicking, hairpin formation, and end processing and joining (recombination, yielding coding joint and signal joint) or target DNA capture and integration (transposition). Shown are schematic diagrams of RAG‐DNA complexes during the transposition reaction. RAG/HMGB1, blue oval; 12RSS with flanking coding DNA, red; 23RSS and flanking coding DNA, yellow; target DNA, green.
- Model for RAG evolution from a transposase to a recombinase (Carmona & Schatz, 2017). In this model, an ancestor Transib transposon which encoded a RAG1‐like (RAG1L) transposase acquired a RAG2‐like (RAG2L) gene to generate the ancestral RAG‐like transposon. In some invertebrates, it remained a transposon, while in jawed vertebrates, the transposon inserted into a gene exon to generate split antigen receptor genes and the RAG1L and RAG2L transposase genes evolved to become the RAG1/RAG2 recombinase genes.
- Sequence alignment of RAG1 and RAG1‐like proteins in the vicinity of RAG1 R848. R848 from jawed vertebrates is highlighted in yellow. Red letters in blue box, relatively well‐conserved residues; white letters on red background, highly conserved residues.
These and other findings support the model that RAG1/RAG2 evolved from a transposon (Thompson, 1995; Fugmann, 2010), a process thought to have begun with an ancient Transib transposon composed of a RAG1‐like gene flanked by terminal inverted repeats (TIRs) resembling RSSs (Kapitonov & Jurka, 2005; Hencken et al, 2012; Liu et al, 2019). Transib is proposed to have acquired a RAG2‐like gene to give rise to the RAG‐like (RAGL) transposon, which was subsequently domesticated for V(D)J recombination in jawed vertebrates (Carmona & Schatz, 2017; Fig 1C). This model predicts the occurrence of evolutionary adaptations in jawed vertebrates to constrain the genome‐destabilizing transposase activities of the RAG enzyme while maintaining DNA cleavage activity.
Recently, two such jawed vertebrate‐specific adaptations were identified: an acidic region in RAG2 and a single amino acid, arginine 848 (R848), in RAG1 (residue numbering according to the mouse RAG proteins) (Zhang et al, 2019). While the RAG2 acidic region was found to suppress transposition in vivo but not in vitro, RAG1 R848 strongly attenuates RAG‐mediated transposition in vitro and almost completely eliminates the reaction in vivo (Zhang et al, 2019). R848 is extremely highly conserved in jawed vertebrate RAG1 proteins but in invertebrate RAGL transposons such as ProtoRAG from amphioxus, this residue is almost invariably methionine (Fig 1D), arguing that this residue likely underwent a change from methionine to arginine early in jawed vertebrate evolution (Zhang et al, 2019; Martin et al, 2020). Eliminating the RAG2 acidic region and mutating RAG1 position 848 from arginine to methionine activate RAG‐mediated transposition in vivo more than 1,000‐fold (Zhang et al, 2019). The molecular mechanisms by which the RAG2 acidic region and RAG1 residue 848 control transposition are not known. Here, we focused on RAG1 position 848 because of the potential to study its mechanism of action biochemically and structurally and to gain broader insight into transposition by DDE transposases/integrases.
After DNA cleavage, transposition by RAG involves release of the hairpin coding flanks to form the signal end complex (SEC) and noncovalent capture of target DNA to form the target capture complex (TCC) (Fig 1B). This is followed by staggered nucleophilic attack on target DNA by the 3′‐OH groups at the ends of the transposon to form the strand transfer complex (STC) (Fig 1B). While multiple STC structures have been reported for DDE transposases/integrases (Richardson et al, 2009; Montano & Rice, 2011; Morris et al, 2016; Ballandras‐Colas et al, 2017; Passos et al, 2017; Arinkin et al, 2019; Ghanim et al, 2019) including recently for RAG (Chen et al, 2020b), no TCC structures have been described for DDE transposases and only one has been reported for a retroviral integrase (Maertens et al, 2010). Here, using a catalytically active, integration‐activated form of mouse RAG1 containing methionine at position 848 (M848), we describe structures of a RAG TCC and of two distinct RAG STCs. The structures reveal that RAG2 helps impose a requirement for two sharp (~ 90°) bends in target DNA in both the TCC and the STC, as also observed in a recent RAG STC structure obtained with R848 RAG1 (Chen et al, 2020b). In both the TCC and the STC, M848 destabilizes target DNA at the bend sites by a mechanism involving base flipping, an activity that R848 RAG1 appears to lack (Chen et al, 2020b), thereby explaining how M848 stimulates transposition and how RAGL transposons were able to overcome the target DNA bending requirement imposed by RAG2. We also identify a jawed vertebrate‐specific extended loop in RAG2 that interacts with target site DNA and demonstrate that it inhibits RAG‐mediated transposition. Our findings help explain key evolutionary transitions—from Transib transposase to RAGL transposase to RAG recombinase—and support an emerging paradigm of transposase‐mediated target DNA distortion as a critical event in transposition (Arinkin et al, 2019).
Results
Two states of the RAG strand transfer complex
To understand the mechanism by which the RAG1 residue at position 848 regulates transposition, we sought to determine the structure of the final intermediates in the transposition reaction using RAG1 containing an R848M alteration and a second mutation, E649V, that provides a further small increase in transposition efficiency (Zhang et al, 2019). We assembled the RAG STC in the presence of Mg2+ using R848M/E649V mouse RAG1 (aa 261–1,008), mouse RAG2 (aa 1–361), the DNA bending cofactor HMGB1 (from human, lacking its C‐terminal acidic tail), and a DNA substrate consisting of a 12RSS and a 23RSS covalently joined to a target DNA molecule so as to mimic the strand transfer product of transposition (Fig EV1A–C). Cryo‐electron microscopy (cryo‐EM) analysis (Fig EV1D–G) yielded a 2.6 Å resolution map of the RAG STC (Fig 2A) that omitted the distal portion of the RSSs including the nonamers, the RAG1 nonamer binding domain, and HMGB1 (Fig EV1E). Difficulty resolving the region encompassing the nonamer binding domain has been reported by others (Ru et al, 2015; Chen et al, 2020b), likely because it is connected to the RAG1 catalytic core by a flexible hinge. In the STC, the DNA resembles a twisted “H”, the lower “legs” of which are composed of RSS (signal) DNA and the top half of which is “U”‐shaped target DNA made up of two upright flank DNA “arms” that are connected to target site DNA through bends of nearly 90° (Fig 2B).
Figure EV1. Assembling the RAG STC and cryo‐EM structure determination.

- Schematic diagram of RAG1 and RAG2 proteins with domains indicated with shading and colors. The portions of the proteins used for STC assembly and cryo‐EM data collection are indicated; the RAG1 protein contained R848M and E649V mutations (pink circles), which activate transposition. ZFA, zinc finger A domain; NBD, nonamer binding domain; CC catalytic core; CTT, C‐terminal tail; AH, acidic hinge; PHD, plant homeodomain.
- Schematic of the RAG STC and the process of complex assembly. The names of the different portions of the DNA as used throughout are indicated. RAG1, RAG2, and HMGB1 (not shown) were incubated together in the presence of Mg2+ with a 12RSS‐containing DNA substrate (red) and a 23RSS‐containing DNA substrate (yellow) to assemble the STC.
- Gel filtration profile shows the purification of the STC.
- 2D class averages from cryo‐EM particles of RAG STC.
- Flowchart of cryo‐EM map reconstruction. Six 3D classes were generated after 3D classification. After multiple steps of data processing as indicated, a 2.6 Å map with C1 symmetry was obtained. The expected position of the NBD/nonamer, which could not be visualized, is indicated with a dashed circle in the final 2.6 Å map.
- Color‐code resolution estimation of RAG STC reconstruction map viewed on the surface and sectioned.
- Unmasked and masked Fourier shell correlation curves, as well as the model versus final map, as indicated. Resolution is read by the cutoff value at FSC = 0.143.
Figure 2. Cryo‐EM structures of the RAG STC.

-
A2.6 Å cryo‐EM map of RAG STC. Protein tetramer and DNA chains are segmented and color‐coded as indicated. DNA in red, 12RSS with its covalently linked target DNA; DNA in yellow, 23RSS with its covalently linked target DNA.
-
BOrthogonal views of the STC DNA density map with RAG protein rendered transparent. Red and yellow, 12RSS and 23RSS and covalently linked target site and flank DNA, respectively.
-
CDiagram of DNA sequence surrounding sites of integration.
-
D, EFront view (defined based on view in (B)) of local maps of target site DNA in paired STC (left) and dynamic STC (right) derived from sub‐classification of the 2.6 Å cryo‐EM map (top panel), and top views of the atomic models built from the two maps (bottom panel).
-
FAtomic model of paired RAG STC. One RAG1‐RAG2 dimer omitted to allow visualization of target site DNA. Cyan arrow, integration site.
-
GSuperimposition of paired RAG STC with the HFC (hairpin‐forming complex) (left) and cartoon model of DNA from RAG HFC (left) and paired STC (right). Orange, target site DNA.
Density for the DNA in the 2.6 Å map was clear except for target site DNA, particularly its central three base pairs (Fig EV2A, Table EV1). Further structural analysis of this region (Fig EV2B and C) revealed a subclass of particles with continuous target site DNA density, yielding a 2.9 Å STC map in which target site DNA is fully base‐paired (Figs 2C and D, and EV2D, E, G and I). A second, larger subclass of particles yielded a 2.7 Å STC map in which central target site DNA density is poor, suggesting a dynamic or unstructured state (Figs 2E, and EV2D, F, H and J). These two maps and the structural models derived from them are almost identical except for the area immediately surrounding target site DNA (Fig EV2K–M) and are hereafter referred to as the paired and dynamic RAG STC structures, respectively. The paired RAG STC structure is very similar (RMSD 0.45 Å) to that of the RAG STC assembled using R848 RAG1 (Chen et al, 2020b; Fig EV3A). Unstructured target DNA as observed in the dynamic STC was not reported in that study. Our findings indicate that while target site DNA can exist in a well ordered, annealed state, it frequently exists in a disordered and presumably dynamic state—a state that methionine at position 848 helps establish, as discussed below.
Figure EV2. Cryo‐EM map of paired‐form and dynamic‐form RAG STC .
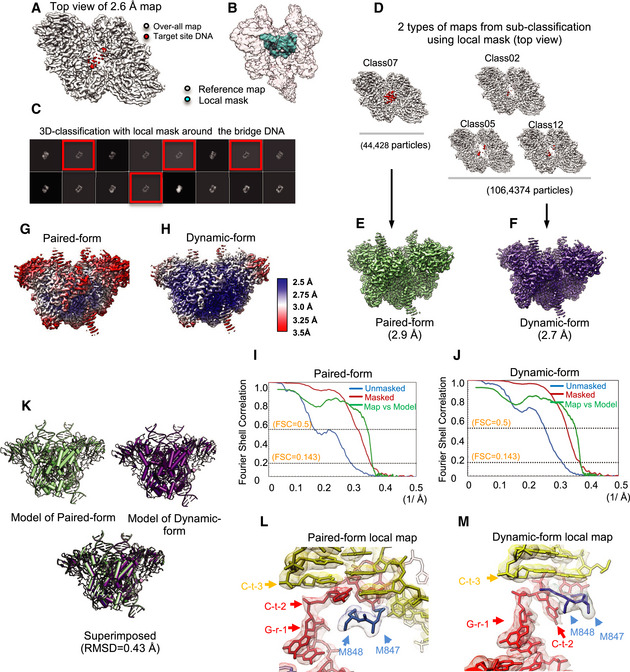
-
ACryo‐EM map of RAG STC (top view) with target site DNA density indicated in red. Target site DNA exhibits weaker density than other portions of the map.
-
BThe mask identifying the region surrounding target site DNA (cyan) is indicated together with the RAG STC map (transparent).
-
CSixteen classes from 3D classification were generated with the local mask. Four classes with better features were selected for further data processing.
-
D–FCryo‐EM map reconstruction of paired‐form and dynamic‐form RAG STC. Four 3D cryo‐EM maps (top view), with target site DNA density indicated in red as in (A), were separated into two groups based on the quality of target site DNA density. The first group, with strong target site DNA density, consisted of class 07. The other three classes with weaker target site DNA density were combined for further processing. This yielded a 2.9 Å paired RAG STC map (E) and a 2.7 Å dynamic RAG STC map (F), which were used for data analysis and model building.
-
G–JColor‐code resolution estimation maps (G and H) and unmasked and masked Fourier shell correlation curves, as well as the model versus final map (I and J) for the paired RAG STC and the dynamic RAG STC depicted as in Fig EV1E and F.
-
KAtomic models of paired and dynamic RAG STC structures, as well as superposition of the two structures. The two structures are extremely similar (RMSD of 0.43 Å).
-
L, MCryo‐EM map density in the vicinity of the integration site for the paired (L) and dynamic (M) RAG STCs, with models depicted within the maps. DNA nomenclature defined in Fig EV3E.
Figure EV3. Structural details of paired RAG STC .

-
ASuperimposition of the paired RAG STC structure determined in this study and the RAG STC structure determined with RAG1 containing R848 (Chen et al, 2020b).
-
B–DStructure of the reaction center of the paired RAG STC, depicted in intact form in (B) and as individual regions surrounding each Mg2+ ion (C and D) to better depict metal ion coordination. Cyan sphere, integration site phosphate; green spheres, Mg2+ ions. In (C), the Mg2+ ion is coordinated by the phosphate of C‐t−2, D600, G601, and E962. In (D), the second Mg2+ is coordinated by the 3′‐OH from flank DNA, D708, and D662. DNA nomenclature defined in (E).
-
ESchematic diagram of DNA after strand transfer with target site DNA fully base‐paired. Yellow and red, 23RSS and 12RSS, respectively; purple, target site DNA; light blue, flank DNA. The nomenclature used for each base is indicated.
-
FSuperimposition of M847 and M848 from the paired RAG STC and M847 and R848 from the RAG STC structure determined with RAG1 containing R848 (Chen et al, 2020b).
-
GSchematic diagram of strand transfer DNA in the paired RAG STC. The major kinks in target DNA exist one base pair in from the edges of the 5 bp target site DNA.
The RAG STC adopts a “closed” conformation similar to that of the HFC
Models of the paired and dynamic STC structures were constructed and reveal the DNA cradled by a central dimer of RAG1 and two monomers of RAG2 (Figs 2F and EV2K). In each RAG1 active site, two Mg2+ ions are surrounded by catalytic carboxylate residues (D600, E662, D708, and E962) and G601, with one Mg2+ in close proximity to the phosphodiester bond linking signal and target DNA (Fig EV3B–D). The RAG STC adopts a compact “closed” conformation remarkably similar to that of the RAG HFC (1.9 Å RMSD, calculated over the protein backbone) (Kim et al, 2018) except in the immediate vicinity of target site DNA (Fig 2G). In this conformation, the constraints imposed on the path of the target DNA flanks create the requirement for the severe bends observed in STC target DNA, formation of which would be expected to impose a substantial barrier to the transposition reaction, as also noted in (Chen et al, 2020b). The structure of DNA in the STC and its close similarity to that in the HFC provides a plausible explanation for why transposition by RAG predominantly generates a 5 bp target site duplication (Agrawal et al, 1998; Zhang et al, 2019), as the cavity between the two flanking DNAs is optimally sized to accommodate 5 bp of DNA (Fig 2G).
Target DNA conformational change related to methionine 848 in RAG STCs
Comparison of the two RAG STC structures provides insight into the mechanism by which RAG1 M848 stimulates transposition. In the RAG STCs, M848 and its neighbor M847 form a hydrophobic patch or platform at the target DNA bend (Fig 3A and B) that should create an unstable local environment for the polar bases and charged phosphate backbone of the nearby DNA. In the paired STC structure, the target base covalently linked to the RSS (C‐t−2) and its paired base (G‐t+2*) rest on the M847/M848 platform, which acts as a wedge to break base stacking on both strands and to facilitate DNA bending (Fig 3A, C and D; DNA naming nomenclature provided in full in Fig EV3E). In the dynamic STC structure, a major structural change is observed: The M847/M848 platform now lies immediately below C‐t−3:G‐t+3* and has disrupted the C‐t−2:G‐t+2* bp (Figs 3E and F, and EV2M). In this new location, M848 displaces the critical bridging base C‐t−2 into an extrahelical position, and its partner base G‐t+2* is no longer visible in the map density (Fig 3E and F, and Movie EV1). Furthermore, no map density is visible for C‐t+1* or the central bp of target site DNA in the dynamic STC map density (Fig 2E). Hence, the M847/M848 hydrophobic wedges contributed by the two RAG1 subunits disrupt base pairing throughout target site DNA in the dynamic STC. Replacing M848 with arginine removes half of the hydrophobic wedge and should compromise the interactions that flip base C‐t−2 and cause target DNA unstacking and melting. Consistent with this, in the R848‐RAG1 STC structure, R848 was positioned similarly to M848 in the paired STC structure (Fig EV3F) and no melting or base flipping in target site DNA was reported (Chen et al, 2020b). Thus, while both R848 and M848 can insert into the kink in fully base‐paired target DNA, M848 has the added capability of destabilizing target site DNA along its length.
Figure 3. Structural details at the integration site of the RAG STC.
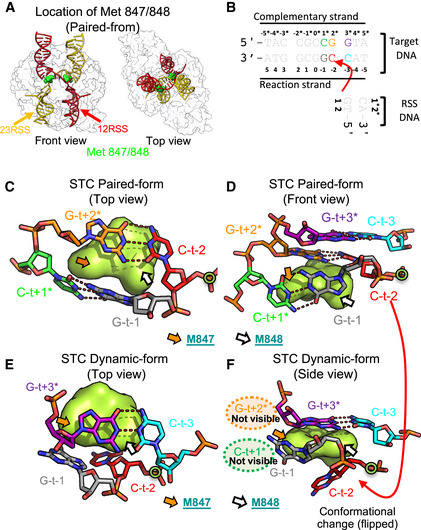
-
ALocation of RAG1 M847 and M848 (surface mode, green) in the RAG STC.
-
BDiagram of DNA sequence surrounding integration site with colors specifying relevant bases as in (C–F).
-
C, DLocal structures of top and front views of paired RAG STC near the 12RSS integration site.
-
E, FLocal structures of top and front views of dynamic RAG STC near the 12RSS integration site.
Identification of a RAG TCC
In the paired RAG STC structure, the target DNA bends are located one bp within target site DNA on each end, not at the sites of strand transfer (Figs 3C–F and EV3G). This allows the 3′‐OH nucleophile on flank DNA to remain close to the scissile phosphate it would attack to perform disintegration, the reverse of the integration reaction (Fig EV4A and B). Consistent with this, RAG, like HIV integrase, is capable of performing disintegration (Mazumder et al, 1994; Melek & Gellert, 2000; Delelis et al, 2008). Using a labeled STC DNA substrate, disintegration was detectable at 4°C in Mg2+ (the STC assembly conditions for structural analysis) and increased in efficiency at higher temperatures or with use of Mn2+ (Fig 4A and B). We therefore considered the possibility that disintegration had occurred in a subset of the cryo‐EM particles. Indeed, a subclass of particles yielded a 3.8 Å resolution cryo‐EM map in which target DNA was intact and separated by a gap from the RSSs (Fig EV4C–E, Table EV1), indicative of disintegration to form the TCC. The TCC structure obtained from these particles closely resembles the STC structure (0.7 Å RMSD calculated over the protein backbone) including “U”‐shaped target DNA containing two bends of nearly 90° (Fig 4C and D), indicating that the RAG proteins are capable of introducing severe DNA bends into noncovalently bound target DNA. This raises the question of how the energetic costs of generating such bends are overcome prior to the integration reaction, which releases strain by introducing breaks in both strands of target DNA. These findings also argue that strand transfer can occur without dramatic conformational changes in the portions of the RAG proteins visible in our structures.
Figure EV4. The RAG target capture complex (TCC).

- Schematic depicting the integration and disintegration reactions. The free 3′‐OH from the RSS DNA or flank DNA serves as the nucleophile that attacks the phosphate in either target DNA or strand transfer DNA.
- The reaction center of the paired RAG STC showing the distance between the 3′‐OH from C‐t−3 and the disintegration scissile phosphate of C‐t−2. The 3′‐OH and phosphate are constrained by coordinating Mg2+ ions. The red arrow indicates the direction for in‐line nucleophilic attack for the disintegration reaction.
- Flowchart of cryo‐EM map reconstruction of RAG TCC. De novo data processing beginning with particle picking. Four 3D classes were generated after 3D classification as indicated. Class 03, with the best resolution and most particles, was selected for further processing. After multiple steps of data processing, 3.3 Å maps were obtained. One sub‐classification of cryo‐EM map was identified to obtain the atomic model of the target capture complex (TCC) by applying the mask surrounding the target site DNA and the integration reaction center. Fourier shell correlation curves and color‐code resolution estimation map are at the bottom.
- Cryo‐EM map density in the vicinity of the 3′‐OH of 23RSS (right) and 12RSS (left) signal DNA of RAG TCC, with models depicted within the maps. The oxygen of 3′‐OH and the phosphate at integration site are colored green and are connected by a green dashed arrow. A gap between the density of RSS signal DNA and target DNA is indicated by the black arrow.
- Cryo‐EM map density of paired RAG STC at the equivalent position in (D) is shown in the same view. The phosphodiester bond formed between the RSS signal 3′‐OH and integration site phosphate is colored green. Black arrow indicates the gap generated in the reaction strand of target DNA after the integration reaction. DNA nomenclature defined in Fig EV3E.
Figure 4. Identification of the RAG TCC.

-
ADiagram of the disintegration reaction and substrate used in (B).
-
BDisintegration reaction (1 h) performed by RAG1 aa 216–1,008 R848M/E649V, RAG2 aa 1–361, and full‐length human HMGB1 with Mg2+ or Mn2+ at the indicated temperatures. Denaturing gel displays the fluorophore‐labeled DNA strand from the RAG STC before (16 nt band, lane 2) and after (37 nt) the disintegration reaction. Lane 1, fluorophore‐labeled 37 nt DNA marker. Representative of 2 independent experiments.
-
C, DCryo‐EM maps of RAG TCC (C) at 3.8 Å and paired STC (D) at 2.9 Å shown in both top and front views with protein colored white, target DNA colored red, and signal DNA colored blue. Insets highlight the different connectivity between signal and target DNA in the TCC and STC.
Target DNA base flipping by methionine 848 in the RAG TCC
Construction of an atomic model for the TCC revealed density consistent with two possible orientations of the M848 side chain at the site where the 23RSS will integrate (Fig 5A and B). In configuration 1, where density is stronger, M848 inserts into target site DNA at the intrahelical position of the C‐t−2 base, flipping C‐t−2 into an extrahelical position and disrupting base pairing and base stacking at the site of target DNA bending (Fig 5A and C–E). In this configuration, the C‐t−2 base resides between the RSS 3′‐OH nucleophile and the target phosphate for integration, which are separated by almost 8 Å (Fig 5A). Hence, configuration 1 of the TCC is not competent for strand transfer. In configuration 2, repositioning of M848 allows C‐t−2 to return to an intrahelical position (Fig 5B), thereby removing the barrier between the 3′‐OH nucleophile and target phosphate and potentially allowing for nucleophilic attack. Only configuration 1 is seen at the site where the 12RSS will integrate into target DNA, raising the possibility of an underlying asymmetry in how RAG bound to the two RSSs engages target DNA in the TCC.
Figure 5. Function of methionine 848 in the RAG TCC.
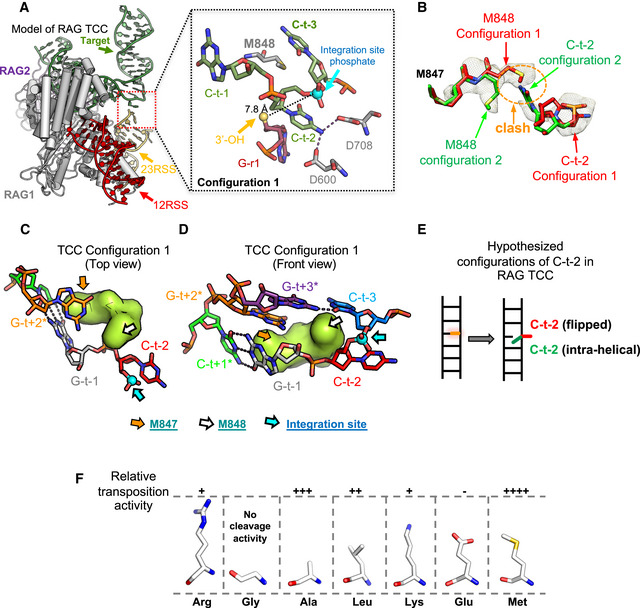
-
ALeft, atomic model of RAG TCC. One RAG1‐RAG2 dimer omitted to allow visualization of target and RSS DNA. Right, expanded view surrounding flipped C‐t−2 base with M848 in configuration 1. C‐t−2 is potentially stabilized by hydrogen bonds with D600 and D708. Blue sphere, integration site phosphate; yellow sphere, RSS 3′‐OH.
-
BDiagram depicting the different configurations of M848 in the RAG TCC. In configuration 1, M848 would clash with C‐t−2 in an intrahelical position. This clash is resolved when M848 adopts configuration 2.
-
C, DLocal structures of top and front views of RAG TCC (M848 configuration 1) near integration site, depicted as in Fig 3C and D.
-
ESpeculative model of DNA configurations in the RAG TCC. Because M848 can adopt two different configurations, C‐t−2 has the possibility of assuming either an intrahelical or flipped configuration.
-
FSchematic of the different amino acid side chains tested for in vitro transposition activity at RAG1 position 848. The relative transposition efficiency of each mutant compared with wild‐type (Arg) is shown above the diagram of each residue (data derived from Zhang et al, 2019). Strongest activity was observed for methionine.
In our previous analysis of different residues at RAG1 position 848, methionine supported transposition most robustly (Fig 5F) and methionine is found at this position in most RAGL transposases identified to date (Fig 1D; Zhang et al, 2019). The structure of the TCC argues that methionine was favored at this position because the length and hydrophobicity of its side chain were particularly well suited for insertion into and disruption of target DNA at the site of bending. This activity would be particularly important given the unusually large bends that RAG induces in target DNA prior to strand transfer and is likely compromised with the positively charged side chain of arginine. Curiously, alanine at position 848 supports higher levels of transposition than leucine despite having a shorter hydrophobic side chain. It is possible that with alanine at position 848, the side chain of the flanking residue methionine 847 is able to compensate partially for the function of methionine 848, and that such compensation is more difficult in the context of the bulky leucine side chain.
RAG2 constrains the path of target DNA and helps impose bending
Transib transposase is thought to be the evolutionary precursor of RAG1/RAG1L proteins and exhibits strong structural similarities to RAG1 despite low sequence similarity (Liu et al, 2019). Unlike retroviral integrases which possess a relatively shallow target DNA binding surface, RAG, HzTransib (from the moth Helicoverpa zea), and other DDE family transposases have a deep target DNA binding pocket that requires substantial target DNA bending to position the scissile phosphates in the transposase active sites (Maertens et al, 2010; Montano et al, 2012; Morris et al, 2016; Passos et al, 2017) (Fig EV5A). However, the nearly 180° change in direction of target DNA observed in the RAG STC and TCC is larger than that observed in the HzTransib STC (~ 150°) (Liu et al, 2019) and is the most extreme observed to date (Chen et al, 2020b). RAG2, which is lacking in Transib transposases, contributes a positively charged surface that extends RAG target flank DNA interactions 8 bp further than with HzTransib (Fig 6A). These interactions closely resemble RAG2‐coding flank DNA interactions in the HFC (Kim et al, 2018; Fig 2G). By establishing the shape of the RAG target DNA binding pocket, RAG2 constrains the trajectory of the target flanks and helps impose the requirement for extreme target DNA bending. In addition, RAG2‐DNA interactions likely stabilize target DNA binding to compensate for the less positively charged target DNA binding surface of RAG1 as compared to HzTransib (Fig 6B and C).
Figure EV5. Deletion of four amino acids from the tip of RAG2 loop 333–342 increases transposition efficiency.

-
ADiverse target DNA conformations and bending angles in transposase and retroviral integrase STC structures. Front and top views of target DNA (red) and RSS/TIR DNA (white) for the paired RAG STC and the indicated transposases or retroviral integrases. Black lines are used to approximate the DNA bends.
-
B–GComparison of RAG2 loop 333–342 between different RAG complexes during the cut‐and-paste transposition. (B) Free RAG complex (mouse) without the DNA substrate; (C) RAG pre‐reaction complex (zebrafish) with intact 12RSS and 23RSS; (D) RAG pre‐reaction complex (zebrafish) with intact 12RSS and 23RSS melted at the catalytic center; (E) RAG hairpin‐forming complex (zebrafish) with nicked 12RSS and 23RSS; (F) RAG TCC (mouse) with target DNA and 12RSS and 23RSS signal DNA; (G) paired RAG STC (mouse) with 12RSS and 23RSS integrated into target DNA. RAG1 and RAG2 are colored white with RAG2 loop 333–342 highlighted in red. DNA is shown in cyan.
-
HExample of plates stained with crystal violet to reveal puromycin‐resistant colonies from the plasmid‐to-genome transposition assay. The RAG2 ∆L mutation (deletion of RAG2 aa 336–339) increases colony numbers, which is a measure of transposition efficiency.
-
ISchematic of GFP fluorescence recombination assay, used for testing RAG V(D)J recombination activity in vivo.
-
JDNA sequence logo obtained from 5 bp target site duplications from 639 plasmid‐to-genome transposition events mediated by RAG1 E649V/R848M and RAG2 1–350 in HEK293T cells (data derived from Zhang et al, 2019). Target site duplications exhibit a GC preference (Zhang et al, 2019) and a pyrimidine/purine step at the edges.
-
KComparison of disintegration activity of WT (green) and RAG1 double mutant (R848M/E649V, purple). Data were fit to Michaelis–Menten equation to draw the curve. Data points represent the mean of 3 biological replicates and are shown ± SEM.
-
L, MDiagram of the valine or glutamic acid at RAG1 position 649 in the structures of dynamic RAG STC (left) or R848 RAG STC (Chen et al, 2020b) (right). S963 and V649 or E649 are shown as stick mode and colored green and cyan, respectively. Protein, target, and signal DNA are colored white, dark‐red, and yellow, respectively. V649 points into the protein core but E649 points toward signal DNA.
Figure 6. Structural comparison of RAG and Transib.
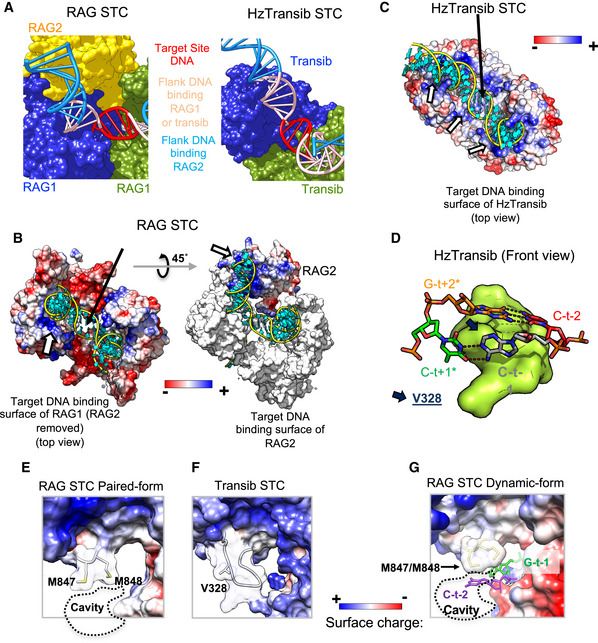
-
ADiagram to show the extent of flank DNA binding for RAG and HzTransib in the STC. Red, target site DNA; light pink, flank DNA bound by both RAG1 and HzTransib; blue, remainder of flank DNA, much of which is contacted by RAG2 in the RAG STC.
-
BLeft, structure of paired RAG STC with RAG2 omitted (top view). RAG1 protein is shown as an electrostatic surface and target DNA in cartoon mode with the backbone colored yellow and base colored cyan. The white arrow indicates the positively charged surface for target DNA binding on RAG1. Right, structure of the paired RAG STC with one RAG2 shown (45° rotation relative to A). RAG1 is shown as a molecular surface and colored white. RAG2 is shown as an electrostatic surface. The white arrow indicates the positively charged surface for target DNA binding on RAG2.
-
CHzTransib STC structure is shown as for RAG in (B). Three white arrows indicate the positively charged surface provided by HzTransib for target DNA binding.
-
DFront view of HzTransib STC near the integration site (PDB:6PR5), depicted as in Fig 3D.
-
E, FProtein electrostatic surface around M847/M848 of RAG1 in the paired STC and equivalent region of HzTransib STC. Loop region rendered transparent. Dashed line, cavity formed due to shorter loop in RAG1.
-
GA similar view of RAG dynamic STC as shown in (E) depicting bases C‐t−2 and G‐t−1 which are flipped and project into the cavity below target site DNA in the RAG STC.
Consistent with its less severe target DNA bending, HzTransib uses a small hydrophobic wedge, made up of V328 (the equivalent of RAG1 M847), to insert into target DNA and disrupt base stacking on only one strand of target DNA (Liu et al, 2019; Fig 6D)—which contrasts with disruption of base stacking on both strands seen with the larger RAG1 M847/M848 wedge (Fig 3C–F). In addition, the loop containing M847/M848 in RAG1 is shorter than the equivalent loop in HzTransib (Fig 6E and F), which leaves a cavity below target site DNA in the RAG but not the HzTransib STC structure (black arrow in Fig 6B and C). Notably, the flipped C‐t−2 base observed in the dynamic RAG STC structure (Fig 3F) protrudes into this cavity (Fig 6G), indicating that RAG, but not HzTransib, is designed to accommodate the target DNA base flipping triggered by RAG1 M848.
Jawed vertebrate‐specific RAG2 loop inhibits transposition
RAG2 contains an extended loop (aa 333–342) that has few interactions with protein and no identified interactions with DNA in existing structures of RAG during RSS binding and cleavage (Fig EV5B–E). It appears to be a jawed vertebrate‐specific adaptation as it is truncated in RAG2‐like transposase proteins from invertebrates including ProtoRAG‐encoded BbeRAG2L (Fig 7A and B). In both our STC and TCC structures, this loop extends downward within the target‐binding pocket to contact target site DNA (Figs 7C, and EV5F and G), a feature also observed in the R848 RAG1 STC structure (Chen et al, 2020b). This interaction raised the possibility that the RAG2 333–342 loop regulates transposition, either positively (e.g., by stabilizing target DNA in the binding pocket) or negatively. To explore this, we deleted the four amino acids at the tip of the loop that are in closest proximity to target DNA (Fig 7A and C) and measured the effects on transposition efficiency using in vivo plasmid‐to‐plasmid and plasmid‐to‐genome transposition assays. In both assays, the RAG2 ΔL mutation increased transposition efficiency twofold to threefold (Figs 7D and E, and EV5H) without significantly altering V(D)J recombination efficiency (Figs 7F and EV5I), arguing that DNA cleavage function was not affected. We conclude that the RAG2 aa 333–342 extended loop arose early in jawed vertebrate evolution in part to provide an additional mechanism to suppress RAG‐mediated transposition. The mechanism by which the loop acts is not known but its poor sequence conservation in jawed vertebrates (Fig 7B) is consistent with the possibility that it interferes sterically with target DNA.
Figure 7. A jawed vertebrate‐specific extended loop on RAG2 contacts target DNA and inhibits transposition.

- Comparison of RAG2 and BbeRAG2L structures. Red color indicates the amino acid 333–342 RAG2 loop and the equivalent loop of BbeRAG2. The four‐amino‐acid deletion, (ΔL in (D)), is indicated with green brackets. Due to areas of low resolution in the BbeRAG2L structure, some regions that are likely to be beta strands cannot be modeled as such.
- Sequence alignment of RAG2 and RAG2‐like proteins in the vicinity of mouse RAG2 loop 333–342. Green shading, four amino acids deleted at tip of loop. Red letters in blue box, relatively well‐conserved residues; white letters on red background, highly conserved residues.
- Cryo‐EM map (transparent) and cartoon model of RAG2 showing contact between the extended loop (red) and target site DNA (yellow; stick model).
- Results of in vivo plasmid‐to-plasmid transposition assay using WT or R848M RAG1 and the indicated forms of RAG2 containing or lacking (ΔL) the four amino acids at the tip of the 333–342 loop, performed as described in (Zhang et al, 2019); (****P < 0.0001). Three biological replicates for each condition except in the last two columns where data from 11 biological replicates are presented.
- Results of in vivo plasmid‐to-genome transposition assay using WT or R848M RAG1 and different forms of RAG2 as indicated. As previously demonstrated (Zhang et al, 2019), R848 (present in WT RAG1) and RAG2 acidic hinge residues 362–383 each potently suppress transposition. Substantial transposition is observed with the combination of RAG1 R848M and RAG2 1–361, which is further increased by ΔL; (***P = 0.001). Four biological replicates for each condition.
- In vivo recombination activity in HEK293T cells of mouse wild‐type RAG1 or RAG1 R848M with RAG2 1–361 or 1–361 ∆L. (P = 0.42 for wild‐type RAG1 group, P = 0.41 for RAG1 R848M group). Three biological replicates for each condition.
Discussion
RAG1 amino acid 848 is inevitably arginine in jawed vertebrates but a hydrophobic residue (usually methionine) in invertebrate RAG1L proteins (Martin et al, 2020), and this difference strongly influences the efficiency of RAG‐mediated transposition (Zhang et al, 2019). With arginine at this position, transposition is powerfully inhibited, particularly in vivo, even when assayed in the context of an activated form of RAG2 lacking its inhibitory acidic region (Zhang et al, 2019). A recent study demonstrated that the RAG STC assembled using R848 RAG1 contains strongly bent target DNA, with R848 and M847 serving as a wedge that inserts at the bend sites (Chen et al, 2020b). Together, these prior findings raised several questions: Is target DNA as strongly bent in the RAG TCC as in the STC? If so, how does RAG overcome the energy barrier that such bending would be predicted to constitute in order to bind target DNA? And, how does methionine at position 848 stimulate RAG‐mediated transposition? The findings reported here help answer these questions.
The RAG TCC structure reveals target DNA in a conformation nearly identical to that in the STC, indicating that RAG can indeed noncovalently bind and bend target DNA into a U shape. The TCC was formed through disintegration of a preassembled strand transfer product DNA substrate, and we cannot rule out the possibility that RAG forms a TCC with less strongly bent target DNA as an intermediate in achieving the fully bent configuration seen in the STC. However, RAG‐DNA complexes adopt a “closed” configuration after nicking (the HFC) and after hairpin formation (the SEC) that closely resembles what we observe in the TCC and STC (Figs 2G, and 4C and D; Ru et al, 2015; Kim et al, 2018). Based on this, we think it likely that target capture occurs in the context of the compact protein configuration established by DNA cleavage and hence requires severe target DNA bending, as observed in our TCC structure. For the Tc1/Mariner family transposase Mos1, target DNA capture and strand transfer are also thought to take place without a major reorganization of the proteins or transposon end DNA (Morris et al, 2016). Our findings strongly support the hypothesis that target DNA bending represents a major barrier to RAG‐mediated transposition (Chen et al, 2020b). Consistent with this, we have thus far not been able to assemble a stable TCC using free (noncovalently linked) target DNA. The previously reported R848 RAG STC structure was also generated using preassembled strand transfer product DNA, but disintegration to form the TCC was prevented by the use of a RAG1 active site mutant (Chen et al, 2020b).
Importantly, the findings reported here provide a mechanism by which the energetic barrier to insertion of target DNA into RAG's deep, tightly constrained binding pocket can be overcome, and in so doing, provide a plausible explanation for how M848 stimulates transposition. In the most prevalent configuration of the TCC observed (configuration 1), M848 inserts deeply into target DNA at both bend sites, flipping the C‐t−2 base out of the helix and disrupting target DNA base stacking and base pairing. By relieving strain on the DNA helix, M848 likely facilitates formation of a TCC in which the target DNA scissile phosphates can approach the RAG active sites. A requirement for additional target DNA dynamics is strongly suggested by the fact that TCC configuration 1 is not compatible with strand transfer and by the observation of a second orientation of the M848 side chain that is consistent with close approach of the 3′ OH to the scissile phosphate. The ability of M848 to disrupt target DNA is reinforced by our identification of the dynamic STC in which C‐t−2 is again flipped out of the helix and the majority of target site DNA is sufficiently unstructured that it has little detectable density in the cryo‐EM map. The dynamic STC was observed more frequently than the paired STC among our cryo‐EM particles, suggesting that in the presence of M848, target DNA disruption is a prevalent state. How the dynamic STC relates to structural changes required for the strand transfer reaction remains to be determined, but we speculate that the highly disrupted state of target site DNA in the dynamic STC reflects conformational changes that occur in the TCC to allow close approach of the 3′ OH to the scissile phosphate for strand transfer.
The structures reported here support a model for RAG‐mediated transposition in which M848 destabilizes target DNA near the sites of bending and strand transfer and thereby facilitates stable target DNA capture and the strand transfer reaction. Multiple prior observations are consistent with this model. First, bubble and hairpin DNA structures are strongly preferred transposition targets for RAG, arguing that single‐stranded character of target DNA facilitates strand transfer (Tsai et al, 2003; Posey et al, 2006), as is the case for other transposases and retroviral integrases (Pribil & Haniford, 2000; Kuduvalli et al, 2001; Yanagihara & Mizuuchi, 2002; Arinkin et al, 2019). Second, RAG strongly prefers GC‐rich target site sequences (Agrawal et al, 1998; Tsai et al, 2003; Zhang et al, 2019), and GC base pairs within GC tracts have a particularly high breathing rate, much higher than that observed for isolated GC base pairs (Dornberger et al, 1999). Third, like HzTransib (Liu et al, 2019), RAG exhibits a pyrimidine–purine preference at the edges of its target site sequence (Fig EV5J); pyrimidine–purine steps are particularly prone to melting and unwinding (Lankas et al, 2003; Johnson et al, 2008). Fourth, hydrophobic side chains other than methionine at RAG1 position 848 can also stimulate transposition while negatively charged Glu strongly suppresses transposition (Zhang et al, 2019; Fig 5F). And fifth, HzTransib, which lacks a hydrophobic residue corresponding to M848, was not observed to melt target site DNA or flip C‐t−2 in the STC or in the model constructed for the TCC (Liu et al, 2019). Similarly, when R848 RAG1 was used to assemble the STC, disordered/melted target DNA was not reported (Chen et al, 2020b). The E649V/R848M mutant RAG1 protein used here performs disintegration somewhat more efficiently than WT RAG1 (Fig EV5K), indicating that increased transposition by this mutant (Zhang et al, 2019) is not due to suppression of disintegration, consistent with prior findings (Chen et al, 2020b). It is not readily apparent from our structures how the RAG1 E649V mutation increases transposition efficiency (Zhang et al, 2019), though this mutation might reduce interactions with the nearby signal DNA (Fig EV5L and M).
The initial proposed step in RAGL transposon evolution, acquisition of RAG2L by Transib, provided extended interactions with flank DNA but also likely imposed constraints on the target DNA binding pocket that required greater target DNA bending. These constraints might have driven selection for the hydrophobic residue that is observed at the equivalent of RAG1 position 848 in essentially all invertebrate RAG1L proteins identified to date (Martin et al, 2020). Much later, when RAG‐mediated transposition gave rise to the first “split” antigen receptor gene early in jawed vertebrate evolution, there was likely strong selective pressure for adaptations that reduced transposition activity while maintaining the cleavage function of RAG. Our findings argue that mutation of position 848 to arginine accomplished this by altering interactions with target DNA and interfering with target DNA distortions important for target DNA capture and strand transfer. Base flipping and melting of target DNA near the sites of integration are a common principle in DNA transposition and retroviral integration (Arinkin et al, 2019), and our findings indicate that evolution selected this step as one of the focal points for domestication of RAG for adaptive immunity. Our finding of a jawed vertebrate‐specific loop in RAG2 that suppresses transposition extends the theme of a multilayered approach to protect the genome from RAG transposon‐mediated insertional mutagenesis (Zhang et al, 2019) and other potential deleterious consequences of transpositional insertion of RSSs into the genome (Hiom et al, 1998; Melek & Gellert, 2000).
Materials and Methods
Plasmid generation
We used the pTT5MP plasmid, a derivative of pTT5 containing maltose‐binding protein (MBP) and a PreScission Protease cleavage site at the C terminus of MBP (Huang et al, 2016) to generate the RAG1 and RAG2 constructs for protein expression in expi293F cells. Mouse gene sequences of RAG1 aa 261–1,008 with mutants R8484M and E649V and RAG2 aa 1–361, used for complex assembling and cryo‐EM data collection, were cloned into pTT5MP separately by In‐Fusion cloning. The constructs used for the in vivo GFP recombination assay and plasmid‐to‐plasmid and plasmid‐to‐genome experiments were pEBB‐RAG1 FL and its mutants (Zhang et al, 2019) and pTT5MP‐RAG2 1–361 with deletion of aa 336K–339M, which was generated by PCR and In‐Fusion cloning base on the construct pTT5MP‐RAG2 1–361.
Protein expression and purification
500 μg of pTT5MP‐RAG1 aa 261–1,008 and pTT5MP‐RAG2 aa 1–361 plasmids was co‐transfected into expi293F cells using the polyethylenimine (PEI, 1 mg/ml) at molar ratio of 1:4. 500 ml of cells containing co‐expressed RAG proteins was collected 3 days after transfection by centrifugation at 500 g and frozen at −80°C. For protein purification, cells were thawed in a room temperature water bath and resuspended in 45 ml lysis buffer (25 mM Tris, pH 7.5, 1 M KCl, 1 mM DTT, EDTA‐free protease inhibitor cocktails (Roche)) and disrupted with 2 passes through an Emulsiflex C3 homogenizer (Avestin). Cell lysate was centrifuged at 192,839 g. (Beckman Coulter Optima LE‐80K Ultracentrifuge, Type 50.2 Ti rotor) for 1 h at 4°C, and the supernatant was mixed with 5 ml pre‐equilibrated amylose resin and then incubated for 2 h with continual rotation at 4°C. The beads and supernatant mixture were loaded on a gravity flow column and washed with 80 ml lysis buffer. Protein was eluted with 10 ml of elution buffer (25 mM Tris, pH 7.5, 0.5 M KCl, 1 mM DTT, 40 mM maltose). The eluted protein was concentrated and further purified by size‐exclusion chromatography (SEC) on a Superdex 200 Increase 10/300 GL column in 20 mM HEPES pH 7.6, 0.5 mM TCEP, 150 mM KCl, and 5 mM MgCl2. SEC peak fractions containing the RAG1/2 complex were collected and pooled, and the protein complex was concentrated to 5–15 μM using an Amicon centrifugal concentrator and stored at −80°C after freezing in liquid nitrogen. Full‐length (FL) histidine‐tagged human HMGB1 (hHMGB1) and histidine‐tagged hHMGB1∆C (aa 1–165 without the acidic C‐terminal region) were expressed as previously described (Bergeron et al, 2006; Huang et al, 2016). HEK293T cells were obtained from ATCC, and expi293F cells were purchased from Thermo.
Strand transfer DNA for cryo‐EM
12RSS and 23RSS substrate DNAs (as depicted in Fig EV1B) were separately assembled by annealing three deoxyoligonucleotides in an equimolar ratio. After annealing, DNAs were frozen at −20°C. Deoxyoligonucleotide sequences were as follows:
12 or 23 RSS flank DNA top strand oligo:
5′‐CTCAGGATAGGGCTAC‐3′;
12RSS signal DNA top strand oligo:
5′‐CACAGTGGTAGTAGGCTGTACAAAAACCtcgaCC‐3′;
12RSS DNA bottom strand oligo:
5′‐GGtcgaGGTTTTTGTACAGCCTACTACCACTGTGCGCCGGTAGCCCTATCCTGAG‐3′;
23RSS signal DNA oligo:
5′‐CACAGTGGTAGTAGGCTGTTGTCTGGCTGTACAAAA ACCtcgaCC‐3′;
23RSS DNA bottom strand oligo:
5′‐GGtcgaGGTTTTTGTACAGCCAGACAACAGCCTACTACCACTGTGCGGCGGTAGCCCTATCCTGAG‐3′
RAG strand transfer complex assembly and purification
MBP‐fused RAG1 261–1,008 and RAG2 1–361 complex was mixed with equal molar 12RSS DNA and 23RSS DNA (see Fig EV1A and B) separately as well as twofold molar excess of hHMGB1∆C in 20 mM HEPES pH 7.6, 0.5 mM TCEP, 5 mM MgCl2, and 150 mM KCl. The sample was incubated at room temperature for 15 min to assemble a STC (strand transfer complex). After the incubation, 10% (v/v) PreScission Protease (0.2 mg/ml) was added and the sample was further incubated at 4°C overnight to remove the MBP tags. The digested mixture was loaded on a Superdex 200 Increase 10/300 GL column and purified by SEC in 20 mM HEPES pH 7.6, 0.5 mM TCEP, 150 mM KCl, and 5 mM MgCl2 to purify the complex from MPB and unassembled DNA and HMGB1. Peak column fractions were collected and concentrated (if necessary) to a protein concentration of 0.3–0.4 mg/ml. The sample was immediately used to prepare cryo‐EM grids.
Disintegration reaction
2 μl of RAG proteins (5 μM) was mixed with an equimolar amount of fluorophore‐labeled 12RSS and non‐labeled 23RSS DNA and a twofold excess of full‐length hHMGB1 in reaction buffer (25 mM HOPS, pH 7.0, 100 mM KCl, 2 mM DTT, 5 mM MgCl2 or MnCl2 depending on the reaction; 16 μl final reaction volume) and incubated at 37°C, 23°C, or 4°C for 1 h. Reactions were stopped by adding 2 μl of 0.5 M EDTA, 1.25 μl 2.5% SDS, and 20 μl formamide, and the sample was incubated at 100°C for 5 min to denature the DNA and cooled on ice. 10 μl of the sample was loaded on a 16% 1× TBE polyacrylamide denaturing gel. After 45 min of electrophoresis at 2 W, gels were imaged using a PharosFX Plus (Bio‐Rad). 12 and 23 RSS DNA used in this experiment are the same as in STC assembly except for the 12 RSS flank DNA top strand, which is replaced by the fluorophore (Alexa 488)‐labeled deoxyoligonucleotide (see Fig 4A): Alexa 488‐5′‐ CTCAGGATAGGGCTAC ‐3′. Disintegration reactions for Fig EV5K were quantitated using ImageJ software. Three independent experiments were done at each reaction time, and the results were fit to the Michaelis–Menten equation by using GraphPad Prism 8.
Cryo‐EM data acquisition
Purified STC (3.5 μl at a concentration of ~ 0.4 mg/ml) was applied to freshly glow‐discharged C‐flat Cu 400 mesh, R2/1 holey carbon grids. Grids were blotted for 4 s in 100% humidity at 4°C and plunge‐frozen in liquid ethane cooled by liquid nitrogen using a Vitrobot Mark IV (Thermo Fisher Scientific). Data were acquired on a Titan Krios G2 electron microscope (Thermo Fisher Scientific) operated at 300 kV in EFTEM mode at Yale University. The slit width of the Gatan Quantum LS energy filter with K2 detector was set to 20 eV. The nominal magnification was set to 130,000, corresponding to 1.05 Å per physical pixel. The dose rate was set to 7.3 electrons per physical pixel per second. Raw movies were saved in super‐resolution mode. The total dose was 73 electrons per Å2, fractionated into 44 frames in 11 s. SerialEM (Mastronarde, 2005) was used for the automated data collection, and five shots were taken in each hole using image shift, with beam tilt compensation. The defocus ranges were set to −0.8 to −1.8 μm and −1.1 to −2.3 μm separately for two grids. The statistics of data acquisition are summarized in Table EV1.
Image processing
In the initial data processing, 3,572 images were collected for the STC. RELION's implementation of MotionCor2 (Zivanov et al, 2018) was used for beam‐induced motion correction and dose weighting. The output aligned images were binned twice, resulting in a final pixel size of 1.05 Å for further data processing. The non‐dose‐weighted aligned images were used for contrast transfer function estimation by CtfFind 4.0. (Rohou & Grigorieff, 2015). Images with resolution worse than 5 Å were discarded. Good images were used for auto‐picking, classification, and reconstruction. Particles from 50 images at difference defocus were manually picked, followed by a round of 2D classification to generate templates for auto‐picking. The auto‐picked particles were subjected to 2D classification in RELION‐3.0 (Kimanius et al, 2016; Zivanov et al, 2018) to remove junk particles. Particle coordinates in good classes were extracted for further manual inspection such that bad particles and images were discarded. After initial 3D classification, good 3D classes were combined and used for gold standard auto‐refinement in RELION‐3.0 with C1 symmetry. Then, a round of CTF refinement and Bayesian polishing was done to further improve the resolution and get a final density map. Resolution estimation was based on the Fourier shell correlation cutoff at 0.143 (FSC = 0.143) between the two half‐maps after a soft mask was applied to mask out the solvent region. The final maps were sharpened by their corresponding negative B factors (−40) within RELION‐3.0. Local resolution variation was estimated by the local resolution module in RELION‐3.0.
The local mask around target site DNA was generated in UCSF Chimera (Pettersen et al, 2004) and applied for a new round of 3D classification (Bai et al, 2015) with the particles generated from auto‐refinement in RELION. Four high‐quality classes were picked from the 16 classifications. Particles from the new good classes were extracted to calculate new density maps. All subsequent refinement steps were the same as initial data processing. One of the classes exhibited good density for target site DNA, and the map was designated paired‐form STC. The other three classes showed similar features but with weak density around target site DNA and were combined together for the recalculation that generated the Dynamic‐form STC density map.
To obtain the structure of the target capture complex (TCC), new de novo data processing was performed with an increased particle picking threshold. Similar processing was applied as for the STC. A similar mask around target site DNA was used to generate 10 classifications. Six of them were processed, and the density maps were compared with the map and atomic model of the STC. One class showed a significant density missing between RSS DNA and target DNA and a connection between flank DNA and target DNA on the reaction strand (Fig EV4C). This class was further processed to obtain the density map of the TCC.
Modeling and refinement
The initial model of the RAG STC was derived from PDB file 5ZDZ (the RAG HFC complex X‐ray crystal structure) (Kim et al, 2018) and was fit into the 2.6 Å cryo‐EM map with UCSF Chimera. The DNA was then re‐built, and the R848 and E649 were manually changed to Met and Val, respectively, in COOT 0.9‐pre (Burnley et al, 2017), and the model was refit into the 2.6, 2.7, and 2.9 Å cryo‐EM map individually. Ca2+ was replaced by Mg2+ (the ion used for assembly of the RAG STC and thought to be used by RAG in vivo) to fit into the map. For the model building of the RAG TCC, the RAG STC P‐form was taken as an initial model to be built into the 3.8 Å EM map of the TCC. Target DNA and signal DNA were generated from the DNA in the STC and modified to fit into the map. Further refinements were performed multiple times using Real_space_refine in phenix‐1.17.1 (Adams et al, 2010) to optimize the structure with secondary structure restraints and Ramachandran restraints. The final models were validated using the PHENIX built‐in validation program, Comprehensive Validation (MolProbity) (Chen et al, 2010), and EMRinger (Barad et al, 2015; Table EV1). All molecular representations were generated in PyMOL (https://www.pymol.org) and UCSF Chimera.
In vivo plasmid‐to‐plasmid transposition assay
The in vivo plasmid‐to‐plasmid transposition assay was performed as described (Chatterji et al, 2006; Zhang et al, 2019). 293T cells were co‐transfected with 4 μg each of the pEBB‐RAG1 full length or R848M mutant and pTT5M‐RAG2 1–361 or RAG2 1–361 ∆L (deletion of four amino acids from K336 to M339), 6 μg donor plasmid (pTetRSS), and 10 μg target plasmid (pECFP‐1) using polyethylenimine. The medium was changed 12 h after transfection, and cells were collected after 48 h. Plasmid DNA was precipitated. 300 ng DNA was transformed into electrocompetent MC1061 Escherichia coli cells, and the cells were plated onto kanamycin or kanamycin–tetracycline–streptomycin (KTS) plates. Transposition efficiency was calculated by dividing the number of colonies obtained on double‐antibiotic plates by the number of colonies obtained on the kanamycin‐alone plate.
In vivo recombination assay
pEBB‐RAG1 full length or R848M mutant and pTT5M‐RAG2 1–361 or RAG2 1–361 ∆L (deletion of four amino acids from K336 to M339) (1 μg) were co‐transfected with 2 μg of pCJGFP32, respectively, into expi293F cells using polyethylenimine (DNA:PEI ratio of 1:3). Cells were collected 72 h after transfection, washed twice with PBS containing 1% FBS, and stained with DAPI (4′,6‐diamidino‐2‐phenylindole). The percentage of live cells expressing GFP was determined by flow cytometry (Corneo et al, 2007).
In vivo plasmid‐to‐genome transposition assay
The in vivo plasmid‐to‐genome transposition assay, 4 μg each of pEBB‐RAG1 or R848M mutant and pTT5M‐RAG2 (1–383, inhibit transposition) or pTT5M‐RAG2 (1–361) or RAG2 1–361 ∆L, and 6 μg of donor pBSK‐12puro23 were co‐transfected into HEK 293T cells. 48 h after transfection, 1 × 106 cells were split into medium containing 0.5 μg/ml puromycin. After 10–12 days of culture, the medium was removed, plates were washed twice with pre‐chilled PBS, and colonies were revealed by staining with crystal violet (Fig EV5H). ImageJ was used for colony counting. Dots with pixel size smaller than 5 were discarded. The final result is summarized as shown in Fig 7E (Zhang et al, 2019).
Statistical analyses
Unpaired two‐tailed t‐tests were used to evaluate the statistical significance of differences between data sets. Where sufficient (> 5) data points were available for testing of normal distribution (as in Fig 7D), the Kolmogorov–Smirnov test of normality was used to ensure that the data did not differ significantly from that which was normally distributed. For Fig 7E and F, no test to determine normal distribution was used.
Data availability
The structural coordinates data from this publication have been deposited to the Protein Data Bank (http://www.rcsb.org/) with accession codes 6XNX, 6XNY, and 6XNZ. The associated density maps were deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb/) with accession codes EMD‐22272, EMD‐22273, and EMD‐22274.
Author contributions
YZ and DGS designed the experiments and wrote the manuscript. YZ prepared the sample for cryo‐EM data collection, processed the data, and performed data analysis. SW collected the cryo‐EM data and provided advice on data processing. YZ performed the plasmid‐to‐plasmid transposition, plasmid‐to‐genome transposition, and disintegration experiments. EC performed in vivo recombination experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Movie EV1
Review Process File
Acknowledgements
We thank G. Ghanim for suggesting the approach for assembly of the STC DNA substrate; K. Zhou for assistance in testing cryo‐EM grids; Q. Lu for generating the RAG2 ΔL construct; S. Z. Zhou for sharing the script for particle picking and many suggestions for structure optimization; C. Liu, Y. Yang, E. Kellogg, and D. Rio for helpful comments on the manuscript and anonymous reviewers for helpful suggestions; and J. Wang and Y. Xiong for critical suggestions regarding model building and structure refinement. This work was supported by the National Institutes of Health grant R01 AI137079 (D.G.S.).
The EMBO Journal (2020) 39: e105857
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal A, Eastman QM, Schatz DG (1998) Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature 394: 744–751 [DOI] [PubMed] [Google Scholar]
- Arinkin V, Smyshlyaev G, Barabas O (2019) Jump ahead with a twist: DNA acrobatics drive transposition forward. Curr Opin Struct Biol 59: 168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Rajendra E, Yang G, Shi Y, Scheres SH (2015) Sampling the conformational space of the catalytic subunit of human gamma‐secretase. Elife 4: e11182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballandras‐Colas A, Maskell DP, Serrao E, Locke J, Swuec P, Jonsson SR, Kotecha A, Cook NJ, Pye VE, Taylor IA et al (2017) A supramolecular assembly mediates lentiviral DNA integration. Science 355: 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad BA, Echols N, Wang RY, Cheng Y, DiMaio F, Adams PD, Fraser JS (2015) EMRinger: side chain‐directed model and map validation for 3D cryo‐electron microscopy. Nat Methods 12: 943–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron S, Anderson DK, Swanson PC (2006) RAG and HMGB1 proteins: purification and biochemical analysis of recombination signal complexes. Methods Enzymol 408: 511–528 [DOI] [PubMed] [Google Scholar]
- Burnley T, Palmer CM, Winn M (2017) Recent developments in the CCP‐EM software suite. Acta Crystallogr D Struct Biol 73: 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona LM, Schatz DG (2017) New insights into the evolutionary origins of the recombination‐activating gene proteins and V(D)J recombination. FEBS J 284: 1590–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterji M, Tsai CL, Schatz DG (2006) Mobilization of RAG‐generated signal ends by transposition and insertion in vivo . Mol Cell Biol 26: 1558–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cui Y, Best RB, Wang H, Zhou ZH, Yang W, Gellert M (2020a) Cutting antiparallel DNA strands in a single active site. Nat Struct Mol Biol 27: 119–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cui Y, Wang H, Zhou ZH, Gellert M, Yang W (2020b) How mouse RAG recombinase avoids DNA transposition. Nat Struct Mol Biol 27: 127–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corneo B, Wendland RL, Deriano L, Cui X, Klein IA, Wong SY, Arnal S, Holub AJ, Weller GR, Pancake BA et al (2007) Rag mutations reveal robust alternative end joining. Nature 449: 483–486 [DOI] [PubMed] [Google Scholar]
- Craig NL (2015) A moveable feast: an introduction to mobile DNA In Mobile DNA III, Craig NL, Chandler M, Gellert M, Lambowitz AM, Rice PA, Sandmeyer SB. (eds), pp 3–39. Washington, DC: ASM Press; [Google Scholar]
- Curry JD, Schulz D, Guidos CJ, Danska JS, Nutter L, Nussenzweig A, Schlissel MS (2007) Chromosomal reinsertion of broken RSS ends during T cell development. J Exp Med 204: 2293–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delelis O, Carayon K, Saib A, Deprez E, Mouscadet JF (2008) Integrase and integration: biochemical activities of HIV‐1 integrase. Retrovirology 5: 114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornberger U, Leijon M, Fritzsche H (1999) High base pair opening rates in tracts of GC base pairs. J Biol Chem 274: 6957–6962 [DOI] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ (2007) DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet 41: 331–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG (2000) The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol 18: 495–527 [DOI] [PubMed] [Google Scholar]
- Fugmann SD (2010) The origins of the Rag genes–from transposition to V(D)J recombination. Semin Immunol 22: 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M (2002) V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem 71: 101–132 [DOI] [PubMed] [Google Scholar]
- Ghanim GE, Kellogg EH, Nogales E, Rio DC (2019) Structure of a P element transposase‐DNA complex reveals unusual DNA structures and GTP‐DNA contacts. Nat Struct Mol Biol 26: 1013–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hencken CG, Li X, Craig NL (2012) Functional characterization of an active Rag‐like transposase. Nat Struct Mol Biol 19: 834–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiom K, Melek M, Gellert M (1998) DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell 94: 463–470 [DOI] [PubMed] [Google Scholar]
- Huang S, Tao X, Yuan S, Zhang Y, Li P, Beilinson HA, Zhang Y, Yu W, Pontarotti P, Escriva H et al (2016) Discovery of an active RAG transposon illuminates the origins of V(D)J recombination. Cell 166: 102–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangam D, Feschotte C, Betran E (2017) Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet 33: 817–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RC, Stella S, Heiss JK (2008) Bending and compaction of DNA by proteins In Protein‐nucleic acid interactions: structural biology, Rice PA, Correll CC. (eds), pp 176–220. London: The Royal Society of Chemistry; [Google Scholar]
- Kapitonov VV, Jurka J (2005) RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol 3: e181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Lapkouski M, Yang W, Gellert M (2015) Crystal structure of the V(D)J recombinase RAG1‐RAG2. Nature 518: 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Chuenchor W, Chen X, Cui Y, Zhang X, Zhou ZH, Gellert M, Yang W (2018) Cracking the DNA code for V(D)J recombination. Mol Cell 70: 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres SH, Lindahl E (2016) Accelerated cryo‐EM structure determination with parallelisation using GPUs in RELION‐2. Elife 5: e18722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Wolf YI, Krupovic M (2020) Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat Rev Genet 21: 119–131 [DOI] [PubMed] [Google Scholar]
- Kuduvalli PN, Rao JE, Craig NL (2001) Target DNA structure plays a critical role in Tn7 transposition. EMBO J 20: 924–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankas F, Sponer J, Langowski J, Cheatham TE III (2003) DNA basepair step deformability inferred from molecular dynamics simulations. Biophys J 85: 2872–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SM (1994) The mechanism of V(D)J joining: lessons from molecular, immunological, and comparative analyses. Adv Immunol 56: 27–150 [DOI] [PubMed] [Google Scholar]
- Little AJ, Matthews AG, Oettinger MA, Roth DB, Schatz DG (2015) The mechanism of V(D)J recombination In Molecular biology of B cells, Alt FW, Honjo T, Radbruch A, Reth M. (eds), pp 13–34. London, UK: Academic Press/Elsevier Limited; [Google Scholar]
- Liu C, Yang Y, Schatz DG (2019) Structures of a RAG‐like transposase during cut‐and‐paste transposition. Nature 575: 540–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens GN, Hare S, Cherepanov P (2010) The mechanism of retroviral integration from X‐ray structures of its key intermediates. Nature 468: 326–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EC, Vicari C, Tsakou‐Ngouafo L, Pontarotti P, Petrescu AJ, Schatz DG (2020) Identification of RAG‐like transposons in protostomes suggests their ancient bilaterian origin. Mob DNA 11: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005) Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152: 36–51 [DOI] [PubMed] [Google Scholar]
- Mazumder A, Engelman A, Craigie R, Fesen M, Pommier Y (1994) Intermolecular disintegration and intramolecular strand transfer activities of wild‐type and mutant HIV‐1 integrase. Nucl Acids Res 22: 1037–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melek M, Gellert M (2000) RAG1/2‐mediated resolution of transposition intermediates: two pathways and possible consequences. Cell 101: 625–633 [DOI] [PubMed] [Google Scholar]
- Montano SP, Rice PA (2011) Moving DNA around: DNA transposition and retroviral integration. Curr Opin Struct Biol 21: 370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano SP, Pigli YZ, Rice PA (2012) The mu transpososome structure sheds light on DDE recombinase evolution. Nature 491: 413–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris ER, Grey H, McKenzie G, Jones AC, Richardson JM (2016) A bend, flip and trap mechanism for transposon integration.Elife 5: e15537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos DO, Li M, Yang R, Rebensburg SV, Ghirlando R, Jeon Y, Shkriabai N, Kvaratskhelia M, Craigie R, Lyumkis D (2017) Cryo‐EM structures and atomic model of the HIV‐1 strand transfer complex intasome. Science 355: 89–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payer LM, Burns KH (2019) Transposable elements in human genetic disease. Nat Rev Genet 20: 760–772 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Posey JE, Pytlos MJ, Sinden RR, Roth DB (2006) Target DNA structure plays a critical role in RAG transposition. PLoS Biol 4: e350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribil PA, Haniford DB (2000) Substrate recognition and induced DNA deformation by transposase at the target‐capture stage of Tn10 transposition. J Mol Biol 303: 145–159 [DOI] [PubMed] [Google Scholar]
- Reddy YV, Perkins EJ, Ramsden DA (2006) Genomic instability due to V(D)J recombination‐associated transposition. Genes Dev 20: 1575–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JM, Colloms SD, Finnegan DJ, Walkinshaw MD (2009) Molecular architecture of the Mos1 paired‐end complex: the structural basis of DNA transposition in a eukaryote. Cell 138: 1096–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N (2015) CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol 192: 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru H, Chambers MG, Fu TM, Tong AB, Liao M, Wu H (2015) Molecular mechanism of V(D)J recombination from synaptic RAG1‐RAG2 complex structures. Cell 163: 1138–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru H, Mi W, Zhang P, Alt FW, Schatz DG, Liao M, Wu H (2018) DNA melting initiates the RAG catalytic pathway. Nat Struct Mol Biol 25: 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinzelle L, Izsvak Z, Ivics Z (2009) Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cell Mol Life Sci 66: 1073–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson PC (2004) The bounty of RAGs: recombination signal complexes and reaction outcomes. Immuno Rev 200: 90–114 [DOI] [PubMed] [Google Scholar]
- Thompson CB (1995) New insights into V(D)J recombination and its role in the evolution of the immune system. Immunity 3: 531–539 [DOI] [PubMed] [Google Scholar]
- Tsai CL, Chatterji M, Schatz DG (2003) DNA mismatches and GC‐rich motifs target transposition by the RAG1/RAG2 transposase. Nucl Acids Res 31: 6180–6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagihara K, Mizuuchi K (2002) Mismatch‐targeted transposition of Mu: a new strategy to map genetic polymorphism. Proc Natl Acad Sci USA 99: 11317–11321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Cheng TC, Huang G, Lu Q, Surleac MD, Mandell JD, Pontarotti P, Petrescu AJ, Xu A, Xiong Y et al (2019) Transposon molecular domestication and the evolution of the RAG recombinase. Nature 569: 79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH (2018) New tools for automated high‐resolution cryo‐EM structure determination in RELION‐3. Elife 7: e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Movie EV1
Review Process File
Data Availability Statement
The structural coordinates data from this publication have been deposited to the Protein Data Bank (http://www.rcsb.org/) with accession codes 6XNX, 6XNY, and 6XNZ. The associated density maps were deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb/) with accession codes EMD‐22272, EMD‐22273, and EMD‐22274.